

Abstract
Adiponectin is a multifunctional cytokine that has a role in regulating inflammation. Here we determined if adiponectin modulates ischemic acute kidney injury. Compared with wild-type mice, adiponectin knockout mice were found to have lower serum creatinine and less tubular damage or apoptosis following ischemia/reperfusion injury. This latter process was associated with decreased Bax and reduced activation of p53 and caspase-3. Targeted disruption of adiponectin was also found to inhibit the infiltration of neutrophils, macrophages, and T cells into the injured kidneys. This was associated with an inhibition of NF-κB activation and reduced expression of the proinflammatory molecules IL-6, TNF-α, MCP-1, and MIP-2 in the kidney after ischemia/reperfusion injury. Wild-type mice engrafted with adiponectin null bone marrow had less kidney dysfunction and tubular damage than adiponectin null mice engrafted with wild-type bone marrow. Conversely, adiponectin null mice engrafted with wild-type bone marrow had similar renal dysfunction and tubular damage compared to wild-type mice engrafted with wild-type bone marrow. In cultured macrophages, adiponectin directly promoted macrophage migration; a process blocked by the PI3 kinase inhibitor, LY294002. Thus, our results show that adiponectin plays a pivotal role in the pathogenesis of acute renal ischemia/reperfusion injury and may be a potential therapeutic target.
Keywords: Chemokine, cytokine, inflammation, apoptosis, ischemia-reperfusion injury, caspase 3, p53, Bax, NF-κB, PI3 kinase
Acute kidney injury (AKI) is a common clinical condition that is associated with high morbidity and mortality1, 2. Ischemia-reperfusion injury (IRI) is a common cause of AKI3, 4. The pathogenesis of IRI is complex and incompletely understood, which involves a number of pathogenic mechanisms that result in acute tubular necrosis/apoptosis and renal dysfunction5, 6. These include adenosine triphosphate (ATP) depletion, generation of reactive species, leukocyte infiltration, production of pro-inflammatory mediators, and induction of tubular apoptosis5–7. There is no effective therapy for this devastating clinical condition except renal replacement. Therefore, a better understanding of the pathogenic mechanisms underlying IRI is essential for ultimately developing effective therapy.
Adiponectin is a multifunctional cytokine that plays an important role in the regulation of energy metabolism and inflammation8, 9. Adiponectin was initially reported to be synthesized exclusively by adipocytes10. However, recent studies have shown that it is also produced by other cell types such as endothelial cells11, macrophages, and lymphocytes11, 12, and epithelial cells13, 14. Circulating adiponectin levels are elevated in patients with chronic kidney disease and high levels of adiponectin predict increased cardiovascular and all-cause mortality and CKD progression15, 16. However, its role in acute kidney injury is unknown.
In this study, we have found that adiponectin is upregulated in the kidney in response to ischemia-reperfusion injury. Therefore, we examined the role of adiponectin in experimental renal ischemia-reperfusion injury using adiponectin knockout (APN-KO) mice. Our results revealed that targeted disruption of adiponectin protects the kidney from ischemia-reperfusion injury by suppressing apoptosis and inflammation.
RESULTS
Adiponectin Is Induced in a Mouse Model of Kidney IRI
We first characterized the induction of adiponectin in the kidney in a mouse model of renal IRI. Using real time RT-PCR, we found that the mRNA level of adiponectin was upregulated in injured kidneys compared with sham-operated controls after 30 minutes of ischemia followed by 24 hours of reperfusion (Figure 1A). To identify the cell types that are responsible for the induction of adiponectin in the kidney, serial sections of kidneys were stained with an anti-adiponectin antibody. Our results revealed that adiponectin protein was mainly induced in the infiltrated inflammatory cells of injured kidneys of WT mice. No positive staining for adiponectin was detected in KO mice (Figure 1B). To further characterize the induction of adiponectin in the inflammatory cells, kidney sections were stained for adiponectin and either F4/80 - a macrophage marker, CD11c - a dentritic cell marker, CD3 - a T cell marker, or MPO - a neutrophil marker. Our results showed that macrophages, dentritic cells, and T cells express adiponectin, but not neutrophils (Figure S1). Western blot analysis was performed to determine which isoforms of adiponectin is induced in the kidney after IRI. Under non-reducing conditions, all isoforms of adiponectin were induced in the kidney following IRI, particularly HMW form (Figure 1C). Under reducing conditions, a major band of 30 KDa was detected in the WT mice, consistent with a full length adiponectin (Figure S2). Of note, adiponectin was not detected in the kidney of KO (Figure S2). We determined if IRI affects serum levels of adiponectin. Our results showed that the serum levels of adiponetin increased significantly after IRI, while adiponectin was not detected in the serum of knockout mice (Figure 1D), confirming the disruption of adiponectin gene.
Figure 1. Adiponectin is upregulated in the kidney after ischemia-reperfusion injury.
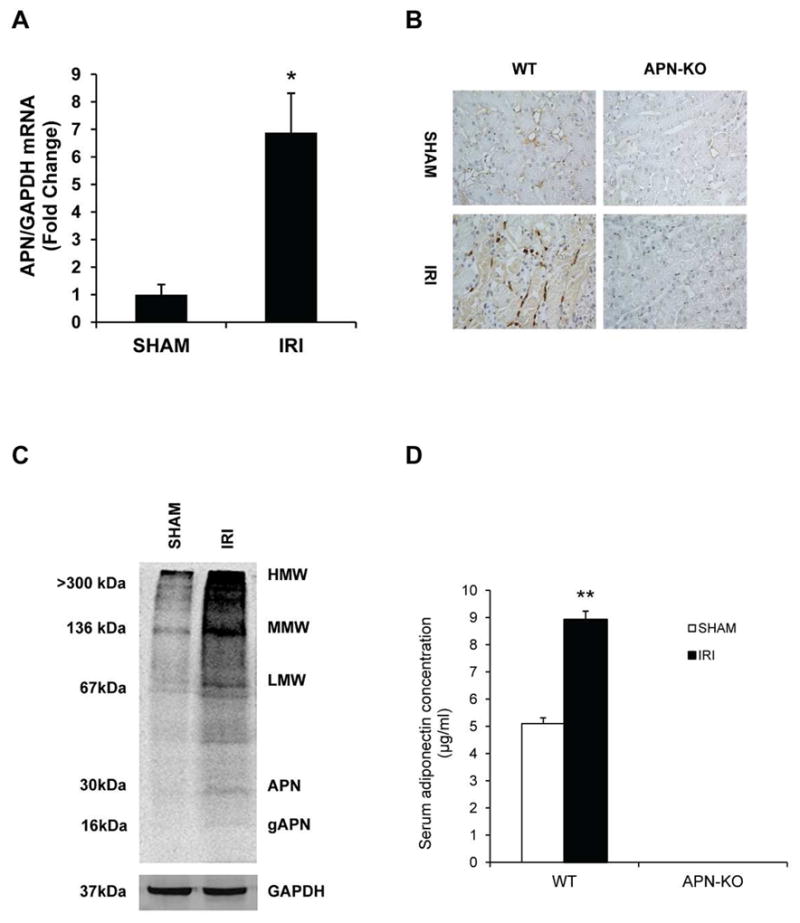
A. Adiponectin mRNA is induced significantly in kidneys of wild type mice after IRI compared with sham-operated mice. ** P < 0.01 vs WT sham. n=4 in each group. B. Representative photomicrographs of kidney sections stained for adiponectin (brown) and counter stained with hematoxylin (blue) (Original magnification: X400). C. Representative Western blot under non-reducing conditions shows that all isoforms of adiponectin protein is induced in IRI kidneys of WT mice. D. Quantification of serum adiponectin levels in sham-operated mice and mice with IRI. ** P < 0.01 vs WT sham. n=5–6 in each group.
APN-KO Mice Are Protected from Kidney IRI
To determine the role of adiponectin in the pathogenesis of kidney IRI, WT and APN-KO mice were subjected to 30 minutes of ischemia followed by 24 hours of reperfusion injury. IRI caused kidney dysfunction in WT mice as reflected by significant elevation of serum creatinine at 24 h after IRI. Kidney function was preserved in APN-KO mice with serum creatinine and BUN markedly lower than WT mice at 24 after IRI (Figure 2A and B). Consistent with the preservation of renal function in APN-KO mice following IRI, there was substantial reduction in kidney histological injury as reflected by less tubular injury, tubular dilation, and intratubular cast formation in APN-KO mice after IRI (Figure 2C and D).
Figure 2. Genetic deficiency of adiponectin protects kidney against IRI.

A. Effect of adiponectin deficiency on serum creatinine in WT and APN-KO mice after sham or IRI. ** P < 0.01 vs WT sham, ## P < 0.05 vs WT IRI; ++ P < 0.05 vs APN-KO IRI. n=5 in each group. B. Effect of adiponectin deficiency on serum urea nitrogen in WT and APN-KO mice after sham or IRI. ** P < 0.01 vs WT sham, ## P < 0.05 vs WT IRI; ++ P < 0.05 vs APN-KO IRI. n=5 in each group. C. Representative photomicrographs of HE staining for kidney sections of WT and APN-KO mice after sham or IRI. D. Quantitative histological assessment of tubular damage after IRI in WT and APN-KO mice. **P < 0.05 vs WT IRI. n=5–6 in each group.
Adiponectin Deficiency Protects Against Apoptotic Cell Death in Kidney IRI
Tubular cell apoptosis has been shown to contribute to the pathogenesis of ischemic kidney injury6, 7, 17, we therefore investigated the role of adiponectin in ischemia-reperfusion induced tubular epithelial cell apoptosis. Our results showed that there was a significant increase in the number of tubular apoptotic cells as assessed by TUNEL staining in kidneys of WT mice following IRI; whereas the number of tubular apoptotic cells was markedly reduced in IRI kidneys of APN-KO mice (Figure 3, A and B).
Figure 3. Adiponectin deficiency protects tubular epithelial cells from apoptosis in IRI kidney.

A. Representative photomicrographs of kidney sections stained for apoptotic cells (brown) and counterstained with methyl green (green) in kidneys of WT and APN-KO mice after sham or IRI. (Original magnification: X400). B. Quantitative analysis of apoptotic cells in kidneys of WT and APN-KO mice after sham or IRI. **P < 0.01 vs WT sham; ## P < 0.01 vs WT IRI; + P < 0.05 vs APN-KO IRI. n=5–6 in each group.
Caspase 3 is the final effector caspase that mediates apoptotic cell death18. As such, we examined the effect of adiponectin deficiency on caspase 3 protein expression in the kidney. Immunohistochemical staining using an antibody against active caspase 3 revealed that active caspase 3 was induced in kidney tubular epithelial cells of WT mice following IRI. However, disruption of adiponectin abolished the induction of active caspase 3 in kidney tubular epithelial cells following IRI (Figure 4, A and B). Consistent with these findings, Western blotting analysis using antibody against active caspase 3 demonstrated that the level of active caspase 3 was significantly higher in kidneys of WT mice after IRI compared with sham-operated mice. In contrast, the induction of active caspase 3 was significantly inhibited in kidneys of APN-KO mice after IRI (Figure 4, C and D). These data indicate that adiponectin deficiency inhibits caspase 3 activation in the kidney following IRI.
Figure 4. Adiponectin deficiency inhibits caspase 3 activation in tubular epithelial cells.

A. Representative photomicrographs of kidney sections stained for cleaved caspase 3 (brown) and counterstained with hematoxylin (blue) in WT and APN-KO mice after sham or IRI. (Original magnification: X400). B. Quantitative analysis of cleaved caspase 3 expression in kidneys of WT and APN-KO mice after sham or IRI. **P < 0.01 vs WT controls; ## P < 0.01 vs WT IRI, ++ P < 0.01 vs APN-KO IRI. n=5–6 in each group. C. Representative Western blots show cleaved caspase 3 protein expression in kidneys of WT and APN-KO mice after sham or IRI. D. Quantitative analysis of cleaved caspase 3 protein expression in kidneys of WT and APN-KO mice after sham or IRI. **P < 0.01 vs WT sham; ## P < 0.01 vs WT IRI, ++ P < 0.01 vs APN-KO IRI. n=5–6 in each group.
Early intracellular events occurring during the apoptotic process comprise mitochondrial changes mediated by members of the Bcl-2 family. Bax is the first identified pro-apoptotic member of Bcl-2 family19. Bax activation results in cytochrome c release and subsequent caspase activation that induces apoptosis20. Therefore, we evaluated if adiponectin deficiency affected Bax protein expression in the kidney. Our results revealed that the expression of Bax was significantly higher in kidney tubular epithelial cells of WT mice after IRI compared with sham-operated mice, but the increase in Bax protein expression was significantly attenuated in kidneys of APN-KO mice after IRI (Figure 5, A and B). These data indicate that adiponectin signaling promotes Bax protein expression in the kidney in response to IRI.
Figure 5. Adiponectin deficiency reduces Bax protein expression and p53 activation in tubular epithelial cells.

A. Representative photomicrographs of kidney sections stained for Bax (brown) and counterstained with hematoxylin (blue) in WT and APN-KO mice after sham or IRI. (Original magnification: X400). B. Quantitative analysis of Bax protein expression density in the kidney of WT and APN-KO mice after sham or IRI. **P < 0.01 vs WT controls; ## P < 0.01 vs WT IRI; ++ P < 0.01 vs APN-KO IRI. n=5–6 in each group. C. Representative photomicrographs of kidney sections stained for phosphor-p53 (brown) and counterstained with hematoxylin (blue) in WT and APN-KO mice after sham or IRI. (Original magnification: X400). D. Quantitative analysis of phosphor-p53 in the kidney of WT and APN-KO mice after sham or IRI. **P < 0.01 vs WT sham; ## P < 0.01 vs WT IRI, ++ P < 0.01 vs APN-KO IRI. n=5–6 in each group.
To explore the mechanisms responsible for Bax activation and upregulation during IRI, we examined whether adiponectin deficiency affects the activation of p53, a transcription factor that has been shown to regulate Bax expression21 and tubular epithelial apoptosis during IRI22. Tissue sections were stained with an antibody against phospho-p53. Our result showed that IRI increased phospho-p53 staining in the nuclei of tubular epithelial cells in kidneys of WT mice. The phospho-p53 staining was significantly reduced in kidneys of APN-KO mice after IRI (Figure 5, C and D). These data suggest that adiponectin signaling activates the p53, resulting in upregulation of Bax and subsequent activation of caspase 3, to promote apoptosis.
Adiponectin Deficiency Impairs Inflammatory Cell Infiltration
Inflammatory cells have been shown to play a critical role in the pathogenesis of ischemia-reperfusion injury23, 24. To examine if adiponectin plays a role in the regulation of inflammatory cell infiltration in kidney IRI, WT and APN-KO mice were subjected to IRI for 24 hours. Our results showed that the number of neutrophils was markedly increased in injured kidneys of WT mice. The number of neutrophils was significantly reduced in IRI kidneys of APN-KO mice (Figure 6, A and B). Similarly, significant infiltration of macrophages and T cells was observed in kidneys of WT mice after IRI compared with sham-operated controls. In comparison, adiponectin deficiency significantly inhibited macrophage and T cell infiltration into kidneys after IRI (Figure 6, C - F). These results indicate that adiponectin promotes inflammatory cell infiltration into the kidney during IRI.
Figure 6. Targeted disruption of adiponectin inhibits infiltration of neutrophils, macrophages, and T cells in the kidney after IRI.

A. Representative photomicrographs of kidney sections stained for MPO (a neutrophils marker) (brown) and counterstained with hematoxylin (blue) in WT and APN-KO mice after sham or IRI. B. Quantitative analysis of MPO+ neutrophils in kidneys of WT and APN-KO mice after sham or IRI. **P < 0.01 vs WT sham; ## P < 0.01 vs WT IRI; ++ P < 0.01 vs APN-KO IRI. n=5–6 in each group. C. Representative photomicrographs of kidney sections stained for F4/80 (a macrophage marker) (brown) and counterstained with hematoxylin (blue) in WT and APN-KO mice after sham or IRI. D. Quantitative analysis of F4/80+ macrophages in kidneys of WT and APN-KO mice after sham or IRI. **P < 0.01 vs WT sham; ## P < 0.01 vs WT IRI; + P < 0.05 vs APN-KO IRI. n=5–6 in each group. E. Representative photomicrographs of kidney sections stained for CD3 (a T lymphocyte marker) (brown) and counterstained with hematoxylin (blue) in WT and APN-KO mice after sham or IRI. F. Quantitative analysis of CD3+ T cells in kidneys of WT and APN-KO mice after sham or IRI. **P < 0.01 vs WT sham; ## P < 0.01 vs WT IRI; ++ P < 0.01 vs APN-KO IRI. n=5–6 in each group.
Adiponectin Mediates the Upregulation of Inflammatory Mediators in the Kidney during IRI
Inflammatory chemokines and cytokines mediate the uptake of inflammatory cell into injured tissue25. We examined the effect of adiponectin deficiency on the expression of known pro-inflammatory molecules that are involved in the pathogenesis of kidney IRI. The mRNA levels of IL-6, TNF-α, MCP-1, and MIP-2 in the kidney were increased significantly in kidneys of WT mice after IRI compared with sham-operated controls. In contrast, the upregulation of IL-6, TNF-α, MCP-1, and MIP-2 was greatly attenuated in IRI kidneys of APN-KO mice (Figure 7). These results suggest that adiponectin is a major regulator of inflammatory cytokine and chemokine expression in the kidney during IRI.
Figure 7. Targeted disruption of adiponectin suppresses gene expression of TNF-α, IL-6 and MCP-1 in the kidney after IRI.

A. Bar graph shows quantitative analysis of TNF-α mRNA expression in kidneys of WT and APN-KO mice after sham or IRI. **P < 0.01 vs WT sham; ## P < 0.01 vs WT IRI; + P < 0.05 vs APN-KO IRI. n=4 in each group. B. Bar graph shows quantitative analysis of IL-6 mRNA expression in kidneys of WT and APN-KO mice after sham or IRI. **P < 0.01 vs WT sham; ## P < 0.01 vs WT IRI; + P < 0.05 vs APN-KO IRI. n=4 in each group. C. Bar graph shows quantitative analysis of MCP-1 mRNA expression in kidneys of WT and APN-KO mice after sham or IRI. **P < 0.01 vs WT sham; ## P < 0.01 vs WT IRI; + P < 0.05 vs APN-KO IRI. n=4 in each group. D. Bar graph shows quantitative analysis of MIP-2 mRNA expression in kidneys of WT and APN-KO mice after sham or IRI. **P < 0.01 vs WT sham; ## P < 0.01 vs WT IRI; + P < 0.05 vs APN-KO IRI. n=4 in each group.
Adiponectin Deficiency Inhibits NF-κB activation during IRI
To explore the molecular mechanisms underlying adiponectin-mediated induction of inflammatory molecules, we examined whether targeted disruption of adiponectin affects the NF-κB signaling pathway, a master regulator of inflammation. Tissue sections were stained with an antibody against p65. Our results showed that p65 staining was significantly increased in kidney inflammatory cells of WT mice after IRI. The p65 staining was markedly reduced in APN-KO mice (Figure 8). These data indicate that adiponectin induces inflammatory molecules through activation of the NF-κB signaling pathway.
Figure 8. Targeted disruption of adiponectin inhibits NF-κB activation in the kidney during IRI.

A. Representative photomicrographs of kidney sections stained for p65 (brown) and counterstained with hematoxylin (blue) in WT and APN-KO mice after sham or IRI. (Original magnification: X400). B. Quantitative analysis of p65 in the kidney of WT and APN-KO mice after sham or IRI. **P < 0.01 vs WT sham; ## P < 0.01 vs WT IRI, ++ P < 0.01 vs APN-KO IRI. n=5–6 in each group.
Adiponectin Deficiency in Bone Marrow-derived Cells Reduces Kidney Injury
To determine the role of adiponectin in bone marrow-derived cells in the pathogenesis of kidney IRI, we generated bone marrow chimeric mice. Eight weeks after bone marrow transplantation, chimeric mice were subjected to kidney IRI. The genotype of bone marrow-derived cells from the chimeric mice was confirmed by PCR of DNA extracted from peripheral blood cells (Figure S3). WT mice engrafted with adiponectin−/− bone marrow cells showed significantly lower serum urea nitrogen and less tubular damage than WT mice engrafted with WT bone marrow at 24 h after IRI (Figure 9). In contrast, APN-KO mice engrafted with WT bone marrow recaptured the phenotypes of WT mice engrafted with WT bone marrow after IRI (Figure 9). These results indicate that bone marrow cell-generated adiponectin contributes to kidney IRI.
Figure 9. Bone marrow cell-produced adiponectin mediates kidney injury.
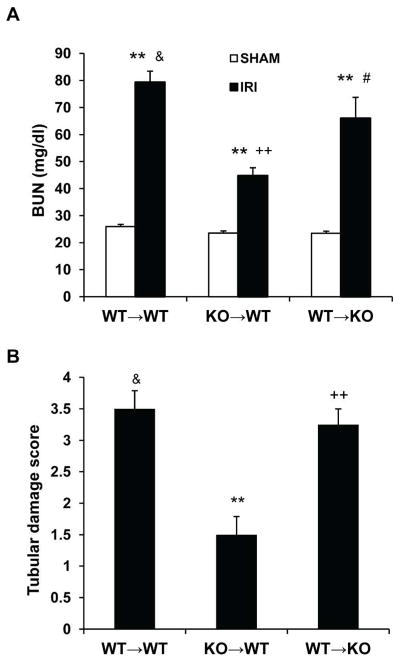
A. Effect of adiponectin deficiency in bone marrow cells on serum urea nitrogen. ** P < 0.01 vs WT→WT sham; ++ P < 0.01 vs WT→WT IRI; # P < 0.05 vs KO→WT; & P > 0.05 vs WT→KO IRI. n=3–4 in each group. B. Quantitative histological assessment of tubular damage. **P < 0.05 vs WT→WT IRI; ++ P < 0.01 vs KO→WT IRI; & P > 0.05 vs WT→KO IRI. n=3–4 in each group.
Adiponectin Promotes Macrophage Migration in vitro
To determine if adiponectin plays a direct role in the regulation of inflammatory cell migration, we examined migration activity of macrophages in presence of adiponectin. We found that adiponectin is capable of inducing RAW 264.7 macrophage migration (Figure 10). To elucidate the mechanisms of adiponectin in the migration of macrophages, we demonstrated that PI3 kinase inhibitor LY-294002 inhibited the migration of macrophages induced by adiponectin (Figure 10), while inhibition of AMP activated protein kinase with compound C did not affect adiponectin-induced cell migration (Data not shown). These data imply that adiponectin promotes migratory activities of macrophages through activation of PI3 kinase.
Figure 10.
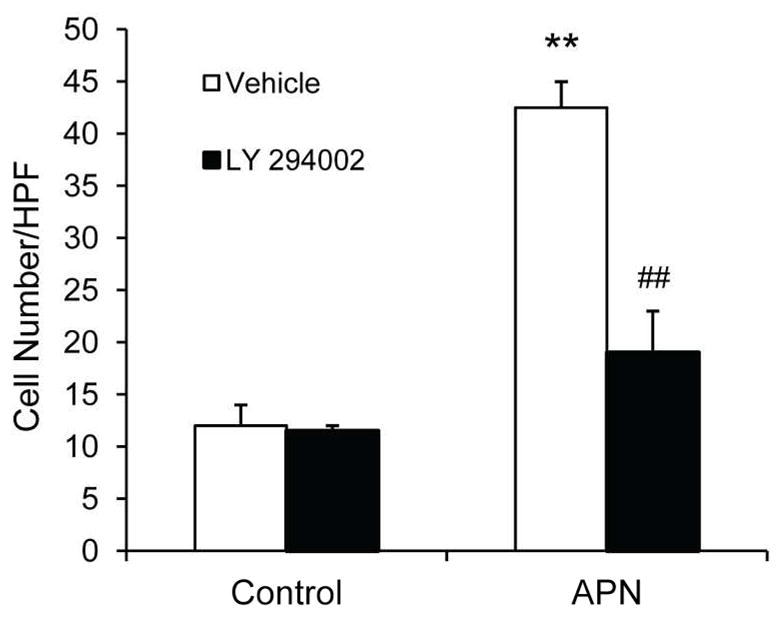
LY 294002 blocks adiponectin-induced migration of Raw 264.7 macrophages. **P < 0.01 vs vehicle controls; ## P < 0.01 vs APN-treated group. n=3 in each group.
DISCUSSION
Adiponectin is a multifunctional cytokine that plays an important role in the regulation of inflammation and energy metabolism. However, its role in kidney injury is unknown. In this study, we have demonstrated that: (1) Adiponectin is induced in the kidney in response to IRI; (2) Targeted disruption of adiponectin protects against kidney IRI and tubular cell apoptosis; (3) Targeted disruption of adiponectin inhibits activation of p53 and caspase 3 and upregulation of Bax; (4) Targeted disruption of adiponectin reduces inflammatory cell infiltration and proinflammatory molecule production in the kidney; (5) Targeted disruption of adiponectin suppresses NF-κB activation. (6) APN promotes macrophage migration through activation of PI3 kinase. These results indicate that adiponectin plays a pivotal role in the pathogenesis of IRI through regulation of inflammation and apoptosis.
Adiponectin was initially reported to be synthesized exclusively by adipocytes10. However, recent studies have shown that it is also produced by other cell types such as endothelial cells11, inflammatory cells, lymphocytes11, 12, and epithelial cells13, 14. Adiponectin shares strong homologies with the complement factor C1q and the pro-inflammatory cytokine TNF-α. Therefore, it belongs to the C1q-TNF-superfamily26. Adiponectin exists as a full-length protein, as well as a proteolytic cleavage fragment, consisting of the globular C-terminal domain termed globular adiponectin. It is thought that a leukocyte elastase, secreted by activated monocytes and/or neutrophils, mediates this cleavage process and generates the globular fragment of adiponectin. Globular adiponectin can trimerize after cleavage, but cannot oligomerize further27. Full-length adiponectin can exist as: a trimer (known as low-molecular-weight adiponectin); a hexamer, which consists of two trimers linked by a disulphide bond (known as middle-molecular weight adiponectin); and a high-molecular-weight 12- to 18-mer28, 29. In this study, we demonstrate that adiponectin is increased in the kidney and circulation following IRI. Subsequently, we investigated the role of adiponectin in kidney IRI using APN-KO mice. Our study shows that adiponectin deficiency preserves renal function and reduces tubular damage in the kidney after IRI. The results from bone marrow chimeric experiments provided further evidence that adiponectin produced by bone marrow-derived cells contributes to IRI of the kidney. These data indicate that adiponectin represents a maladaptive response that promotes IRI of the kidney.
Following ischemia-reperfusion, tubular epithelial cells are unable to maintain adequate intracellular ATP for essential processes, which lead to cell injury and cell death by necrosis, apoptosis, or autophagy-associated cell death6, 30. The relative contribution of each form of cell death to injury is variable and depends on the severity of the insults. In response to apoptotic signals, various enzymes are activated in a pathway-specific manner and the classical caspase activation chain reaction is set in motion31. Mammals have mainly two distinct apoptosis signaling pathways, the death receptor pathway and the mitochondrial pathway. In the mitochondrial pathway, the B-cell lymphoma-2 (Bcl-2)-family of proteins play a crucial role. The Bcl-2 family comprises three subfamilies, namely, an anti-apoptotic family, pro-apoptotic family, and pro-apoptotic BH3-only protein family. Bax was the first identified pro-apoptotic member of the Bcl-2 protein family19. Bax is strictly regulated through both transcription and post-transcription mechanisms32. The expression of Bax is upregulated by the tumor suppressor protein p53. Bax upregulation triggers cytochrome c release from mitochondria, which consequentially induces a chain reaction leading to caspase 3 activation and apoptosis. Inhibition of apoptosis may have therapeutic potential for ischemia-reperfusion injury. In the present study, we show that targeted disruption of adiponectin prevents p53 activation, Bax induction, caspase 3 activation, and apoptosis. These data indicate that adiponectin induces apoptosis through p53 mediated Bax induction that results in caspase 3 activation.
Since the discovery of adiponectin, much has been said and even more has been hypothesized regarding its role in health and disease. However, its biological activities remain incompletely understood. Adiponectin is a cytokine that has been reported to possess both pro- and anti-inflammatory properties. The role of adiponectin in regulating inflammatory responses appears to be tissue and context specific33–37. It was initially thought that adiponectin has anti-inflammatory property38. However, accumulating evidence indicates that adiponectin exerts pro-inflammatory effect. Experimental studies have shown that adiponectin by itself activates NF-κB and promote inflammatory cytokine production39, 40. Clinical studies have demonstrated that adiponectin levels are elevated in patients with chronic kidney disease and elevated serum adiponectin levels predict mortality and CKD progression15, 16. In an attempt to uncover mechanisms underlying the protection from ischemic kidney injury in APN-KO mice, we evaluated the expression of several cytokines and chemokines that are known to be involved in the pathogenesis of kidney IRI. We demonstrate for the first time that targeted disruption of adiponectin inhibits the gene expression of pro-inflammatory cytokines – IL-6 and TNF-α and chemokines – MCP-1 and MIP-2, which are associated with a reduction in infiltration of neutrophils, macrophages, and T cells into the kidney after ischemia-reperfusion. Furthermore, we demonstrate that targeted disruption of adiponectin suppresses NF-κB activation in inflammatory cells. These data indicate that adiponectin exerts a pro-inflammatory effect in the kidney through activation of NF-κB in response to ischemia-reperfusion.
In this study, we demonstrate for the first time that adiponectin directly promotes migration of macrophages. PI3 kinase is a cellular lipid kinase that converts phosphatidylinositol-4,5-bisphosphate to phosphatidylinositol-3,4,5-trisphosphate, a second messenger involved in a variety of cell functions including cell migration41, 42. We show that adiponectin-induced migration of macrophages was blocked by LY294002, a PI3 kinase inhibitor. These results indicate that the effect of adiponectin on macrophage migration is mediated by PI3 kinase signaling pathway.
In contrast to our study, Cheng et al. reported that administration of adiponectin protects the kidney from IRI43. They show that administration of adiponectin reduces renal injury. However, recombinant human adiponectin expressed in and purified from E. coli was used in that study. It is not clear which isoform of adiponectin was purified from E coli in that study. Tsao et al. have reported that adiponectin from E coli can only forms trimers and hexamer, while adiponectin from HEK293T cells can form trimers, haxamers, and HMW complexes29. Evidence indicates that different isoforms from different preparation may have distinct functions44, 45. Consistent with this notion, studies have shown that both HMW adiponectin and globular adiponectin activate NF-κB in inflammatory cells39 and endothelial cells with varying potency46, while mutated adiponectin that is unable to form HMW complexes cannot activate NF-κB in inflammatory cells39. Therefore, the function of adiponectin is isoform and cell type dependent. In our study, we found that serum adiponectin levels are significantly elevated in response to IRI, which is consistent with published studies showing that adiponectin levels increases with impaired renal function47, 48. In contrast, Cheng et al. reported that serum adiponectin levels decreased after IRI.
In summary, our study defines a novel mechanism by which adiponectin participates in renal IRI. In response to injury, the upregulated adiponectin leads to recruitment of circulating inflammatory cells into the kidney and induction of apoptosis, which play a critical role in the pathogenesis of IRI. These data suggest that inhibition of adiponectin could represent a novel therapeutic approach for acute kidney injury.
METHODS
Animals
The animal experiments were conducted according to the guidelines of laboratory animal care and were approved by the Institutional Animal Care and Use Committee of the Baylor College of Medicine. Wild type (WT) C57BL/6 mice were purchased from the Jackson Laboratory (Bar Harbor, ME). The APN-KO mice on a background of C57BL/6 were generated through homologous recombination, which removed the translation initiation codon containing exon 2 as previously described49. The APN-KO mice were backcrossed into C57BL/6 mice for six generations. Male WT mice and APN-KO mice at 8 to 12 weeks old age, weighting about 20 to 30 grams, were anesthetized by intraperitoneal injection of ketamine (80mg/kg) and xylazine (10 mg/kg). Kidneys were exposed through flank incision and were subjected to ischemia by clamping renal pedicles with non-traumatic microaneurism clamps. After 30 minutes, the clamps were removed and blood reflow was conformed. Body temperature was maintained at 36.5–37.5°C throughout the procedure. Sham control mice underwent identical surgical procedure but without pedicle clamping. Animals were sacrificed at 24 hours after reperfusion. Kidneys were perfused and harvested.
Serum Adiponectin Measurement
Serum adiponectin was measured using a mouse adiponectin ELISA kit (R&D Systems Inc., Minneapolis, MN) according to the manufacturer’s instruction.
Measurement of Renal Function
Serum creatinine was measured using a creatinine assay kit (BioAssay Systems, Hayward, CA) according to the manufacturer’s instruction. Blood urea nitrogen was determined fluorometrically as described50.
Renal Morphology
Kidney tissue was fixed in 10% buffered formalin, embedded in paraffin, and cut at 4-Lm thickness. After deparaffinization and rehydration, sections were stained with hematoxylin and eosin. Tissue damage was examined in a blinded manner and scored according to the percentage of damaged tubules: 0, no damage; 1, less than 25% damage; 2, 25%–50% damage; 3, 50%–75% damage; and 4, more than 75% damage as reported51.
Immunohistochemistry
Immunohistochemical staining was performed on paraffin sections. Antigen retrieval was performed with antigen unmasking solution (Vector Laboratories, Burlingame, CA). Endogenous peroxidase activity was quenched with 3% H2O2. After blocked with 5% normal serum, slides were incubated with primary antibodies in a humidified chamber overnight. After washing, slides were incubated with appropriate secondary antibodies and ABC solution sequentially according to the ABC kit (Vector Laboratories, Burlingame, CA). Slides were then visualized by incubation in DAB solution for an appropriate period of time. Nuclear staining was performed with hematoxylin. The slides were dehydrated, cleared, and mounted. The images from these slides were acquired and analyzed by NIS Element software with Nikon microscope image system.
Apoptosis Detection
TUNEL assay was performed to evaluate apoptosis using ApopTagR plus Peroxidase in Situ Apoptosis Detection Kit (Millipore, Billerica, MA) according to manufacturer’s instruction. The number of TUNEL-positive cells per high-power field were counted and analyzed in a blinded fashion.
Quantitative Real-Time RT-PCR
Total RNA was extracted from kidney tissues with TRIzol reagent (Invitrogen). Aliquots (1 μg) of total RNA were reverse transcribed using SuperScript II reverse transcriptase. Real-time PCR was performed using IQ SYBR green supermix reagent (Bio-Rad, Herculus, CA) with a Bio-Rad real-time PCR machine according to the manufacturer’s instructions. The comparative Ct method (ΔΔCt) was used to quantify gene expression, and the relative quantification was calculated as 2−ΔΔCt. The expression levels of the target genes were normalized to GAPDH level in each sample. The primer sequences were: adiponectin - forward, 5′-GCAGAGATGGCACTCCTGGA-3′, reverse, 5′-CCCTTCAGCTCCTGTCATTCC-3′; IL-6 - forward, 5′-AGGATACCACTCCCAACAGACCTG-3′, reverse, 5′-CTGCAAGTGCATCATCGTTGTTCA-3′; TNF-α-forward, 5′-CATGAGCACAGAAAGCATGATCCG-3′ reverse, 5′-AAGCAGGAATGAGAAGAGGCTGAG-3′; MCP-1 - forward, 5′-TCACCTGCTGCTACTCATTCACCA-3′, reverse, 5′-TACAGCTTCTTTGGGACACCTGCT-3′; MIP-2 - forward, 5′-AAAGTTTGCCTTGACCCTGAAGCC-3′, reverse, 5′-TCCAGGTCAGTTAGCCTTGCCTTT-3′; GAPDH - forward, 5′-CCAATGTGTCCGTCGCGTGGATCT-3′, reverse, 5′-GTTGAAGTCGCAGGAGACAACC-3′.
Western Blot Analysis
Protein was extracted using RIPA buffer containing cocktail proteinase inhibitors and quantified with a Bio-Rad protein assay. For non-reducing conditions, 2-mercaptoethanol and DTT were excluded from the sample buffer. An equal amount of protein was separated on SDS-polycrylamide gels in Tris/SDS buffer system, and then transferred onto nitrocellulose membranes. The membranes were incubated with primary antibodies overnight followed by incubation with appropriate fluorescence-conjugated secondary antibodies. The proteins of interest were analyzed using an Odyssey IR scanner, and signal intensities were quantified using NIH Image/J software.
Bone Marrow Transplantation
Bone marrow transplantation was performed as described previously52. Briefly, bone marrow cells (5×106) were injected into irradiated WT or APN-KO mice through tail vein. After transplantation, mice were allowed to recuperate for 8 weeks prior to induction of kidney IRI.
Migration Assay
Transwell migration assay was performed using a modification of a 24-well chemotaxis assay. RAW 264.7 cells were loaded into top chambers (8 LM pore filter, Corning Costar). Adiponectin and LY 294002 were then added into lower chamber at a final concentration of 5 Lg/ml and 10 LM respectively. After 4 hours of incubation, nonmigrated cells in the upper chamber were removed with a cotton swab, and migrated cells were fixed with 4% paraformaldehyde and stained with 0.1% crystal violet and 2% ethanol for 5 minutes at RT. The number of migrated cells per membrane was counted in 10 random high-power fields with an inverted microscope.
Statistical Analysis
All data were expressed as mean ± SEM. Multiple group comparisons were performed by One-way ANOVA followed by the Bonferroni procedure for comparison of means. Comparisons between two groups were analyzed by the two-tailed t test. P<0.05 was considered statistically significant.
Supplementary Material
Acknowledgments
We thank Dr. William E. Mitch for helpful discussion. This work was supported in part by a National Institutes of Health grant - HL92958, an American Heart Association grant -11BGIA7840054, and a grant from Dr. and Mrs. Harold Selzman (to YW), National Institutes of Health grants - HL51586 and P30DK79638 (to LC).
Footnotes
Disclosures
None
References
- 1.Chertow GM, Burdick E, Honour M, et al. Acute kidney injury, mortality, length of stay, and costs in hospitalized patients. Journal of the American Society of Nephrology : JASN. 2005;16:3365–3370. doi: 10.1681/ASN.2004090740. [DOI] [PubMed] [Google Scholar]
- 2.Palevsky PM. Epidemiology of acute renal failure: the tip of the iceberg. Clin J Am Soc Nephrol. 2006;1:6–7. doi: 10.2215/CJN.01521005. [DOI] [PubMed] [Google Scholar]
- 3.Mehta RL, Pascual MT, Soroko S, et al. Spectrum of acute renal failure in the intensive care unit: the PICARD experience. Kidney international. 2004;66:1613–1621. doi: 10.1111/j.1523-1755.2004.00927.x. [DOI] [PubMed] [Google Scholar]
- 4.Liano F, Pascual J. Epidemiology of acute renal failure: a prospective, multicenter, community-based study. Madrid Acute Renal Failure Study Group. Kidney international. 1996;50:811–818. doi: 10.1038/ki.1996.380. [DOI] [PubMed] [Google Scholar]
- 5.Bonventre JV, Yang L. Cellular pathophysiology of ischemic acute kidney injury. The Journal of clinical investigation. 2011;121:4210–4221. doi: 10.1172/JCI45161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sharfuddin AA, Molitoris BA. Pathophysiology of ischemic acute kidney injury. Nature reviews Nephrology. 2011;7:189–200. doi: 10.1038/nrneph.2011.16. [DOI] [PubMed] [Google Scholar]
- 7.Devarajan P. Update on mechanisms of ischemic acute kidney injury. Journal of the American Society of Nephrology : JASN. 2006;17:1503–1520. doi: 10.1681/ASN.2006010017. [DOI] [PubMed] [Google Scholar]
- 8.Tilg H, Moschen AR. Adipocytokines: mediators linking adipose tissue, inflammation and immunity. Nature reviews Immunology. 2006;6:772–783. doi: 10.1038/nri1937. [DOI] [PubMed] [Google Scholar]
- 9.Karbowska J, Kochan Z. Role of adiponectin in the regulation of carbohydrate and lipid metabolism. J Physiol Pharmacol. 2006;57 (Suppl 6):103–113. [PubMed] [Google Scholar]
- 10.Scherer PE, Williams S, Fogliano M, et al. A novel serum protein similar to C1q, produced exclusively in adipocytes. J Biol Chem. 1995;270:26746–26749. doi: 10.1074/jbc.270.45.26746. [DOI] [PubMed] [Google Scholar]
- 11.Wolf AM, Wolf D, Avila MA, et al. Up-regulation of the anti-inflammatory adipokine adiponectin in acute liver failure in mice. Journal of hepatology. 2006;44:537–543. doi: 10.1016/j.jhep.2005.08.019. [DOI] [PubMed] [Google Scholar]
- 12.Crawford LJ, Peake R, Price S, et al. Adiponectin is produced by lymphocytes and is a negative regulator of granulopoiesis. Journal of leukocyte biology. 2010;88:807–811. doi: 10.1189/jlb.1109723. [DOI] [PubMed] [Google Scholar]
- 13.Katsiougiannis S, Kapsogeorgou EK, Manoussakis MN, et al. Salivary gland epithelial cells: a new source of the immunoregulatory hormone adiponectin. Arthritis Rheum. 2006;54:2295–2299. doi: 10.1002/art.21944. [DOI] [PubMed] [Google Scholar]
- 14.Miller M, Cho JY, Pham A, et al. Adiponectin and functional adiponectin receptor 1 are expressed by airway epithelial cells in chronic obstructive pulmonary disease. Journal of immunology. 2009;182:684–691. doi: 10.4049/jimmunol.182.1.684. [DOI] [PubMed] [Google Scholar]
- 15.Becker B, Kronenberg F, Kielstein JT, et al. Renal insulin resistance syndrome, adiponectin and cardiovascular events in patients with kidney disease: the mild and moderate kidney disease study. Journal of the American Society of Nephrology : JASN. 2005;16:1091–1098. doi: 10.1681/ASN.2004090742. [DOI] [PubMed] [Google Scholar]
- 16.Jorsal A, Tarnow L, Frystyk J, et al. Serum adiponectin predicts all-cause mortality and end stage renal disease in patients with type I diabetes and diabetic nephropathy. Kidney international. 2008;74:649–654. doi: 10.1038/ki.2008.201. [DOI] [PubMed] [Google Scholar]
- 17.Sanz AB, Santamaria B, Ruiz-Ortega M, et al. Mechanisms of renal apoptosis in health and disease. Journal of the American Society of Nephrology : JASN. 2008;19:1634–1642. doi: 10.1681/ASN.2007121336. [DOI] [PubMed] [Google Scholar]
- 18.Thornberry NA, Lazebnik Y. Caspases: enemies within. Science. 1998;281:1312–1316. doi: 10.1126/science.281.5381.1312. [DOI] [PubMed] [Google Scholar]
- 19.Oltvai ZN, Milliman CL, Korsmeyer SJ. Bcl-2 heterodimerizes in vivo with a conserved homolog, Bax, that accelerates programmed cell death. Cell. 1993;74:609–619. doi: 10.1016/0092-8674(93)90509-o. [DOI] [PubMed] [Google Scholar]
- 20.Wolter KG, Hsu YT, Smith CL, et al. Movement of Bax from the cytosol to mitochondria during apoptosis. The Journal of cell biology. 1997;139:1281–1292. doi: 10.1083/jcb.139.5.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Miyashita T, Krajewski S, Krajewska M, et al. Tumor suppressor p53 is a regulator of bcl-2 and bax gene expression in vitro and in vivo. Oncogene. 1994;9:1799–1805. [PubMed] [Google Scholar]
- 22.Kelly KJ, Plotkin Z, Vulgamott SL, et al. P53 mediates the apoptotic response to GTP depletion after renal ischemia-reperfusion: protective role of a p53 inhibitor. Journal of the American Society of Nephrology : JASN. 2003;14:128–138. doi: 10.1097/01.asn.0000040596.23073.01. [DOI] [PubMed] [Google Scholar]
- 23.Kinsey GR, Li L, Okusa MD. Inflammation in acute kidney injury. Nephron Exp Nephrol. 2008;109:e102–107. doi: 10.1159/000142934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jang HR, Rabb H. The innate immune response in ischemic acute kidney injury. Clin Immunol. 2009;130:41–50. doi: 10.1016/j.clim.2008.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chung AC, Lan HY. Chemokines in renal injury. Journal of the American Society of Nephrology : JASN. 2011;22:802–809. doi: 10.1681/ASN.2010050510. [DOI] [PubMed] [Google Scholar]
- 26.Shapiro L, Scherer PE. The crystal structure of a complement-1q family protein suggests an evolutionary link to tumor necrosis factor. Current biology : CB. 1998;8:335–338. doi: 10.1016/s0960-9822(98)70133-2. [DOI] [PubMed] [Google Scholar]
- 27.Waki H, Yamauchi T, Kamon J, et al. Generation of globular fragment of adiponectin by leukocyte elastase secreted by monocytic cell line THP-1. Endocrinology. 2005;146:790–796. doi: 10.1210/en.2004-1096. [DOI] [PubMed] [Google Scholar]
- 28.Waki H, Yamauchi T, Kamon J, et al. Impaired multimerization of human adiponectin mutants associated with diabetes. Molecular structure and multimer formation of adiponectin. J Biol Chem. 2003;278:40352–40363. doi: 10.1074/jbc.M300365200. [DOI] [PubMed] [Google Scholar]
- 29.Tsao TS, Murrey HE, Hug C, et al. Oligomerization state-dependent activation of NF-kappa B signaling pathway by adipocyte complement-related protein of 30 kDa (Acrp30) J Biol Chem. 2002;277:29359–29362. doi: 10.1074/jbc.C200312200. [DOI] [PubMed] [Google Scholar]
- 30.Hotchkiss RS, Strasser A, McDunn JE, et al. Cell death. The New England journal of medicine. 2009;361:1570–1583. doi: 10.1056/NEJMra0901217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hengartner MO. The biochemistry of apoptosis. Nature. 2000;407:770–776. doi: 10.1038/35037710. [DOI] [PubMed] [Google Scholar]
- 32.Youle RJ, Strasser A. The BCL-2 protein family: opposing activities that mediate cell death. Nature reviews Molecular cell biology. 2008;9:47–59. doi: 10.1038/nrm2308. [DOI] [PubMed] [Google Scholar]
- 33.Kumada M, Kihara S, Ouchi N, et al. Adiponectin specifically increased tissue inhibitor of metalloproteinase-1 through interleukin-10 expression in human macrophages. Circulation. 2004;109:2046–2049. doi: 10.1161/01.CIR.0000127953.98131.ED. [DOI] [PubMed] [Google Scholar]
- 34.Fayad R, Pini M, Sennello JA, et al. Adiponectin deficiency protects mice from chemically induced colonic inflammation. Gastroenterology. 2007;132:601–614. doi: 10.1053/j.gastro.2006.11.026. [DOI] [PubMed] [Google Scholar]
- 35.Xydakis AM, Case CC, Jones PH, et al. Adiponectin, inflammation, and the expression of the metabolic syndrome in obese individuals: the impact of rapid weight loss through caloric restriction. J Clin Endocrinol Metab. 2004;89:2697–2703. doi: 10.1210/jc.2003-031826. [DOI] [PubMed] [Google Scholar]
- 36.Pini M, Gove ME, Sennello JA, et al. Role and regulation of adipokines during zymosan-induced peritoneal inflammation in mice. Endocrinology. 2008;149:4080–4085. doi: 10.1210/en.2008-0327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pini M, Sennello JA, Chan L, et al. Adiponectin deficiency does not affect the inflammatory response to endotoxin or concanavalin a in mice. Endocrinology. 2006;147:5019–5022. doi: 10.1210/en.2006-0855. [DOI] [PubMed] [Google Scholar]
- 38.Ouchi N, Walsh K. Adiponectin as an anti-inflammatory factor. Clin Chim Acta. 2007;380:24–30. doi: 10.1016/j.cca.2007.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Haugen F, Drevon CA. Activation of nuclear factor-kappaB by high molecular weight and globular adiponectin. Endocrinology. 2007;148:5478–5486. doi: 10.1210/en.2007-0370. [DOI] [PubMed] [Google Scholar]
- 40.Awazawa M, Ueki K, Inabe K, et al. Adiponectin enhances insulin sensitivity by increasing hepatic IRS-2 expression via a macrophage-derived IL-6-dependent pathway. Cell Metab. 2011;13:401–412. doi: 10.1016/j.cmet.2011.02.010. [DOI] [PubMed] [Google Scholar]
- 41.Van Haastert PJ, Devreotes PN. Chemotaxis: signalling the way forward. Nature reviews Molecular cell biology. 2004;5:626–634. doi: 10.1038/nrm1435. [DOI] [PubMed] [Google Scholar]
- 42.Andrew N, Insall RH. Chemotaxis in shallow gradients is mediated independently of PtdIns 3-kinase by biased choices between random protrusions. Nature cell biology. 2007;9:193–200. doi: 10.1038/ncb1536. [DOI] [PubMed] [Google Scholar]
- 43.Cheng CF, Lian WS, Chen SH, et al. Protective effects of adiponectin against renal ischemia-reperfusion injury via prostacyclin-PPARalpha-heme oxygenase-1 signaling pathway. J Cell Physiol. 2012;227:239–249. doi: 10.1002/jcp.22726. [DOI] [PubMed] [Google Scholar]
- 44.Sun Y, Xun K, Wang C, et al. Adiponectin, an unlocking adipocytokine. Cardiovasc Ther. 2009;27:59–75. doi: 10.1111/j.1755-5922.2008.00069.x. [DOI] [PubMed] [Google Scholar]
- 45.Neumeier M, Weigert J, Schaffler A, et al. Different effects of adiponectin isoforms in human monocytic cells. J Leukoc Biol. 2006;79:803–808. doi: 10.1189/jlb.0905521. [DOI] [PubMed] [Google Scholar]
- 46.Tomizawa A, Hattori Y, Kasai K, et al. Adiponectin induces NF-kappaB activation that leads to suppression of cytokine-induced NF-kappaB activation in vascular endothelial cells: globular adiponectin vs. high molecular weight adiponectin. Diab Vasc Dis Res. 2008;5:123–127. doi: 10.3132/dvdr.2008.020. [DOI] [PubMed] [Google Scholar]
- 47.Stenvinkel P. Adiponectin in chronic kidney disease: a complex and context sensitive clinical situation. J Ren Nutr. 2011;21:82–86. doi: 10.1053/j.jrn.2010.10.019. [DOI] [PubMed] [Google Scholar]
- 48.Zoccali C, Mallamaci F, Tripepi G, et al. Adiponectin, metabolic risk factors, and cardiovascular events among patients with end-stage renal disease. J Am Soc Nephrol. 2002;13:134–141. doi: 10.1681/ASN.V131134. [DOI] [PubMed] [Google Scholar]
- 49.Ma K, Cabrero A, Saha PK, et al. Increased beta -oxidation but no insulin resistance or glucose intolerance in mice lacking adiponectin. J Biol Chem. 2002;277:34658–34661. doi: 10.1074/jbc.C200362200. [DOI] [PubMed] [Google Scholar]
- 50.May RC, Kelly RA, Mitch WE. Mechanisms for defects in muscle protein metabolism in rats with chronic uremia. Influence of metabolic acidosis. J Clin Invest. 1987;79:1099–1103. doi: 10.1172/JCI112924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Brooks C, Wei Q, Cho SG, et al. Regulation of mitochondrial dynamics in acute kidney injury in cell culture and rodent models. J Clin Invest. 2009;119:1275–1285. doi: 10.1172/JCI37829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen G, Lin SC, Chen J, et al. CXCL16 recruits bone marrow-derived fibroblast precursors in renal fibrosis. Journal of the American Society of Nephrology : JASN. 2011;22:1876–1886. doi: 10.1681/ASN.2010080881. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.