

Abstract
We have evaluated the frequency of KRAS gene mutations during the critical transition from villous adenoma to colorectal carcinoma to assess whether the adenomas contain a KRAS mutation more frequently than carcinomas. We analyzed sporadic villous and tubulovillous adenomas, in situ carcinomas, and primary colorectal carcinomas from multiple patients. The cancers were further evaluated for mucinous status and microsatellite instability. Standard PCR molecular techniques were used for KRAS and microsatellite analyses. A KRAS mutation was found in 61.9% of 134 adenomas, 67.8% of 84 in situ carcinomas, and just 31.6% of 171 carcinomas. Our study clearly demonstrates that tubulovillous and villous adenomas, as well as both the benign and malignant parts of in situ carcinomas, are statistically more likely to contain a somatic KRAS gene mutation than colorectal carcinomas. This difference is confined to the non-mucinous and the microsatellite stable tumors. Our data support the possibility that non-mucinous and microsatellite stable carcinomas with wild-type KRAS gene may have had a mutation in the KRAS gene during their earlier stages, with the mutation lost during further growth.
Keywords: KRAS gene, colorectal adenomas, colorectal carcinoma, microsatellite instability, tubulovillous adenoma, villous adenoma
Introduction
Colorectal cancer cells demonstrate a number of genetic changes, and it is now accepted that the accumulation of these changes, rather than a specific sequence, determines the development of the malignancy. Some of these genetic changes can be detected within colorectal adenomas, which are the precursor lesions for many colorectal cancers [1]. Current theories regarding tumor progression suggest that there is a process of clonal evolution involving progressively more biologically aggressive subpopulations, with the sequential accumulation of mutations in a number of genes [2]. The retention of all new mutations from one subpopulation to the next is implied in this concept. Therefore, it is assumed that once a molecular genetic change has occurred, it is retained throughout the progression of the benign adenoma into a colorectal cancer.
Somatic KRAS gene mutations are important in the carcinogenesis process [3], and they occur in approximately one-third of colorectal cancers [4]. Our previous studies [5], as well as the studies of others, have demonstrated the presence of KRAS mutations in adenomas, and have suggested, interestingly, that there may be a higher frequency of this mutation in villous adenomas than in carcinomas. Were this observation found to be consistent within a larger study group, the finding would provide a further clue concerning the molecular biology of colorectal cancer [6]. To clarify this issue, we have evaluated the frequency of KRAS mutations during the critical transition from villous adenoma to colorectal carcinoma by analyzing villous and tubulovillous adenomas, in situ carcinomas, and primary colorectal carcinomas from multiple patients. We also assessed the mucinous status and microsatellite instability for each carcinoma and its relationship to KRAS mutation.
Materials and methods
Our research has involved a series of studies evaluating particular groups of patients with colorectal neoplasms with respect to molecular genetic abnormalities, both germ line and somatic. Each individual study was approved by the hospital Institutional Review Board, as was this analysis. All samples were archived material from the Department of Pathology. Clinical material primarily reflects a suburban community of middle economic level, with substantial representations from various minority groups (Asian, African-American) of both middle and low economic status. Histological slides stained with hematoxylin and eosin (H&E) and paraffin blocks were available for all cases. One clinical pathologist reviewed all histological slides and indicated the areas for molecular study. Criteria for differentiation of adenomas followed the World Health Organization criteria with respect to villous component: tubular adenomas, <20%; tubulovillous adenomas, 20 – 80%; and villous adenomas, >80%. Mucinous carcinoma was diagnosed when at least 50% of the tumor contained secretory mucin. Right side colonic segments were defined as cecum, ascending, hepatic flexure, transverse; and the left side was considered the splenic flexure, descending, sigmoid and rectum. We defined in situ carcinomas (ISCs) as tumors in which both a benign component and a malignant component were present (Figure 1). We limited our study to in situ cancers with villous or tubulovillous benign areas and did not include the uncommon ISCs with only benign tubular areas.
Figure 1.
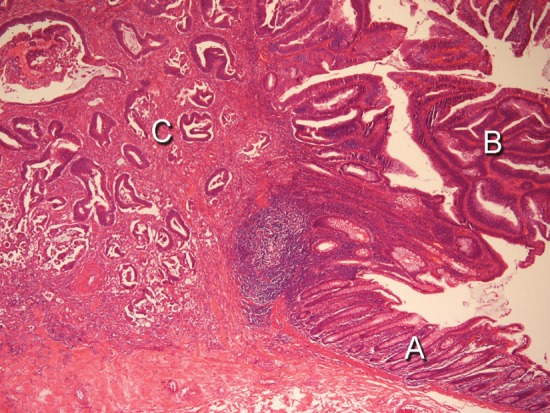
An in situ carcinoma. Hematoxylin and Eosin stain, 400X power. A: normal colonic mucosa. B: residual villous adenoma. C: in situ carcinoma.
Our series of studies contained patients who had numerous tumors removed over intervals spanning as much as 20 years. Other patients were determined to have germ line mutations found most frequently in Ashkenazi Jews, including the APC*I1307K, MSH21906G>C*, and BLMAsh. In this current analysis, we excluded patients with known germ line mutations and patients with multiple tumors, on the assumption that the development of mutations in tumors from these individuals might occur through mechanisms unrelated to the process occurring in isolated sporadic cases. Patients were included only if they were found, over time, to have the following: a maximum of only one or two other adenomas for the tubulovillous and villous group, and a maximum of only one other adenoma for the in situ carcinoma and the carcinoma groups.
DNA extraction and purification
All tissue specimens were formalin-fixed and paraffin-embedded. Histological slides stained with H&E were examined and the area of relevant tissue was identified and marked, as was an area of normal tissue. Consecutive unstained slides from the paraffin block were then prepared and the corresponding areas were isolated under a dissecting microscope by manual dissection. The paraffin wax was removed by xylene and ethanol washes. The cellular material was lysed in a proteinase potassium buffer solution. DNA was isolated and purified using the Qiagen QIAmp Tissue Kit (Qiagen Inc., Valencia, CA).
Sequence analysis of the KRAS gene
The codon 12/13 region of exon 1 in the KRAS gene was amplified using the primer set 5’-AAGGCCTGCTGAAAATGACTG-3’ sense and 5-GGTCCTGCACCAGTAATATGCA-3’. Hot-start PCR was performed in 50 μl volumes with AmpliTAq Gold polymerase and ABI reagents (Applied Biosystems, Foster City, CA) using 100 ng of DNA, 50 pmols of primer, and 2.0 mM MgCl2 on a GeneAmp PCR system 9700 (Applied Biosystems, Foster City, CA). PCR consisted of an initial 8 minutes of denaturation at 94°C, followed by a total of 40 cycles of a 30 second denaturation at 94°C, a 30 second annealing, and a one minute elongation at 72°C, with a final 30 minute extension at 72°C. The annealing temperature was stepped down at 62°C, 60°C, and 58°C for 5, 20, and 15 cycles respectively. The post-PCR products were quality checked by agarose gel and then purified using the QIAquick PCR Purification Kit (Qiagen Inc., Valencia, CA) prior to sequencing. The sequencing reactions were done in 20 μl volumes using 0.5X BigDye Terminator Cycle Sequencing Reagents (Applied Biosystems, Foster City, CA), 5.0 pmol of the reverse Ki-ras primer, and 1.0 μl of the purified PCR reaction. Reactions are run on a GeneAmp PCR System 9700 (Applied Biosystems, Foster City, CA) for 25 cycles using 2 minutes of extension time. The sequencing reaction fragments were cleaned by isopropanol precipitation. Sequencing products were separated by capillary electrophoresis with an ABI 3130 Genetic Analyzer and the data was processed with Sequencing Analysis (Applied Biosystems, Foster City, CA) software (Figure 2).
Figure 2.
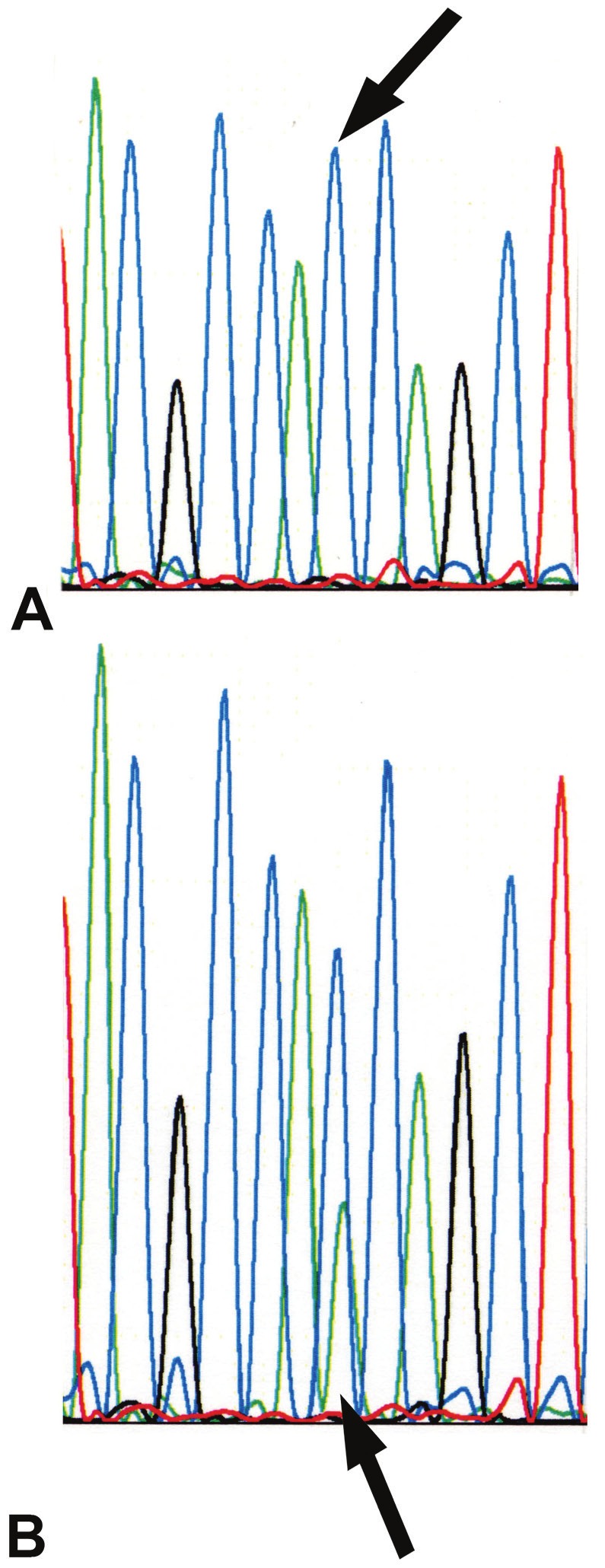
KRAS DNA sequence analysis. A: DNA from normal tissue. The arrow indicates the wild-type pattern with a single peak at the second nucleotide position of codon 12. B: DNA from an ascending colon carcinoma. The arrow indicates a heterozygous mutant peak under the wild-type peak.
Microsatellite instability (MSI) analysis
MSI was detected using the Bethesda panel of markers that include two mononucleotide markers Bat25 and Bat26, and three dinucleotide markers D2S123, D5S346, and D17S250. In all primer sets the forward primer contains a 5’ fluorescent label while the reverse primer contains a 5’- GTGTCTT tail. PCR was performed in 30 μl volumes with AmpliTaq polymerase and ABI reagents (Applied Biosystems, Foster City, CA) using 100 ng of DNA, 8 pmols of primer, and 1.5 mM MgCl2 on a GeneAmp PCR System 9700 (Applied Biosystems, Foster City, CA). PCR consisted of an initial denaturation at 94°C, followed by 35 cycles of a 30 second denaturation at 94°C, a 30 second annealing at 55°C, and a 60 second elongation at 72°C, with a final 30 minute extension at 72°C. PCR products were separated by capillary electrophoresis with an ABI 3130 Genetic Analyzer and the data was processed with GeneMapper (Applied Biosystems, Foster City, CA) software. A tumor was defined as MSI-high if two or more of the five markers had a changed allele pattern, and is referred to as “MSI”.
Statistical analysis
The main analysis focuses on whether the frequency of KRAS gene mutation differs for adenomas, in situ carcinomas, and carcinomas, controlling for mucin status and microsatellite instability for carcinomas. The t-test was used for two group comparisons, and analysis of variance was used for three group comparisons regarding mean age by group. The chi-square test was used to assess differences in proportions for characteristics by tumor group. We derived odds ratios for KRAS status for each group in a case-case analysis of adenoma or in-situ cancer compared to cancer. These case-case odds ratios were also derived controlling for mucinous status and microsatellite instability using the Mantel-Haenszel procedure with test for heterogeneity. Ninety-five percent confidence intervals were derived for the odds ratios [7].
Results
Selection of study cases
Our series of studies included a total of 914 tumors, and the excluded group consisted of 525 tumors from 406 patients (Table 1). A higher percentage of carcinomas, rather than adenomas or in situ cancers, were excluded, reflecting that excluded patients had germ line mutations or multiple tumors, both features potentially resulting in a greater likelihood of a cancer. The incidence of a KRAS mutation was statistically more common in the excluded adenomas than the excluded carcinomas ([odds ratio] OR = 2.19, [confidence interval] CI = 1.39-3.44, p </= 0.0006), and also more common in the carcinomatous portion of the ISCs than in the carcinomas (OR = 2.26, CI = 1.47-3.49, p </= 0.0002) (Table 1). For all excluded tumors, 80.6% of the KRAS mutations were in codon 12, 17.0% were in codon 13 and 2.4% were other. Of the 166 mutations in codon 12, 143 (86.1%) were in the second position and 23 (13.9%) were in the first position. Of the 35 mutations in codon 13, only one was in the first position, all others were in the second position. With our exclusion criteria, the final study group consisted of a total of 389 tumors, with 134 (34.4%) tubulovillous and villous adenomas, 84 (21.6%) in situ carcinomas, and 171 (44%) carcinomas. Clinical characteristics of age, gender and location for these three study groups of tumors are shown in Table 2.
Table 1.
Total, excluded, and study groups of tumors. Percentages with a KRAS gene mutation indicated for the tumor types of the excluded group
| Group | Total # studied | TV/Vil* (% mutated) | ISC† (% mutated) | Cancer |
|---|---|---|---|---|
| Total | 914 | 241 | 205 | 468 |
| Excluded | 525 | 107 (49.5) | 121 (50.4) | 297 (31.0) |
| Study | 389 | 134 | 84 | 171 |
TV/vil = tubulovillous and villous adenomas.
ISC = in situ carcinoma.
Table 2.
Clinical characteristics of study patients and their tumors
| Adenomas | Cancer part of ISCs | Cancer | |
|---|---|---|---|
| No. (%) | No. (%) | No. (%) | |
| Characteristic | |||
| Number | 134 | 84 | 171 |
| Age (mean) | 66.9 | 66.9 | 69.2 |
| S.D | 11.5 | 12.6 | 12.5 |
| Gender | |||
| Male | 59 (44) | 38 (45.2) | 75 (44) |
| Female | 75 (56) | 46 (54.8) | 96 (56) |
| Location | |||
| Right | 66 (49.3) | 61 (72.6) | 93 (54.4) |
| Left | 68 (50.7) | 23 (27.4) | 78 (45.6) |
| Microsatellite status | |||
| Stable | 73 (92.4) | 141 (84.4) | |
| Unstable | 6 (7.6) | 26 (15.6) | |
| Mucin status | |||
| Mucinous | 21 (25) | 31 (18.4) | |
| Non-mucinous | 63 (75) | 137 (81.6) |
Adenomas
The adenoma study group contained 34 tubulovillous and 100 villous adenomas. A comparison between these two subtypes of adenomas showed no statistical difference in mean age of the patient when the tumor was removed (64.7 vs 67.6 years; p=0.20), percentage of patients of male gender (50% vs 58%; p=0.42), percentage of tumors from the left colon (64.7% vs 46%; p=0.06), or the percentage of tumors with a KRAS gene mutation (67.6% vs 60.0% p=0.43). Therefore, these two groups were combined for further analysis.
A KRAS mutation was detected in 83 (61.9%) of the 134 adenomas. One adenoma had two separate KRAS mutations for a total of 84 mutations. Of the 69 tumors with mutations in codon 12, there were 12 mutations in the first position (c.34G>A = 8, c.34G>C = 1, and c.34G>T = 3); and 57 mutations in the second position (c.35G>A = 23, c.35G>C = 1, c.35G>T = 33). One of the 15 mutations in codon 13 was in the first position (c.37G>T), and 14 were in the second position, (c.38G>A). For the adenomas, there was no difference between tumors with or without a KRAS mutation by gender (for females, p = 0.84); or by age (67.8 vs 65.4, p = 0.24). A total of 66 adenomas (49.3%) were from the right colon and 68 (50.7%) were from the left colon. A KRAS mutation was less likely to be found in an adenoma from the left colon (54.4%) than from the right colon (69.7%), but this was not statistically different (p = 0.07). The distribution of the adenomas and percentage with a KRAS mutation is shown in Table 3. Of the 134 adenomas, 86 (64.2%) were tested for MSI. Only one (1.2%) adenoma demonstrated microsatellite instability and it contained only wild type KRAS gene.
Table 3.
Numbers of tumors by colon segment and percentage with a KRAS gene mutation
| Tumor type | Cecum | Ascend | Transv | Right* | Descend | Sigmoid | Rect | Left* |
|---|---|---|---|---|---|---|---|---|
| # (%) | # (%) | # (%) | # (%) | # (%) | # (%) | # (%) | # (%) | |
| Adenoma | 15 (87) | 22 (68) | 22 (55) | 7 (86) | 12 (75) | 43 (53) | 12 (33) | 1 (100) |
| In situ cancer | 28 (71) | 17 (82) | 6 (50) | 10 (80) | 2 (50) | 17 (47) | 3 (100) | 1 (0) |
| Cancer | 39 (38) | 30 (37) | 22 (23) | 2 (0) | 7 (14) | 63 (29) | 4 (75) | 4 (25) |
Tumor labeled only as right or left colon.
In situ carcinomas
A KRAS mutation was detected in the benign portions of in situ tumors (ISCs) in 50 of 84 (59.5%) ISCs, while the malignant portion contained a KRAS mutation in 57 of 84 ISC (67.8%) (p = 0.052). There were 61 (72.6%) ISCs found in the right colon and 23 (27.4%) from the left colon. The distribution of the ISCs with percentages of KRAS mutations is shown in Table 3. Both the benign and malignant parts of 24 of 84 (28.6%) ISCs contained only wild type KRAS genes. The mutational changes in the two parts of the other 60 ISCs are shown in Table 4.
Table 4.
KRAS gene findings in 84 in situ carcinomas by benign and malignant portions
| Benign / Malignant | Number (%) |
|---|---|
| Wild / wild | 24 (28.6) |
| Same mutation | 43 (51.2) |
| Different mutations | 4 (4.8) |
| Mutated / wild | 3 (3.6) |
| Wild / mutated | 10 (11.9) |
Four ISCs demonstrated a mutation in the first codon position of codon 12: c.34G>T = 3, c.34G>A = 1. The second position of the codon 12 contained a mutation in 39 lesions: c.35G>A = 21, c.35G >T = 11; and codon 13: c.38G>A = 7. There was no difference between the ISCs with or without a KRAS gene mutation in the carcinomatous portion when controlling for age (68.0 vs 64.4, p = 0.22); or gender (56.1% for females with a mutated ISC, p = 0.71). A KRAS mutation was less likely to be found in the carcinomatous portion of an ISC from the left colon (52.2%) than from the right colon (73.8%), but this was not statistically different, (p = 0.06). A total of 79 (94%) ISCs were assayed for MSI within the malignant part of the tumor, and microsatellite instability was detected in six (7.6%). In 5 of these 6 tumors the KRAS gene was wild type, and one demonstrated a KRAS mutation. All 84 ISCs were evaluated for mucin within the carcinomatous area and 21 (25%) were mucinous. Eleven of the mucinous ISCs contained a KRAS mutation (52.4%), and 46 of the 63 non-mucinous ISCs (73%) contained a KRAS mutation. This difference did not reach statistical significance, (p = 0.08).
Carcinomas
A KRAS gene mutation was found in 54 (31.6%) of 171 carcinomas. There were 93 (54.4%) carcinomas from the right side and 78 (45.6%) from the left side. The distribution of the cancers and percentage with a KRAS mutation is shown in Table 3. There were 44 mutations in codon 12, 8 were in the first position (c.34G>A = 3, c.34G>C = 1, c.34G>T = 4), and 36 mutations in the second position (c.35G>A = 14, c.35G>C = 5, c.35G>T = 17). There were 9 mutations in codon 13, all in the second position c.38G>A. One carcinoma contained a mutation in codon 19, c.57G>T.
There was no difference between the carcinomas with or without a KRAS gene mutation when controlling for gender (53.7% for females with a mutated cancer, p = 0.66), or location (29.5% of left side carcinomas had a mutation and 33.3% of the carcinomas from the right side, p = 0.59); or age (68.4 vs 69.6, p = 0.58). There were only 5 carcinomas occurring in patients less than 45 years of age at the time of diagnosis, and two contained a KRAS mutation while three did not. There was no difference in the percentages of cancers with a KRAS mutation across three age distributions of </= 59 years (35.1%), 60-74 years (30.1%), and >74 years (30%) (p = 0.84).
A total of 167 cancers were assessed for microsatellite instability, and 141 (84.4%) cancers were microsatellite stable while 26 (15.6%) demonstrated MSI. A KRAS mutation was more frequently seen in cancers that were microsatellite stable (35.5%) than in those with MSI (11.5%), (p = 0.02). A total of 168 cancers (98.2%) were assessed for the presence of mucin, and 31 cancers (18.4%) contained mucin while 137 (81.6%) did not. Of the 26 (15.6%) cancers with microsatellite instability, 25 were tested for mucin and 9 (36%) contained mucin and 16 (64%) did not. A higher percentage of cancers with MSI contained mucin (36%) than did microsatellite stable cancers (15%), (p = 0.01). Mucinous cancers were more likely to have a KRAS mutation (45.2%) than non-mucinous cancers (29.2%), but this difference did not reach statistical significant (p = 0.08).
Comparisons among tumor types
There was no difference in the percentage of tumors with a KRAS mutation between the adenomas (61.9%) and the ISCs (67.8%), (OR = 0.77, CI = 0.43-1.37, p = 0.37). However, a KRAS gene mutation was significantly less frequent in the carcinomas (31.6%) than in either the adenomas or in the in situ tumors (Table 5). The difference in frequency of KRAS mutation between adenomas plus ISCs compared to carcinomas is also statistically significant, (OR = 3.9, CI = 2.5-5.9, p < 0.0001).
Table 5.
KRAS gene mutation for each tumor type, with or without mucin or MSI
| Tumor Type | Odds Ratio In Situ versus Cancer | 95% Confi-dence Interval | P-value | Interaction effect | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
|
| ||||||||||
| Adenomas* | In situ cancer† | Cancer‡ | ||||||||
|
| ||||||||||
| Ki-ras | No. | Mutated | No. | Mutated | No. | Mutated | ||||
| n (%) | n (%) | n (%) | ||||||||
| Total | 134 | 83 (61.9) | 84 | 57 (67.8) | 171 | 54 (31.6) | 4.6 | 2.6-8.0 | <0.0001§ | |
| Mucinous status | ||||||||||
| Non-mucinous | - | - | 63 | 46 (73.0) | 137 | 40 (29.2) | 6.6 | 3.4-12.9 | <0.0001§ | 0.01 |
| Mucinous | - | - | 21 | 11 (52.4) | 31 | 14 (45.2) | 1.3 | 0.4-4.1 | 0.6 | |
| Microsatellite status | ||||||||||
| Microsatellite stable | - | - | 73 | 52 (71.2) | 141 | 50 (35.5) | 4.5 | 2.4-8.3 | <0.0001§ | 0.4 |
| Microsatellite unstable | - | - | 6 | 1 (16.7) | 26 | 3 (11.5) | 1.5 | 0.1-18.0 | 0.7 | |
0Adenomas vs. Cancers: O.R. = 3.5, C.I. = 2.2 – 5.7 p = <0.0001.
5 in situ cancers not assayed for microsatellite instability.
3 cancers not assayed for mucin.
denotes statistical significance.
A statistically significant difference was found between the frequency of a KRAS mutation in non-mucinous ISCs (73%) and non-mucinous carcinomas (29.2%), (OR = 6.6, CI = 3.4-12.8, p < 0.0001). There was no statistical difference between mucinous ISCs and mucinous carcinomas regarding the frequency of KRAS mutation (OR = 1.3, CI = 0.4-4.1, p = 0.6) (Table 5).
The percentage of microsatellite stable ISCs (carcinomatous portion) with a KRAS mutation (71.2%) is statistically greater than the percentage found in microsatellite stable carcinomas (35.5%) (OR = 4.5, CI = 2.4-8.3, p < 0.0001). The numbers of ISCs and carcinomas found to be microsatellite unstable was small, 6 and 26 respectively. There was a slightly greater percentage of unstable ISCs with KRAS mutation than unstable carcinomas, but this was not statistically significant (OR =1.5, CI =0.1-18.0, p =0.7) (Table 5). Overall odds ratios are higher in the adenomas and the in situ carcinomas than in the carcinomas. However, this relationship is confined to the non-mucinous and the microsatellite stable tumors. In the mucinous and microsatellite unstable tumors, there is much less difference in the incidence of KRAS mutation between the tumor types, with test of heterogeneity 0.01 for mucin status and 0.4 for microsatellite status.
Discussion
Numerous studies have reported the frequency of KRAS gene mutations in colorectal cancer. The largest series was the “RASCAL” study of 2721 colorectal cancer cases reporting an incidence of 37.7% [4]. Similar frequencies were reported subsequently [8-10]. Information on the frequency of KRAS gene mutation in adenomas is more limited, particularly with respect to adenomas with villous features. The largest study reported a KRAS mutation in 78 (32.8%) of 238 tubulovillous and villous adenomas. This study, however, included only 30 villous adenomas, with 12 (40%) having a mutation [11]. A smaller study reported a KRAS gene mutation in 25 of 49 (51%) adenomas with moderate dysplasia [12], while another small study found a KRAS mutation rate of 15.9% (7 of 44) in a mixture of tubulovillous and villous adenomas [13]. None of these three articles included comparable data on carcinomas. We previously reported a frequency of 52.5% (21 of 40) for KRAS gene mutations in tubulovillous and villous adenomas, and we suggested the possibility of a higher incidence of KRAS mutation in villous adenomas than in colorectal carcinomas [5]. By comparison, the frequency of KRAS mutations in tubular adenomas is quite low [1]. We previously reported a frequency of only 9.2% in 553 tubular adenomas [14]; consequently, we have not included data on tubular adenomas in this report.
The frequency of KRAS gene mutation in carcinomas was the same for both our excluded group and our study group. However, the frequency of KRAS mutations was borderline lower for adenomas (p </= 0.054) and lower for ISCs (p </= 0.013) in the excluded group than for the study group, possibly suggesting that other molecular mechanisms may be more critical for tumorigenesis in the excluded adenomas and ISCs.
KRAS mutations in carcinomas with mucinous features
In the RASCAL study, mucinous status was reported for 790 (60%) of the accumulated cases, and mucin was present in 252 (31.9%) cancers [4]. A KRAS mutation was present in 44% of the mucinous cancers and in 35.1% of the non-mucinous cancers. This is statistically significant, with p =0.0161, (O.R. = 1.45, C.I. = 1.07-1.97). Similar results have been reported in other, smaller studies [15], indicating that KRAS gene mutations are more common in mucinous cancers than in cancers without mucin. Our data on carcinomas are similar, but did not reach statistical significance. In contrast, our mucinous ISCs had a lower incidence of KRAS mutation than the non-mucinous ISCs, but this also did not reach statistical significance.
KRAS gene mutations in cancers with microsatellite instability
Microsatellite instability (MSI-H) is detected in 10% to 15% of sporadic colorectal cancers [16], and studies have shown a lower frequency of KRAS gene mutation in colon carcinomas with microsatellite instability than in microsatellite stable cancers [17]. Our data support this finding. There have been several reports showing that microsatellite unstable carcinomas are frequently mucinous [18,19]. As a KRAS mutation is more common with mucinous carcinomas, and mucin is more common with MSI-H tumors, one would anticipate a higher incidence of KRAS mutation in MSI-H tumors, but this is not the usual finding. It is paradoxical that KRAS mutation is actually more common with microsatellite stable tumors. Perhaps the reports detailing carcinomas with MSI and mucin included patients for whom MSI developed through several different mechanisms – germ line as well as somatic – and that the correlation between MSI and mucin is not the same for all mechanisms. It is also possible that other unknown, or additional molecular genetic changes in the tumors may overwhelm the relationship between mucinous feature and KRAS mutation.
Our study clearly demonstrates that tubulovillous and villous adenomas, as well as both the benign and malignant parts of in situ carcinomas, are statistically more likely to contain a somatic KRAS gene mutation than colorectal carcinomas. However, approximately 70% of colorectal carcinomas lack a mutation in the KRAS gene. It is possible that carcinomas with wild-type KRAS develop through a process devoid of KRAS mutations; for example, the serrated adenoma pathway involving BRAF gene mutations and MSI. However, MSI is detected in just 15% of all colorectal carcinomas and this pathway does not explain all KRAS wild-type colorectal cancer. Furthermore, the frequency of KRAS mutations in our microsatellite stable carcinomas was half the frequency found in adenomas or microsatellite stable ISCs.
Mechanisms leading to the marked differences in the frequency of KRAS mutation could arise on the level of the tumor, the cell, or the allele. For example: 1) A precursor adenoma with a KRAS mutation might fail to progress as frequently as those adenomas without the mutation. The reported frequency of tubulovillous and villous adenomas found on routine colonoscopic screening in a large study of men and women (ages 50-66 years) without a family history of CRC was reported to be 2.4%, while the frequency of CRC detection was 0.88%, or two to three times less frequent [20]. This crosss-ectional data suggests that not all adenomas advance to carcinomas, and the difference in frequency is similar to the difference in KRAS mutation rates we determined. This would suggest that an equal percentage of adenomas with and without KRAS gene mutation are destined to progress to a carcinoma. If this were true, it would imply there is no particular value of a KRAS mutation to the neoplastic process. Furthermore, in the cited study, the frequency of finding an ISC was significantly lower than that of cancers, suggesting further that ‘failure to progress’ is probably not the explanation for our findings.
2) On the cellular level, initially there could be two distinct populations of tumor cells in an adenoma, and progression beyond a certain point might involve loss of the KRAS mutated cells and growth of the cells with wild-type KRAS. Indeed, studies have demonstrated that not all areas of a colorectal tumor necessarily demonstrate identical KRAS results [21]. With regard to cellular growth, a study of melanomas reported that malignant cells with BRAF gene mutations do not outgrow the cells with wild-type BRAF, and one suggested explanation is that the cells with BRAF mutation undergo senescence [22]. Nonetheless, a colorectal tumor with both KRAS wild-type and mutated populations of cells is an infrequent finding, suggesting that two competing clones of tumor cells with one mutated and one wild is also an unlikely explanation for the large difference between adenomas and carcinomas with respect to KRAS mutations.
3) On the molecular level, the development of additional random changes might result in the loss of the allele with the KRAS mutation during the late stages of a tumor becoming a carcinoma. This mechanism is speculative. It implies that early in carcinogenesis all tumor cells in a particular tumor may contain a KRAS mutation, without a competing wild-type clone. The KRAS mutation confers a selective growth advantage to the cells at this point in tumorigenesis, and it is a ‘driver’ mutation. Using a mathematical model, Bozic et al showed that the selective advantage of a ‘driver’ mutation is very low, 0.4% [23]. As pointed out by others, this translates into a high ‘extinction rate’ for those mutations that confer a selective growth advantage: more than 99% of ‘driver’ mutations that emerge during tumor development will become extinct [24]. We would then postulate that at a later stage in the neoplastic process, the allele with the KRAS mutation is lost from the clonal cells, with retention of the wild type allele.
In conclusion, our data show that tubulovillous and villous adenomas, as well as both the benign and malignant parts of in situ carcinomas, are more likely to contain a somatic KRAS gene mutation than are colorectal carcinomas. This difference is confined to the non-mucinous and the microsatellite stable carcinomas. These epidemiological data support the possibility that non-mucinous and microsatellite stable carcinomas with wild-type KRAS gene may have had a mutation in the KRAS gene during their earlier stages, with the mutation lost during further growth of the tumor.
Acknowledgements
The authors wish to thank Dr. Errol Berman for review of histological slides, Dr. Ann Zauber for statistical assistance, and Dr. Tim Bishop for discussions regarding the findings. We also thank The Nussbaum Foundation of Saint Barnabas Medical Center and the Isermann Family Foundation for financial support.
References
- 1.Vogelstein B, Fearon ER, Hamilton SR, Kern SE, Presinger AC, Leppert M, Nakamura Y, White R, Smits AM, Bos JL. Genetic alterations during colorectal-tumor development. N Engl J Med. 1988;319:525–532. doi: 10.1056/NEJM198809013190901. [DOI] [PubMed] [Google Scholar]
- 2.Nowell PC. Tumor progression: a brief historical perspective. Semin Cancer Biol. 2002;12:261–266. doi: 10.1016/s1044-579x(02)00012-3. [DOI] [PubMed] [Google Scholar]
- 3.Li J, Mizukami Y, Zhang X, Won-Seok J, Chung DC. Oncogenic K-ras stimulates Wnt signaling in colon cancer through inhibition of GSK-3B. Gastroenterology. 2005;128:1907–1918. doi: 10.1053/j.gastro.2005.02.067. [DOI] [PubMed] [Google Scholar]
- 4.Jervoise H, Andreyev N, Norman AR, Cunningham D, Oates JR, Clarke PA. Kirsten ras mutations in patients with colorectal cancer: the muticenter “RASCAL” study. J Natl Cancer Inst. 1998;90:675–684. doi: 10.1093/jnci/90.9.675. [DOI] [PubMed] [Google Scholar]
- 5.Zauber NP, Sabbath-Solitare M, Marotta SP, Zauber AG, Bishop DT. Molecular changes in the Ki-ras and APC genes in colorectal adenomas and carcinomas arising in the same patient. J Pathol. 2001;193:303–309. doi: 10.1002/1096-9896(2000)9999:9999<::AID-PATH813>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 6.Stratton MR, Campbell PJ, Futreal PA. The cancer genome. Nature. 2009;458:719–724. doi: 10.1038/nature07943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Greenland S, Rothman KJ. Introduction to stratified analysis. In: Rothman KJ, Greenland S, Lash TL, editors. Modern Epidemiology. 3rd edition. Philadelphia: Lippincott Williams & Wilkins; 2008. pp. 258–282. [Google Scholar]
- 8.Karapetis CS, Khambata-Ford S, Jonker DJ, O’Callaghan CJ, Tu D, Tebbutt NC, Simes RJ, Chalchal H, Shapiro JD, Robitaille S, Price TJ, Shepherd L, Heather-Jane A, Langer C, Moore MJ, Zalcberg JR. K-ras Mutations and Benefit from Cetuximab in Advanced Colorectal Cancer. N Engl J Med. 2008;359:1757–1765. doi: 10.1056/NEJMoa0804385. [DOI] [PubMed] [Google Scholar]
- 9.Ogino S, Meyerhardt JA, Irahara N, Niedzwiecki D, Hollis D, Saltz LB, Mayer RJ, Schaefer P, Whittom R, Hantel A, Benson AB, Goldberg RM, Bertagnolli MM, Fuchs CS. KRAS mutation in stage III colon cancer and clinical outcome following intergroup trial CALGB 89803. Clin Cancer Res. 2009;15:7322–7329. doi: 10.1158/1078-0432.CCR-09-1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cusem EV, Kohne CH, Lang I, Folprecht G, Nowacki MP, Cascinu S, Shchepotin I, Maurel J, Cunningham D, Tejpar S, Schlichting M, Zubel A, Celik I, Rougier P, Ciardiello F. Cetuximab plus irinotecan, fluorouracil and leucovorin as first-line treatment for metastatic colorectal cancer: updated analysis of overall survival according to tumor KRAS and BRAF mutation status. J. Clin. Oncol. 2011;29:2011–2019. doi: 10.1200/JCO.2010.33.5091. [DOI] [PubMed] [Google Scholar]
- 11.Einspahr JG, Martinez ME, Jiang R, Hsu CH, Bhattacharrya AK, Ahnen DJ, Jacobs ET, Houlihan PS, Webb CR. Associations of Ki-ras proto-oncogene mutation and p53 gene overexpression in sporadic colorectal adenomas with demographic and clinicopathologic characteristics. Cancer Epidemiol Biomarkers Prev. 2006;15:1443–1450. doi: 10.1158/1055-9965.EPI-06-0144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ohnishi T, Tomita N, Monden T, Ohue M, Yana I, Takami K, Yamamoto H, Yagyu T, Kikkawa N, Shimano T, Monden M. A detailed analysis of the role of K-ras gene mutation in the progression of colorectal adenoma. Br J Cancer. 1997;75:341–347. doi: 10.1038/bjc.1997.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Barry ELR, Baron JA, Grau MV, Wallace K, Waile RW. K-ras mutations in incident sporadic colorectal adenomas. Cancer. 2006;106:1036–1040. doi: 10.1002/cncr.21721. [DOI] [PubMed] [Google Scholar]
- 14.Zauber NP, Sabbath-Solitare M, Marotta S, Zauber AG, Foulkes W, Chan M, Turner F, Bishop DT. Clinical and genetic findings in an Ashkenazi Jewish population with colorectal neoplasms. Cancer. 2005;104:719–729. doi: 10.1002/cncr.21230. [DOI] [PubMed] [Google Scholar]
- 15.Ogino S, Brahmandam M, Cantor M, Namgyal C, Kawasaki T, Kirkner G, Meyerhardt JA, Loda M, Fuchs CS. Distinct molecular features of colorectal carcinoma with signet ring cell component and colorectal carcinoma with mucinous component. Mod Pathol. 2006;19:59–68. doi: 10.1038/modpathol.3800482. [DOI] [PubMed] [Google Scholar]
- 16.Jass JR. Towards a molecular classification of colorectal cancer. Int J Colorectal Dis. 1999;14:194–200. doi: 10.1007/s003840050211. [DOI] [PubMed] [Google Scholar]
- 17.Jass JR, Biden KG, Cummings MC, Simms LA, Walsh M, Schoch E, Meltzer SJ, Wright C, Searle J, Young J, Leggett BA. Characterization of a subtype of colorectal cancer combining features of the suppressor and mild mutator pathways. J Clin Pathol. 1999;52:455–460. doi: 10.1136/jcp.52.6.455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Colomer A, Erill N, Vidal A, Calvo M, Roman R, Verdu M, Cordon-Cardo C, Puig X. A novel logistic model based on clinicopathological features predicts microsatellite instability in colorectal carcinomas. Diagn Mol Pathol. 2005;14:213–223. doi: 10.1097/01.pas.0000177800.65959.48. [DOI] [PubMed] [Google Scholar]
- 19.Jenkins MA, Hayashi S, O’shea A, Burgart LJ, Smyrk TC, Shimizu D, Waring PM, Ruszkiewicz AR, Pollett AF, Redston M, Barker MA, Baron JA, Casey GR, Dowty JG, Giles GG, Limburg P, Newcomb P, Young JP, Walsh MD, Thibodeau SN, Lindor NM, Lemarchand L, Gallinger S, Haile RW, Potter JD, Hopper JL, Jass JR. Pathology features in Bethesda Guidelines predict colorectal cancer microsatellite instability: a population-based study. Gastroenterology. 2007;133:48–56. doi: 10.1053/j.gastro.2007.04.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Regula J, Rupinski M, Kraszewska E, Polkowski M, Pachlewski J, Orlowska J, Nowacki MP, Butruk E. Colonoscopy in colorectal cancer screening for detection of advanced neoplasia. N Engl J Med. 2006;355:1863–1672. doi: 10.1056/NEJMoa054967. [DOI] [PubMed] [Google Scholar]
- 21.Losi L, Baisse B, Bouzourene H, Benhattar J. Evolution of intratumoral genetic heterogeneity during colorectal cancer progression. Carcinogenesis. 2005;26:916–922. doi: 10.1093/carcin/bgi044. [DOI] [PubMed] [Google Scholar]
- 22.Lin J, Goto Y, Murata H, Sakaizawa K, Uchiyama A, Saida T, Takata M. Polyclonality of BRAF mutations in primary melanoma and the selection of mutant alleles during progression. Brit J Cancer. 2011;104:464–468. doi: 10.1038/sj.bjc.6606072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bozic I, Antal T, Ohtsuki H, Carter H, Kim D, Chen S, Karchin R, Kinzler KW, Vogelstein B, Nowak MA. Accumulation of driver and passenger mutations during tumor progression. Proc Natl Acad Sci USA. 2010;107:18545–18550. doi: 10.1073/pnas.1010978107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Regoes RR. Population genetics meets cancer genomics. Proc Natl Acad Sci USA. 2010;107:18241–18242. doi: 10.1073/pnas.1013177107. [DOI] [PMC free article] [PubMed] [Google Scholar]