

Abstract
Highly complex molecular networks, which play fundamental roles in almost all cellular processes, are known to be dysregulated in a number of diseases, most notably in cancer. As a consequence, there is a critical need to develop practical methodologies for constructing and analysing molecular networks at a systems level. Mathematical models built with continuous differential equations are an ideal methodology because they can provide a detailed picture of a network’s dynamics. To be predictive, however, differential equation models require that numerous parameters be known a priori and this information is almost never available. An alternative dynamical approach is the use of discrete logic-based models that can provide a good approximation of the qualitative behaviour of a biochemical system without the burden of a large parameter space. Despite their advantages, there remains significant resistance to the use of logic-based models in biology. Here, we address some common concerns and provide a brief tutorial on the use of logic-based models, which we motivate with biological examples.
Introduction
The emergence of molecular biology has produced a vast literature on the cellular function of individual genes and their protein products. It has also generated massive amounts of molecular interaction data derived from high-throughput methods as well as more classical low-throughput methods, such as immunoprecipitation, immunoblotting, and yeast two-hybrid systems. From this accumulation of interaction data, researchers can now attempt to reconstruct and analyse the highly complex molecular networks involved in cellular function.
Intracellular molecular networks are known to be highly dysregulated in a number of diseases, most notably in cancer, and targeted molecular inhibitors have emerged as a leading anti-cancer strategy. Despite promising pre-clinical studies, many targeted inhibitors are beset by harmful off-target effects and/or lower than expected efficacy in the clinic. The large number of off-target effects associated with molecular inhibitors was recently termed the “whack a mole problem”1 because inhibiting one molecular target often results in the activation of another non-targeted molecule. It is increasingly clear that the inability of many targeted therapies to keep a disease in check is related to the complex interactions and emergent, non-linear behaviours found in intracellular networks. As a consequence, there is a critical need to develop practical methodologies for constructing and analysing molecular networks at a systems level.
The objective of systems biology is to integrate experimental data with theoretical methods to build predictive models of complex biological processes across a variety of spatial and temporal scales. Two very different paradigms of system biology are frequently used to construct and analyse network models of molecular interactions inferred from experimental data: structural network analysis methods and mathematical models based on differential equations. A third increasingly important network analysis paradigm in systems biology is the application of logic-based methods to generate predictive output.2,3 Although qualitative in nature, logic-based methods have the capacity to provide insights into the dynamics of highly complex gene regulatory and signal transduction networks without the burden of large parameter spaces.
Understanding the networks associated with neoplastic diseases offers especially difficult challenges. Fundamental problems in understanding the transition from the normal to near normal to dysplastic to neoplastic to metastatic states of cancer progression can theoretically be modelled by longitudinal comparisons of networks in which, as progression occurs, certain molecular interactions are rendered stronger (for instance through gene amplification) or lost (through mutation, deletion, down-regulation, or methylation). Logic models provide a framework in which these types of network comparisons are possible. Multi-state logic models can simulate signal amplification and random order asynchronous logic models can simulate the heterogeneous response in a population of cells to diverse stimuli. Logic-models are also well suited for performing in silico molecular perturbations, which could be used to predict a population level response to a targeted therapy or a combination of therapies. In this review, we provide a tutorial on the use of logic-based methods as well as a discussion of their limitations, using biologically motivated examples.
Modelling intracellular networks
Typically, knowledge of molecular interactions is summarized in diagrams of varying complexity, commonly known as interaction networks.4 In an interaction network diagram, each node represents a molecule and a line drawn between two nodes represents a molecular interaction, also referred to as an edge in graph theory. If the nature of an interaction between two nodes is known (e.g., which molecule is the regulator that activates or inhibits the other molecule), the edge is said to have directionality. If a correlation between the activities or expression levels of two nodes is known but the causal relationship underlying their interaction is not, the edge is said to lack directionality.
Structural network methods
Structural network analysis provides a picture of the correlations between molecules in very large networks. In structural network models, which are usually derived from high-throughput genomic or proteomic experimental methods, the directionality of interactions in the network is generally not known and it is the static correlations in expression patterns that are important. The primary objective of these methods is to infer functional patterns in large networks using statistical methods.5 These methods are also used to construct species specific interactomes.4 A limitation of these methods, however, is that they generally provide only a static view of molecular interactions in a network at single point in time. Additionally, the current experimental methods used to generate data for structural network models are extremely noisy,6 which further limits the predictive power of this method.
Differential equation methods
On the other hand, systems of ordinary differential equations (ODEs) are frequently used to model biochemical reactions involved in gene and protein regulation. In these models, information about the mechanistic nature of the interaction is essential and edges between species must be directional. ODE models are built from underlying biophysical principles, such as biochemical rate laws and the conservation of mass and energy. Consequently, ODE models have the capacity to be highly predictive.7–9 This predictive power translates into the ability to generate a dynamic view of the concentration of each interacting species in the network over time as well as the ability to identify biologically realistic steady states.8 The predictive power of ODE systems is dependent, however, on large numbers of kinetic parameters that are rarely known with any degree of certainty. These powerful methods are, therefore, limited both by the enormous parameter spaces involved in even a relatively simple network and by their need for detailed mechanistic knowledge a priori.
Logic-based methods
Logic-based network models were pioneered in the biomedical sciences by Kauffman 10,11 and represent a compromise between structural analysis and ODE methods in terms of precision and complexity.7 While logic models do not require mechanistic knowledge of interactions, they do require knowledge of edge directionality. In their simplest form, logic-based models permit each biochemical species (represented as nodes in a network) to be in one of two discrete states: ON or OFF. The state of a logic network evolves in a dynamic fashion as nodes in the network are switched ON and OFF according to the state of other nodes in the network, until the network settles into an unchanging state, often referred to as an attractor.3 Logic-based models with only two binary states are generally known as Boolean models. While there is no explicit notion of time in a logic model, each round of updating can be considered an arbitrary time unit.
Logic-based models approximate biochemical regulation
The assumption that a molecule can have only two possible states is a simplification of biological complexity. It is a reasonable regulatory approximation, however, given the switch-like sigmoidal relationship often observed between an affector molecule and its target molecule (Fig. 1A, B).3,12–14 It is important to emphasize that when a molecule’s node is OFF in a discrete logic model, it does not imply that the molecule has zero concentration in the system. Instead, it implies that the molecule is not present at a high enough level to induce a change in the molecules it directly regulates.14 Wh en a molecule’s node is ON in a logic model, it means the molecule has reached a threshold of functional activation that is high enough to affect the state of the molecules it directly regulates. More specifically, a target molecule will remain OFF in a logic model until its activator reaches a specific threshold of activity (Fig. 1B). Likewise, a target molecule will remain ON in a logic model until its inhibitor reaches a specific threshold of activity (Fig. 1A).14 As a consequence, logic models can only provide qualitative approximations of molecular regulation. While this represents a limitation of the methodology, in reality, the majority of experimental data available on molecular regulations are also qualitative in nature.15
Fig. 1.
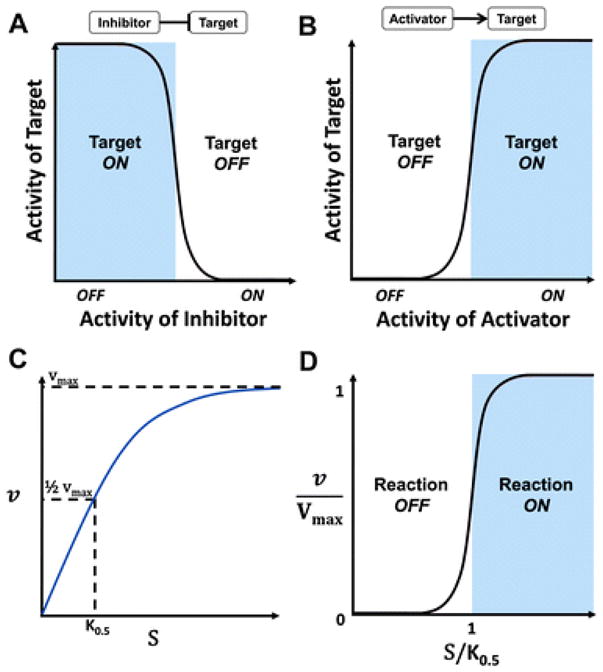
Logic models approximate molecular interactions. The motivation behind logic models comes from the sigmoidal relationship observed between regulatory molecules and their target molecules. This relationship approximates a Boolean switch and can be thought of as having two states: saturated (ON) and non-saturated (OFF). (A) An inhibitor regulates a target molecule in a logic model: the target remains ON until the inhibitor’s activity surpasses a threshold of activation. (B) An activator regulates a target molecule in a logic model: the target remains OFF until the activator’s activity surpasses a threshold of activation. In these models, ON means the molecule has reached a threshold of functional activation that is high enough to affect the state of other molecule(s) it directly regulates, while OFF means the molecule is not present at a high enough level to affect the molecules it directly regulates. Critically, OFF does not necessarily mean the molecule has a zero concentration. (C) A standard hyperbolic saturation curve measuring reaction velocity, v, as substrate concentration, S, increases. K0.5 is the substrate concentration needed to reach half the maximum velocity, Vmax. (D) A plot of v/Vmax vs. S/K0.5 (the specific substrate concentration) takes the form of a sigmoidal saturation curve approximating a Boolean- ≪1 like switch. When S/K0.5 1, the reaction is effectively OFF. When S/K0.5is ≫1, the reaction is effectively ON.
More complex logic-based methods have been developed, such as multi-state and fuzzy logic methods, which permit nodes to be in more than two discrete states. In addition, logic models that allow node states to vary continuously between states (e.g., from 0 to 1) have also been developed.7,12,16,17 Although theoretically able to more precisely simulate biochemical regulation,16 these more complicated approaches require parameter value estimates that are rarely known and, in some cases, are difficult to correlate with biophysical chemistry theory. Thus, discrete two-state logic models (Boolean models) are an intuitive and predictive method for describing biochemical interactions without requiring prior knowledge of complex mechanistic details of reaction kinetics (needed for ODE systems) or degrees of membership (needed for multi-state fuzzy logic systems).
Moreover, Boolean models can produce the same qualitative output as more quantitatively precise ODE models, when well-constructed. For example, Albert and Othmer18 used a Boolean model of the genetic regulation of segmentation patterns in Drosophila to produce results that were in close agreement with an earlier ODE model of the same system.19 Fauré et al.15 analysed a simple Boolean model of cell cycle regulation and found qualitative steady state agreement with a complex ODE model.20 More recently, Akman et al.21 demonstrated that a series of Boolean models produce the same qualitative output as a series of ODE models of circadian clock regulation. In addition, Boolean models are well suited for the testing of hypothesized regulatory mechanisms18,22 and for helping to direct future experiments. 23–26 They are also useful for performing a preliminary network analysis25 prior to developing more detailed experimental or theoretical models. For all these reasons, the development and analysis of two-state Boolean models will be the primary focus of this review.
A basis for the Boolean approximation is also found in chemical reaction kinetics
In addition to the switch-like regulatory dependence described above, another switch-like behaviour in biochemical systems is related to the substrate concentration (S) needed to reach half the maximum velocity (Vmax) of a reaction, commonly represented by K0.5. Enzymologists routinely use saturation curves depicting how reaction velocity (v) varies with S (Fig. 1C) to estimate the K0.5of a reaction. In reality, v is not dependent on S but is, instead, dependent on the specific substrate concentration, defined as the ratio of S to K0.5( S/K0.5). The well-known constant Km is the substrate concentration needed to reach K0.5under Michaelis –Menten (MM) kinetics.27 While K0.5is sometimes called the “apparent Km”, K0.5is not restricted to kinetic mechanisms that follow the MM approximation.
The standard MM expression for the velocity (vMM) of a non-reversible enzyme catalysed reaction is presented in eqn (1). This expression is similar to the generalized expression for the velocity (v) of any enzyme catalysed reaction presented in eqn (2), where S is converted to a product. Here, n refers to the Hill coefficient describing the degree of cooperativity in the reaction (i.e., positive, negative, or no cooperativity). Dividing both sides of eqn (2) by Vmax and factoring K0.5out of the right -hand side, gives eqn (3), which is now in the form commonly used to normalize reaction velocities (v′). Eqn (3) also resembles a standard Hill equation.
| (1) |
| (2) |
| (3) |
In eqn (3), when S/K0.5 = 1, then v′ = 1/2. Consequently, there are two distinct regions where the normalized reaction velocity responds in a characteristic way to S/K0.5(Fig. 1D). When S/K0.5 ≪1, then S < (0.01 × K0.5), meaning the enzyme is not saturated and the reaction rate is linear with respect to the substrate concentration. In contrast, when S/K0.5 ≫ 1, then S > (100 × K0.5), meaning the enzyme is saturated, the reaction rate is independent of the substrate concentration, and the reaction has reached (or is very near to) its maximum velocity. Thus, the specific substrate concentration serves as the on-off switch of a reaction. In logical terms, when the node representing S is OFF(or 0), then the specific substrate concentration will be less than 1 and the reaction cannot not proceed. Likewise, when the node representing S is ON (or 1), then the specific substrate concentration will be greater than 1 and the reaction will proceed at a rate near Vmax( Fig. 1D).
The use of logic to describe chemical change is not limited to the rate of biochemical reactions. Molecular substrates have been described as computational devices that process physical and chemical inputs into outputs according to Boolean logic.28 Molecular logic operations are achieved by leveraging observable chemical changes to create a computational device. These devices, for example, can be used to solve arithmetic or logic operations by exploiting changes in the conformations of chemical components. In the laboratory, molecular logic functions have been developed that rely on charge transfer, which affects the fluorescent state of a molecule.29,30 Logical functions have also been created to exploit charge transfers in cascades of coupled enzymes.31,32 In fact, molecular logic operations serve as the basis for many nanosensors currently used in the basic sciences, industry, and medicine.28
Predictive logic-based models in the literature
Although the theoretical underpinnings of logic models provide qualitative approximations of molecular and biochemical regulation, in reality these models can only generate predictive output when the logic model is well constructed. Several examples of well-constructed logic models providing good agreement with experimental data exist in the literature.16–18,26,33–36
For example, Li et al.34 constructed a Boolean model of the genetic network controlling the cell cycle in Saccharomyces cerevisiae. The authors found that 86% of the 2048 possible states in the network settled into a steady state (also known as a stable attractor) that corresponded to the G1 stationary phase of the cell cycle. Their analysis suggested that the regulatory network controlling the yeast cell cycle is resistant to stochastic perturbations. The authors interpreted these findings to mean that robustness in the underlying network is advantageous for the organism because, under normal conditions, there is a high probability the regulatory network dynamics will settle into the G1 state regardless of the current state of the network. Once in the G1 state, the network will remain in that state until a significant external signal perturbs the network and initiates another round of cellular division. Subsequent work by Davidich and Bornholdt 37 used a similar Boolean approach to study the cell cycle regulation in Schizosaccharomyces pombe. These authors found that the majority of S. pombe network states settle into a steady state corresponding to the G1 stationary phase, which is in agreement with the results of the S. cerevisiae model. However, they also found significant differences in the regulatory network of S. pombe compared to S. cerevisiae, which yielded very different network dynamics.
Using a somewhat different logic-based approach, Bolouri and colleagues25,38 constructed an a priori gene regulatory network of endomesoderm specification control in sea urchin embryos. The network was logic-based and generated a series of testable predictions. Using computational methodologies and large-scale perturbation analyses, the authors iteratively tested and revised their model by comparing model output to biological readouts. Their use of a regulatory network to logically map inputs and outputs for cis-regulatory elements identified system level properties that would not otherwise have been observable. From this information, the authors were able to draw important conclusions about the developmental features of endomesoderm specification.
In addition to gene regulatory networks, logic models can also be used to model signal transduction networks. Li et al.26 developed a Boolean model of the signal transduction network controlling abscisic acid regulation of stomatal closure in plants. The authors employed a network construction approach that inferred indirect molecular relationships from data to build the sparsest logic network possible that was compatible with available experimental data. A random order asynchronous Boolean approach was then used to simulate the heterogeneity in a population of cells. The model results were in good agreement with previous experimental findings and generated novel predictions about the conditions likely to have the strongest effect on stomatal closure. In a subsequent manuscript, which also serves as an excellent tutorial, Albert et al.12 contrasted the asynchronous approach used in the Li et al.26 model with a continuous piecewise Boolean model of the same system that allowed node states to vary between 0 and 1. They reported that the asynchronous discrete Boolean model produced the same qualitative results as the continuous piecewise Boolean model.
For additional examples of predictive logic-based models in the literature, we direct the reader to comprehensive reviews by Morris et al.7 and Albert et al.12 Both reviews provide detailed case studies of logic model implementations and demonstrate the variety of ways logic-based models can be applied to answer biological questions.
Time in a logic model
Each time a network is updated in a standard two-state Boolean model, signals are transferred according to logic functions in a synchronous and deterministic manner. In these models, all nodes are updated instantaneously in the same time step so that the state of the network is always fully determined by the state of all nodes in the previous time step. Thus, the underlying assumption is that all molecular interactions in the network take the same arbitrary amount of time to complete. In reality, the time it takes for molecular interactions to complete varies widely.
An alternate updating method involves asynchronous updating where one node is selected at random and instantly updated according to the current state of the network. In these models, the next state of the network is non-deterministic. The random and instantaneous updating of a node is repeated many times with each random update representing a time step in the model. This non-deterministic updating scheme is thought to more closely resemble biological variation by eliminating temporal uniformity in the model.2,12,14,39,40 Typically, implementation of this type of scheme involves running a large number of model simulations to calculate a probability that any given node will end up ON or OFF for any set of initial conditions. 12,26 Synchronous and asynchronous updating methods are discussed in more detail in later sections of this review.
Building predictive logic-based models
Creating a logic model is relatively straight-forward. Building a logic model that can generate predictive output that can be leveraged by experimentalists, however, requires considerably more effort. Specifically, building predictive logic-based models entails two primary steps: building a detailed interaction network and translating the interaction network into a logic network. The development of a logic network includes the careful derivation of the logic functions that will drive the network’s dynamics.
Build an interaction network by surveying the literature
The first step in developing a logic model is to construct an interaction network of the system to be modelled (see, for example, Fig. 2A). To do so will typically require a thorough literature or database search. This is a critical step and should be performed by, or in close collaboration with, someone who is well acquainted with the biology of the system. Once the interactions involved in the network have been identified, it is often desirable to perform a node reduction to reduce network complexity, especially for very large networks. The formulation of any theoretical model requires sound judgments about which approximations are appropriate for simplifying model complexity without losing essential elements of the underlying mechanism(s).41 This is certainly true of logic models where decisions must be made about whether some complex interactions can be lumped into a smaller subset of nodes and interactions (Fig. S1, ESI†). In general, the objective is to use the simplest network possible that still agrees with known experimental data. This may be done manually or with the assistance of computational algorithms.26,42,43 If automated tools are used, it is useful to validate that the reduced network generated includes a suitable amount of complexity for the system and problem considered.
Fig. 2.

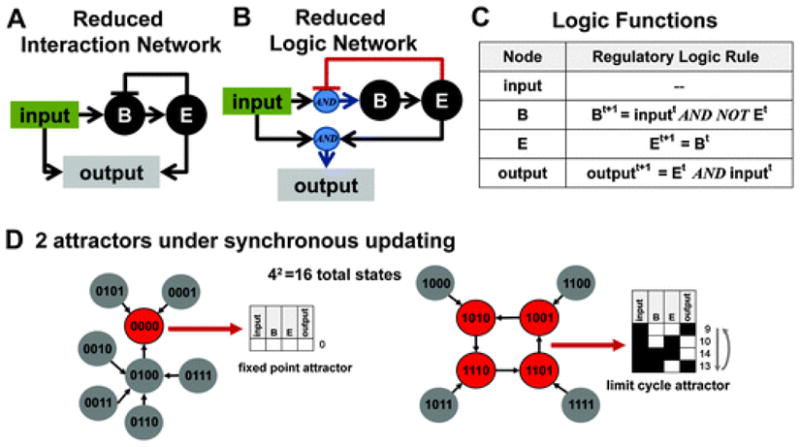
Hypothetical network example with 12 nodes. (A) An interaction network diagram summarizing a hypothetical network with 12 nodes where input is the only input node, output is the only output node, and A–J are internally regulated nodes. (B) A logic network diagram explicitly identifying how multiple signals (edges in the diagram) will be integrated to produce a response in nodes Band output, the only nodes with more than one regulator. (C) A table explicitly listing the logic functions used in the hypothetical logic model. The information in this table is equivalent to the information available in (B), and both (B) and (C) contain more information than (A). (D) Each unique network state in a logic network can be identified in a number of ways: a graphical table where white = OFF and black = ON, a binary representation where 0 = OFF and 1 = ON, or a decimal value that is equivalent to the binary representation. Each of these states will give rise to another state based on the logic functions (B–C) that control signal transfer in the network. (E) Eventually, all states will settle into an attractor, a state from which it is not possible to escape without an external perturbation to the system. In the graphs, attractor states are colored red and non-attractor states are colored grey. An example of a limit cycle and a fix point attractor are highlighted. In this model, there are a total of 212(4096) possible states that settle in to 1 of 10 attractors. Blue circles indicate AND interactions.
A common manual approach is to eliminate redundant linear regulations. For example, in Fig. 2A, nodes B and Care both activators of E because an even number of inhibitions produces an activating regulation. In contrast, E is an inhibitor of B because there is one inhibitory regulation between E and B. A reduced version of this network is presented in Fig. 3. As shown in Fig. S2 (ESI†), both forms of the network produce the same qualitative output.
Fig. 3.
Hypothetical network example reduced to only 4 nodes. (A) An interaction network diagram summarizing an alternate form of the network in Fig. 2A. Here all linear regulations have been collapsed into single regulations to reduce model complexity. (B) A logic network diagram explicitly identifying how multiple signals (edges in the diagram) will be integrated to produce a response in nodes Band output, the only nodes with more than one regulator. (C) A table explicitly listing the logic functions used in the reduced hypothetical model. The information in this table is equivalent to the information available in (B), and both (B) and (C) contain more information than (A). (D) Eventually, all states will settle into an attractor. In the graphs, attractor states are colored red and non-attractor states are colored grey. In this model, there are 24(16) possible states that settle in to 1 of 2 attractors, which, because of their small size, can be easily visualized. The binary numbers on the nodes represent the state of each node in the network in the following order input, B, E, and output. Blue circles indicate AND interactions.
Translate the interaction network into a logic network
Ultimately, the interaction network must be translated into a set of logic functions (also referred to as transfer functions or logic gates) that will be used to transfer information (or signals) between nodes each time the model is updated. Logic functions often contain one or more Boolean operators. The AND and OR operators are used to define how distinct signals acting on the same node (which may be stimulatory and/or inhibitory) will be processed. The NOT operator is used to negate the state of the node it precedes. The derivation of logic functions is discussed in detail in the next section.
A justification from the literature (or evidence from experimental data) should be provided to support each logic function. When individual logic functions qualitatively agree with experimental data, it is more likely that the model, as a whole, will be predictive. Ideally, a table or appendix summarizing each logic function’s rationale will be included with all published models (see Table 1for an example). 15,17,18,26
From an interaction network diagram alone, it is not possible to infer how multiple signals acting on the same node should be processed. Therefore, the use of descriptive logic network diagrams is recommended to graphically depict a logic model. The information contained in the full set of logic functions should be equivalent to the information contained in a logic network diagram (compare Fig. 2B to C and Fig. 3B to C). The use of descriptive diagrams to represent a biological network is not a novel concept.2,44 Albert and colleagues18 proposed the use of “pseudo-nodes” and “complementary pseudo-nodes” to clarify the functional nature of edge interactions in a logic network diagram. In a large-scale Boolean model of EGFR and ERB2 signalling, Samaga et al.36 used a graphical representation where AND interactions were depicted as small blue circles and all other interactions were assumed to be OR interactions. More recently, Morris et al.7 presented logic network diagrams that used “logic gate” notation similar to that used in an engineering diagram.
Throughout this review, we have adopted a modified version of the notation used by Morris et al.7 Our notation graphically illustrates how multiple edges regulating the same node will be integrated by explicitly identifying where AND and OR operators are used in the network. In addition, all activating interactions in our logic network diagrams are indicated with a black arrow and all inhibiting interactions are indicated with a red line and a blunt edge. Regardless of the graphical method used, the use of diagrammatic logic networks is strongly encouraged to remove ambiguity from interaction networks.
Deriving logic functions: the importance of characterizing each interaction
When translating a set of interactions into a logic model, the implicit assumptions underlying all logic functions must be carefully considered. We recommend the construction of truth tables for each logic function to confirm the logical output of each function is in agreement with experimental data (or that of a hypothesized regulatory mechanism). A truth table provides the logical output of all possible combinations of input values a logic function may receive. In Boolean models with only two discrete states, there are 2rpossible combinations of regulatory inputs in a truth table, where r is the number of regulators (or edges) leading into the regulated node. We have provided truth tables for two biologically motivated network examples in S1 (ESI†) for reference.
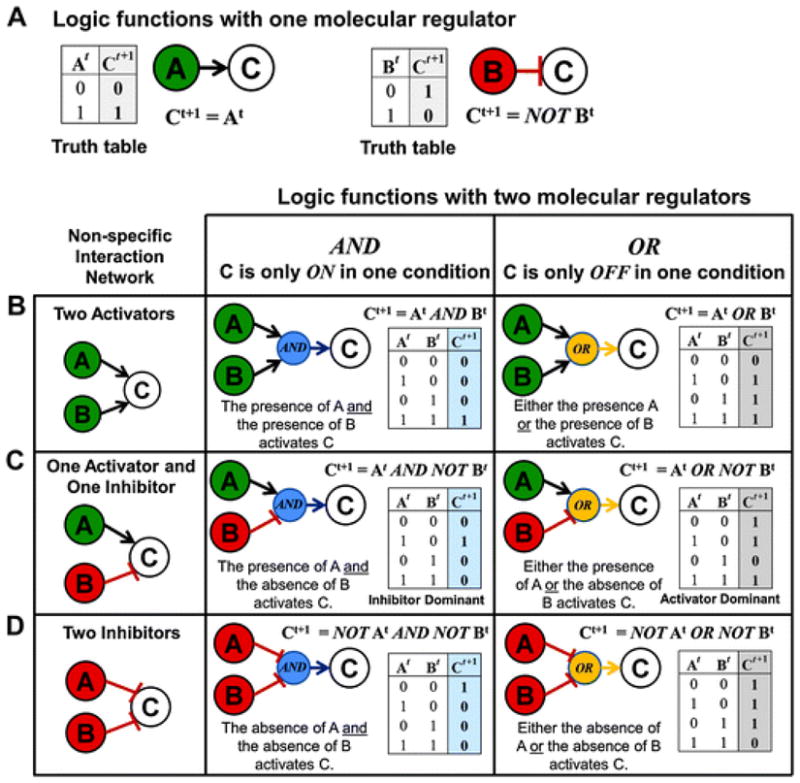
For nodes with one regulator, derivation of the logic function is straightforward: the next state of the regulated node is fully determined by the current state of its only regulator (Fig. 4A). An example of the two ways a single molecule can regulate another molecule in a Boolean model is presented in Fig. 4A, along with corresponding truth tables. In the case where Cis activated by A, the logic rule is represented as Ct+1= At, which means the value of C in the next arbitrary time step (t+ 1) will be the current value of A. In the case where Cis inhibited by B, the rule is represented as Ct+1 = NOT Bt, which means the value of C in the next arbitrary time step (t+ 1) will be the inverse of the current value of B. Importantly, because C will always be ON whenever B is OFF in this inhibition example, the implicit meaning of this function is that of constitutive activation of C in the absence of B. If this turned out to be an inappropriate assumption for the of regulation of C, then additional activators of C would need to be added to the model in order to generate a more complex and accurate regulatory logic function for C.
Fig. 4.
Logic Functions. Each node in a logic model has a corresponding logic function that controls its regulation each time the model is updated. (A) Derivation of logic functions for nodes regulated by only one node: the next state of the regulated node (at time t+ 1) is fully determined by the current state of the regulating node (at time t). Ct+1indicates the value of Cat the next time step. (B –D) When multiple nodes regulate the same node, derivation of logic functions is more complex. Three node networks are presented where Cis regulated by two nodes: A and/or B. In the left hand column, typical representations of interaction diagrams are presented with regulation by (B) two activators, (C) an activator and an inhibitor, or (D) two inhibitors. The corresponding logic functions when an AND or an OR relationship is used, respectively, are presented in the next two columns. In each example, a more precise logic network is depicted that clearly indicates how the two nodes will be integrated to produce a response in C. A truth table is provided that illustrates the output (Ct+1) generated for all possible input conditions. 0 indicates (OFF) and 1 indicates (ON). For clarity, activating nodes are colored green and inhibiting nodes are colored red. Blue circles indicate AND interactions and yellow circles indicate OR interactions.
For nodes that have multiple regulators, the development of a logic function can be more challenging. In a logic model, there are three possible mechanisms by which two nodes can regulate another node: regulation by two activators (Fig. 4B), regulation by an activator and an inhibitor (Fig. 4C), or regulation by two inhibitors (Fig. 4D). The simple 3 node interaction networks presented in the first column of Fig. 4B–D clearly indicate that 2 nodes regulate another node but do not provide precise information about how the signals will be integrated to produce a response in the regulated node. In contrast, the second and third columns of Fig. 4B–D provide logic network diagrams and truth tables illustrating the logical output produced when an AND or OR operator is used, respectively. In general, the use of an AND operator with 2 regulators results in the regulated node turning ON in one of the four possible input conditions. In contrast, the use of an OR operator with two regulators results in the regulated node turning ON in three of the four possible input conditions. Although the examples provided are simple, there are important underlying assumptions that should be emphasized.
AND NOT: inhibitor wins
When an activator and an inhibitor are joined by an AND operator (Fig. 4C: AND column), the function is referred to as an AND NOT function. In the logic function Ct+1 = At AND NOT B t, the inhibitor is dominant because the state of C is OFF in 3 of the 4 possible input conditions. The presence of the inhibitor also trumps the presence of the activator when both are ON(refer to the last row in the truth table).
OR NOT: activator wins
Similarly, when an activator and an inhibitor are joined with an OR operator ( Fig. 4C: OR column), the function is referred to as an OR NOT function. In this case, the activator is dominant because the state of Cis ON in three of the four input conditions. The presence of the activator trumps the presence of the inhibitor when both are ON(refer to the last row in the truth table). Importantly, when both the activator and inhibit or are OFF, C will turn ON (refer to the first row in the truth table). The implicit meaning of Ct+1 = At OR NOT Bt, therefore, is that C becomes activated when both regulators are absent or below their functional threshold, even if C was OFF in the previous time step. At first glance this may seem counter intuitive and biologically implausible. However, what the OR NOT function actually simulates is the condition where the regulated node is ubiquitously expressed at a functionally active level such that it can only be deactivated by the presence of a direct inhibitor and, importantly, this direct inhibitor can be overridden by an activator (refer to the truth table). Later, we will consider a biological example where OR NOT is the correct function to model a biological interaction.
To be OR NOT to be?
As illustrated in Fig. 4 and described above, when more than one node regulates the same target node, the AND and OR logical operators produce very different outcomes. It is important to understand the underlying assumptions of all logic function included in a logic model to ensure they provide a reasonable biological approximation.40 Of course, the exact nature of an interaction may not be known ( e.g., what is necessary and what is sufficient for a molecular activation). In these cases, logic models can also be used to test hypothetical interactions and compare if results match experimental data.
Logical models have one or more fixed states known as “attractors”
The network state of a logic model can be uniquely represented in a variety ways, including by binary or decimal notation (Fig. 2D). In synchronously updated Boolean logic models (where all nodes are updated at the same instant each time step), each state deterministically gives rise to another state according to the model’s logic functions. Eventually, all states will settle into one or more stable states known as attractors. If an attractor consists of a series of states that oscillate in a cycle, the attractor is called a cycle attractor or a limit cycle. If an attractor consists of a single fixed state (which will be the case when a state always gives rise to itself), the attractor is called a fixed point attractor.3,45 Examples of each type of attractor a re presented in Fig. 2E and 3D. In asynchronously updated Boolean logic models (where a single node is selected randomly and updated instantly), the next state of the network is non-deterministic. Nevertheless, the network will eventually settle into an attractor regardless of the initial state of the network.2,14,40
In small to moderate sized network models that have been well constructed, attractors often empirically agree with biological phenotypes.3,34 In the model previously discussed of yeast cell cycle regulation developed by Li et al.,34 for example, there were 11 nodes which generated 2048 possible network states and 7 attractors. The vast majority of all possible states (1764 out of 2048) settled into a fixed point attractor that represented the stationary phase of the cell cycle. The authors found that the trajectory of states (also referred to as a basin of attraction45) leading to this point attractor followed expected molecular changes observed during cell cycle progression.
In any binary Boolean logic model consisting of n nodes, there are 2npossible network states. In the hypothetical 12 node network presented in Fig. 2 there are 4096 possible states and in the synchronous form of this model there are 10 attractors ( Fig. 2E). In a 21 node model we recently developed, there were 2 097 152 possible states that settled into 1 of 52 possible attractors (data not shown). Clearly, as the network size increases, it becomes difficult to draw biological inferences from the full attractor space. In addition, for large-scale networks, the enumeration of all attractors quickly becomes computationally intractable owing to the exponential relationship between the number of nodes in the network and the number of possible network states.46 One approach for analysing large attractor spaces is to measure the robustness of each attractor. 47–49 Unfortunately, these methods are generally more useful to a theoretician than a biologist. Another approach for analysing large state spaces with many attractors is to use asynchronous updating methods.12,26 These methods can generate a probability that a given network node will be ON or OFF under a particular set of conditions. Because asynchronous methods represent a repeated sampling of many different timescales, 14,40,50 they are also useful for modelling the heterogeneity of a population of cells.26,51 Asynchronous methods can also facilitate in silico molecular perturbations (such as knock-downs or constitutive activations) to generate output that is readily comparable to biological data.26
Finally, not all stable attractors in a deterministic synchronous logic model remain stable under nondeterministic asynchronous update conditions. The random perturbations associated with an asynchronously updated model result in the disappearance of some synchronous attractors when the assumption of timescale uniformity is removed.3,14,45 Thus, the asynchronous method is more likely to identify attractors that are robust to the typical stochastic variations observed in molecular interactions52,53 under physiological conditions.
A biologically motivated example
In Fig. 5, we present a 10 node network of the regulation of cellular proliferation and apoptosis. This model is adapted from the Boolean model used by Ribba et al. to control cell division and cell death in a multiscale model of colorectal cancer.54 For simplicity, our version of the model eliminates linear regulations. In the original Ribba model, for example, P53 activated BAX and BAX, in turn, activated apoptosis. BAX was eliminated in our model so that P53 is a direct activator of apoptosis. In this example, the linear node reduction results in the same qualitative network behaviour and output. The interaction network for this model is presented in Fig. 5A and the logic network is depicted in Fig. 5B. There are 4 input nodes representing the signals a cell may respond to in the model: Growth Factor, Over-population, Hypoxia, and DNA Damage. The output nodes are proliferation and apoptosis. The 4 internally regulated nodes are MYC, P27, Cyc-CDK, and P53. The logic functions used in this model are listed in Table 1along with a biological justification for each function and a statement of any relevant assumptions. Truth tables for each logic function are provided in S1 (ESI†).
Fig. 5.
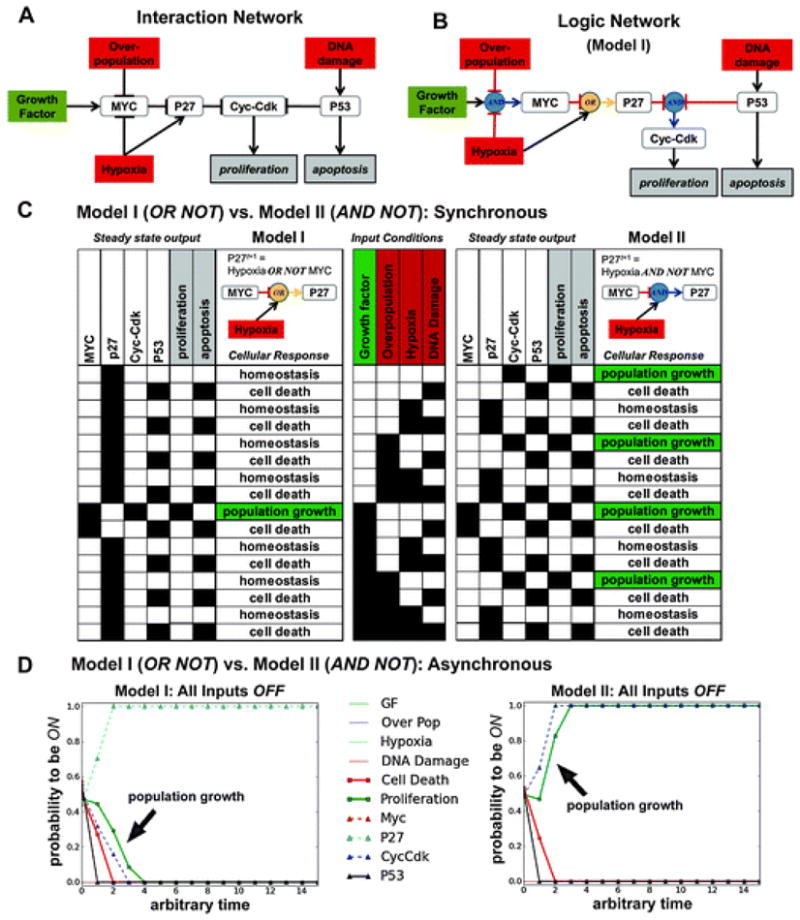
A Model of Normal Cell Proliferation and Apoptosis: OR NOT vs. AND NOT. (A) An interaction network diagram depicting regulation of cellular proliferation and apoptosis based on a model published by Ribba et al.54 Overpopulation, Growth Factor, Hypoxia, and DNA Damage are the input nodes and proliferation and apoptosis are the output nodes. The four internally regulated nodes are colored white. (B) Logic network describing the logic functions used in Model I (see also Table 1). (C) The 10 nodes in this network produce a total of 210 (1024) possible states, each of which settles into 1 of 16 possible attractors (identified as steady state output in the table), under a synchronous updating scheme. The 16 possible input conditions are displayed in the center table. On the left, Model I output is presented (with P27 regulated by OR NOT) and, on the right, Model II output is presented (with P27 regulated by AND NOT). (D) Comparison between asynchronous attractors produced by Model I and Model II when all input nodes are OFF. This input condition corresponds to the first row in the synchronous scheme’s table. An asynchronous random order update was repeated 200 times for 15 time steps. Each curve shows the average value of a node after 200 simulations for each time point. Blue circles indicate AND interactions and yellow circles indicate OR interactions.
In this model there are 1024 (210) possible network states and 16 (24) possible input conditions (Fig. 5C). For the logic network referred to as Model I (Fig. 5B), under synchronous updating each of the 1024 possible states eventually settle into one of 16 fixed point attractors corresponding to one of three biological states (Fig. 5C, left). In Model I, when all input signals are OFF, apoptosis and proliferation are also OFF in the attractor, indicating cellular homeostasis (first row in table). When all four input signals are ON, the combination of these signals leads to apoptosis ON and cell death (last row in the table). The only input condition leading to proliferation ON in Model I is Growth Factor ON and all other inputs OFF.
In another version of this model referred to as Model II, the logical operator controlling the regulation of P27 by MYC inhibition and Hypoxia activation is changed to AND NOT from OR NOT( Fig. 5C, right). This seemingly small change produces very different output. In Model II, under synchronous updating when all input signals are OFF, all states settle into an attractor that leads to proliferation ON and population growth (first row in table), which is not the behaviour expected from a normal cell. Moreover, there are now 4 input conditions that give rise to proliferation ON, including the state where all input signals are OFF. In this network example, the use of OR NOT for the regulation of P27, rather than AND NOT, is essential for obtaining the expected readout.
Under asynchronous updating, Model I and II settle into the same attractors each model produced under synchronous updating (Fig. 5D). Empirically, this will be the case when only point attractors are possible under synchronous updating because no regulatory feedback loops are included in the network. For more complex networks with multiple feedback loops, the attractor space produced by synchronous and asynchronous updating methods will not always be identical.
Oscillations in Boolean models
Logic models have been used to model oscillations in a number of biological systems.18,21,24 Oscillations play important roles in many biological processes including the cell cycle, circadian rhythms, developmental processes, and the cellular response to stress.55 It is easy to generate sustained oscillations (which are, by definition, a cycle attractor) with a synchronous logic model. All that is required is the presence of a negative feedback between one or more nodes. Even a single node can generate oscillations (Fig. 6A).56 In contrast, to generate sustained oscillations in chemical systems with an ODE model requires at least 3 distinct species represented by2 or more ODEs. 57,58 Physically realistic sustained oscillations are possible with only 2 chemical species when delay differential equations are used because these equations introduce an explicit time delay into the system.59,60 It must be emphasized that simply generating oscillations in a mathematical model of any type does not imply the underlying mechanism driving the oscillations in the model is equivalent to that driving the experimentally observed oscillations.
Fig. 6.
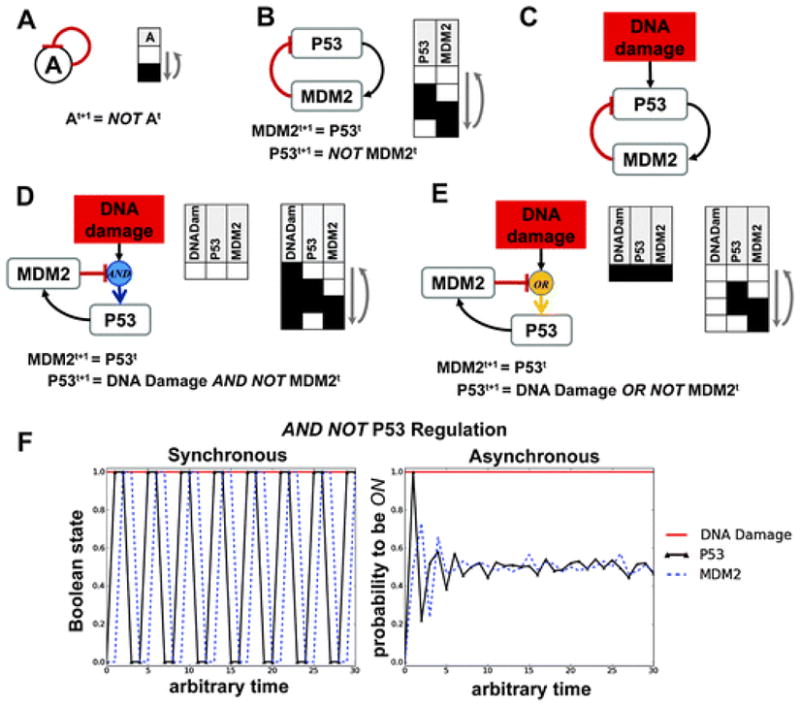
Regulatory feedback and oscillations in logic models. (A) A single node will produce an oscillating attractor if a negative auto-feedback is present. (B) Two nodes will produce an oscillating attractor if a negative feedback is present. In this example P53 activates MDM2, which inhibits P53. (C) A more realistic representation of the P53/MDM2 circuit includes activation of P53 by DNA Damage. This representation of the network does not identify whether an AND NOT or an OR NOT should be used. (D) When P53 is regulated by an AND NOT function, two synchronous attractors are possible. (E) When P53 is regulated by an OR NOT function, two synchronous attractors are possible. (F) Based on biological data, the AND NOT appears to be the correct regulation because the presence of DNA Damage induces functional levels of P53 that give rise to oscillations in the attractor. In the absence of DNA Damage, all nodes in the attractor are below their functional activation threshold (OFF). In the plots a synchronous update scheme and an asynchronous update scheme (with 200 simulations) are compared for the logic model presented in (D) when DNA Damage is ON and the other 2 nodes are OFF at the start of the simulation. Blue circles indicate AND interactions and yellow circles indicate OR interactions.
Given the ease with which oscillations can be generated with a logic model, it is essential to ensure that the implicit assumptions underlying the logic functions in a model are appropriate for the oscillatory system modelled. The protein P53 has been dubbed the “guardian of the genome” for its role in maintaining genome integrity and tumour suppression in normal cells.61 P53 plays critical regulatory roles in both cell cycle progression and apoptosis. In response to stress, such as DNA damage from ionizing radiation, P53 protein is known to mediate the transcription of MDM2, which targets P53 for degradation via ubiquitin ation.55,62–64 In response to DNA damage, this antagonistic relationship induces important cellular oscillations in P53 and MDM2 expression, which have been described as a digital behaviour.55
If we first consider the simple 2 node network in Fig. 6B consisting of only P53 and MDM2nodes, we can see that, because only one edge leads into each node, the interaction network is equivalent to its representation as a logic network (using the logic network notation described previously). In this simple network, all 4 possible states make up the limit cycle attractor. Thus, if no other inputs are added to this network, it will perpetually oscillate between these 4 states. A slightly more realistic model includes adding a DNA Damage node as the input signal (Fig. 6C). Because two nodes now regulate P53 (an activator and an inhibitor), we must decide whether AND NOT or OR NOT is appropriate for the regulation of P53. In the AND NOT case (Fig. 6D), 4 of the 8 possible network states settle into a fixed point attractor where everything is OFF. The remaining 4 states settle into a limit cycle where P53 and MDM2oscillate and DNA Damage is fixed to ON. In contrast, in the OR NOT case ( Fig. 6E), 4 of the 8 possible states settle into a point attractor where everything is ON and the remaining 4 states settle into a limit cycle where P53 and MDM2oscillate and DNA Damage is fixed to OFF. Because there is nothing in the network to regulate DNA Damage, it will never oscillate in either version of model.
P53 and MDM2 proteins are known to exist at low endogenous levels in the absence of DNA damage.55 We, therefore, consider them to be at levels below their functional threshold in the absence of DNA damage. However, when DNA damage is present, P53 becomes functionally activated,55 which triggers functional MDM2 expression levels and oscillations. Thus, we conclude that the AND NOT function is appropriate for this regulation (Fig. 6D). All model results presented in Fig. 6D, Erelied on a synchronous updating scheme. In Fig. 6F, the synchronous and asynchronous updating schemes are compared for the AND NOT form of the model using the following initial conditions: P53 OFF, MDM2 OFF, and DNA Damage ON. In the synchronous approach, it can clearly be seen that DNA Damage stays constant, while P53 and MDM2 oscillate out of phase between 0 and 1 (or OFF and ON). In the asynchronous model, which represents and average of 200 simulations using a random update order, over time both P53 and MDM2are roughly 50% likely to be ON. This is the expected probability when all 200 simulations settle into a limit cycle attractor where P53and MDM2 oscillate, which is also suggestive of the average signal across a population of cells exposed to DNA damage.
Expanding the biologically motivated example to include MDM2 and AKT regulation
In the previous model of cellular proliferation and apoptosis (Fig. 5) only fixed point attractors were possible because no regulatory feedback loops were present in the network model. If the feedback loop in Fig. 6D is added to the network in Fig. 5, then the 8 fixed point attractors with DNA Damage ON become cycle attractors with P53 and MDM2 oscillations (data not shown). In addition to adding an MDM2node, we expanded Model I (Fig. 5) to include 6 additional nodes (Fig. 7A). An AKT node was included because AKT related signalling plays an important role in activating cellular growth and inhibiting apoptosis. In addition, PTEN, a powerful tumour suppressor and upstream inhibitor of AKT activation was included.65 RAS, FOXO, BAD, and BAX were also added. Truth tables for each logic function are provided in S1 (ESI†).
Fig. 7.
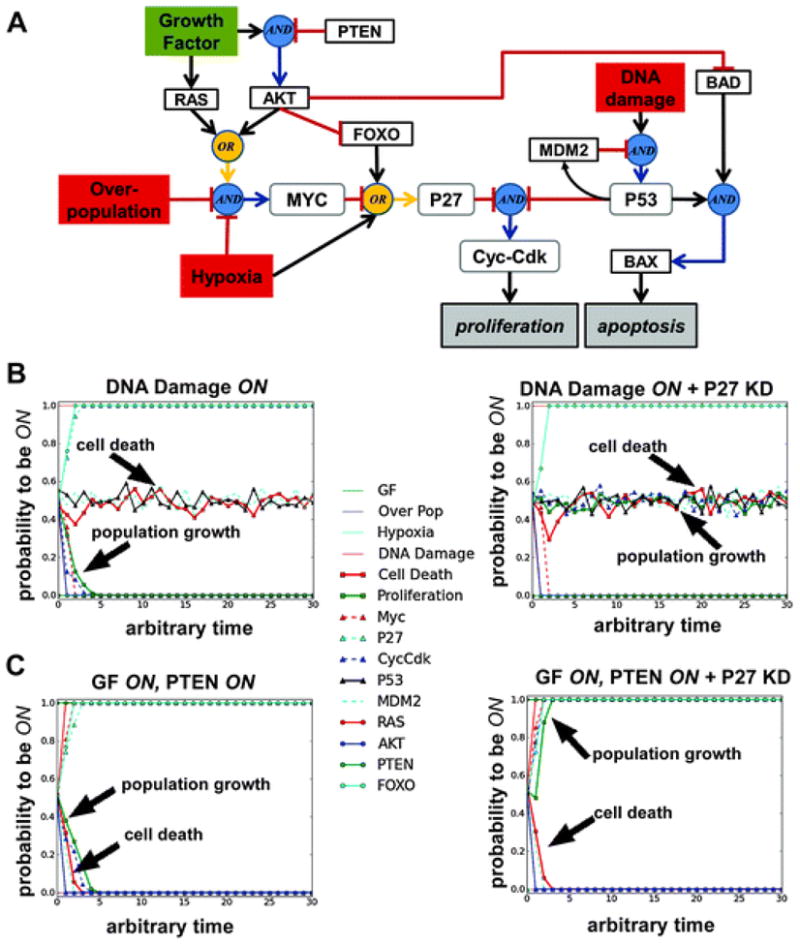
An Expanded Model of Normal Cell Proliferation and Apoptosis. (A) A logic model diagram of a more detailed version of Model I in Fig. 5. Specifically, growth factor activation of MAPK and PI3K signaling pathways is incorporated via RAS and AKT, respectively. In addition, PTEN’s downstream inhibition of AKT is included as is AKT’s influence on cell cycle progression and apoptosis. (B) Control and in silico perturbations of this model were performed using asynchronous simulations. Comparison between asynchronous attractors with and without P27knock down (KD) for the initial condition: DNA Damage ON and all other nodes randomize d. (C) Comparison between asynchronous attractors with and without P27 KD for the initial condition: Growth Factor (GF) ON, PTEN ON, and all other nodes randomized. All simulations with P27 KD began with P27 OFF and kept it fixed to this state for the duration of the simulation. All asynchronous simulations were performed 200 times to get the probability each node would be ON under the test conditions. Blue circles indicate AND interactions and yellow circles indicate OR interactions.
In Fig. 7B, C, we performed asynchronous simulations using two distinct conditions: (1) DNA Damage ON and all other inputs OFF and (2) Growth Factor ON, PTEN ON, and all other inputs OFF. We also perturbed each of these conditions by preventing P27 from turning ON in the simulations, which effectively served as an in silico knock down (KD) of this node. In the DNA Damage ON condition, the result is as expected: all states settle into attractors where there is 0% probability for population growth (proliferation) and approximately 50% probability for cell death (apoptosis). This intermediate cell death probability, which results from the fact that P53, MDM2, and apoptosis nodes are oscillating in the attractor, is representative of an average between the oscillating states. A very different outcome is found, however, when the DNA Damage ON condition is tested with the P27 KD. In this case, proliferation also has approximately 50% probability to be ON (Fig. 7B).
When both Growth Factor(which should trigger proliferative signals) and PTEN(which should suppress proliferative signals) are ON, then cellular homeostasis (or quiescence) occurs because all states end up in an attractor where both population growth (proliferation) and cell death (apoptosis) have a 0% probability to be ON. When the same conditions are tested with the P27KD, however, population growth is 100% likely to be ON in the attractor (Fig. 7C). While these results are not surprising given the importance of P27 in regulating cell cycle progression and proliferation, they do demonstrate how models of this type may be used to make predictions related to network dynamics that can then be verified experimentally.
The use of Boolean models in dynamical systems
We have seen how Boolean models can be used to simulate network dynamics and have also discussed that Boolean models are capable of qualitative agreement with more mechanistically precise ODE models. It must be stressed, however, that a Boolean model will not always be appropriate for modelling network dynamics. In complex biochemical pathways, the time evolution of the concentration of interacting species is governed by nonlinear feedback loops in which the output of a pathway is not proportional to its input.66 Examples of network behaviour that a two-state Boolean model cannot approximate include retroactive signalling,67,68 load-induced modulation,69 and bifurcations associated with nonlinear equations. 70 In the case of retroactivity and load-induced modulation, which involve upstream signal propagation in covalently modified signalling cascades, a two-state Boolean model is too qualitative to predict this behaviour because these signalling processes arise from enzyme sequestration mechanisms that are out of reach of two-state Boolean models. In the case of bifurcations, which occur when the qualitative behaviour of the solution of a nonlinear system changes as a parameter changes, two-state Boolean models are unable to predict bifurcations because they lack parameters. It is possible that multistate or continuous piecewise logic models (which are parameter driven) are capable of predicting bifurcations. To our knowledge, however, this has not yet been explored systematically.
In a recent paper by Batchelor et al.,71 the mechanisms regulating P53 response to different perturbations were investigated. The authors employed ODE models as part of an analysis into the amplitude, duration, and frequency of individual p53 pulses in response to varying amounts of ultra violet radiation. While a synchronous Boolean model would not be able to elucidate mechanisms driving the degrees of response in a system like this, multistate fuzzy logic16 and continuo us piecewise logic models12 have been used for similar purposes in other systems. In addition, asynchronous Boolean models may be used for these types of responses. In the model of guard cell aperture closure previously discussed,26 the authors chose to use an asynchronous Boolean model to generate a probability of stomate closure because stomate aperture responses are known to be graded and cannot be represented as simply open or closed. While the predictive power of an ODE model is preferred for a dose dependent response, an ODE model may not be practical for modelling such responses in large networks for computational and parameter space reasons. In such cases when a binary response is not sufficient, the use of more complex logic-based methods, such as asynchronous, multistate, fuzzy, or piecewise models, may be a reasonable alternative.
Conclusions
Logic-based models are predictive tools that can be leveraged in the absence of reliable parameter information or mechanistic details needed for more quantitatively precise methods, such as ODE models. Importantly, the predictive power of logic methods is dependent on the nature of the logic network model constructed. In this review, we have pointed out important factors to consider when building predictive logic-based models. We have emphasized the importance of using descriptive logic network diagrams and provided biologically motivated example networks. Most significantly, we have emphasized the need to properly characterize the nature of all interactions in the network and to understand the implicit meaning of logic functions used to integrate multiple input signals.
As we have seen, the use of AND and OR logical operators produce very different results for the same input conditions (Fig. 4B–D, Fig. 5C, D and Fig. 6D, E). We strongly encourage the creation of truth tables to verify that the output of each logic function is reasonable and in qualitative agreement with experimental data, if available. When the nature of the interaction modelled by a logic function is not known (e.g., whether an activator will trump an inhibitor, if both are active, or vice versa), then the logic model can be used to test hypothesized mechanisms for the uncertain interaction. The use of “incomplete truth tables”, a computational approach for analysing the effect of logical uncertainty in a logic network, has also been proposed for these cases.72
Despite their advantages, resistance to the use of logic-based models in biology exists. Some resistance is related to the idea that a molecule’s state can be reduced to discrete ON and OFF values. In actuality, experimental molecular states are often qualitatively described in binary terms. Genes may be characterized as up-regulated or down-regulated in microarray experiments and proteins are often referred to as activated or inactivated to indicate their functional state. Given the stochastic variation in gene and protein expression across cells, biological molecular networks are remarkably robust.73 The presence of growth factors in the local environment, for example, will almost invariably result in the induction of proliferative pathways within a population of cells, despite the heterogeneity in the molecular expression across individual cells in the population. This deterministic output from a given cellular input has been compared to cellular digital computation.74 Fundamentally, the basis of digital readouts are 0’s and 1’s–at least at the computational level. Another point of concern with logic-based Boolean models is that time is unrelated to physiological time and can provide only a qualitative chronology of molecular activations.3 While this is true, Boolean models can provide qualitative predictive values, which allow biomedical scientists to gain unique insights into molecular network dynamics that may otherwise be out of reach.
For those interested in using logic models to study large networks, the use of asynchronous updating is generally recommended.2,45 A variety of algorithms exist for introducing asynchronous updates in a logic model.45 For most purposes, the repeated random order asynchronous45 update method (which is similar to a statistical Monte Carlo simulation) will be sufficient. This is the algorithm used for the asynchronous simulations in this review. Some attractors found with the simpler synchronous updating scheme may be artifacts of uniform timescales. In contrast, an asynchronous scheme introduces stochastic variation in timescales. Moreover, asynchronous methods can produce qualitative readouts that are more representative of biological readouts (Fig. 5D and Fig. 7B, C) and can easily facilitate in silico perturbations, such as knock downs and constitutive activations.
We view logic models as complementary to other network analysis methods in systems biology and consider them to be an important tool for making biological inferences about the dynamics of intracellular networks. A number of software tools for logic-based network analysis are available.12,43,72,75 The appropriate software tool to use will depend on the nature of the network model and objectives of the analysis.7 For asynchronous simulations and in silico molecular perturbation studies, we recommend Boolean Net, 12 which is a relatively easy to use open source tool developed in Python. Needless to say, the implementation of logic-based models requires computational and mathematical proficiency. As a consequence, collaboration between integrative biologists and computational scientists will play a pivotal role in the successful development, analysis, and interpretation of logic-based models.
Importantly, logic-based models are also a powerful approach for constructing models of biological networks that can ultimately be integrated into multiscale models–models that consider the integration between different scales and phenomena in a biological system or process–to provide an integrative view of biological systems.76 In the literature, multiscale models of cancer growth have been developed that account for the cellular, genetic, and environmental factors regulating tumour growth.54,77 These models have implemented genetic and signalling networks as Boolean models to regulate cell cycle progression where the response to signals from the intracellular gene network determines whether a cell will proliferate or die and, therefore, directly influences the cellular and the extracellular tissue level of the model.
In conclusion, it is never feasible to create a model that is an exact replica of a complex system and, as a consequence, compromises must be made between the predictive power of a model and the complexity of a model. The discrete nature of a Boolean model sacrifices quantitative dynamics for qualitative dynamics. In exchange, a parameter-free modelling framework can be used to investigate complex intracellular networks.
Supplementary Material
Table 1.
Justifications for logic functions used in Fig. 5, Model I. A number of literature citations are provided in the table55,78–85
| Regulatory logic rule | Justification |
|---|---|
| MYCt+1 = Growth Factor AND NOT Overpopulation AND NOT Hypoxia | Both overpopulation and hypoxia inhibit MYC expression. Hypoxia initiates the TGF-β pathway and promotes phosphorylation of SMAD’s MH2 domain, which inhibits MYC expression (Yagi, Furuhashi et al., 2002; Feng, Liang et al., 2002). Overpopulation activates the Wnt signaling pathway, releasing the APC destruction complex and promoting degradation of β-catenin, a transcriptional activator of MYC (Su, Fu et al. 2008). In contrast, growth factors activate MYC transcription via RAS signaling (Lewis, Shapiro, 1998). Our logic rule assumes that MYC activation (via growth factor induced signaling) is only possible when neither inhibitor (Overpopulation and Hypoxia) is active. |
| P27t+1 = Hypoxia OR NOTMYC t | Hypoxia activates P27, a potent cell cycle inhibitor, by initiating the TGF-β pathway and promoting phosphorylation of SMAD’s MH2 domain, which plays a role in maintaining P27 expression (Yagi, Furuhashi et al.2002; Feng, Liang et al. 2002). MYC, when active, inhibits transcription of P27 by binding to its promoter (Yang, Sheng, 2001). We assume there are endogenous levels of active P27 present in the absence of hypoxia. The OR NOT logic rule ensures that when present, Hypoxia’s activation of P27 dominates MYC’s inhibition. |
| Cyc-Cdkt+1 = NOT P27tAND NOT P53t | Cyclin dependent kinases (Cyc-Cdk) are inhibited by both P27 and P21. P21 is activated by P53 in response to DNA damage signals. For simplicity we have excluded P21 from this regulation (Baldin, V., J. Lukas, et al., 1993; Coqueret, 2003). |
| P53t+1 = DNA Damage | P53 is transcriptionally activated in the presence of DNA Damage (Lahav et al. 2004). |
| Proliferationt+1 = Cyc-Cdkt | Cyclin dependent kinases (Cyc-Cdk) are required for cell progression in the G1 phase. They act to inhibit the Rb protein by phosphorylation, which is an inhibitor of cell cycle progression and proliferation. (Baldin, V., J. Lukas, et al., 1993; Coqueret, 2003). |
| Apoptosist+1 = P53t | P53 transcriptionally activates BAX, which is an upstream activator of apoptosis (Basu and Halder, 1998). |
Insight, innovation, integration.
The adaptations that living systems continually make are carried out through the transmission of information across large gene regulatory and protein signal transduction networks. These intracellular networks exhibit an enormous range of adaptive behaviours, termed emergent properties. The study of networks from first principles is challenging because quantitative data needed to estimate model parameters are seldom available. Here we show how parameter-free logic-based models are intuitive, predictive, and robust tools for qualitatively describing the integration of complex biochemical interactions without prior knowledge of mechanistic details. Logic models can provide robust predictions of emergent behaviours in networks and can be used to help biomedical scientists unravel fundamental properties of molecular networks.
Acknowledgments
This work was partially supported by the University of Michigan Center for Computational Medicine & Bioinformatics Pilot Grant 2010. MLW acknowledges support from the Rackham Merit Fellowship, NIH T32 CA140044, and the Breast Cancer Research Foundation. SDM acknowledges support from the Burroughs Wellcome Fund, Breast Cancer Research Foundation, and NIH R01 CA77612. NC and SS were partially supported by the NIH R25 DK088752. SS also acknowledges support from NIH R01 DK053456. The authors thank Tanya Salyers for helpful discussions and an algorithm for the enumeration of synchronous attractors.
Footnotes
Electronic supplementary information (ESI) available. See DOI: 10.1039/c2ib20193c
Contributor Information
Michelle L. Wynn, Email: mlwynn@umich.edu.
Nikita Consul, Email: nikitac@mit.edu.
Sofia D. Merajver, Email: smerajve@umich.edu.
Santiago Schnell, Email: schnells@umich.edu.
References
- 1.McCormick F. Curr Opin Genet Dev. 2011;21:29–33. doi: 10.1016/j.gde.2010.12.002. [DOI] [PubMed] [Google Scholar]
- 2.Thomas R. J Theor Biol. 1973;42:563–585. doi: 10.1016/0022-5193(73)90247-6. [DOI] [PubMed] [Google Scholar]
- 3.Thomas R, D’Ari R. Biological feedback. CRC Press; Boca Raton, Fla: 1990. [Google Scholar]
- 4.Vidal M, Cusick ME, Barabási AL. Cell. 2011;144:986–998. doi: 10.1016/j.cell.2011.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Albert R, Barabási AL. Rev Mod Phys. 2002;74:47–97. [Google Scholar]
- 6.Reguly T, Breitkreutz A, Boucher L, Breitkreutz BJ, Hon GC, Myers CL, Parsons A, Friesen H, Oughtred R, Tong A, Stark C, Ho Y, Botstein D, Andrews B, Boone C, Troyanskya OG, Ideker T, Dolinski K, Batada NN, Tyers M. J Biol. 2006;5:11. doi: 10.1186/jbiol36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Morris MK, Saez-Rodriguez J, Sorger PK, Lauffenburger DA. Biochemistry. 2010;49:3216–3224. doi: 10.1021/bi902202q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Beard DA. Wiley Interdiscip Rev: Syst Biol Med. 2011;3:136–146. doi: 10.1002/wsbm.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Crampin EJ, Schnell S, McSharry PE. Prog Biophys Mol Biol. 2004;86:77–112. doi: 10.1016/j.pbiomolbio.2004.04.002. [DOI] [PubMed] [Google Scholar]
- 10.Kauffman SA. J Theor Biol. 1969;22:437–467. doi: 10.1016/0022-5193(69)90015-0. [DOI] [PubMed] [Google Scholar]
- 11.Kauffman SA. The Origins of Order: Self-organization and Selection in Evolution. Oxford University Press; New York: 1993. [Google Scholar]
- 12.Albert I, Thakar J, Li S, Zhang R, Albert R. Source Code Biol Med. 2008;3:16. doi: 10.1186/1751-0473-3-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Glass L, Kauffman SA. J Theor Biol. 1973;39:103–129. doi: 10.1016/0022-5193(73)90208-7. [DOI] [PubMed] [Google Scholar]
- 14.Thomas R. Syst Biol. 2006;153:140–153. doi: 10.1049/ip-syb:20050101. [DOI] [PubMed] [Google Scholar]
- 15.Fauré A, Naldi A, Chaouiya C, Thieffry D. Bioinformatics. 2006;22:e124–e131. doi: 10.1093/bioinformatics/btl210. [DOI] [PubMed] [Google Scholar]
- 16.Aldridge BB, Saez-Rodriguez J, Muhlich JL, Sorger PK, Lauffenburger DA. PLoS Comput Biol. 2009;5:e1000340. doi: 10.1371/journal.pcbi.1000340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang R, Shah MV, Yang J, Nyland SB, Liu X, Yun JK, Albert R, Loughran TP., Jr Proc Natl Acad Sci U S A. 2008;105:16308–16313. doi: 10.1073/pnas.0806447105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Albert R, Othmer HG. J Theor Biol. 2003;223:1–18. doi: 10.1016/s0022-5193(03)00035-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.von Dassow G, Meir E, Munro EM, Odell GM. Nature. 2000;406:188–192. doi: 10.1038/35018085. [DOI] [PubMed] [Google Scholar]
- 20.Novák B, Tyson JJ. J Theor Biol. 2004;230:563–579. doi: 10.1016/j.jtbi.2004.04.039. [DOI] [PubMed] [Google Scholar]
- 21.Akman OE, Watterson S, Parton A, Binns N, Millar AJ, Ghazal P. J R Soc Interface. 2012;9:2365–2382. doi: 10.1098/rsif.2012.0080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pandey S, Wang RS, Wilson L, Li S, Zhao Z, Gookin TE, Assmann SM, Albert R. Mol Syst Biol. 2010;6:372. doi: 10.1038/msb.2010.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Achcar F, Camadro JM, Mestivier D. BMC Syst Biol. 2011;5:51. doi: 10.1186/1752-0509-5-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schlatter R, Schmich K, Avalos Vizcarra I, Scheurich P, Sauter T, Borner C, Ederer M, Merfort I, Sawodny O. PLoS Comput Biol. 2009;5:e1000595. doi: 10.1371/journal.pcbi.1000595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Davidson EH, Rast JP, Oliveri P, Ransick A, Calestani C, Yuh CH, Minokawa T, Amore G, Hinman V, Arenas-Mena C, Otim O, Brown CT, Livi CB, Lee PY, Revilla R, Schilstra MJ, Clarke PJC, Rust AG, Pan Z, Arnone MI, Rowen L, Cameron RA, McClay DR, Hood L, Bolouri H. Dev Biol. 2002;246:162–190. doi: 10.1006/dbio.2002.0635. [DOI] [PubMed] [Google Scholar]
- 26.Li S, Assmann SM, Albert R. PLoS Biol. 2006;4:e312. doi: 10.1371/journal.pbio.0040312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cornish-Bowden A. Fundamentals of Enzyme Kinetics. 3. Portland Press; London: 2004. [Google Scholar]
- 28.de Silva AP, Uchiyama S. Nat Nanotechnol. 2007;2:399–410. doi: 10.1038/nnano.2007.188. [DOI] [PubMed] [Google Scholar]
- 29.de Silva AP, Gunaratne HQ, Gunnlaugsson T, Huxley AJM, McCoy CP, Rademacher JT, Rice TE. Chem Rev. 1997;97:1515–1566. doi: 10.1021/cr960386p. [DOI] [PubMed] [Google Scholar]
- 30.de Silva PA, Gunaratne NHQ, McCoy CP. Nature. 1993;364:42–44. [Google Scholar]
- 31.Baron R, Lioubashevski O, Katz E, Niazov T, Willner I. Org Biomol Chem. 2006;4:989–991. doi: 10.1039/b518205k. [DOI] [PubMed] [Google Scholar]
- 32.Baron R, Lioubashevski O, Katz E, Niazov T, Willner I. Angew Chem, Int Ed. 2006;45:1572–1576. doi: 10.1002/anie.200503314. [DOI] [PubMed] [Google Scholar]
- 33.Davidson EH, Rast JP, Oliveri P, Ransick A, Calestani C, Yuh CH, Minokawa T, Amore G, Hinman V, Arenas-Mena C, Otim O, Brown CT, Livi CB, Lee PY, Revilla R, Rust AG, Pan Z, Schilstra MJ, Clarke PJC, Arnone MI, Rowen L, Cameron RA, McClay DR, Hood L, Bolouri H. Science. 2002;295:1669–1678. doi: 10.1126/science.1069883. [DOI] [PubMed] [Google Scholar]
- 34.Li F, Long T, Lu Y, Ouyang Q, Tang C. Proc Natl Acad Sci U S A. 2004;101:4781–4786. doi: 10.1073/pnas.0305937101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Oliveri P, Carrick DM, Davidson EH. Dev Biol. 2002;246:209–228. doi: 10.1006/dbio.2002.0627. [DOI] [PubMed] [Google Scholar]
- 36.Samaga R, Saez-Rodriguez J, Alexopoulos LG, Sorger PK, Klamt S. PLoS Comput Biol. 2009;5:e1000438. doi: 10.1371/journal.pcbi.1000438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Davidich MI, Bornholdt S. PLoS One. 2008;3:e1672. doi: 10.1371/journal.pone.0001672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brown CT, Rust AG, Clarke PJ, Pan Z, Schilstra MJ, De Buysscher T, Griffin G, Wold BJ, Cameron RA, Davidson EH, Bolouri H. Dev Biol. 2002;246:86–102. doi: 10.1006/dbio.2002.0619. [DOI] [PubMed] [Google Scholar]
- 39.Chaves M, Albert R, Sontag ED. J Theor Biol. 2005;235:431–449. doi: 10.1016/j.jtbi.2005.01.023. [DOI] [PubMed] [Google Scholar]
- 40.Garg A, Di Cara A, Xenarios I, Mendoza L, De Micheli G. Bioinformatics. 2008;24:1917–1925. doi: 10.1093/bioinformatics/btn336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Aris R. Appl Math Model. 1977;1:386–394. [Google Scholar]
- 42.Aho AV, Garey MR, Ullman JD. SIAM J Comput. 1973;1:131–137. [Google Scholar]
- 43.Kachalo S, Zhang R, Sontag E, Albert R, DasGupta B. Bioinformatics. 2008;24:293–295. doi: 10.1093/bioinformatics/btm571. [DOI] [PubMed] [Google Scholar]
- 44.Kohn KW, Aladjem MI. Mol Syst Biol. 2006;2:2006.0002. doi: 10.1038/msb4100044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Saadatpour A, Albert I, Albert R. J Theor Biol. 2010;266:641–656. doi: 10.1016/j.jtbi.2010.07.022. [DOI] [PubMed] [Google Scholar]
- 46.Zhao Q. IEEE Trans Neural Netw. 2005;16:1715–1716. doi: 10.1109/TNN.2005.857944. [DOI] [PubMed] [Google Scholar]
- 47.Aldana M, Cluzel P. Proc Natl Acad Sci U S A. 2003;100:8710–8714. doi: 10.1073/pnas.1536783100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kauffman S, Peterson C, Samuelsson B, Troein C. Proc Natl Acad Sci U S A. 2003;100:14796–14799. doi: 10.1073/pnas.2036429100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Willadsen K, Wiles J. J Theor Biol. 2007;249:749–765. doi: 10.1016/j.jtbi.2007.09.004. [DOI] [PubMed] [Google Scholar]
- 50.Chaves M, Sontag ED, Albert R. Syst Biol. 2006;153:154–167. doi: 10.1049/ip-syb:20050079. [DOI] [PubMed] [Google Scholar]
- 51.Wu M, Yang X, Chan C. PLoS One. 2009;4:e8040. doi: 10.1371/journal.pone.0008040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Turner TE, Schnell S, Burrage K. Comput Biol Chem. 2004;28:165–178. doi: 10.1016/j.compbiolchem.2004.05.001. [DOI] [PubMed] [Google Scholar]
- 53.Grima R, Schnell S. Essays Biochem. 2008;45:41–56. doi: 10.1042/BSE0450041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ribba B, Colin T, Schnell S. Theor Biol Med Model. 2006;3:7. doi: 10.1186/1742-4682-3-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lahav G, Rosenfeld N, Sigal A, Geva-Zatorsky N, Levine AJ, Elowitz MB, Alon U. Nat Genet. 2004;36:147–150. doi: 10.1038/ng1293. [DOI] [PubMed] [Google Scholar]
- 56.Ferrell JE, Jr, Tsai TYC, Yang Q. Cell. 2011;144:874–885. doi: 10.1016/j.cell.2011.03.006. [DOI] [PubMed] [Google Scholar]
- 57.Epstein IR, Pojman JA. An Introduction to Nonlinear Chemical Dynamics: Oscillations, Waves, Patterns, and Chaos. Oxford University Press; New York: 1998. [Google Scholar]
- 58.Murray JD. Mathematical Biology. 3. Springer; New York: 2001. [Google Scholar]
- 59.Epstein IR. Int Rev Phys Chem. 1992;11:135–160. [Google Scholar]
- 60.Roussel MR. J Phys Chem. 1996;100:8323–8330. [Google Scholar]
- 61.Lane DP. Nature. 1992;358:15–16. doi: 10.1038/358015a0. [DOI] [PubMed] [Google Scholar]
- 62.Ciliberto A, Novak B, Tyson JJ. Cell Cycle. 2005;4:488–493. doi: 10.4161/cc.4.3.1548. [DOI] [PubMed] [Google Scholar]
- 63.Michael D, Oren M. Semin Cancer Biol. 2003;13:49–58. doi: 10.1016/s1044-579x(02)00099-8. [DOI] [PubMed] [Google Scholar]
- 64.Wee KB, Surana U, Aguda BD. PLoS One. 2009;4:e4407. doi: 10.1371/journal.pone.0004407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hay N. Cancer Cell. 2005;8:179–183. doi: 10.1016/j.ccr.2005.08.008. [DOI] [PubMed] [Google Scholar]
- 66.Drazin PG. Nonlinear Systems. Cambridge University Press; Cambridge, England: 1992. [Google Scholar]
- 67.Ventura AC, Sepulchre JA, Merajver SD. PLoS Comput Biol. 2008;4:e1000041. doi: 10.1371/journal.pcbi.1000041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wynn ML, Ventura AC, Sepulchre JA, García HJ, Merajver SD. BMC Syst Biol. 2011;5:156. doi: 10.1186/1752-0509-5-156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jiang P, Ventura AC, Sontag ED, Merajver SD, Ninfa AJ, Del Vecchio D. Sci Signaling. 2011;4:ra67. doi: 10.1126/scisignal.2002152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tyson JJ, Chen KC, Novak B. Curr Opin Cell Biol. 2003;15:221–231. doi: 10.1016/s0955-0674(03)00017-6. [DOI] [PubMed] [Google Scholar]
- 71.Batchelor E, Loewer A, Mock C, Lahav G. Mol Syst Biol. 2011;7:488. doi: 10.1038/msb.2011.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Klamt S, Saez-Rodriguez J, Lindquist JA, Simeoni L, Gilles ED. BMC Bioinf. 2006;7:56. doi: 10.1186/1471-2105-7-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kitano H. Nat Rev Genet. 2004;5:826–837. doi: 10.1038/nrg1471. [DOI] [PubMed] [Google Scholar]
- 74.Bornholdt S. J R Soc, Interface. 2008;5:S85–S94. doi: 10.1098/rsif.2008.0132.focus. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gonzalez AG, Naldi A, Sánchez L, Thieffry D, Chaouiya C. Biosystems. 2006;84:91–100. doi: 10.1016/j.biosystems.2005.10.003. [DOI] [PubMed] [Google Scholar]
- 76.Wynn ML, Merajver SD, Schnell S. Adv Exp Med Biol. 2012;736:179–189. doi: 10.1007/978-1-4419-7210-1_9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Jiang Y, Pjesivac-Grbovic J, Cantrell C, Freyer JP. Biophys J. 2005;89:3884–3894. doi: 10.1529/biophysj.105.060640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Baldin V, Lukas J, Marcote MJ, Pagano M, Draetta G. Genes Dev. 1993;7:812–821. doi: 10.1101/gad.7.5.812. [DOI] [PubMed] [Google Scholar]
- 79.Basu A, Haldar S. Mol Hum Reprod. 1998;4:1099–1109. doi: 10.1093/molehr/4.12.1099. [DOI] [PubMed] [Google Scholar]
- 80.Coqueret O. Trends Cell Biol. 2003;13:65–70. doi: 10.1016/s0962-8924(02)00043-0. [DOI] [PubMed] [Google Scholar]
- 81.Feng XH, Liang YY, Liang M, Zhai W, Lin X. Mol Cell. 2002;9:133–143. doi: 10.1016/s1097-2765(01)00430-0. [DOI] [PubMed] [Google Scholar]
- 82.Lewis TS, Shapiro PS, Ahn NG. Adv Cancer Res. 1998;74:49–139. doi: 10.1016/s0065-230x(08)60765-4. [DOI] [PubMed] [Google Scholar]
- 83.Su Y, Fu C, Ishikawa S, Stella A, Kojima M, Shitoh K, Schreiber EM, Day BW, Liu B. Mol Cell. 2008;32:652–661. doi: 10.1016/j.molcel.2008.10.023. [DOI] [PubMed] [Google Scholar]
- 84.Yagi K, Furuhashi M, Aoki H, Goto D, Kuwano H, Sugamura K, Miyazono K, Kato M. J Biol Chem. 2002;277:854–861. doi: 10.1074/jbc.M104170200. [DOI] [PubMed] [Google Scholar]
- 85.Yang W, Shen J, Wu M, Arsura M, FitzGerald M, Suldan Z, Kim DW, Hofmann CS, Pianetti S, Romieu-Mourez R, Freedman LP, Sonenshein GE. Oncogene. 2001;20:1688–1702. doi: 10.1038/sj.onc.1204245. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.