

Abstract
While much work has been devoted to nanoscale assembly of functional materials, selective reversible assembly of components in the nanoscale pattern at selective sites has received much less attention. Exerting such a reversible control of the assembly process will make it possible to fine-tune the functional properties of the assembly and to realize more complex designs. Herein, by taking advantage of different binding affinities of biotin and desthiobiotin toward streptavidin, we demonstrate selective and reversible decoration of DNA origami tiles with streptavidin, including revealing an encrypted Morse code “NANO” and reversible exchange of uppercase letter “I” with lowercase “i”. The yields of the conjugations are high (> 90%) and the process is reversible. We expect this versatile conjugation technique to be widely applicable with different nanomaterials and templates.
Programmable assembly of functional materials at the nanometer scale with designed patterns has received increasing attention in scientific research and engineering, because of its potential applications in electronics, photonics, and medicine.1 An excellent example of recent success in programmable assembly of nanomaterials was the development of structural DNA1a and DNA origami1b as a template for assembly of functional components such as nanoparticles,2 quantum dots,3 carbon nanotubes,4 and proteins.5 More recent advances in design6, large scale patterning,7 and assembly of DNA structures in 1D,8 2D,9 and 3D,10 offer a path towards practical applications.11 While tremendous progress has been made in the assembly of these components with excellent spatial resolution,12 a significant gap remains in our ability to achieve selectively reversible assembly of certain component(s) at specific site(s); exerting such a level of control of the assembly process will make it possible to fine-tune the functional properties of the real time assembly and to realize more complex designs by replacing one component of the assembly with another in subsequent assembly steps. Reversible processes are common in biology as well as in large-scale engineering systems with many important functions. For example, the reversible process in protein synthesis makes it possible to proof-read and correct errors in incorporation of wrong amino acids with a similar structure.
Despite such examples of programmable, reversible control of the processes both in biology and in bulk-scale engineering systems, very few processes in nanoscale assembly demonstrate similar controllability.13 One successful strategy to achieve selective, reversible nanoscale binding is through the use of complementary DNA, invasive DNA strands, or toe-hold DNA designs.14 These techniques have been widely applied for use in DNA walkers and DNA motors that follow preprogrammed paths.15 In addition, the use of catalytic DNA or DNAzymes for proof-reading and error corrections of DNA-templated assembly of nanoparticles have been demonstrated.13b While the technique is useful, the use of DNA hybridization-based conjugation is limited to buffered conditions similar to that found in the cellular environment. In contrast, covalent conjugation techniques form stable linkages but typically suffer from lower yields and require specific reaction conditions. Consequently there are few examples of completely reversible bonding under mild conditions.
Herein we demonstrate for the first time the use of a combination of biotinylated and desthiobiotinylated DNA to achieve selective and reversible binding of streptavidin protein on a DNA origami template. Applications in reversible encoding and decoding of a nanoscale Morse code message on a DNA origami template, as well as in the reversible transformation of a DNA origami image are also presented.
The biotin-avidin conjugation pair is widely used in nanoscale science and technology today, thanks to the conjugation's exceptional stability and selectivity while requiring only mild conditions.16 As a result, it is commonly used to functionalize nanomaterials such as metal nanoparticles, quantum dots, carbon nanotubes, and proteins, as well as on a variety of surfaces, many of which are available commercially. A significant shortcoming of this conjugation, however, is its near irreversibility. Due to the extraordinarily strong binding affinity of the interaction (Kd ~ 10−15 M), a combination of high temperature, extreme pH, and other chaotropic elements are required to disrupt it.16a,17 Since those disruption forces often destabilize or even destroy the materials on which the conjugation pair reside (double stranded DNA, proteins, nanoparticles) such a reaction is incompatible for many systems for which dynamic and reversible assembly and disassembly would be needed.
Desthiobiotin, with a binding affinity to streptavidin ~1000 times weaker than biotin (Kd ~ 10−11 M), has long been used with solid supports to purify biotin-binding proteins under mild conditions by competition with free biotin in solution,17a,18 but to our knowledge has not yet been exploited for nanoscale assembly. To demonstrate its utility in selective and reversible assembly, we first compared the binding of biotinylated and desthiobiotinylated DNA strands with streptavidin.
To illustrate the binding and release of streptavidin from the functionalized DNA, either biotinylated or desthiobiotinylated DNA strands were incubated with an excess of streptavidin for 2 hours. The mixtures were then treated with 5 mM biotin overnight and analyzed by a gel shift assay. As shown in Figure 1B, upon incubation with streptavidin, both biotinylated and desthiobiotinylated DNA strands in Lanes 1 and 2 transformed into discrete, higher molecular weight products (see Lanes 3 and 4), indicating specific conjugation of the streptavidin with both DNA strands. In contrast, subsequent treatment of both conjugates with excess biotin at room temperature resulted in a different migration of the bands on the gel. While addition of excess biotin did not change the position of the biotinylated DNA strand, the addition under the same condition resulted in a complete reversal of the conjugated high molecular band to the lower molecular weight of free desthiobiotinylated DNA. These results reveal the competitive binding mechanism whereby biotin specifically replaces the weaker binding desthiobiotin from streptavidin, effectively reversing the conjugation process. The process was repeated by recovering the free desthiobiotinylated DNA strand and again incubating with free streptavidin, confirming that the binding is fully reversible and that no observable degradation of desthiobiotin occurred during the process.
Figure 1.
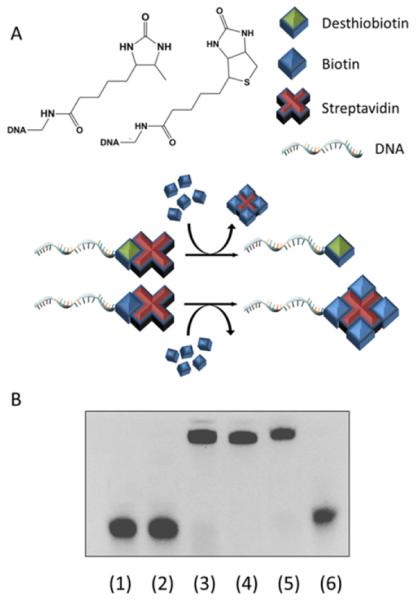
A) Chemical structures and schematic illustrations of the competitive binding between biotinylated and desthiobiotinylated DNA strands toward biotin; B) Native gel shift assay showing biotinylated and desthiobiotinylated DNA strands before (Lanes 1,2 respectively), after incubation with streptavidin (Lanes 3,4 respectively), and then after treatment with excess biotin (Lanes 5,6 respectively).
Having demonstrated full reversibility of the desthiobiotinylated DNA system binding to streptavidin, we then explored its application in reversible nanoscale assemblies for message encryption. In our everyday lives, important messages are often encrypted before transmission and then reversed to reveal the original message to protect the privacy of the communications. To illustrate the nanoscale control of binding and unbinding of proteins to a nanoscale template, and to use biotin as a key to decrypt an encrypted message at the nano-scale, we took advantage of the rectangular 100 × 70 nm2 DNA origami tile as first described by Rothemund.1b Since each of the 226 strands are uniquely indexed on the tile, site-specific modification on the DNA origami tile is a simple process of picking and functionalizing the desired strands based on a diagram of the staple strands.5b The resulting protein patterns on each tile can be revealed by obtaining height images under atomic force microscope.
Invented in the 1840's, Morse code was the prevalent language of telecommunication for most of the 19th and into the 20th century. Following the dots and dashes that make up Morse code, we constructed a message using proteins while simultaneously “encrypting” the message with excess proteins (Figure 2A). To design the nano-Morse codes, a “primary” pattern or masked message is first defined by selecting a series of staple strands that gives the best representation of the original design. Within this pattern of sequences, it is then possible to incorporate a “secondary” design, or hidden message or code. The secondary design consists of sequences that are modified with biotin while the remaining staple strands of the primary design are functionalized with desthiobiotin. As a result, the primary design will be visible after streptavidin incubation and the secondary design or decoded message will be revealed after incubation with biotin.
Figure 2.

Design of the encrypted nano-Morse code tile with corresponding code translation. An illustration of the encrypted and decrypted Morse code designs displaying the nonsense coded message “OOOO” and the hidden message “NANO” A) AFM images of tiles displaying encrypted message “OOOO” and (B) decrypted Morse code message “NANO” after biotin addition. Scale bar = 70 nm.
To realize the encrypted Morse code, we designed an origami tile consisting of 9 desthiobiotinylated and 15 biotinylated staple strands to encode the word “NANO.” In the current design, each row represents one letter, with two proteins representing a dash and a single protein corresponding to a dot within the Morse code (Figure 2). Translated, the encrypted message generated by incubating the tile with streptavidin represents a nonsensical encrypted message (Figure 2A). However, upon subsequent addition of the “decoder,” the original intended message “NANO” is revealed (Figure 2B).
Based on the above design, a combination of biotin- and desthiobiotin- modified staple strands representing specific positions within the origami scaffold, the modified staple strands, along with unmodified strands were annealed from 90 °C to 20 °C over 5 hours to form the desired origami structure. After removing excess staple strands, the formed tiles were allowed to incubate with an excess of streptavidin overnight. Subsequent high resolution AFM reveals a 4×3 array of dashes (each consisting of two proteins) (Figure 2D), as predicted in our scheme. The measured height of the DNA tile and protein are 1.7 nm and 4 nm respectively, in excellent agreement with the known dimensions of these biomolecules.
Next, to demonstrate selective release of protein from the origami template and thus “decrypt” the nanoscale Morse code, the protein-decorated tiles were treated with an excess of biotin, initiating the competitive binding between the free biotin and desthiobiotin bound streptavidin. The AFM images in Figure 2E show that the addition of biotin was able to reveal the hidden Morse code message “NANO.”
AFM analysis of eight well-formed, encrypted nano-Morse code tiles similar to those shown in Figure 2D reveal that 90% of the streptavidin had bound correctly. Interestingly, the percentage of the streptavidin correctly bound to biotin-modified staple strands was ~95% while the same tiles only had ~84% bound correctly to desthiobiotinylated modified strands. This discrepancy is likely due to the weaker binding affinity of desthiobiotin to streptavidin than biotin. Further more, even with minimal tapping force between the AFM probe and the sample, it is conceivable that some proteins were removed through tip interactions. Because of the differences in affinity for streptavidin, more desthiobiotin-bound streptavidin would be dislodged than the biotin-bound proteins. Following incubation with biotin to displace desthiobiotin-bound streptavidin, an analysis of 8 well-formed tiles similar to that shown in Figure 2E shows that approximately 96% of proteins are correctly bound, confirming reversible assembly in high yield.
To further demonstrate the versatility and reversibility of the method described here, we designed an “I” pattern, again made from a combination of biotin and desthiobiotin modifications (Figure 3A). Upon initial addition of streptavidin, the capital I pattern should be visible under AFM. Upon incubation with an excess of biotin in solution, the stronger binding between the biotin-streptavidin replaces and removes the streptavidin bound to desthiobiotin on the surface and subsequently, the expected lower case “i” becomes visible. Finally, we demonstrate the availability of the newly unmasked desthiobiotin sites by washing away the excess biotin and protein and incubate further with streptavidin, which is now free to bind once again to the newly exposed desthiobiotin moieties on the surface of the DNA tile.
Figure 3.
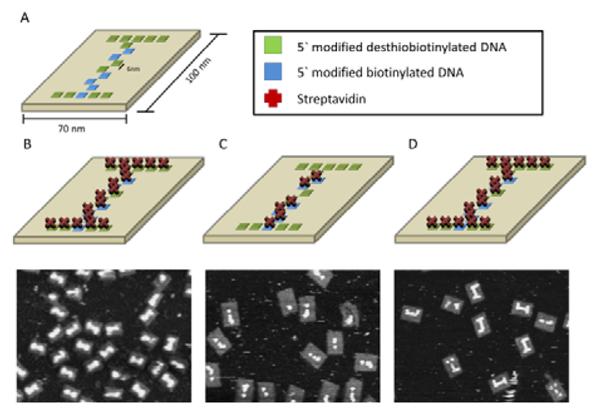
(A) Design of an uppercase “I” and lower case “i” as revealed by atomic force microscopy. (B) First incubation of functionalized tiles with streptavidin, revealing the capital “I” feature. (C) Subsequent incubation with excess biotin selectively removes streptavidin bound to desthiobiotin modifications, revealing the lower case “i.” (D) After washing, newly introduced streptavidin is allowed to bind to free desthiobiotin sites, recovering the capital “I” pattern on the DNA tile. Scale bar = 70 nm.
Furthermore, due to the small separation between biotin and desthiobiotin modifications in this design, we were able to investigate whether the spacing between conjugated streptavidin would hinder the binding or release of the protein from desthiobiotin in the presence of excess biotin. AFM data suggests that the close proximity of the binding sites were not a factor, as the initial incubation of the tiles shows a majority of well-formed patterns on the DNA tiles. Following overnight incubation with biotin at 4 °C, more than 90% of the tiles were successfully converted to a lower case “i” pattern, again suggesting the efficient and competitive release of proteins attached to desthiobiotin. For the final step, we demonstrate the availability of desthiobiotin sites for subsequent functionalization by incubating again with streptavidin after removing released protein and biotin. AFM imaging shows greater than 70% of the desthiobiotin sites available on the tiles were bound to streptavidin, demonstrating the reversible nature of the conjugation.
In conclusion, we have taken advantage of the different binding affinities of biotin and desthiobiotin toward streptavidin and demonstrated for the first time selective and reversible decoration of DNA origami tiles with streptavidin. In the first demonstration of such a versatile approach, we showed that an encrypted Morse code message could be revealed after addition of biotin to replace desthiobiotin-bound streptavidin. In a further demonstration of reversibility, a pattern of letter “I” can be replaced with “i” first, and then restored back to “I”. The yields of the conjugations are high (> 90%) and the process is reversible. The competitive replacement of proteins demonstrated here is not limited to the biotin/desthiobiotin interaction and the basic technique presented here should find wide applications within the fields of nanoeletronics, photonics, and biomedicine where it is desirable for researchers to create nanoscale assemblies with programmable capture and release of nanomaterials.
Supplementary Material
ACKNOWLEDGMENT
This work has been supported by the National Science Foundation Center for Nanoscale Chemical-Electrical-Mechanical Manufacturing Systems (Nano-CEMMS) under NSF Award no. 0749028 (CMMI). Atomic Force Microscopy was carried out at the Center for Microanalysis of Materials, University of Illinois, which is supported by the U.S. DoE under grant DEFG02-91-ER45439. N.W. was funded at University of Illinois at Urbana Champaign by NIH National Cancer Institute Alliance for Nanotechnology in Cancer `Midwest Cancer Nanotechnology Training Center Grant R25 CA154015A.
Footnotes
Supporting Information Materials, methods, and experimental data. This material is available free of charge via the Internet at http://pubs.acs.org
The authors declare no competing financial interests.
REFERENCES
- (1).(a) Seeman NC. Nature. 2003;421:427. doi: 10.1038/nature01406. [DOI] [PubMed] [Google Scholar]; (b) Rothemund PWK. Nature. 2006;440:297. doi: 10.1038/nature04586. [DOI] [PubMed] [Google Scholar]; (c) Lu Y, Liu J. Acc. Chem. Res. 2007;40:315. doi: 10.1021/ar600053g. [DOI] [PubMed] [Google Scholar]; (d) Xing H, Wong NY, Xiang Y, Lu Y. Curr. Opin. Chem. Biol. 2012;16:429. doi: 10.1016/j.cbpa.2012.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).(a) Ding BQ, Deng ZT, Yan H, Cabrini S, Zuckermann RN, Bokor J. J. Am. Chem. Soc. 2010;132:3248. doi: 10.1021/ja9101198. [DOI] [PubMed] [Google Scholar]; (b) Pal S, Deng ZT, Ding BQ, Yan H, Liu Y. Angew. Chem. Int. Ed. 2010;49:2700. doi: 10.1002/anie.201000330. [DOI] [PubMed] [Google Scholar]; (c) Acuna GP, Bucher M, Stein IH, Steinhauer C, Kuzyk A, Holzmeister P, Schreiber R, Moroz A, Stefani FD, Liedl T, Simmel FC, Tinnefeld P. ACS Nano. 2012;6:3189. doi: 10.1021/nn2050483. [DOI] [PubMed] [Google Scholar]; (d) Pal S, Varghese R, Deng ZT, Zhao Z, Kumar A, Yan H, Liu Y. Angew. Chem. Int. Ed. 2011;50:4176. doi: 10.1002/anie.201007529. [DOI] [PubMed] [Google Scholar]; (e) Zhao WA, Gao Y, Kandadai SA, Brook MA, Li YF. Angew. Chem. Int. Ed. 2006;45:2409. doi: 10.1002/anie.200600061. [DOI] [PubMed] [Google Scholar]; (f) Lee JH, Wernette DP, Yigit MV, Liu J, Wang Z, Lu Y. Angew. Chem. Int. Ed. 2007;46:9006. doi: 10.1002/anie.200702569. [DOI] [PubMed] [Google Scholar]
- (3).(a) Wang RS, Nuckolls C, Wind SJ. Angew. Chem. Int. Ed. 2012;51:11325. doi: 10.1002/anie.201206389. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ko SH, Gallatin GM, Liddle JA. Adv. Funct. Mater. 2012;22:1015. [Google Scholar]
- (4).(a) Maune HT, Han SP, Barish RD, Bockrath M, Goddard WA, Rothemund PWK, Winfree E. Nat. Nanotechnol. 2010;5:61. doi: 10.1038/nnano.2009.311. [DOI] [PubMed] [Google Scholar]; (b) Zhao W, Gao Y, Brook MA, Li YF. Chem. Commun. 2006;3582 doi: 10.1039/b606518j. [DOI] [PubMed] [Google Scholar]
- (5).(a) Derr ND, Goodman BS, Jungmann R, Leschziner AE, Shih WM, Reck-Peterson SL. Science. 2012;338:662. doi: 10.1126/science.1226734. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Chhabra R, Sharma J, Ke YG, Liu Y, Rinker S, Lindsay S, Yan H. J. Am. Chem. Soc. 2007;129:10304. doi: 10.1021/ja072410u. [DOI] [PubMed] [Google Scholar]; (c) Kuzyk A, Laitinen KT, Torma P. Nanotechnology. 2009;20:23505. doi: 10.1088/0957-4484/20/23/235305. [DOI] [PubMed] [Google Scholar]; (d) Wong NY, Zhang C, Tan LH, Lu Y. Small. 2011;7:1427. doi: 10.1002/smll.201100140. [DOI] [PubMed] [Google Scholar]; (e) Kuzuya A, Kimura M, Numajiri K, Koshi N, Ohnishi T, Okada F, Komiyama M. ChemBioChem. 2009;10:1811. doi: 10.1002/cbic.200900229. [DOI] [PubMed] [Google Scholar]; (f) Nakata E, Liew FF, Uwatoko C, Kiyonaka S, Mori Y, Katsuda Y, Endo M, Sugiyama H, Morii T. Angew. Chem. Int. Ed. 2012;51:2421. doi: 10.1002/anie.201108199. [DOI] [PubMed] [Google Scholar]; (g) Lee JH, Wong NY, Tan LH, Wang ZD, Lu Y. J. Am. Chem. Soc. 2010;132:8906. doi: 10.1021/ja103739f. [DOI] [PubMed] [Google Scholar]
- (6).(a) Douglas SM, Marblestone AH, Teerapittayanon S, Vazquez A, Church GM, Shih WM. Nucl. Acids Res. 2009;37:5001. doi: 10.1093/nar/gkp436. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Castro CE, Kilchherr F, Kim D-N, Shiao EL, Wauer T, Wortmann P, Bathe M, Dietz H. Nat Methods. 2011;8:221. doi: 10.1038/nmeth.1570. [DOI] [PubMed] [Google Scholar]
- (7).(a) Hung AM, Micheel CM, Bozano LD, Osterbur LW, Wallraff GM, Cha JN. Nat. Nanotechnol. 2010;5:121. doi: 10.1038/nnano.2009.450. [DOI] [PubMed] [Google Scholar]; (b) Endo M, Sugita T, Rajendran A, Katsuda Y, Emura T, Hidaka K, Sugiyama H. Chem. Commun. 2011;47:3213. doi: 10.1039/c0cc05306f. [DOI] [PubMed] [Google Scholar]
- (8).(a) Li Z, Liu MH, Wang L, Nangreave J, Yan H, Liu Y. J. Am. Chem. Soc. 2010;132:13545. doi: 10.1021/ja106292x. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Dutta K, Fujimoto T, Inoue M, Miyoshi D, Sugimoto N. Chem. Commun. 2010;46:7772. doi: 10.1039/c0cc00710b. [DOI] [PubMed] [Google Scholar]
- (9).(a) Liu WY, Zhong H, Wang RS, Seeman NC. Angew. Chem. Int. Ed. 2011;50:264. doi: 10.1002/anie.201005911. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zhao Z, Liu Y, Yan H. Nano Lett. 2011;11:2997. doi: 10.1021/nl201603a. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Yun JM, Kim KN, Kim JY, Shin DO, Lee WJ, Lee SH, Lieberman M, Kim SO. Angew. Chem. Int. Ed. 2012;51:912. doi: 10.1002/anie.201106198. [DOI] [PubMed] [Google Scholar]
- (10).(a) Douglas SM, Dietz H, Liedl T, Hoegberg B, Graf F, Shih WM. Nature. 2009;459:414. doi: 10.1038/nature08016. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Han DR, Pal S, Nangreave J, Deng ZT, Liu Y, Yan H. Science. 2011;332:342. doi: 10.1126/science.1202998. [DOI] [PubMed] [Google Scholar]; (c) Andersen ES, Dong M, Nielsen MM, Jahn K, Subramani R, Mamdouh W, Golas MM, Sander B, Stark H, Oliveira CLP, Pedersen JS, Birkedal V, Besenbacher F, Gothelf KV, Kjems J. Nature. 2009;459:73. doi: 10.1038/nature07971. [DOI] [PubMed] [Google Scholar]
- (11).Endo M, Katsuda Y, Hidaka K, Sugiyama H. Angew. Chem. Int. Ed. 2010;49:9412. doi: 10.1002/anie.201003604. [DOI] [PubMed] [Google Scholar]
- (12).Rinker S, Ke Y, Liu Y, Chhabra R, Yan H. Nat. Nanotechnol. 2008;3:418. doi: 10.1038/nnano.2008.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).(a) Voigt NV, Torring T, Rotaru A, Jacobsen MF, Ravnsbaek JB, Subramani R, Mamdouh W, Kjems J, Mokhir A, Besenbacher F, Gothelf KV. Nat. Nanotechnol. 2010;5:200. doi: 10.1038/nnano.2010.5. [DOI] [PubMed] [Google Scholar]; (b) Liu JW, Wernette DP, Lu Y. Angew. Chem. Int. Ed. 2005;44:7290. doi: 10.1002/anie.200501815. [DOI] [PubMed] [Google Scholar]; (c) Yoon HC, Hong MY, Kim HS. Langmuir. 2001;17:1234. [Google Scholar]; (d) Gu HZ, Chao J, Xiao SJ, Seeman NC. Nat. Nanotechnol. 2009;4:245. doi: 10.1038/nnano.2009.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Numajiri K, Kimura M, Kuzuya A, Komiyama M. Chem. Commun. 2010;46:5127. doi: 10.1039/c0cc00044b. [DOI] [PubMed] [Google Scholar]
- (15).(a) Lund K, Manzo AJ, Dabby N, Michelotti N, Johnson-Buck A, Nangreave J, Taylor S, Pei R, Stojanovic MN, Walter NG, Winfree E, Yan H. Nature. 2010;465:206. doi: 10.1038/nature09012. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Sherman WB, Seeman NC. Nano Lett. 2004;4:1203. [Google Scholar]; (c) Gu H, Chao J, Xiao S-J, Seeman NC. Nature. 2010;465:202. doi: 10.1038/nature09026. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Douglas SM, Bachelet I, Church GM. Science. 2012;335:831. doi: 10.1126/science.1214081. [DOI] [PubMed] [Google Scholar]
- (16).(a) Savage D, Mattson G, Desai S, Nielander G, Morgensen S, Conklin E. Avidin-Biotin Chemistry: A Handbook. Pierce Chemical Company; Rockford, IL: 1992. [Google Scholar]; (b) Meyer R, Niemeyer CM. Small. 2011;7:3211. doi: 10.1002/smll.201101365. [DOI] [PubMed] [Google Scholar]
- (17).(a) Finn FM, Titus G, Hofmann K. Biochemistry. 1984;23:2554. doi: 10.1021/bi00307a003. [DOI] [PubMed] [Google Scholar]; (b) Hirsch JD, Eslamizar L, Filanoski BJ, Malekzadeh N, Haugland RP, Beechem JM. Anal. Biochem. 2002;308:343. doi: 10.1016/s0003-2697(02)00201-4. [DOI] [PubMed] [Google Scholar]
- (18).(a) Green N, Anfinsen C, Edsall J, Richards F. Advances in Protein Chemistry. Vol. 29. Academic Press; New York: 1975. [Google Scholar]; (b) Wu SC, Wong SL. Anal. Biochem. 2004;331:340. doi: 10.1016/j.ab.2004.03.056. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.