

Abstract
Objectives
Neuropsychiatric manifestations and brain atrophy of unknown etiology are common and severe complications of systemic lupus erythematosus (SLE). An autoantibody that binds to N-methyl-D-aspartate (NMDA) receptor NR2 has been proposed as a key factor in the etiology of central nervous system (CNS) SLE. This hypothesis was supported by evidence suggesting memantine (MEM), an uncompetitive NMDA receptor antagonist, prevents behavioral dysfunction and brain pathology in healthy mice immunized with a peptide similar to an epitope on the NR2 receptor. Given that SLE is a chronic condition, we presently examine the effects of MEM in MRL/lpr mice, which develop behavioral deficits alongside SLE-like disease.
Methods
A broad behavioral battery and 7-Tesla MRI were used to examine whether prolonged treatment with MEM (~25 mg/kg b.w. in drinking water) prevents CNS involvement in this spontaneous model of SLE.
Results
Although MEM increased novel object exploration in MRL/lpr mice, it did not show other beneficial, substrain-specific effects. Conversely, MEM was detrimental to spontaneous activity in control MRL +/+ mice and had a negative effect on body mass gain. Similarly, MRI revealed comparable increases in the volume of periventricular structures in MEM-treated groups.
Conclusions
Sustained exposure to MEM affects body growth, brain morphology, and behavior primarily by pharmacological, and not autoimmunity-dependant mechanisms. Substrain-specific improvement in exploratory behavior of MEM-treated MRL/lpr mice may indicate that the NMDA system is merely a constituent of a complex pathogenenic cascade. However, it was evident that chronic administration of MEM is unable to completely prevent the development of a CNS SLE-like syndrome.
Keywords: memantine, glutamate receptor, CNS lupus, autoimmunity, brain atrophy, MRI, animal model
Introduction
Systemic lupus erythematosus (SLE) is a severe autoimmune disease that primarily affects skin, kidneys, and joints. In many SLE patients however, neuropsychiatric manifestations and brain atrophy also occur at different phases of disease development (1;2). The lack of insight into pathogenic mechanisms has necessitated the development of animal models, which show significant validity and usefulness in studying CNS involvement (3).
Two classes of SLE animal models were established over the past decades. Inbred strains of NZB, NZB/W, BXSB and MRL mice spontaneously develop a systemic autoimmune disease, which steadily progresses over their lifespan. Conversely, “induced models” of SLE develop an acute autoimmune response to a systemically administered auto-antigen (4). More recently, immunization of healthy BALB/c mice with a pentapeptide (DWEYS) was shown to generate serum anti-DNA antibodies, which cross-react with an NMDA receptor in the brain (5). Autoantibody binding resulted in neurodegeneration and broad deficits in behavior, including altered emotional reactivity (6) and memory (7). The pathogenicity of anti-NMDA antibodies was proposed to be mediated by enhanced postsynaptic transmission and excitotoxicity (8). Consistent with this notion, both behavioral deficits and demise of central neurons in an induced model of CNS SLE were prevented by the noncompetitive NMDA receptor antagonist memantine (MEM) (6;7). This effect appears to be the result of stabilized mitochondrial permeability (8), and not due to the inhibition of autoantibody binding to the NMDA receptor (7). However, inconsistencies among recent clinical reports and the fact that anti-NMDA receptor antibodies are detected in merely ~35% of SLE patients bring into question whether a dysfunctional NMDA system fully accounts for CNS manifestations in SLE (9;10).
The “spontaneous” MRL model has been used for more than two decades (11-14) and proven instrumental in documenting bona fide neurodegeneration of central neurons and cytotoxicity of cerebrospinal fluid (CSF) in SLE-like disease (15;16). In particular, MRL/MpJ-Faslpr/J (MRL/lpr) and MRL/MpJ (MRL +/+) mice spontaneously develop lupus-like manifestations (e.g., high serum levels of autoantibodies, skin lesions, lymph node and spleen enlargement, renal inflammation), but differ in their onset. While MRL/lpr mice exhibit high serum levels of autoantibodies and pro-inflammatory cytokines within the first two months of life, congenic MRL +/+ mice develop similar symptoms much later (17). Alterations in exploration, spatial learning, and emotional reactivity represent key features of the “autoimmunity-associated behavioral syndrome” (AABS) in the MRL/lpr substrain (18). Impaired performance in several paradigms have suggested that altered emotional reactivity and spatial learning are consequences of an accelerated form of SLE-like disease (12;13;19-21). Furthermore, in comparison to the MRL +/+ substrain, MRL/lpr mice show blunted responsiveness to palatable stimuli, impaired spontaneous and exploratory activity, and increased anxiety-related behavior (20;22-24). Behavioral changes in lupus-prone mice are accompanied by infiltration of mononuclear cells into the choroid plexus and meninges, neuronal loss in limbic and cortical areas, as well as retarded brain growth and ventricular enlargement (15;25-29). Similar to anti-NMDA antibodies in the peptide-induced model of CNS SLE (6;7), brain-reactive antibodies of the IgG class seem to account for CSF cytoxicity towards mature and immature neurons in vitro (16;30;31).
The blood-brain barrier (BBB) is breached in diseased MRL/lpr mice, as evidenced by an upregulation of cell adhesion molecules in periventricular regions, widespread perivascular leakage (26), and infiltration of immunocytes into the choroid plexus and multiple regions of brain parenchyma (28;32;33). One may assume that “spontaneous” and “induced” models of CNS SLE (where the BBB is breached chemically) share comparable neuropathogenic mechanisms when autoantibodies cross the BBB. This notion is supported by elevated levels of anti-NMDA antibodies in both serum (34) and CSF of diseased MRL/lpr mice (ms in preparation). If the NMDA hypothesis of CNS SLE is indeed true and MEM can be used as a therapeutic modality (7), then prolonged treatment should have beneficial effects in the “spontaneous” CNS SLE model too. Taken together, the present study examines whether prolonged administration of MEM prevents the constellation of behavioral deficits and brain atrophy in the spontaneous MRL/lpr model (18;35;36).
Methods and Materials
Mice
To avoid the potential confounding effects of estrus cycling on behavioral performance, male mice, which develop a comparable disease to MRL/lpr females (17), were used. Twenty-four males from MRL/lpr and MRL +/+ substrains were purchased from The Jackson Laboratory (Bar Harbor, ME) at 4 weeks of age. Animals were matched for body weight and assigned into four groups (n=12 mice/group) according to substrain and treatment. Mice were habituated over five days and singly-housed under standard laboratory conditions (light: 8 A.M.–8 P.M., room temperature ~22°C, humidity ~62%, regular rodent chow and tap water ad libitum, bedding changed every 3–4 days). Two MRL/lpr mice died prematurely, thus reducing the sample size to N=46. Body weight was recorded on a weekly basis and wet spleen weight (an index of autoimmune status) was measured on an analytical scale at sacrifice. All protocols were performed in accordance with the rules and regulations of the Canadian Council of Animal Care and approved by the local Animal Research Ethics Board.
Drug Administration
To avoid confounding behavioral effects of injection-induced stress, memantine-hydochloride (MEM, lot M9292, Sigma-Aldrich, St. Louis, MO) was dissolved in tap water and mice were allowed to drink it ad libitum from leak-proof bottles (Ancare, Bellmore, NY). Based on body size and daily fluid intake (6–8 ml), a single mouse ingested between 20–30 mg/kg b.w. daily, which was previously shown to fall within the therapeutic dose range in a mouse model of Alzheimer’s disease (37). The other half of male mice were provided with drinking tap water (vehicle, Veh). Treatment started at 5 weeks of age, and persisted over 9 weeks: MEM was given 5 weeks prior to behavioral testing, continued throughout the testing period (10–14 weeks of age), and terminated 2 days before sacrifice. The rationale for such a design was based on previous findings indicating CNS involvement begins around 8 weeks of age (26;38) and antedates systemic manifestations evident approximately 4 months after birth (17).
Behavioral Testing
A single test from our behavioral battery was given nightly to cohorts from each group in the order described below.
Sucrose Preference Test
Impaired preference for palatable stimulation is proposed to model anhedonia, the second core symptom of depression (39). Indeed, in the MRL model, this paradigm reveals a deficit in central reward circuits, and not changes in peripheral sensory input (24). The 60-min sucrose preference test was performed in the evening hours, as described earlier (23). To determine the dose-response in a linear manner, 1-8% solutions were provided to mice and the consumption of sucrose mass was calculated for each concentration.
Spontaneous nocturnal activity
As described earlier (12), spontaneous nocturnal activity was assessed from 6 P.M.-8 A.M. by measuring distance and time traversed in computerized activity boxes (VersaMax, AccuScan Instruments Inc., Columbus, OH).
Open Field/Novel Object Test
The novel object test was used to assess anxiety-like behavior and exploratory drive in a conflict (approach-avoidance) setting (20). Each mouse was gently placed in a corner of a square table (160 × 160 cm, elevated ~50 cm) with a blue, steel cylinder (H=12 cm) in its centre. The test lasted 30 min (performed daily from 6–10 P.M.) and behavior was videotaped with an overhead hard drive-based video camera. The table was cleaned with a mild solution of glass cleaner between trials. EthoVision XT 5 tracking software (Noldus, Leesburg, VA) was used to measure moving distance, moving time, “thigmotaxis”, or time spent along the perimeter (thigmotaxic zone was defined as a 16cm-wide band along table edges). Time spent exploring the cylinder was assessed using a 3-point tracking feature, with snout as the reference point during object sniffing, climbing, or biting.
Climbing Test
Spontaneous climbing is a behavioral pattern proposed to be controlled by the dopamine system (40;41). Moreover, several lines of evidence suggest aberrant dopaminergic neurotransmission in autoimmune MRL/lpr mice (42-45). We further examine this notion and the effects of MEM by employing a brief climbing test. Mice were placed in a rectangular box (H=28 cm, W=26 cm, D=9 cm) made of wire-mesh and videotaped for 10 min. Duration and frequency of climbing, rearing, and grooming were scored using the Observer XT software package (Noldus, Leesburg, VA).
Step-down test
Mouse readiness to escape from an elevated platform placed in an unfamiliar, brightly-lit, and spacious environment is proposed to reflect an anxious response, which differs in MRL substrains (20). Each mouse was gently placed on a wire-mesh covering a rectangular glass box. The time to step-down onto a black surface with all four paws was recorded in a 5-min trial. Step-down latency was assessed from video recordings using a stopwatch.
Rotarod test
Muscle strength and acquisition of sensorimotor coordination were assessed using Rotarod test (EZRod version 1.20, Accuscan Instruments inc., Columbus OH). Three daily trials were performed over two days, with the latency and speed at fall recorded under the following parameters: duration of trial=5 min, maximal speed=20 RPM, time to maximal speed=15 s.
Beam walking
Being sensitive to motor cortex damage (46), walking on a narrow beam is often used to test psychomotor coordination in rodents. “Shaping protocol” and other details were reported previously (47). In the present study, a single test was recorded and traversing time was analyzed with Observer XT scoring software (Noldus Information Technology, Leesburg, VA).
Forced swim test
Increased floating in a no-escape situation is proposed to reflect depressive-like behavior (48). In the present study, each mouse was gently lowered into a circular swimming pool (dia. 183 cm) along the inner side of the wall. Floating time during the 10-min test was measured by EthoVisionXT software (Noldus, Leesburg, VA) using swimming velocity <2.5 cm/s as the criterion for floating.
Morris Water Maze
Using the same swimming pool described above, we measured spatial learning and memory formation, known to be affected in MRL/lpr mice (12;19). Mice were trained in four, 2-min cue trials (Day 1), with the platform above water surface and a blue cylinder placed on the top. On Day 2, the platform was hidden in the NW quadrant and 4 acquisition trials were performed daily over 4 days. To examine whether a spatial learning strategy was employed, a 2-min probe trial was carried out on Day 6. “Cognitive flexibility” was measured in 4 reversal trials. All behaviors were measured with EthoVisionXT software (Noldus Information Technology, Leesburg, VA). Latency to find the platform, distance traversed, and swimming speed were recorded. The time spent in the NW quadrant was measured in the probe trial.
Tissue collection and MRI analysis
At sacrifice, body weight and wet spleen weight were recorded on analytical scale. Tissue preparation and MRI recording with a multi-channel 7.0-T MRI scanner (Varian Inc., Palo Alto, CA) were performed as described in detail elsewhere (35). The custom alignment procedure (49) was used to compute the volume of sixty-two structures in each of the specimens based on a 3D anatomical MRI atlas of the mouse brain (50). Two-way analysis of variance was performed using the software package R (http://www.r-project.org/) for each anatomical structure with substrain, treatment, and substrain:treatment interaction as factors. For comparison, a three-way ANCOVA was also performed which included body weight (covariate), substrain (factor), treatment (factor), and all interactions between the three. Each F-value obtained in the analysis was corrected for multiple comparisons across the 62 structures using the false discovery rate method (51).
In addition to the analysis of anatomical structure volumes, whole brain maps of local volume differences were created by applying the aforementioned statistical procedures on a point-by-point basis throughout the brain (52;53). This procedure allowed for the direct 3D visualization of brain regions affected by each factor. For this analysis, the transformation data was smoothed with a 0.5 mm Gaussian kernel and the significance threshold established based on a 5% false discovery rate.
Statistical analysis
The specimen weight and behavioural results were analyzed by analysis of variance (ANOVA) with Substrain and Treatment as between-group factors, and Age, Sucrose Concentration, and Time as within-group factors. Student’s t-test was used in post-hoc analysis. Pearson’s and Spearman correlations were used to assess associations between variables. Graphs show mean values ± SEM and significant differences of p ≤ 0.05, p < 0.01 and p < 0.001 are indicated by *, **, and ***, respectively. All computations were performed using the SPSS 16 statistical package (SPSS Inc. Chicago, IL).
Results
Although MRL/lpr mice were heavier before MEM treatment commenced (Substrain: F(1,43) = 10.309, p = .003), they were lighter than MRL +/+ mice at the end of the study (Substrain: F(1,42) = 16.538, p< .001; Figure 1A). This effect was largely accounted for by prolonged exposure to MEM in both experimental and control groups (Treatment: F(1,42) = 9.518, p = .004). As expected, splenomegaly (a peripheral marker of disease severity) was confirmed in MRL/lpr mice (Substrain: F(1,42) = 88.528, p< .001; Figure 1B). Despite a trend for reduced spleen weight in drug-treated groups (Treatment: F(1,42) = 3.248, p = .079), immunosuppressive effect of MEM seems unlikely given its association with overall growth impairment, as shown by a significant correlation between spleen size and body mass within the MRL/lpr group (r20 = .582, .p = .004).
Figure 1.
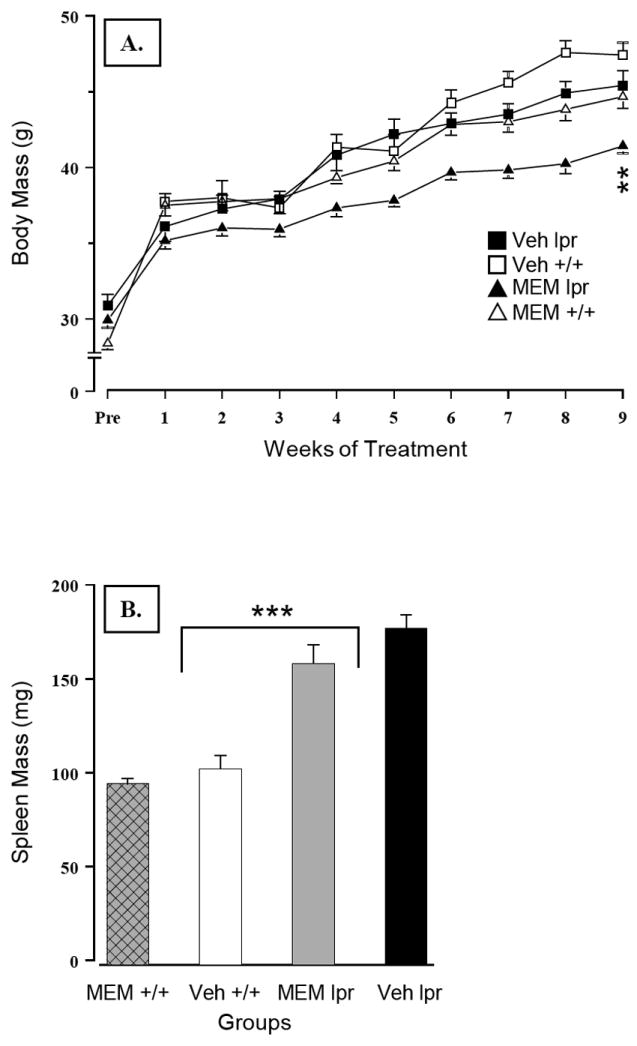
(A) Detrimental effects of sustained exposure to MEM on growth, as evidenced by retarded body mass gain in MEM-treated groups. (B) Autoimmune status in MRL/lpr groups was confirmed by splenomegaly. MEM-treated mice showed a trend for decreased spleen mass, but this effect significantly correlated with the overall impairment in body growth (p = .004). Note: This and other graphs show mean values ± SEM. Mean differences of p≤ .05, p< .01 and p< .001 significance levels are indicated by *, **, and ***, respectively.
Blunted responsiveness to sucrose in MRL/lpr mice was confirmed by a lower slope of the regression line and raw concentration-intake data analysis (Substrain × Concentration: F(3,132) = 7.995, p< .001, Figure 2A). However, chronic MEM treatment increased performance of both substrains (Treatment: F(1,44) = 5.941, p = .019), suggesting a pharmacological, but not an immunomodulatory effect. Conversely, spontaneous activity in the MRL +/+ control group was significantly reduced by MEM, as evidenced by a shorter distance traversed (Substrain × Treatment: F(1,43) = 6.113, p = .004, Figure 2B) and reduced movement time (Substrain × Treatment: F(1,43) = 4.34, p = .043; data not shown). More interestingly, the novel object test revealed an effect that could be immunonomodulatory in nature. Namely, without affecting the performance of the control group, MEM treatment increased MRL/lpr exploration of a novel object (Substrain × Treatment: F(1,43) = 4.138, p = .048, Figure 3A). Other measures, such as moving distance, moving time, and thigmotaxis were not affected. Taken together, the results from the novel object test suggest exploratory drive and/or olfaction (rather than anxiety-related behaviours) were altered by sustained NMDA receptor blockade in autoimmune MRL/lpr mice. In the wire-mesh box, MRL/lpr climbed less frequently than MRL +/+ mice (Substrain: F(1,43) = 15.624, p< .001; data not shown). However, the time they spent climbing the wall was not reduced by MEM, in contrast to MEM-treated MRL +/+ controls (Substrain by Treatment: F(1,43) = 4.402, p = .042; Figure 3B). No significant group differences were detected with respect to rearing and grooming frequency or duration. In the step-down test, MEM treatment failed to reduce longer step-down latency in the MRL/lpr group (Substrain: F(1,42) = 6.385, p = .012; Figure 4A).
Figure 2.
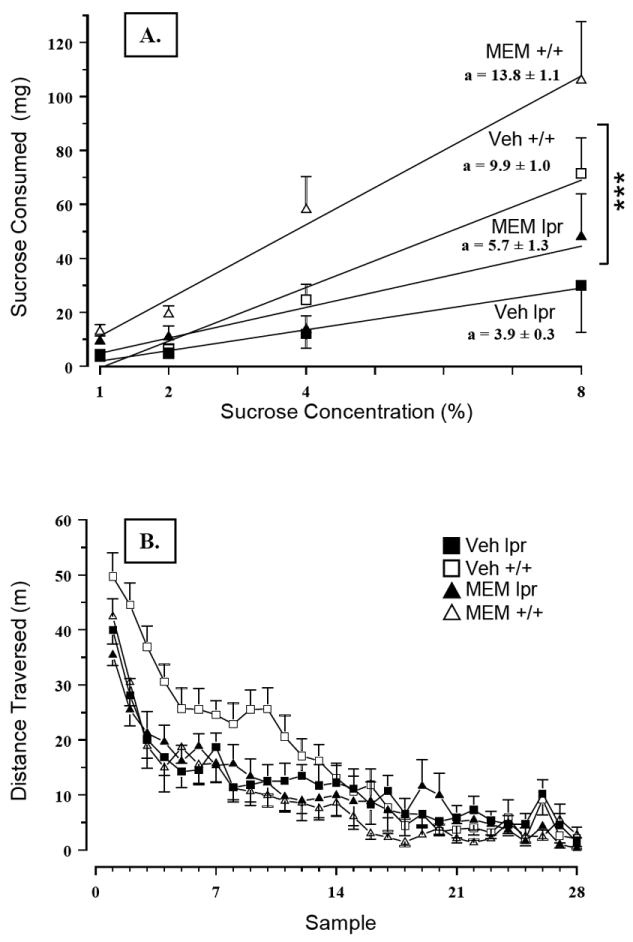
(A) Chronic treatment with MEM improved responsiveness to sucrose in both MRL substrains. Although beneficial, this pharmacological effect is clearly independent of immunological status. (B) As expected, dissimilar spontaneous activity levels were confirmed by comparing Veh-treated MRL/lpr and MRL +/+ groups. However, MEM significantly affected performance in the MRL +/+ group, as evidenced by impaired novelty-induced hyperactivity and shorter distances traversed and movement time (data not shown) during the night phase.
Figure 3.
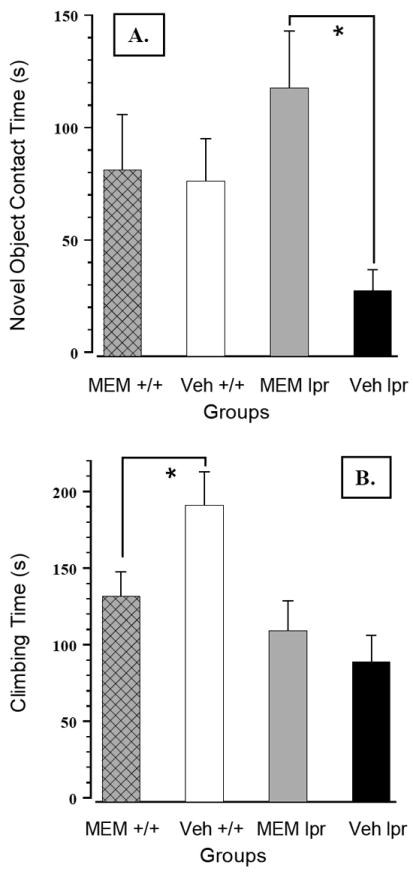
(A) Following prolonged treatment with MEM, MRL/lpr mice spent significantly more time exploring the novel object. Considering this effect was substrain-specific, the results suggest MEM may be capable of inhibiting unknown immunopathogenic circuit(s) in autoimmune mice. (B) Conversely, sustained exposure to MEM decreased climbing time exclusively in MRL +/+ controls, which (when untreated) spent more time climbing the mesh wall in comparison to diseased MRL/lpr mice.
Figure 4.
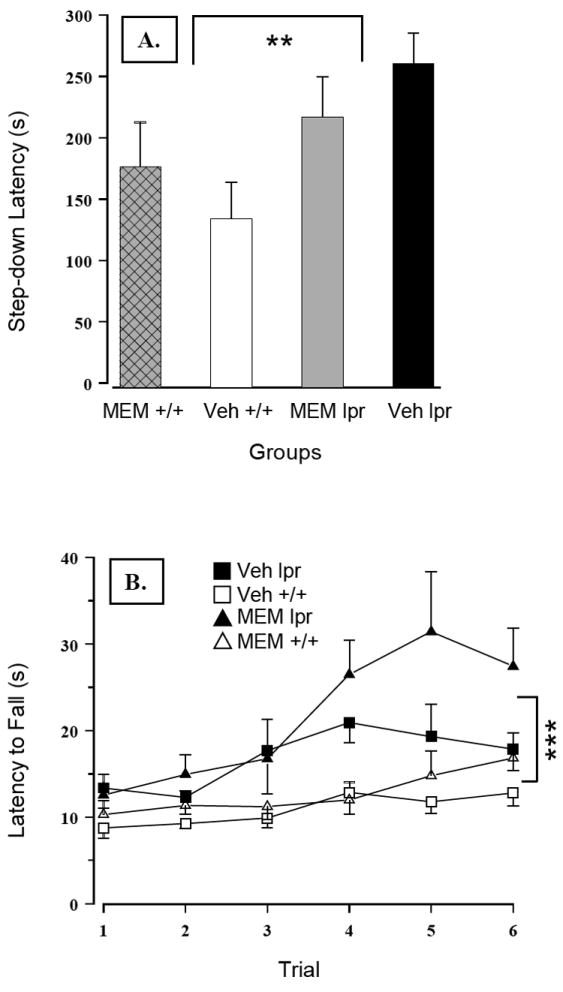
(A) Increased step-down latency in MRL/lpr mice was not affected by prolonged administration of MEM. (B) Performance of MRL/lpr mice in the Rotarod test was consistently better than in control mice. However, significant negative correlations between body mass and fall speed revealed that smaller animals (in this case MRL/lpr mice) were generally better performers on Rotarod than heavy mice. Nevertheless, these results demonstrated that the diseased MRL/lpr group does not exhibit deficits in movement coordination and muscle strength. More interestingly, sustained MEM administration improved sensorimotor learning over the testing period in both groups.
As shown in Figure 4B, MRL/lpr mice exhibited no deficits in muscle strength or motor coordination when tested in the Rotarod test. Conversely, their performance was better than control mice when fall latency (Substrain: F(1,42) = 24.154, p< .001), or speed at fall were considered (Substrain: F(1,42) = 19.341, p< .001). Moreover, MEM treatment increased fall latency in both groups (Treatment: F(1,42) = 4.672, p = .036). However, significant negative correlations between body mass and fall speed (even within the group of untreated mice; falling speed rho20 = - .585, p = .004), suggested that smaller mice were generally better performers on Rotarod than heavy mice. In the beam walking test, MEM did not reduce longer traversing time in the MRL/lpr group (Substrain: F(1,42) = 3.993, p = .05; Figure 5A). Similarly, it was ineffective in reducing immobility of MRL/lpr mice exposed to the forced swim test (Substrain: F(1,42) = 5.329, p = .026; Figure 5B). In the Morris water maze, MEM failed to reduce longer latencies of MRL/lpr mice to locate the platform in cue and reversal trials (Substrain: F(1,42) = 15.306, p< .001; Figure 6A). On the other hand, treatment increased latencies in both substrains when tested in cue trials (Treatment: F(1,43) = 8.168, p = .042; Figure 6B) and the probe trial (Treatment: F(1,42) = 6.916, p = .012; 7 - 10s on average, data not shown). As shown earlier (12), during “reversal learning”, longer search time in the MRL/lpr group was associated with increased perseveration of a previously learned response (Substrain: F(1,42) = 6.783, p = .013; Figure 6B).
Figure 5.
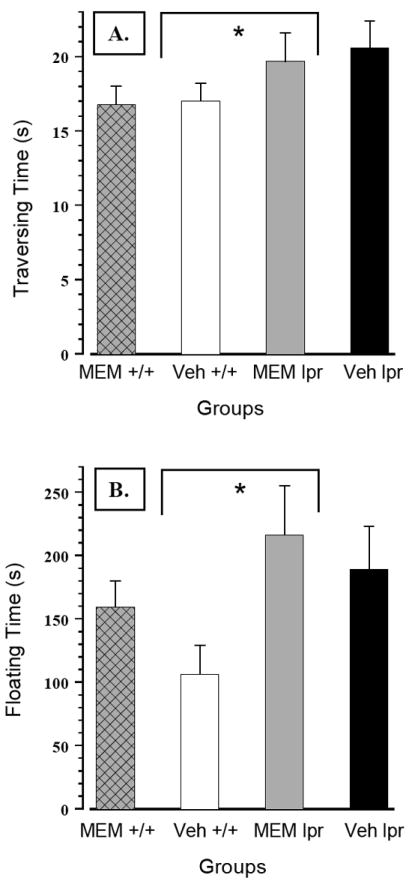
(A) In the beam-walking test, sustained MEM treatment was not effective in reducing longer traversing time in the MRL/lpr substrain. (B) Similarly, it was completely ineffective in reducing increased immobility of MRL/lpr mice in the forced swim test.
Figure 6.
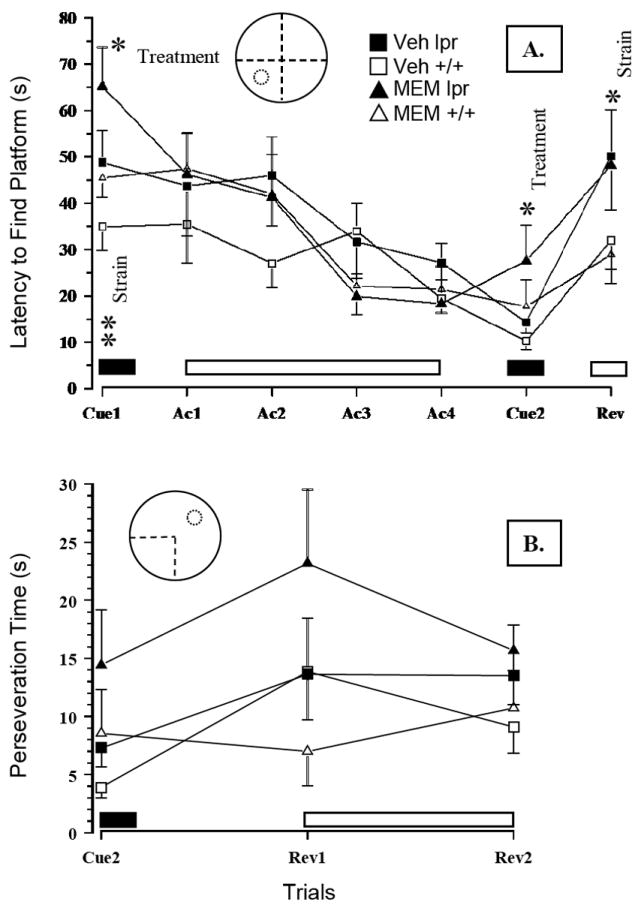
(A) Prolonged drug treatment of MRL/lpr mice failed to reduce increased latencies to locate the platform in cue and reversal trials. Conversely, MEM comparably increased the latency in cue trials and the time spent in the south-west quadrant during the probe trial (data not shown). (B) During “reversal learning”, longer search time in the MRL/lpr group was associated with increased perseveration of the previously learned response and could not be abolished with MEM treatment. Note: solid and open blocks indicate when the escape platform was either visible or invisible, respectively. Abbreviations: Cue – cue trials, Ac – acquisition trials, Rev – reversal trials.
Two-way analysis of variance with substrain and treatment as factors reproduced previously reported substrain effects on regional anatomical volumes (35). Brain volume was found to be highly related to body weight at sacrifice, such that a significant proportion of inter-individual variation could be explained. Including body weight as the first factor in a three-way ANCOVA with substrain and treatment and all cross terms as factors, body weight was found to be a significant in all brain regions (FDR < 5%). In addition, the effect of treatment after accounting for body weight and substrain was significant in 53 out of 62 regions. Table 1 shows the volume of anatomical structures identified in the atlas for each substrain and treatment group. Also shown is the effect of treatment for each region, computed using the ANCOVA model and accounting for body weight. The effect of treatment is a relatively uniform increase in volume across much of the brain that did not differ between the substrains. No evidence supporting a differential effect of treatment between the two substrains was found. Regressing body weight against substrain and treatment showed a significant effect of treatment that did not differ by substrain, such that MEM-treated mice were 2.3 g lighter on average (p < 0.01). Recognizing that the association among brain volume, body weight, and treatment could lead to a false association between treatment and brain volume, we examined the spatial pattern of volume change associated with these factors. The results of applying the same, three-way ANCOVA procedure at every point in the brain are shown in Figure 7. For each factor the subset of statistically significant points at a FDR or 5% are shown in colour. The colour scale shows the effect size on volume. Since no interaction between substrain and treatment survived correction for multiple comparisons, the treatment effect was averaged for the two substrains. As seen in Figure 7, treatment led to a pattern of brain volume increases that was different from that of substrain and body weight. The areas enlarged by MEM treatment were mainly periventricular. Body weight was associated with a pattern of increase that was largely uniform throughout the brain, although larger increases were observed in the temporal lobe.
Table 1.
Brain region volumes (mean ± standard deviation) for each substrain and treatment group. The effect of treatment (estimated from the ANCOVA model) is expressed as a percentage of MRL +/+ volume for that structure. Also, shown is the significance of the treatment expressed as a false discovery rate to account for the multiple comparisons. Regions are sorted from most to least significant treatment effect.
| region | untreated MRL+/+ mean ± SD (mm3) | untreated MRL/lpr mean ± SD (mm3) | treated MRL+/+ mean ± SD (mm3) | treated MRL/lpr mean ± sd (mm3) | treatment effect (%) | treatment effect significance |
|---|---|---|---|---|---|---|
| dentate gyrus of hippocampus | 4.39 ± 0.17 | 4.12 ± 0.18 | 4.57 ± 0.25 | 4.12 ± 0.16 | 5.8 | 0.0073 |
| cerebellar peduncle inferior | 0.99 ± 0.03 | 0.93 ± 0.04 | 1.01 ± 0.05 | 0.94 ± 0.03 | 2.6 | 0.0089 |
| cerebral aqueduct | 1.56 ± 0.05 | 1.46 ± 0.07 | 1.62 ± 0.09 | 1.46 ± 0.06 | 4.2 | 0.0089 |
| cerebral peduncle | 2.92 ± 0.12 | 2.77 ± 0.13 | 3.01 ± 0.15 | 2.76 ± 0.11 | 5.0 | 0.0089 |
| colliculus inferior | 5.26 ± 0.23 | 4.86 ± 0.32 | 5.38 ± 0.26 | 4.89 ± 0.21 | 4.6 | 0.0089 |
| colliculus superior | 9.32 ± 0.40 | 8.40 ± 0.47 | 9.59 ± 0.56 | 8.52 ± 0.40 | 4.8 | 0.0089 |
| corpus callosum | 15.90 ± 0.82 | 14.95 ± 0.66 | 16.47 ± 0.78 | 14.77 ± 0.65 | 5.5 | 0.0089 |
| habenular commissure | 0.81 ± 0.03 | 0.77 ± 0.03 | 0.83 ± 0.04 | 0.76 ± 0.03 | 4.2 | 0.0089 |
| hippocampus | 19.80 ± 0.88 | 18.60 ± 0.77 | 20.43 ± 1.12 | 18.52 ± 0.84 | 5.3 | 0.0089 |
| internal capsule | 3.08 ± 0.13 | 2.90 ± 0.12 | 3.18 ± 0.18 | 2.89 ± 0.11 | 4.9 | 0.0089 |
| mammillary bodies | 1.31 ± 0.04 | 1.24 ± 0.05 | 1.35 ± 0.06 | 1.24 ± 0.05 | 3.9 | 0.0089 |
| midbrain | 22.80 ± 1.05 | 20.87 ± 1.35 | 23.41 ± 1.40 | 20.99 ± 0.89 | 4.4 | 0.0089 |
| olfactory bulbs | 19.15 ± 0.64 | 18.06 ± 0.48 | 19.70 ± 0.98 | 18.37 ± 0.58 | 3.3 | 0.0089 |
| periaqueductal grey | 9.46 ± 0.37 | 8.68 ± 0.44 | 9.77 ± 0.53 | 8.72 ± 0.38 | 4.4 | 0.0089 |
| posterior commissure | 1.15 ± 0.05 | 1.08 ± 0.04 | 1.18 ± 0.06 | 1.08 ± 0.04 | 4.5 | 0.0089 |
| stratum granulosum of hippocampus | 2.42 ± 0.10 | 2.29 ± 0.07 | 2.51 ± 0.12 | 2.26 ± 0.09 | 5.4 | 0.0089 |
| stria medullaris | 1.15 ± 0.04 | 1.09 ± 0.04 | 1.19 ± 0.07 | 1.08 ± 0.04 | 4.8 | 0.0089 |
| stria terminalis | 1.41 ± 0.06 | 1.33 ± 0.05 | 1.45 ± 0.08 | 1.32 ± 0.05 | 4.9 | 0.0089 |
| thalamus | 16.87 ± 0.83 | 15.75 ± 0.60 | 17.59 ± 1.11 | 15.78 ± 0.60 | 5.8 | 0.0089 |
| fourth ventricle | 1.07 ± 0.04 | 1.01 ± 0.04 | 1.10 ± 0.05 | 1.01 ± 0.04 | 3.6 | 0.0096 |
| arbor vita of cerebellum | 7.91 ± 0.35 | 7.50 ± 0.39 | 8.08 ± 0.47 | 7.64 ± 0.39 | 3.4 | 0.01 |
| cerebellar peduncle superior | 1.00 ± 0.05 | 0.93 ± 0.06 | 1.03 ± 0.06 | 0.94 ± 0.04 | 3.8 | 0.01 |
| cuneate nucleus | 1.19 ± 0.04 | 1.13 ± 0.04 | 1.22 ± 0.06 | 1.12 ± 0.04 | 3.3 | 0.01 |
| facial nerve cranial nerve 7 | 1.08 ± 0.04 | 1.02 ± 0.04 | 1.11 ± 0.05 | 1.02 ± 0.04 | 3.9 | 0.01 |
| fasciculus retroflexus | 1.64 ± 0.06 | 1.56 ± 0.06 | 1.68 ± 0.08 | 1.55 ± 0.06 | 3.9 | 0.01 |
| fimbria | 3.68 ± 0.18 | 3.49 ± 0.13 | 3.81 ± 0.23 | 3.43 ± 0.17 | 5.7 | 0.01 |
| fornix | 1.39 ± 0.06 | 1.32 ± 0.05 | 1.43 ± 0.07 | 1.31 ± 0.05 | 4.1 | 0.01 |
| cerebellar peduncle middle | 1.76 ± 0.06 | 1.68 ± 0.05 | 1.81 ± 0.09 | 1.67 ± 0.07 | 4.2 | 0.011 |
| mammilothalamic tract | 0.72 ± 0.03 | 0.68 ± 0.03 | 0.74 ± 0.04 | 0.68 ± 0.03 | 4.1 | 0.011 |
| pons | 29.69 ± 1.18 | 27.43 ± 1.27 | 30.34 ± 1.38 | 27.71 ± 0.95 | 3.3 | 0.011 |
| pre para subiculum | 2.47 ± 0.12 | 2.33 ± 0.16 | 2.50 ± 0.12 | 2.31 ± 0.09 | 3.8 | 0.011 |
| optic tract | 1.31 ± 0.06 | 1.24 ± 0.05 | 1.35 ± 0.07 | 1.24 ± 0.05 | 4.3 | 0.012 |
| third ventricle | 2.06 ± 0.08 | 1.95 ± 0.07 | 2.13 ± 0.11 | 1.94 ± 0.09 | 4.8 | 0.013 |
| anterior commissure pars posterior | 1.60 ± 0.06 | 1.51 ± 0.05 | 1.64 ± 0.08 | 1.50 ± 0.06 | 3.8 | 0.013 |
| anterior commissure pars anterior | 1.67 ± 0.07 | 1.55 ± 0.07 | 1.70 ± 0.08 | 1.55 ± 0.05 | 3.3 | 0.015 |
| interpedunclar nucleus | 1.26 ± 0.05 | 1.19 ± 0.05 | 1.29 ± 0.06 | 1.19 ± 0.05 | 3.4 | 0.015 |
| ventral tegmental decussation | 1.04 ± 0.04 | 0.98 ± 0.04 | 1.06 ± 0.05 | 0.97 ± 0.04 | 3.7 | 0.016 |
| bed nucleus of stria terminalis | 1.94 ± 0.08 | 1.82 ± 0.07 | 1.98 ± 0.11 | 1.81 ± 0.07 | 3.7 | 0.017 |
| lateral olfactory tract | 1.75 ± 0.06 | 1.65 ± 0.04 | 1.79 ± 0.08 | 1.64 ± 0.06 | 3.2 | 0.018 |
| subependymale zone rhinocele | 0.60 ± 0.02 | 0.57 ± 0.02 | 0.61 ± 0.03 | 0.56 ± 0.02 | 3.5 | 0.018 |
| globus pallidus | 3.61 ± 0.16 | 3.40 ± 0.15 | 3.69 ± 0.21 | 3.40 ± 0.13 | 3.5 | 0.019 |
| fundus of striatum | 1.36 ± 0.06 | 1.30 ± 0.05 | 1.40 ± 0.07 | 1.28 ± 0.05 | 4.1 | 0.02 |
| cerebellar cortex | 47.96 ± 1.83 | 46.68 ± 2.39 | 49.03 ± 2.46 | 46.40 ± 2.12 | 3.5 | 0.025 |
| medial septum | 2.60 ± 0.11 | 2.47 ± 0.08 | 2.67 ± 0.15 | 2.45 ± 0.11 | 3.7 | 0.025 |
| hypothalamus | 8.84 ± 0.45 | 8.39 ± 0.40 | 9.14 ± 0.51 | 8.44 ± 0.45 | 4.3 | 0.027 |
| pontine nucleus | 2.03 ± 0.07 | 1.88 ± 0.05 | 2.08 ± 0.11 | 1.86 ± 0.09 | 3.8 | 0.027 |
| inferior olivary complex | 0.91 ± 0.04 | 0.86 ± 0.04 | 0.93 ± 0.05 | 0.85 ± 0.03 | 3.5 | 0.034 |
| medial lemniscus medial longitudinal fasciculus | 2.59 ± 0.12 | 2.39 ± 0.09 | 2.65 ± 0.13 | 2.40 ± 0.10 | 3.2 | 0.036 |
| superior olivary complex | 1.29 ± 0.06 | 1.21 ± 0.06 | 1.31 ± 0.05 | 1.20 ± 0.04 | 2.4 | 0.038 |
| medulla | 55.80 ± 2.18 | 51.39 ± 2.09 | 56.74 ± 2.57 | 51.75 ± 2.15 | 2.2 | 0.044 |
| striatum | 23.16 ± 1.14 | 21.63 ± 0.92 | 23.78 ± 1.32 | 21.32 ± 1.31 | 3.9 | 0.049 |
| basal forebrain | 6.83 ± 0.28 | 6.48 ± 0.23 | 6.97 ± 0.35 | 6.44 ± 0.24 | 2.8 | 0.049 |
| nucleus accumbens | 4.69 ± 0.23 | 4.42 ± 0.17 | 4.81 ± 0.24 | 4.39 ± 0.19 | 3.2 | 0.049 |
| cerebral cortex frontal lobe | 42.50 ± 2.29 | 40.42 ± 1.35 | 43.94 ± 2.17 | 39.55 ± 2.43 | 5.5 | 0.051 |
| corticospinal tract pyramids | 2.33 ± 0.13 | 2.15 ± 0.08 | 2.38 ± 0.12 | 2.15 ± 0.10 | 3.4 | 0.051 |
| cerebral cortex parieto temporal lobe | 70.18 ± 3.76 | 67.87 ± 2.63 | 71.80 ± 3.31 | 66.93 ± 3.44 | 3.7 | 0.07 |
| lateral septum | 3.50 ± 0.17 | 3.30 ± 0.12 | 3.59 ± 0.24 | 3.22 ± 0.20 | 4.3 | 0.07 |
| cerebral cortex entorhinal cortex | 8.89 ± 0.29 | 8.61 ± 0.30 | 9.04 ± 0.45 | 8.51 ± 0.34 | 2.6 | 0.088 |
| lateral ventricle | 3.05 ± 0.17 | 2.96 ± 0.13 | 3.15 ± 0.20 | 2.82 ± 0.19 | 6.0 | 0.1 |
| olfactory tubercle | 3.82 ± 0.20 | 3.66 ± 0.12 | 3.90 ± 0.20 | 3.61 ± 0.14 | 1.5 | 0.25 |
| amygdala | 15.05 ± 0.65 | 14.54 ± 0.74 | 15.31 ± 0.79 | 14.01 ± 0.70 | 2.3 | 0.41 |
| cerebral cortex occipital lobe | 5.83 ± 0.17 | 5.56 ± 0.26 | 5.85 ± 0.23 | 5.34 ± 0.26 | 1.9 | 0.77 |
Figure 7.
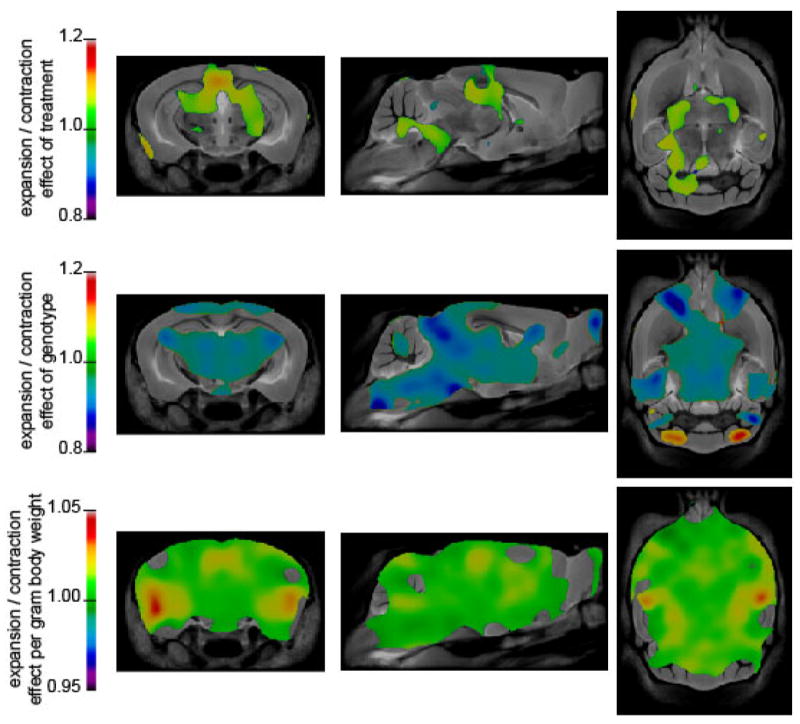
Coronal, sagittal, and transverse sections are shown with a colour overlay corresponding to the effect of treatment, substrain, or body weight on local brain volume. The first row shows the relative difference brain volume associated with treatment after accounting for body weight and substrain. The second row displays the relative size of MRL/lpr mice compared to MRL +/+ after account for body weight. The third row demonstrates the effect of body weight expressed in units of fractional volume increase per gram. Regions that are coloured were significant at the false discovery rate of 5%.
Discussion
We previously established that the two MRL substrains differ in responsiveness to neurotransmitter modulators, such as quinpirole, amphetamine, sertraline, and risperidone (42;44;54). In the present study, we observed discrepancies in responsiveness to MEM when nocturnal activity and climbing behavior were considered. Although these findings may indicate dissimilar activity of the NMDA receptor system, the fact that MEM also binds with similar potency to other receptors (55;56) suggests detrimental effects in control mice and lack of responsiveness in MRL/lpr mice could also be mediated by other neurotransmitters. Such a mechanism would be particularly relevant to the dopaminergic system, where MEM may act as a receptor inhibitor and blocker of endocrine function (57).
Based on the neuroprotective effects of MEM in a peptide-induced model of CNS SLE (6;7) and clinical studies demonstrating a relationship between circulating anti-NR2 antibodies and neuropsychiatric manifestations (58;59), we expected that prolonged administration of MEM would prevent or attenuate the constellation of behavioral deficits and brain atrophy in the spontaneous, MRL/lpr model of CNS SLE. However, the present results do not support the hypothesis that autoimmunity-associated behavioral dysfunction and brain pathology are mediated exclusively by changes to the NMDA system. Namely, save the increase in novel object exploration, prolonged exposure to MEM did not result in other beneficial effects in diseased MRL/lpr mice. More frequently, chronic MEM treatment produced comparable behavioral effects in both MRL substrains, as well as enlargement of brain volume. One may hypothesize that lack of more restorative effects represents the consequence of insufficient MEM dosage. However, significant effects on brain structure and function after the 9-week treatment are inconsistent with this possibility. Indeed, a more viable explanation is that SLE-like disease and CNS involvement in MRL/lpr mice are more severe and complex than modeled in the pentapeptide-immunized mice (5). In other words, the NMDA system seems to act as one of multiple targets that account for the constellation of behavioral abnormalities in SLE patients and lupus-prone mice. Recent clinical reports are consistent with this possibility. In particular, levels of serum anti-NR2 antibodies are found to be associated with depressive mood, but not with cognitive dysfunction in CNS SLE patients (60). Without the intention to anthropomorphize the current results, one may assume that the capacity of MEM to increase novel object exploration in MRL/lpr mice and inability to prevent their “cognitive inflexibility” are in accordance with the clinical findings above. Another clinical study found that anti-NR2 antibodies are detected in the sera of 35% of SLE patients, but also failed to associate their presence with cognitive dysfunction (61). Moreover, Kozora and colleagues failed to identify any significant relationships between serum anti-NR2 antibodies and global cognitive / memory indices, or with depression. (62). The current lack of broad support for the anti-NMDA hypothesis and generalized behavioral dysfunction is further evidenced by a recent clinical trial in which prolonged MEM treatment largely failed to improve general cognitive function, with the exception of controlled oral word association (63). Similarly, other clinical studies could not confirm the proposed relationship between serum anti-DNA and anti-NR2 receptor antibodies (64;65), or the importance of serum anti-NR2 antibodies in the induction of CNS SLE (66).
It was documented by our group that, when challenged with stimulants of dopamine release, MRL/lpr mice fail to increase sucrose intake (44) and behaviourally respond as control MRL +/+ mice (43;54). Therefore, the observed increase in sucrose intake (“anti-anhedonic” effect) in mice treated with MEM deserves particular attention. As mentioned above, MEM may affect the dopamine receptor system in a region- and cell-specific manner (67-69). Therefore, direct stimulation of post-synaptic D2high receptors (57) in structures such as nucleus accumbens may be more effective than stimulation of dopaminergic pre-synaptic neurons (44). Whichever mechanism underlies MEM-induced increase in sucrose intake, it is clear that it does not depend on NMDA receptor blockade during lupus-like disease. Along the same line, despite significant negative correlations between body mass and Rotarod performance, reduced body weight and improved Rotarod performance in MEM-treated mice seem in concordance with reported effects of MEM on ingestive behavior (70;71) and sensorimotor capacity (72).
Substantial within-group variability in brain volume was associated with body weight and large enough to mask some of the morphological differences between substrains, as well as treatment-induced differences. Although incorporating body weight as a covariate allowed for the assessment of main effects, the observation that MEM reduces body weight complicates the interpretation of these results. A reduction in body weight caused by MEM treatment leads to a brain volume that is larger than expected. After accounting for body weight, we also observed a pattern of morphological change associated with MEM that was different from that associated with body weight alone. We interpret this as direct effect of MEM on brain morphology that we were underpowered to detect without including body weight as covariate.
Taken together, the obtained results suggest that effects of sustained exposure to MEM are largely pharmacological in nature, showing little restorative effect on behavior and brain morphology of autoimmune MRL/lpr mice. If the NMDA receptors were chronically blocked, then improved exploratory behavior in the MEM-treated MRL/lpr group suggests that the NMDA system is but one of multiple pathogenic circuits. Given the poor benefit / risk ratio, this study represents the first line of experimental evidence that does not support chronic administration of MEM in the treatment of CNS SLE.
Acknowledgments
We thank Sultan Chaudhry for technical assistance. This work was supported by funds from the National Institute of Health (1R21 AR49163-01) and the Canadian Institutes of Health Research (grant MOP 38065).
Reference List
- 1.Hanly JG. Neuropsychiatric lupus. Rheum Dis Clin North Am. 2005 May;31(2):273–98. vi. doi: 10.1016/j.rdc.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 2.Appenzeller S, Bonilha L, Rio PA, Min LL, Costallat LT, Cendes F. Longitudinal analysis of gray and white matter loss in patients with systemic lupus erythematosus. Neuroimage. 2007 Jan 15;34(2):694–701. doi: 10.1016/j.neuroimage.2006.09.029. [DOI] [PubMed] [Google Scholar]
- 3.Alexander JJ, Quigg RJ. Systemic lupus erythematosus and the brain: what mice are telling us. Neurochem Int. 2007 Jan;50(1):5–11. doi: 10.1016/j.neuint.2006.08.005. [DOI] [PubMed] [Google Scholar]
- 4.Mozes E, Alling D, Miller MW, Payne SM, Zinger H, Via CS, et al. Genetic analysis of experimentally induced lupus in mice. Clin Immunol Immunopathol. 1997 Oct;85(1):28–34. doi: 10.1006/clin.1997.4423. [DOI] [PubMed] [Google Scholar]
- 5.DeGiorgio LA, Konstantinov KN, Lee SC, Hardin JA, Volpe BT, Diamond B. A subset of lupus anti-DNA antibodies cross-reacts with the NR2 glutamate receptor in systemic lupus erythematosus. Nat Med. 2001 Nov;7(11):1189–93. doi: 10.1038/nm1101-1189. [DOI] [PubMed] [Google Scholar]
- 6.Huerta PT, Kowal C, DeGiorgio LA, Volpe BT, Diamond B. Immunity and behavior: Antibodies alter emotion. Proc Natl Acad Sci U S A. 2006 Jan 17;103(3):678–83. doi: 10.1073/pnas.0510055103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kowal C, DeGiorgio LA, Nakaoka T, Hetherington H, Huerta PT, Diamond B, et al. Cognition and immunity; antibody impairs memory. Immunity. 2004 Aug;21(2):179–88. doi: 10.1016/j.immuni.2004.07.011. [DOI] [PubMed] [Google Scholar]
- 8.Faust TW, Chang EH, Kowal C, Berlin R, Gazaryan IG, Bertini E, et al. Neurotoxic lupus autoantibodies alter brain function through two distinct mechanisms. Proc Natl Acad Sci U S A. 2010 Oct 26;107(43):18569–74. doi: 10.1073/pnas.1006980107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lauvsnes MB, Omdal R. Systemic lupus erythematosus, the brain, and anti-NR2 antibodies. J Neurol. 2011 Sep 10; doi: 10.1007/s00415-011-6232-5. [DOI] [PubMed] [Google Scholar]
- 10.Hanly JG. New insights into central nervous system lupus: a clinical perspective. Curr Rheumatol Rep. 2007 Apr;9(2):116–24. doi: 10.1007/s11926-007-0005-2. [DOI] [PubMed] [Google Scholar]
- 11.Forster MJ, Popper MD, Retz KC, Lal H. Age differences in acquisition and retention of one-way avoidance learning in C57BL/6NNia and autoimmune mice. Behav Neural Biol. 1988;49:139–51. doi: 10.1016/s0163-1047(88)90462-1. [DOI] [PubMed] [Google Scholar]
- 12.Sakic B, Szechtman H, Keffer M, Talangbayan H, Stead R, Denburg JA. A behavioral profile of autoimmune lupus-prone MRL mice. Brain Behav Immun. 1992;6:265–85. doi: 10.1016/0889-1591(92)90048-s. [DOI] [PubMed] [Google Scholar]
- 13.Hess DC, Taormina M, Thompson J, Sethi KD, Diamond B, Rao R, et al. Cognitive and neurologic deficits in the MRL/lpr mouse: a clinicopathologic study. J Rheumatol. 1993;20:610–7. [PubMed] [Google Scholar]
- 14.Walker SE, Wright DC, O’Sullivan FX, Johnson GC, Besch-Williford CL, Vogelweid CM. Memory, learning ability, and neuropathologic status of mice with systemic lupus erythematosus. Ann N Y Acad Sci. 1997 Aug 14;823:107–15. doi: 10.1111/j.1749-6632.1997.tb48383.x. [DOI] [PubMed] [Google Scholar]
- 15.Sakic B, Szechtman H, Denburg JA, Gorny G, Kolb B, Whishaw IQ. Progressive atrophy of pyramidal neuron dendrites in autoimmune MRL-lpr mice. J Neuroimmunol. 1998;87(1-2):162–70. doi: 10.1016/s0165-5728(98)00085-x. [DOI] [PubMed] [Google Scholar]
- 16.Maric D, Millward JM, Ballok DA, Szechtman H, Barker JL, Denburg JA, et al. Neurotoxic properties of cerebrospinal fluid from behaviorally impaired autoimmune mice. Brain Res. 2001;920(1-2):183–93. doi: 10.1016/s0006-8993(01)03060-8. [DOI] [PubMed] [Google Scholar]
- 17.Theofilopoulos AN. Murine models of lupus. In: Lahita RG, editor. Systemic lupus erythematosus. 2. New York: Churchill Livingstone; 1992. pp. 121–94. [Google Scholar]
- 18.Sakic B, Szechtman H, Denburg JA. Neurobehavioral alteration in autoimmune mice. Neurosci Biobehav Rev. 1997;21(3):327–40. doi: 10.1016/s0149-7634(96)00018-8. [DOI] [PubMed] [Google Scholar]
- 19.Sakic B, Szechtman H, Denburg SD, Carbotte RM, Denburg JA. Spatial learning during the course of autoimmune disease in MRL mice. Behav Brain Res. 1993;54:57–66. doi: 10.1016/0166-4328(93)90048-u. [DOI] [PubMed] [Google Scholar]
- 20.Sakic B, Szechtman H, Talangbayan H, Denburg SD, Carbotte RM, Denburg JA. Disturbed emotionality in autoimmune MRL-lpr mice. Physiol Behav. 1994;56(3):609–17. doi: 10.1016/0031-9384(94)90309-3. [DOI] [PubMed] [Google Scholar]
- 21.Vogelweid CM, Wright DC, Johnson JC, Hewett JE, Walker SE. Evaluation of memory, learning ability, and clinical neurologic function in pathogen-free mice with systemic lupus erythematosus. Arthritis Rheum. 1994;37:889–97. doi: 10.1002/art.1780370617. [DOI] [PubMed] [Google Scholar]
- 22.Sakic B, Szechtman H, Denburg SD, Denburg JA. Immunosuppressive treatment prevents behavioral deficit in autoimmune MRL-lpr mice. Physiol Behav. 1995;58(4):797–802. doi: 10.1016/0031-9384(95)00135-6. [DOI] [PubMed] [Google Scholar]
- 23.Sakic B, Denburg JA, Denburg SD, Szechtman H. Blunted sensitivity to sucrose in autoimmune MRL-lpr mice: a curve-shift study. Brain Res Bull. 1996;41(5):305–11. doi: 10.1016/s0361-9230(96)00190-6. [DOI] [PubMed] [Google Scholar]
- 24.Ballok DA, Szechtman H, Sakic B. Taste responsiveness and diet preference in autoimmune MRL mice. Behav Brain Res. 2003 Mar 18;140(1-2):119–30. doi: 10.1016/s0166-4328(02)00276-0. [DOI] [PubMed] [Google Scholar]
- 25.Alexander EL, Murphy ED, Roths JB, Alexander GE. Congenic autoimmune murine models of central nervous system disease in connective tissue disorders. Ann Neurol. 1983;14:242–8. doi: 10.1002/ana.410140211. [DOI] [PubMed] [Google Scholar]
- 26.Vogelweid CM, Johnson GC, Besch-Williford CL, Basler J, Walker SE. Inflammatory central nervous system disease in lupus-prone MRL/lpr mice: comparative histologic and immunohistochemical findings. J Neuroimmunol. 1991;35:89–99. doi: 10.1016/0165-5728(91)90164-3. [DOI] [PubMed] [Google Scholar]
- 27.Denenberg VH, Sherman GF, Rosen GD, Morrison L, Behan PO, Galaburda AM. A behavior profile of the MRL/Mp lpr/lpr mouse and its association with hydrocephalus. Brain Behav Immun. 1992;6:40–9. doi: 10.1016/0889-1591(92)90058-v. [DOI] [PubMed] [Google Scholar]
- 28.Sakic B, Maric I, Koeberle PD, Millward JM, Szechtman H, Maric D, et al. Increased TUNEL-staining in brains of autoimmune Fas-deficient mice. J Neuroimmunol. 2000;104(2):147–54. doi: 10.1016/s0165-5728(99)00277-5. [DOI] [PubMed] [Google Scholar]
- 29.Ballok DA, Millward JM, Sakic B. Neurodegeneration in autoimmune MRL-lpr mice as revealed by Fluoro Jade B staining. Brain Res. 2003 Feb 28;964(2):200–10. doi: 10.1016/s0006-8993(02)03980-x. [DOI] [PubMed] [Google Scholar]
- 30.Sakic B, Kirkham DL, Ballok DA, Mwanjewe J, Fearon IM, Macri J, et al. Proliferating brain cells are a target of neurotoxic CSF in systemic autoimmune disease. J Neuroimmunol. 2005 Dec;169(1-2):68–85. doi: 10.1016/j.jneuroim.2005.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sidor MM, Sakic B, Malinowski PM, Ballok DA, Oleschuk CJ, Macri J. Elevated immunoglobulin levels in the cerebrospinal fluid from lupus-prone mice. J Neuroimmunol. 2005 Aug;165(1-2):104–13. doi: 10.1016/j.jneuroim.2005.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Farrell M, Sakic B, Szechtman H, Denburg JA. Effect of cyclophosphamide on leucocytic infiltration in the brain of MRL/lpr mice. Lupus. 1997;6(3):268–74. doi: 10.1177/096120339700600310. [DOI] [PubMed] [Google Scholar]
- 33.Zameer A, Hoffman SA. Increased ICAM-1 and VCAM-1 expression in the brains of autoimmune mice. J Neuroimmunol. 2003 Sep;142(1-2):67–74. doi: 10.1016/s0165-5728(03)00262-5. [DOI] [PubMed] [Google Scholar]
- 34.Gao HX, Sanders E, Tieng AT, Putterman C. Sex and autoantibody titers determine the development of neuropsychiatric manifestations in lupus-prone mice. J Neuroimmunol. 2010 Dec 15;229(1-2):112–22. doi: 10.1016/j.jneuroim.2010.07.020. [DOI] [PubMed] [Google Scholar]
- 35.Sled JG, Spring S, van Eede M, Lerch JP, Ullal S, Sakic B. Time course and nature of brain atrophy in the MRL mouse model of central nervous system lupus. Arthritis Rheum. 2009 Jun;60(6):1764–74. doi: 10.1002/art.24523. [DOI] [PubMed] [Google Scholar]
- 36.Gulinello M, Putterman C. The MRL/lpr mouse strain as a model for neuropsychiatric systemic lupus erythematosus. J Biomed Biotechnol. 2011;2011:207504. doi: 10.1155/2011/207504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Minkeviciene R, Banerjee P, Tanila H. Memantine improves spatial learning in a transgenic mouse model of Alzheimer’s disease. J Pharmacol Exp Ther. 2004 Nov;311(2):677–82. doi: 10.1124/jpet.104.071027. [DOI] [PubMed] [Google Scholar]
- 38.Ma X, Foster J, Sakic B. Distribution and prevalence of leukocyte phenotypes in brains of lupus-prone mice. J Neuroimmunol. 2006 Oct;179(1-2):26–36. doi: 10.1016/j.jneuroim.2006.06.023. [DOI] [PubMed] [Google Scholar]
- 39.Muscat R, Willner P. Suppression of sucrose drinking by chronic mild unpredictable stress: a methodological analysis. Neurosci Biobehav Rev. 1992;16(4):507–17. doi: 10.1016/s0149-7634(05)80192-7. [DOI] [PubMed] [Google Scholar]
- 40.Costall B, Eniojukan JF, Naylor RJ. Dopamine agonist action in mesolimbic, cortical and extrapyramidal areas to modify spontaneous climbing behaviour of the mouse. Psychopharmacology (Berl) 1985;86(4):452–7. doi: 10.1007/BF00427907. [DOI] [PubMed] [Google Scholar]
- 41.Depoortere R, Bardin L, Auclair AL, Kleven MS, Prinssen E, Colpaert F, et al. F15063, a compound with D2/D3 antagonist, 5-HT 1A agonist and D4 partial agonist properties. II. Activity in models of positive symptoms of schizophrenia. Br J Pharmacol. 2007 May;151(2):253–65. doi: 10.1038/sj.bjp.0707159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sakic B, Lacosta S, Denburg J, Szechtman H. Altered neurotransmission in brains of autoimmune mice: pharmacological and neurochemical evidence. J Neuroimmunol. 2002 Aug;129(1-2):84–96. doi: 10.1016/s0165-5728(02)00171-6. [DOI] [PubMed] [Google Scholar]
- 43.Ballok DA, Earls AM, Krasnik C, Hoffman SA, Sakic B. Autoimmune-induced damage of the midbrain dopaminergic system in lupus-prone mice. J Neuroimmunol. 2004 Jul;152(1-2):83–97. doi: 10.1016/j.jneuroim.2004.04.003. [DOI] [PubMed] [Google Scholar]
- 44.Anderson KK, Ballok DA, Prasad N, Szechtman H, Sakic B. Impaired response to amphetamine and neuronal degeneration in the nucleus accumbens of autoimmune MRL-lpr mice. Behav Brain Res. 2006;166:32–8. doi: 10.1016/j.bbr.2005.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chun S, McEvilly R, Foster JA, Sakic B. Proclivity to self-injurious behavior in MRL-lpr mice: implications for autoimmunity-induced damage in the dopaminergic system. Mol Psychiatry. 2007 Sep 4; doi: 10.1038/sj.mp.4002078. [DOI] [PubMed] [Google Scholar]
- 46.Feeney DM, Gonzales A, Law WA. Amphetamine, haloperidol and experience interact to affect rate of recovery after motor cortex injury. Science. 1982;217(4562):855–7. doi: 10.1126/science.7100929. [DOI] [PubMed] [Google Scholar]
- 47.Sakic B, Szechtman H, Stead R, Denburg JA. Joint pathology and behavioral performance in autoimmune MRL-lpr mice. Physiol Behav. 1996;60(3):901–5. doi: 10.1016/0031-9384(96)00065-0. [DOI] [PubMed] [Google Scholar]
- 48.Porsolt RD, Bertin A, Jalfre M. “Behavioural despair” in rats and mice: strain differences and the effects of imipramine. Eur J Pharmacol. 1978;51:291–4. doi: 10.1016/0014-2999(78)90414-4. [DOI] [PubMed] [Google Scholar]
- 49.Lerch JP, Sled JG, Henkelman RM. MRI phenotyping of genetically altered mice. Methods Mol Biol. 2011;711:349–61. doi: 10.1007/978-1-61737-992-5_17. [DOI] [PubMed] [Google Scholar]
- 50.Dorr AE, Lerch JP, Spring S, Kabani NJ, Henkelman RM. High resolution three dimensional brain atlas using an average magnetic resonance image of 40 adult C57Bl/6J mice. Neuroimage. 2008 Aug 1;42(1):60–9. doi: 10.1016/j.neuroimage.2008.03.037. [DOI] [PubMed] [Google Scholar]
- 51.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. Journal of the Royal Statistical Society Series B. 1995;57:289–300. [Google Scholar]
- 52.Gaser C, Volz HP, Kiebel S, Riehemann S, Sauer H. Detecting structural changes in whole brain based on nonlinear deformations-application to schizophrenia research. Neuroimage. 1999 Aug;10(2):107–13. doi: 10.1006/nimg.1999.0458. [DOI] [PubMed] [Google Scholar]
- 53.Nieman BJ, Flenniken AM, Adamson SL, Henkelman RM, Sled JG. Anatomical phenotyping in the brain and skull of a mutant mouse by magnetic resonance imaging and computed tomography. Physiol Genomics. 2006 Jan 12;24(2):154–62. doi: 10.1152/physiolgenomics.00217.2005. [DOI] [PubMed] [Google Scholar]
- 54.Chun S, McEvilly R, Foster JA, Sakic B. Proclivity to self-injurious behavior in MRL-lpr mice: implications for autoimmunity-induced damage in the dopaminergic system. Mol Psychiatry. 2008 Nov 15;13(11):1043–53. doi: 10.1038/sj.mp.4002078. [DOI] [PubMed] [Google Scholar]
- 55.Rammes G, Rupprecht R, Ferrari U, Zieglgansberger W, Parsons CG. The N-methyl-D-aspartate receptor channel blockers memantine, MRZ 2/579 and other amino-alkyl-cyclohexanes antagonise 5-HT(3) receptor currents in cultured HEK-293 and N1E-115 cell systems in a non-competitive manner. Neurosci Lett. 2001 Jun 22;306(1-2):81–4. doi: 10.1016/s0304-3940(01)01872-9. [DOI] [PubMed] [Google Scholar]
- 56.Buisson B, Bertrand D. Open-channel blockers at the human alpha4beta2 neuronal nicotinic acetylcholine receptor. Mol Pharmacol. 1998 Mar;53(3):555–63. doi: 10.1124/mol.53.3.555. [DOI] [PubMed] [Google Scholar]
- 57.Seeman P, Caruso C, Lasaga M. Memantine agonist action at dopamine D2High receptors. Synapse. 2008 Feb;62(2):149–53. doi: 10.1002/syn.20472. [DOI] [PubMed] [Google Scholar]
- 58.Omdal R, Brokstad K, Waterloo K, Koldingsnes W, Jonsson R, Mellgren SI. Neuropsychiatric disturbances in SLE are associated with antibodies against NMDA receptors. Eur J Neurol. 2005 May;12(5):392–8. doi: 10.1111/j.1468-1331.2004.00976.x. [DOI] [PubMed] [Google Scholar]
- 59.Kowal C, DeGiorgio LA, Lee JY, Edgar MA, Huerta PT, Volpe BT, et al. Human lupus autoantibodies against NMDA receptors mediate cognitive impairment. Proc Natl Acad Sci U S A. 2006 Dec 26;103(52):19854–9. doi: 10.1073/pnas.0608397104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lapteva L, Nowak M, Yarboro CH, Takada K, Roebuck-Spencer T, Weickert T, et al. Anti-N-methyl-D-aspartate receptor antibodies, cognitive dysfunction, and depression in systemic lupus erythematosus. Arthritis Rheum. 2006 Aug;54(8):2505–14. doi: 10.1002/art.22031. [DOI] [PubMed] [Google Scholar]
- 61.Hanly JG, Robichaud J, Fisk JD. Anti-NR2 glutamate receptor antibodies and cognitive function in systemic lupus erythematosus. J Rheumatol. 2006 Aug;33(8):1553–8. [PubMed] [Google Scholar]
- 62.Kozora E, West SG, Maier SF, Filley CM, Arciniegas DB, Brown M, et al. Antibodies against N-methyl-D-aspartate receptors in patients with systemic lupus erythematosus without major neuropsychiatric syndromes. J Neurol Sci. 2010 Aug 15;295(1-2):87–91. doi: 10.1016/j.jns.2010.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Petri M, Naqibuddin M, Sampedro M, Omdal R, Carson KA. Memantine in Systemic Lupus Erythematosus: A Randomized, Double-Blind Placebo-Controlled Trial. Semin Arthritis Rheum. 2011 Mar 31; doi: 10.1016/j.semarthrit.2011.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Husebye ES, Sthoeger ZM, Dayan M, Zinger H, Elbirt D, Levite M, et al. Autoantibodies to a NR2A peptide of the glutamate/NMDA receptor in sera of patients with systemic lupus erythematosus. Ann Rheum Dis. 2005 Aug;64(8):1210–3. doi: 10.1136/ard.2004.029280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ganor Y, Goldberg-Stern H, Lerman-Sagie T, Teichberg VI, Levite M. Autoimmune epilepsy: distinct subpopulations of epilepsy patients harbor serum autoantibodies to either glutamate/AMPA receptor GluR3, glutamate/NMDA receptor subunit NR2A or double-stranded DNA. Epilepsy Res. 2005 Jun;65(1-2):11–22. doi: 10.1016/j.eplepsyres.2005.03.011. [DOI] [PubMed] [Google Scholar]
- 66.Steup-Beekman G, Steens S, van BM, Huizinga T. Anti-NMDA receptor autoantibodies in patients with systemic lupus erythematosus and their first-degree relatives. Lupus. 2007;16(5):329–34. doi: 10.1177/0961203307078224. [DOI] [PubMed] [Google Scholar]
- 67.Peeters M, Maloteaux JM, Hermans E. Distinct effects of amantadine and memantine on dopaminergic transmission in the rat striatum. Neurosci Lett. 2003 Jun 12;343(3):205–9. doi: 10.1016/s0304-3940(03)00398-7. [DOI] [PubMed] [Google Scholar]
- 68.Shearman E, Rossi S, Szasz B, Juranyi Z, Fallon S, Pomara N, et al. Changes in cerebral neurotransmitters and metabolites induced by acute donepezil and memantine administrations: a microdialysis study. Brain Res Bull. 2006 Mar 31;69(2):204–13. doi: 10.1016/j.brainresbull.2005.12.001. [DOI] [PubMed] [Google Scholar]
- 69.Costall B, Naylor RJ. Neuropharmacological studies on D145 (1,3-dimethyl-5-aminoadamantan) Psychopharmacologia. 1975 Jul 23;43(1):53–61. doi: 10.1007/BF00437615. [DOI] [PubMed] [Google Scholar]
- 70.Bisaga A, Danysz W, Foltin RW. Antagonism of glutamatergic NMDA and mGluR5 receptors decreases consumption of food in baboon model of binge-eating disorder. Eur Neuropsychopharmacol. 2008 Nov;18(11):794–802. doi: 10.1016/j.euroneuro.2008.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Brennan BP, Roberts JL, Fogarty KV, Reynolds KA, Jonas JM, Hudson JI. Memantine in the treatment of binge eating disorder: an open-label, prospective trial. Int J Eat Disord. 2008 Sep;41(6):520–6. doi: 10.1002/eat.20541. [DOI] [PubMed] [Google Scholar]
- 72.Joo IS, Hwang DH, Seok JI, Shin SK, Kim SU. Oral administration of memantine prolongs survival in a transgenic mouse model of amyotrophic lateral sclerosis. J Clin Neurol. 2007 Dec;3(4):181–6. doi: 10.3988/jcn.2007.3.4.181. [DOI] [PMC free article] [PubMed] [Google Scholar]