

Abstract
Experimental and clinical studies indicate that cells of the innate and adaptive immune system have both anti- and pro-tumor activities. This dual role of the immune system has led to a conceptual shift in the role of the immune system's regulation of cancer, in which immune-tumor cell interactions are understood as a dynamic process that comprises at least five phases: immunosurveillance, immunoselection, immunoescape, oncotraining, and oncopromotion. The tumor microenvironment shifts immune cells to perform functions more in tune with the tumor needs (oncotraining); these functions are related to chronic inflammation and tissue remodeling activities. Among them are increased proliferation and survival, increased angiogenesis and vessel permeability, protease secretion, acquisition of migratory mesenchymal characteristics, and self-renewal properties that altogether promote tumor growth and metastasis (oncopromotion). Important populations in all these pro-tumor processes are M2 macrophages, N2 neutrophils, regulatory T cells, and myeloid derived suppressor cells; the main effectors molecules are CSF-1, IL-6, metalloproteases, VEGF, PGE-2, TGF-β, and IL-10. Cancer prognosis correlates with densities and concentrations of protumoral populations and molecules, providing ideal targets for the intelligent design of directed preventive or anticancer therapies.
1. Introduction
Somatic cells are in constant risk of cancer transformation and organisms are endowed with surveillance mechanisms carried out by the immune system to control the generation of cancer cells. These mechanisms are (i) controlling infection by oncogenic pathogens, (ii) resolving local inflammation to prevent the establishment of a tumorigenic chronic inflammatory microenvironment, and (iii) eliminating potentially transformed cells. The AIDS pandemic and laboratory recombinant technologies have provided plenty of support to the Burnet and Thomas hypothesis of immunosurveillance (Figure 1) [1, 2]. However, increasing understanding of the relationship between the immune system and cancer points out that there is more than one side to this tale, with more recent evidence supporting a role for the immune system in promoting oncogenesis and tumor growth [3, 4]. This duality displayed by the immune system has led to the concept of an “immunological shift” in cancer, in which immune and transformed cells interact in a dynamic process comprising at least five phases: immunosurveillance, immunoselection, immunoescape, oncotraining, and oncopromotion. The first phase represents a functional immune system engaging in protective functions that successfully eliminates aberrant/malignant cells. In the second phase, an equilibrium is reached between tumor cells and immune cells; this phase depends on the mutational rate of the transforming cell, which creates rapidly proliferating clones, resistant to death and/or self-renewal capacities; thus, the immunosurveillance function is incapable of eliminating all aberrant cells and instead selects clones with increasingly tumorigenic properties. With time, the mutational rate/immune selection process allows cells to develop mechanisms to evade immunosurveillance resulting in an equilibrium shift favoring tumor growth. The immunologically shaped tumor clones take advantage of some immune functions to create a microenvironment in which immune cells are switched from anti- to pro-tumoral activities, in a collective mechanism referred to as oncotraining. Among the immune cell functions hijacked by the tumor stroma is the capacity to stimulate immune regulatory activities, turning off classical phagocytic and cytotoxic immune responses while promoting tissue remodeling functions. Together, these processes result in an oncopromoting phase favoring tumor growth, local invasion, and metastasis.
Figure 1.

Cancer immunosurveillance activity. Each immune cell fulfills unique and redundant functions to achieve tumor cell elimination. Among the anti-tumor activities found in the tumor microenvironment are cytotoxicity mediated by CD8+ T and NK cells, phagocytosis by M1 macrophages, cytolysis induced by mast cells, and humoral responses by B cells. Dendritic cells are primed by tumor antigens, which are then presented to T and B cells for adaptive responses. These activities are coordinated by a variety of molecules secreted by the immune and tumor cells directly to the tumor environment or to circulation where it serves to recruit additional immune populations to the tumor site (see text for details). Red dashed arrows represent direct anti-tumoral effects, blue dashed arrows indirect tumor cell elimination. Dotted arrows represent recruitment of other cell populations.
The term immunoediting has recently been coined to describe the dual role of the immune system in cancer. However, this term mostly reflects the differences between tumors developing in immunocompetent or immunodeficient mice [5], and therefore the role of the immune system in selecting tumor clones with evolutionarily advantages. Immunoediting is divided into three stages, (1) elimination, (2) equilibrium, and (3) escape, mainly reflecting the close relationship between immunogenic properties of the cancer cell and the responses they trigger. The last two stages concern the selection of clones in which the cancer immunogenic determinants have been edited to remain invisible to immunosurveillance mechanisms. However, the immune response does more than selection and can directly participate in every step of the carcinogenic process even directly switching off immunosurveillance. Considering the steady increase in the frequency of cancers observed in recent years, an in-depth understanding of the mechanisms of immunological shift is needed. In this paper, we present evidence regarding the pro-tumoral roles of the immune system and discuss how the immune system can be instructed by the tumor stroma to exhibit cancer-promoting functions.
2. Anti-Tumoral Immune Responses
Dendritic cells (DCs), macrophages, and mast cells (MCs) constitutively reside in physiologically normal tissues acting as sentinels that monitor the microenvironment in search of stress signals; when tissue homeostasis is compromised they release cytokines, chemokines, reactive oxygen species (ROS), and bioactive mediators, which among many other functions induce mobilization and infiltration of other leucocytes to the injured site in the process of inflammation. In an inflamed tissue, innate immune cells perform diverse and redundant tasks when activated; for instance, both MCs and granulocytes release their preformed granules to kill or inactivate invasive agents, while macrophages, neutrophils, and DCs carry out phagocytosis. NK cells recognize and kill either virus-infected or malignantly transformed cells. Also, all of these populations act as antigen-presenting cells (APCs), although this is the main function of DCs. Macrophages and DCs, that have engulfed the aggressor, mobilize to lymphoid organs to present antigens to adaptive immune cells and the combined action of innate and adaptive immunity leads to the elimination of stress stimuli and resolution of tissue damage.
MCs are present throughout all tissues in which they are traditionally known to function in the first stages of inflammation and during allergic responses. MCs are activated after ligand binding via the Fcγ, complement and/or pathogen-associated molecular patterns (PAMP) receptors, releasing bioactive molecules such as histamine, proteases, lipid mediators, cytokines, and chemokines. These molecules are required for direct pathogen killing, recruitment of immune cells, increased angiogenesis, vascular permeability, and degradation of the injured tissue. To date, the participation of MCs in immunosurveillance is controversial, with some of their bioactive molecules reported to have direct anti-tumor activities; for instance, connective tissue MCs are enriched for granules containing tryptase and chondroitin sulphate that may promote a strong local inflammatory response and inhibit metastasis, respectively [6]. Glycoprotein interactions between tumor cells and the tissues they invade are essential for metastasis. Chondroitin sulphate expressed in the tumor stroma competes with these interactions stopping tumor cells from leaving the primary tumor site [7]. Also, histamine is thought to increase the synthesis of prostacyclin, a potent antimetastatic factor in endothelial cells. MC-released TNF-α, IL-1, and IL-6 inhibit tumor growth and angiogenesis in melanoma [8, 9]. MC-derived chemokines recruit phagocytic and cytotoxic cells, and the MC-mediated recruitment and survival of eosinophils had a tumor regression effect in a mouse melanoma model [10]. Interestingly, a direct MC phagocytic activity against tumor cells was observed in invasive ductal breast cancer samples [11].
Neutrophils are the most abundant circulating polymorphonuclear (PMN) granulocyte; they search for chemotactic signals to direct them to sites of infection or injury. Although neutrophils' half-lives are only of a few hours, they survive much longer in an inflammatory microenvironment. Like MCs, this lineage protects against invading microorganisms and assists in wound healing through releasing of a wide variety of effector molecules stored in cytoplasmic granules, including proteases such as neutrophil elastase, cathepsin G, proteinase-3, metalloproteases 8 and 9 (MMP-8 and -9), antimicrobial molecules such as defensin peptides and ROS, and a number of cytokines (TNF-α, IL-1β, IL-8, IL-12, among others) [12, 13]. Whether PMNs are able to effectively target their cytotoxic effects on tumor cells is still controversial. Several reports support a polarization of neutrophils to adopt a highly active anti-tumor profile, in which they produce high amounts of proinflammatory cytokines [14–16]. These anti-tumor “N1” neutrophils synthesize higher levels of TNF-α, MIP-1α, H2N2, and NO2 and show cytotoxic activity against tumor cells both in vivo and in vitro [17]. However, Gregory and Houghton argue that the increase in cytotoxic activity does not represent a transcriptional switch but rather shows that neutrophils can be activated to various degrees in response to different stimuli [18].
Basophils and eosinophils represent approximately 4% of blood PMNs and play essential roles in allergic and antiparasitic responses; they are not usually present in tissues but are recruited to inflammatory sites. The main content of basophil granules are histamine, chondroitin sulphate, and proteases, while eosinophil granules contain mostly basic major protein, eosinophil cationic protein, peroxidases, hydrolases, and phospholipases. Although both cell types are recruited to tumor stroma, the functions in this environment have not been elucidated yet. Epidemiologic studies have found an inverse relationship between cancer and both allergic disease and parasitic infections, suggesting a protective function for the granulocytes activated by these stimuli [19]. Similarly, an inverse association has been found between IgE levels, the predominant antibody isotype present in allergic and antiparasite responses, and cancer [20]. High- and low-affinity IgE receptors (FcεRI and CD23, respectively) are present in several populations of the immune system, suggesting several possibilities for anti-tumorigenic functions.
Macrophages are the main phagocytic cell lineage of the immune system and are classified according to the type of response in which they participate [21]. Classically activated (M1) macrophages are activated in response to a microenvironment enriched with Th1 cytokines (IFN-γ, GM-CSF, IL-12, ROI, RNI, iNOS, and CXCL10). In these cells, IFN-γ and Toll-like receptor (TLR) ligands activate the NFκB signaling pathway turning on a Th1 transcriptional program characterized by high expression of MHC/HLA, IL-12, TNF-α, ROS, and NO. Alternatively activated (M2) macrophages are formed in response to Th2 cytokines (IL-4, IL-10, IL-13, M-CSF, CCL2, CCL5, CCL22, and HIF-1α) and are characterized by the expression of JMJD3, arginase-1, YM, and FIZZ1 genes and secretion of IL-4, IL-10, and IL-13 upon activation, an expression/secretion profile more in tune with tissue remodeling activities. Macrophages often constitute the most abundant innate immune lineage in the tumor mass and their phagocytic activity remains one of the most important immune anti-tumor functions.
NKs have the ability to recognize and lyse a variety of abnormal cells, including tumor, virus infected, antibody bound, allogeneic, and stressed cells. Recognition of target cells by NKs is achieved through inhibitory or activating receptors expressed on plasma membrane and the lysis of the targets occurs only when activating signals outweigh inhibitory signals. There are three known inhibitory receptor families on NK cells surface that recognize MHC-I/HLA molecules: killer cell immunoglobulin-like receptor (KIR) in humans, Ly49 in mice, and CD94/NKG2A present in both human and mice. In agreement with the hypotheses of “missing-self” and “induced-self”, these receptors are very important to the NK anticancer response, since low amounts or absence of MHC-I/HLA molecules tends to be a characteristic of cells undergoing malignant transformation or viral infection [22, 23]. NK cells have two lytic mechanisms: (i) release of granzyme and perforin and (ii) induction of apoptosis through release of TNF ligands. Additionally, NK cells have a number of other anti-tumor activities: IFN-γ release inhibits tumor cell proliferation in vitro; release of antiangiogenic factors and DC activation can elicit a T-cell-mediated response [24].
DCs are constitutive residents of skin and mucous membranes where they rapidly respond to microenvironmental signals, turning into mature DCs capable of antigen capture and cross-priming to naïve B and T lymphocytes. DCs collect tumor antigens either from phagocytized tumor cells or through a direct mechanism of capture from living tumor cells; hence they are essential initiators of anti-tumor adaptive immune responses. Tumor-derived antigens presented to B and T lymphocytes trigger high-affinity responses with the capacity to generate immunological memory. Upon activation, CD8 cytotoxic T cells (CTLs) directly eliminate tumor cells while CD4 T helper cells (Th) stimulate B cells supporting both humoral and cytotoxic responses. Antibodies against tumor antigens may directly inhibit tumor cells or mark them as targets of a complement or antibody-mediated cellular activities. Th cells can be classified into four main subtypes: Th1, which mainly secrete IFN-γ, TNF-α, and IL-12, Th2 secrete IL-4, IL-5, and IL-13, Th17 secrete IL-17 and IL-22, and regulatory T cells (Tregs) that secrete IL-10 and TGF-β (transforming growth factor-beta) [25]. Macrophages are also activated by IFN-γ; thus Th1 responses are defined by cytotoxic and phagocytic activity. Th1, together with Th17, responses are important to establish an inflammatory microenvironment. A significant increase in neoplasias is observed in patients who are CD8+ T cell deficient, supporting the key role of CTLs in tumor surveillance.
Tregs are perhaps the most important immunomodulatory population. Tregs turn off inflammatory and humoral responses after the trigger signal has been eliminated, thus preventing chronic immune stimulation and autoimmunity [26]. IL-10 and TGF-β, the most important Treg-secreted cytokines, have a powerful immunosuppressive activity; these cytokines induce arrest in cell cycle of cytotoxic T cells and block DCs maturation, among many other functions [27].
3. The Role of Chronic Inflammation in the Pro-Tumoral Immune Response
Tumor initiation and progression are governed by intrinsic mechanism, such as aberrant expression of oncogenes or tumor suppressor genes. Increased evidence points to a critical role for external signals mainly given by immune cells infiltrating chronically inflamed tissue. Injured tissues trigger immune cell recruitment and cytokines, growth factors and other factors secreted by immune cells often promote oncogenic changes, thus creating a positive loop between inflammation and cancer (see Figure 2) [28]. Multiple examples of persistent infections support the crucial role of chronic inflammation in oncogenic processes. In gastric cancer associated with Helicobacter pylori infection, oncogenic transformation evolves through progressive stages starting with an inflammatory gastritis, followed by metaplasia, dysplasia, and finally cancer [29]. Similar events have been related to liver cancer associated with hepatitis B and C virus infection. Also, inflammatory autoimmune processes, such as Bowel's disease and prostatitis, trigger the appearance of colorectal and prostate cancer, respectively [3]. An important example of a mutagenic factor frequently enriched in an inflammatory microenviroment is ROS (e.g., oxygen ions and peroxides), which results from the oxidative stress induced by phagocytic cells. ROS are highly reactive molecules that damage DNA augmenting the cell mutation rate, thus favoring the appearance of clones with oncogenic properties or selecting cancer clones with more malignant characteristics [30, 31].
Figure 2.
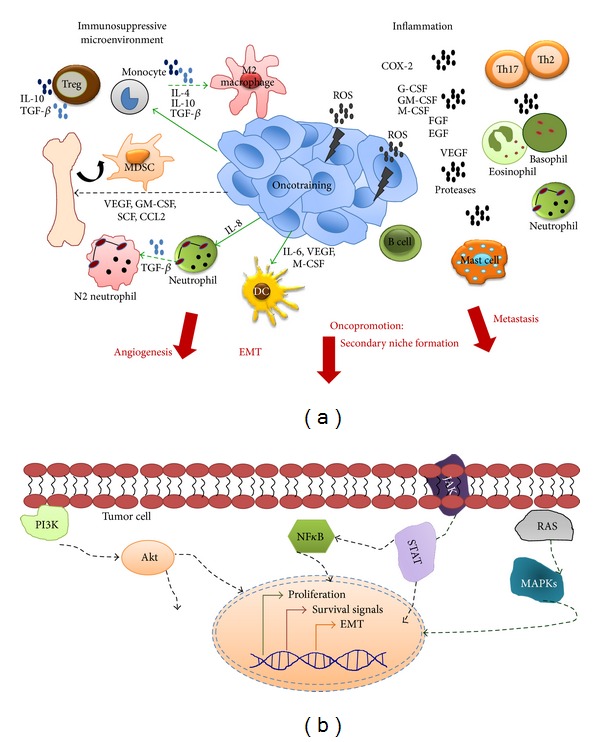
Pro-tumoral activities of the immune system. (a) Soluble factors secreted by tumor and immune cells create a microenvironment in which arriving and local immune cells are (i) inactivated creating immunosuppressive conditions, (ii) maintaining inflammation, and/or (iii) switched from anti- to pro-tumoral activities. Tregs, M2 macrophages, N2 neutrophils, and MDSCs are among the most important immunosuppressive populations and IL-10 and TGF-β the main cytokines contributing to this microenvironment. A chronic inflammatory microenvironment contributes to oncogenesis and tumor growth through secretion of mutagenic (e.g., ROS) or inflammatory molecules (e.g., COX-2). Almost all innate immune populations contribute to inflammation plus Th17 and Th2 T and B cells. The anti- to pro-tumoral switch refers to a mechanism in which the tumor microenvironment reprograms or trains the immune cells to perform activities more in tune with the tumor needs (oncopromotion). Polarization to M2 macrophages and N2 neutrophils are perhaps the most studied examples of this process. Among the important molecules for inflammation, oncotraining, and oncopromotion are G-CSF, GM-CSF, and M-CSF (for immune cell recruitment), VEGF (for angiogenesis), proteases (matrix degradation), and TGF-β (for EMT). Overall this mechanism contributes to tumor growth, invasion, formation of distant pro-tumoral niches and metastasis (oncopromotion). (b) Intrinsic changes in tumor cells in response to the tumor microenvironment. Signaling from receptors to growth factors, interleukins and other inflammatory molecules activate many pathways. Among the most important are MAPKs and STATs triggering proliferation (e.g., in response to FGF, EGF, HGF and some cytokines), NFκB and PI3K triggering cell survival (e.g., in response to interleukins). Also, one of the most critical mechanisms contributing to tumor malignancy is the transition from epithelial to a more mobile mesenchymal phenotype (EMT) (see text for a detailed explanation).
Around 95% of adult cancers are of epithelial origin (carcinomas); epithelium is the outermost layer of organs and functions as a protector barrier against environmental aggressors—chemical (e.g., tobacco components), physical (sun ultraviolet radiation), or biological (infections). An early response of damaged tissues is production of IL-8 by the epithelial cell itself, which together with histamine and TNF-α, secreted by resident mast cells and macrophages allows neutrophil extravasation to the injured sites, initiating inflammation [32]. Epithelial cells of the gastric mucosa secrete IL-8 in response to Helicobacter pylori infection. Interestingly, this is not just a response to the bacteria gastric colonization but to the presence of the most virulent bacterial factor, the oncoprotein CagA [29].
The leukocyte infiltrate varies in amount, composition, and distribution and each of its components favors cancer at distinct levels. As early as in the 18th century Rudolf Virchow observed that tumors usually have a large leukocyte infiltration. Among malignant characteristics promoted by inflammatory mediators are increased cell proliferation and survival, angiogenesis and vessels permeability, loss of anchorage dependence, and acquisition of a migratory mesenchymal phenotype, the so-called epithelial-mesenchymal transition (EMT), which promotes cancer cell invasiveness and metastasis [33, 34]. Also, tumor cells gain stemness characteristics (e.g., self-renewal properties), which often correlates with treatment resistance and high frequency of relapse. Most of these inflammatory pro-tumoral processes are characteristics of tissue repair mechanisms; as it was originally postulated by Virchow: “cancer is a healing process that never stops.”
Among the inflammatory factors promoting proliferation are TGF-β, fibroblast growth factor (FGF), and epithelial growth factor (EGF). TGF-β is synthesized by mast cells, macrophages, and lymphocytes as an inactive precursor that is activated by proteases in inflammatory microenvironments. TGF-β promotes mesenchymal cell proliferation and facilitates tumor invasion and metastasis of cells that have gone through EMT [35, 36]. The FGF family is a mitogenic trigger of fibroblasts, endothelial and epithelial cells, especially during a healing process. Overexpression of Her2/neu, a member of the EGF receptor family, contributes to over 50% of human breast cancer cases and might also participate in gastric cancer and aggressive forms of uterine cancer [37–39]. Activated macrophages are an important source of both FGF and EGF. IL-6 has also been depicted as a proliferative factor in several types of cancers, and it has been associated with poor prognosis in prostate cancer [40, 41]. Several chemotactic factors such as G-CSF, GM-CSF, and M-CSF can also induce proliferation of innate immune cells at inflammatory sites [42, 43].
Several cytokines, chemokines, growth factors, and TLR ligands trigger signaling cascades that activate the NFκB transcription factor. NFκB is a master regulator of immune responses; it triggers expression of proinflammatory cytokines, adhesion molecules, enzymes, such as cyclooxygenase 2 (COX-2), iNOS, metalloproteases, and angiogenic factors [44]. The NFκB pathway is also an important mediator of cell survival by inducing BCL-2 and Bcl-xL protein expression [45–47]. Nowadays, NFκB and PI3K are recognized as the main mediators of cell survival at inflammatory sites. PI3K is activated by receptors with, or associated with, tyrosine kinase activities (IL-1, -2, -3, -4, -6, and EGF receptors). Activated PI3K phosphorylates and converts phosphatidylinositol (4,5) bisphosphate (PIP2) into phosphatidylinositol (3,4,5) triphosphate (PIP3), which recruits protein kinases such as Akt to membrane-bound signaling complexes. Akt is an important inhibitor of pro-apoptotic proteins, such as BAD and caspase-9, among many other functions [48].
Growth factors often target endothelial cells promoting formation of new blood and lymphatic vessels and fulfilling a critical role for tumor maintenance and growth. Vessel formation helps the arrival or exit of O2, nutrients, inflammatory factors, and immune cells. Some examples of angiogenic factors are vascular endothelial growth factor (VEGF), EGF, FGF, and hepatocyte growth factor (HGF) [49]. VEGF expression is induced by hypoxic conditions and its principal regulator is the hypoxia inducible factor (HIF-1). HIF-1 is a complex of the α and β subunits; although both subunits are constitutively expressed, the α subunit is constitutively hydroxylated, ubiquitinated, and targeted to the proteasome for degradation in response to O2. Therefore, hypoxic conditions suppress HIF-1α degradation, promoting stability of the HIF-1 transcriptional complex and resulting in VEGF expression [50, 51]. VEGF promotes the formation of new capillaries by triggering endothelial cell proliferation from preexisting vessels or attracts bone marrow endothelial precursors triggering their differentiation and proliferation [52, 53]. VEGF is also a vasodilator that augments vessel permeability helping the interchange of molecules and cells between the tumor and distant sites. Thus, angiogenesis also facilitates tumor metastasis. VEGF concentration in serum and the number of tumor vessels correlate with cancer prognosis [54]. Currently, VEGF inhibitors are used as therapeutic agents in clinical trials against several neoplasias [3].
Protective barrier functions conferred by epithelial cells depend on cell polarization and formation of protein structures that permit intimate attachments between neighboring cells. These properties are critical for tissue integrity allowing epithelial cells a strong anchorage. One of the most important events in tumor progression is the loss of epithelial features (e.g., cell junction structures, such as E-cadherin, keratin 18, occludins and claudins), and acquisition of more mobile mesenchymal properties (the EMT). It is thought that EMT facilitates detachment of the tumor cell in the primary tumor site, migration and colonization of secondary organs [55, 56]. Inherited mutations in E-cadherin are associated with familial forms of gastric and breast cancer [57, 58]. Moreover, oncogenic pathogens often target this protein [59, 60]: Helicobacter pylori oncoprotein CagA interacts with E-cadherin blocking its binding to β-catenin; as a result, β-catenin accumulates in nucleus where it transactivates gene p21WAF1 important for cell cycle progression, and CDX1, which is associated with intestinal metaplasia [61, 62]. CagA-induced E-cadherin loss also correlates with gain of the mesenchymal markers vimentin and fibronectin [63]. Adenovirus 5 destroys cell junctions through interactions with CAR receptor to promote viral exit [64]. Similar mechanisms have been proposed for HPV16 and HBV supporting the importance of E-cadherin loss and EMT [65, 66]. Some inflammatory triggers of EMT are TNF-α, TGF-β, IL-6, FGF, and EGF [36, 67, 68].
Epithelial cells are attached to a basement membrane and extracellular matrix (ECM) of connective tissue, and degradation of both by proteolytic cascades is a key mechanism for tumor cell invasion of surrounding tissues and eventual metastasis. Some of the most important inflammatory proteases are metalloproteases-2 and -9 (MMP-2, MMP-9); serine-, aspartic-, and cystein-proteases (urokinase-like plasminogen activator or uPA, cathepsin D and B, respectively) [69, 70]. MMP-9 is overexpressed in lobulillar breast cancer [71]. Cathepsin B is found at the tumor invasive border supporting its importance for tumor spread [72]. Proteases also activate growth factors and interleukin zymogens. TGF-β and plasminogen zymogens are among the most important uPA targets, and high uPA levels are associated with bad prognosis in breast and ovarian cancers [73–75]. In agreement, ECM degradation and cell invasion are reduced by inhibition of cysteine proteases [76, 77]. In murine models of cervix, skin, and pancreatic cancers, VEGF is sequestered by ECM components and angiogenesis only occurs in the presence of metalloproteases [78, 79]. Inhibition of MMP-9 expression by bisphosphonates significantly reduces metastasis to bone in a breast cancer model [80]. Thus, increased expression of proteases and concomitant basement membrane/ECM degradation, EMT, and angiogenesis, all together facilitate tumor metastasis and are poor prognosis markers.
The “seed and soil” hypothesis postulated by Steven Paget in 1889 proposed that tumor cells are systematically released to circulation from the primary site and distributed throughout the body; however, they generate secondary tumors only upon arrival to specific organs. This hypothesis is currently understood as the release of soluble factors by the primary tumor creating the optimal conditions for tumor growth at distant sites. Many functions are fulfilled for those fertilizing factors, for example, recruitment of innate immune cells to secondary sites. In a mouse model of breast cancer, RANKL is secreted to circulation by regulatory T cells, promoting migration of RANK-expressing tumor cells to the bone [81]. Also, VEGFR-1+ and VLA-4+ hematopoietic progenitors migrate from bone marrow to specific tissues prone to secondary growth [82, 83]. In summary, the arrival of soluble factors and different cell populations helps to create a permissive microenvironment for tumor cell colonization, the premetastatic niche, where a secondary tumor will be formed upon arrival of circulating cancer cells.
Proof of the immune cells pro-tumoral role is the positive correlation between their densities at tumor sites and disease prognosis, for example, macrophage and MC numbers correlate with tumor vascularization [84, 85]. In a murine model, attenuation of innate immune cell infiltration prevented the switch from a premalignant to a malignant lesion [86]; also, mice lacking MCs exhibit decreased tumor growth [87]. A similar effect is achieved by blocking macrophage chemotaxis to the tumor with an M-CSF receptor inhibitor [88]. CSF1-deficient mice also show a decreased breast tumor growth and a reduced metastasis to lungs [89]. In humans, CSF1 levels have been shown to correlate with macrophage, tumor density, and poor prognosis [90]. Taken together, all these data argue for an important pro-tumoral role of inflammatory cells infiltrating the tumor stroma. In agreement, COX-2 is an inflammatory protein that is upregulated in several cancers in which it has been associated with bad prognosis [91, 92]. Individuals with excessive blood clotting are frequently treated with periodical amounts of COX-2 inhibitors; these individuals have shown lower rates of breast, colon, lung, and prostate cancer [93, 94].
In summary, the immune system is of great help to control cancer through immunosurveillance mechanisms, but it can also trigger cancer promoting mechanisms, and the fine line between both activities is not well defined. In a recent study in mice, injection of flagellin, a TLR5 and NAIP5 ligand, was sufficient to clear tumor cells in a macrophage- and CD8+ T cell-dependent manner [95]. It is possible that chronic—rather than acute—inflammation, as it occurs in response to persistent infections, is more in tune with cancer promotion. Therefore, inflammation is recognized as the seventh hallmark of cancer [30].
4. Cancer Immunological Shift
4.1. Macrophage Oncotraining
There is increasing evidence supporting that the tumor environment instructs cells of the innate and adaptive immune system to shift from anti- to pro-tumoral activities. In this oncotraining process, immune cells entering the tumor stroma lose important anti-tumor activities (e.g., cytotoxicity and phagocytosis), while other immune processes are promoted (e.g. tissue repair activities such as increased cell proliferation and survival, angiogenesis, and EMT) (Figure 2). Tumor-associated macrophages (TAMs) can be phenotypically and functionally divided into two subtypes: M1 or classically activated (by Th1 cytokines) and M2 or alternatively activated (by Th2 cytokines) also referred to as killer or healer, respectively [3]. M1 TAMs have immunostimulatory activities; they are accomplished antigen-presenting cells and secrete Th1 cytokines promoting T cell cytotoxic functions, while M2 TAMs are immune suppressors that in homeostatic conditions participate in tissue maintenance and regeneration in case of damage. M2 TAMs also produce CCL22 that attracts Treg cells, which help to maintain an immunosuppressive environment. A shift from M1 to M2 activities normally occurs in an infection episode upon pathogen eradication, and it is thought that in tumoral microenvironments M1 macrophages are often redirected towards M2 functions.
M2 TAMs promote angiogenesis (by secreting VEGF, angiopoietins 1 and 2, GM-CSF, and EGF), invasion (by secreting proteases MMP-1, -2, -9, cathepsin B and D), and chronic inflammation (by secreting COX-2) [96]. They also protect tumor cells from chemotherapy-induced apoptosis [97]. High densities of M2 TAMs significantly correlate with cancers of poor prognosis; in histological sections of invasive tumors M2 TAMs are preferentially located in areas of basement membrane degradation and increased protease secretion [98–101]. In agreement, knockout mice for the primary tumor macrophage chemoattractant, CSF-1, have a slow tumor growth, low progression to invasive stages, and reduced metastasis [89]. CSF-1 levels are also associated with poor prognosis in patients with breast, ovarian, endometrial, prostate, liver, and colon cancers, and also in several hematological malignancies [97, 102–104].
M2 macrophages also inhibit effector activities of several immune populations, such as M1 macrophages, NK, and cytotoxic T cells, by suppressing the expression of IFN-γ and IL-12. IL-10 and TGF-β are the main immunomodulatory cytokines and are important effector molecules of M2 macrophages [105]. M2s are normally activated by the resident gut microflora preventing autoimmune colitis [106, 107]. Resident flora-activated M2s secrete TGF-β promoting differentiation of T cells into Tregs and triggering apoptosis of activated T cells [108, 109]. Prostaglandin-2 (PGE-2) is secreted by M2 TAMs activated by COX-2 in an inflammatory microenvironment, and PGE-2 also controls differentiation and activation of Tregs [110].
It is documented that TAMs exhibit a high level of plasticity and can shift from M1 to M2 and vice versa, according to the environmental conditions [105]. Condeelis and Pollard showed that there is direct communication between macrophages and tumor cells through receptor-ligand interactions, mainly EGFR-EGF and CXCR4-CSF-1 [111]. Taking all these data together support a model in which macrophages arrive to the tumor site, in which their differentiation into M2 pro-tumor cells is promoted. In this scenario, the cells in the tumor stroma help to create an environment in which immune cells are reprogrammed and shifted to functions more in tune with the tumor needs. The consequences of this pro-tumoral shift are immune cells triggering the tumor malignant characteristics, such as increased growth, invasiveness, and metastasis (immune-cell-induced oncopromotion) (see Figure 2). Hence, several authors have proposed to direct anti-tumor therapies against immune modulators of oncotraining and oncopromotion. In this example, drugs directed against M2 macrophages should include inhibiting CSF-1, TGF-β, or PGE-2 [112].
4.2. Oncotraining of Other Innate Immune Populations
Neutrophils are attracted to the inflammatory site by IL-8 released from the epithelial cell itself in response to oncogenic stress, for instance Helicobacter pylori oncoprotein CagA translocation or expression of an oncogenic RAS mutant [113]. Similar to macrophages, neutrophils are polarized in the tumor microenvironment from an anti-“N1” to a pro-tumoral “N2” phenotype, and this shift is mainly regulated by TGF-β. Neutrophil depletion with antibodies in experimental models reduced angiogenesis, tumor growth, and metastasis [114]. Accordingly, high densities of neutrophils in renal cell and bronchioalveolar carcinomas are associated with bad prognosis [115, 116]. Interestingly, IL-8 levels also correlate with neutrophil density and reduced survival [113].
Although neutrophils secrete ROS and matrix degrading proteases, it is not clear how they promote tumor growth. Granulocytes are released from bone marrow as mature cells, but in inflammatory processes their myelocytes and promyelocytes precursors also exit to peripheral circulation. Kowanetz and colleagues showed that lung cancer cells secrete G-CSF and mobilize immature cells to premetastatic niches [117]. In these niches, granulocytes secrete prokinectin 2 to attract tumor cells and MMP-9 and induce neighbor cells to secrete VEGF to facilitate the arrival of tumor cells [43]. Granulocytes promote tumor cell proliferation in response to growth factor PDGF [118]. In models of lung adenocarcinoma and mesothelioma, TGF-β favors neutrophil N2 accumulation, induces arginase-1 expression, and inhibits TNF-α, CCL3, and ICAM-1 production. N2 neutrophil elimination allows an increase in the activity of cytotoxic T lymphocytes [17].
Many tumors are surrounded by mast cells brought to the tumor by SCF (stem cell factor) and other inflammatory chemoattractants, and MCs secrete inflammatory cytokines that in some cases favor tumor growth, angiogenesis, and metastasis [119]. MCs participate in tissue remodeling: activating NFκB through inflammatory mediators, which increases cell survival, and also through suppression of T and NK cell cytotoxic activities [120]. In experimental models, MCs play a decisive role triggering the angiogenic switch preceding malignant transformation, and MC-induced angiogenesis also favors tumor progression in human cancers [121]. Tumor progression stops or is reduced in the absence of MCs [122]. Thus, mast cells and N2 neutrophils have also been proposed as a directed target for cancer therapy [123, 124].
The tumor stroma can also suppress immune effector functions without promoting pro-tumoral activities. Different studies support that hypoxia, accumulation of extracellular adenosine and lactate, VEGF, M-CSF, and IL-6, all together, cooperate to inhibit DCs antigen-presenting activity [125–127]. DCs isolated from early stages of mouse ovarian cancer are immunocompetent, but in advanced tumors they fail to activate T cells, correlating with high levels of expression of PDL1, HIF-1α, and A2B adenosine receptor [128]. DCs and M2 TAMs have elevated levels of arginase activity [122]. TGF-β-enriched microenvironments are also critical for DCs immunosuppression. DCs with high expression of IDO (indoleamine 2,3-dioxygenase) favor the recruitment of T cells; however, the local TGF-β induces their differentiation into Tregs, generating an immunosuppressive microenvironment that favors tumor progression [43, 129]. According to these data, depletion of DCs at early time points of tumorigenesis favors tumor progression, but it has a therapeutic effect in advanced tumors.
4.3. Oncotraining of Adaptive Immune Cells
70% of solid tumors contain high densities of B lymphocytes, and in preneoplastic lesions (e.g., hyperplasia) they tend to be the most abundant population [130]. The presence of lymphoid follicles in some tumors indicates that the tumor stroma is a center of B cell activation. Furthermore, a positive correlation has been found between the number of B cells secreting IgG or IgM and poor prognosis [131, 132]. To date, it is not clear whether this is a bonafide pro-tumoral mechanism or it is just an indirect effect of the inflammatory microenvironment [133].
A large percentage of solid tumors also have high densities of T cells correlating with either good or bad prognosis [134]. It is proposed that the ratio of CD4+ to CD8+ T cells is a reliable indicator of prognosis: CD4/CD8 values >1 correlate with poor prognosis and ≤1 with a better prognosis [135]. IFN-γ secreted by CD8+ T cells helps M1 macrophage polarization and also triggers cytotoxic (by the CD8+ T cell itself) and phagocytic (by the M1 macrophage) activities [4]. Th1 CD4+ T cells secrete Th1 cytokines enhancing cytotoxic/phagocytic activities, while Th2 CD4+ T cells secrete pro-inflammatory cytokines (IL-4, IL-5, IL-6, and IL-13), which inhibit Th1 responses and along with Th17 CD4+ T cells promote inflammatory conditions [33]. Treg cells negatively regulate all types of immune responses, innate and adaptive, Th1, Th2, or Th17 [136]. Therefore, CD8 or CD4 Th1 cells favor anti-tumor activities, while Th2, Th17, and Treg populations favor pro-tumor and/or immunosuppressive conditions.
Tregs are characterized by the expression of CD4, CD25, and FOXP3 and two populations have been reported: (i) natural Tregs that mature in thymus and (ii) inducible Tregs, whose differentiation into Tregs is promoted in TGF-β-enriched inflammatory environments [137]. The importance of Tregs as pro-tumoral enhancers is based on in vivo depletion experiments using anti-CD25 antibodies, in which tumor regression was observed [138]. Tregs exert their immunomodulatory role by secreting TGF-β and IL-10 immunosuppressive cytokines, and through cell-cell interactions, mainly via CTLA4. IL-10 and TGF-β arrest cell cycle of cytotoxic T cells and block DCs maturation [27]. There is accumulative data that IL-10 is an inhibitor of the JAK/ STAT and NFκB signaling pathways and thereby of expression of IFN-γ, IL-2, IL-3, TNF-α, and GM-CSF immunostimulatory cytokines [139, 140]. CTLA4 is a T cell inhibitor. T cell activation occurs upon interaction of the TCR and antigenic peptides presented by MHC/HLA molecules in APCs plus CD28 and CD80 or CD86 costimulatory interactions. CTLA4 is also a ligand of CD80 and CD86, thus it competes CD28-CD80/86 interactions inhibiting T cell activation [136]. The CTL4 antagonist, ipilimumab, is currently being used in cancer therapy [141, 142].
4.4. Myeloid-Derived Suppressor Cells Oncopromotion Mechanisms
Myeloid-derived suppressor cells (MDSC) are a heterogeneous population of macrophages, granulocytes, DCs, and other early myeloid precursors, which are powerful suppressors of immune cells. Precursor cells with the same phenotype as MDSCs are continuously generated in bone marrow of healthy individuals, in which they differentiate into mature myeloid cells without immunosuppressive activity [143]. Large numbers of CD34+ myeloid precursors are present in peripheral blood of patients with head and neck carcinoma and in murine models of lung cancer [144–146]. MDSCs have the ability to suppress innate and T-cell-adaptive immune responses and also to promote tumor angiogenesis, invasion, and metastasis. Two main MDSC populations have been characterized in mice: monocytic MDSCs (CD11b+ Gr-1lo) and polymorphonuclear MDSCs (CD11b+ Gr-1hi), also known as granulocytic MDSCs. In cancer patients MDSC are typically Linneg CD11b+ CD33+ CD34+ CD14neg HLA-DRneg and can vary in their expression of CD15 and other markers [147–149]. Manipulating G-CSF concentration in the tumor microenvironment results in increased accumulation of granulocytic MDSC and tumor growth (high G-CSF) or vice versa [150].The presence of these populations in cancer patients and animal models supports the idea that the tumor microenvironment favors their recruitment and their varied phenotype suggests that different tumors are likely to recruit different subtypes of MDSCs.
The activation and expansion of MDSCs is influenced by factors released by tumor and stromal cells. Within the tumor microenvironment, COX-2, prostaglandins, SCF, CCL2, GM-CSF, M-CSF, VEGF, CXCL5, calcium-binding pro-inflammatory proteins S100A8 and S100A9, and TNF, all favor the chemotaxis and expansion of immunosuppressive MDSCs [151]. STAT3 is arguably the master regulator of MDSCs survival and proliferation, probably through augmenting transcription of BCL-xL, cyclin D1, MYC, and survivin. Persistent activation of STAT3 in myeloid progenitors prevents their differentiation into mature cells and together with the induction of proliferation favors their continuous presence in tumor microenvironments [143, 152].
MDSCs immunosuppressive function is activated by IFN-γ, TLR ligands, IL-13, IL-4, and TGF-β, which trigger STAT3, STAT6, STAT1, and NFκB signaling pathways [153, 154]. Once activated, MDSCs inhibit T cell activation through an RNS (reactive-nitrogen-species-) dependent mechanism of nitrating the T cell antigen receptor (TCR), which dissociates CD3-γ from the TCR, preventing antigen/MHC peptide recognition by T cells. MDSCs downregulate arginase synthase and upregulate nitric oxide synthase depleting of arginine and cysteine and increasing nitric oxide, which inhibits T cell activation and proliferation [43, 143, 154, 155]. MDSCs also interrupt T cell migration to lymph nodes by releasing ADAM17, which downregulates the homing receptor CD62L (L-selectin) on T cells [156]. They also inhibit migration of effector CD8+ T cells to the tumor by peroxynitrite modification of the chemoattractant CCL2 [157]. MDSCs expand natural Treg cells and promote differentiation of inducible Tregs through their production of IL-10 and TGF-β, and through CD40-CD40 ligand interactions [158–160]. MDSCs also inhibit NK cell activity through membrane bound TGF-β1, resulting in inhibition of IFN-γ and NKG2D expression [161]. MDSCs tumor densities inversely correlate with the anti-tumor activity of cytotoxic T and NK cells [162]. Adoptive transfer experiments of MDSCs into tumor-bearing hosts promote their differentiation into M2 TAMs [43, 163]. MDSCs can also differentiate into DCs, but whether these DCs are immunosuppressed is not currently known [164].
In addition to inhibiting anti-tumor immune responses, MDSCs trigger oncopromotion through facilitating angiogenesis, tissue remodeling, and helping to create premetastatic niches [165]. MDSCs infiltrate the primary site of melanoma promoting cancer cell dissemination by inducing EMT [166]. TGF-β, EGF, and HGF are required by MDSCs to induce EMT in cancer cells in in vitro assays [166]. MDSCs support the growth of tumor-initiating cells and induce resistance to apoptosis of premalignant cells from the intestinal epithelium [167]. In a murine model of pancreatic cancer, Kras oncogene expression at the beginning of the transformation program correlates with accumulation of MDSCs, IL-6, and IL-11 expression, STAT3 activation, increased proliferation, and resistance to apoptosis [168]. In agreement with all these data, a direct correlation between tumor size and the density of MDSCs in tumors have also been found [169] and MDSCs high frequencies in blood have shown correlation with poor prognosis in patients with breast and colorectal cancer [170–172].
Ostrand-Rosenberg et al. described a communication network between MDSCs, macrophages, and DCs that promotes and maintains an immunosuppressive microenvironment [173]. This network is critical for oncotraining of tumor arriving immune populations, tipping the balance towards tumor promotion. This communication occurs through inflammatory mediators, mainly IL-1β, IL-6, IL-10, PGE-2, and TGF-β. MDSCs are also expanded during transplantation and their activity could participate in preventing graft rejection as well as graft-versus-host disease [152, 174]. This is interesting because of the high frequency of malignancies arising in transplanted patients. Because MDSCs seem to play a central role in immunoescape, oncotraining, and oncopromotion, different therapeutic strategies are currently being explored directly targeting these cells.
5. Conclusions
Experimental and clinical studies indicate that cells of the innate and adaptive immune system have dual roles in cancer: the traditional immunosurveillance role and a cancer-promoting role. Immune cell metabolites help to create an optimal environment for tumor growth in which increased proliferation and survival of tumor cells is favored. Immune cells also promote angiogenesis and vessel permeability nourishing and oxygenating the tumor; protease secretion triggering ECM degradation, which together with the high tumor vascularization favor the detachment and exit of tumor cells to distant sites. Immune mediators also help to create the premetastatic niche in which secondary tumors will be formed. Specific immune cell populations have also the capacity to downregulate immunosurveillance mechanisms promoting immunosuppressive environments. In agreement, tumor densities of inflammatory cells correlate with diseases prognosis. All together, these observations support that the tumor stroma shifts immune cells to perform functions more in tune with the tumor needs, a process that is better described as oncotraining, while all pro-tumoral activities resulting from oncotraining are better framed as a mechanism of oncopromotion. More research needs to be done to have a clearer picture of the mechanisms of immunoescape, oncotraining, and oncopromotion, but the important populations in these processes, M2 macrophages, N2 neutrophils, Tregs, and MDSCs, and their main modulators, CSF-1, IL-6, metalloproteases, VEGF, PGE-2, TGF-β and IL-10, provide ideal targets for the intelligent design of directed preventive or anticancer therapies.
Several strategies are envisioned, either by harnessing immunosurveillance or antagonizing mechanisms of oncotraining and oncopromotion. Since plasticity is a common feature of immune cells often exploited by tumors to their own benefit, reprogramming of these populations switching back from pro-tumoral or immunosuppressive to immunosurveillance functions can potentially lead to tumor clearance. In that scenario, a chimeric antibody against IL-6 is in phase II trials in ovarian cancer treatment. Oxaliplatin increases the expression of calreticulin on tumor cells, enhancing phagocytosis, antigen presentation, and ultimately their removal. Adjuvant therapy with bacterial immunostimulatory products has also been helpful. In animal models, antagonistic antibodies against IL-4 or agonistic anti-CD40 favor anti-tumor Th1 functions. Also, the T cell stimulant IL-2 is currently used to treat metastatic renal cancer and melanoma. Alternatively, M2 macrophages, N2 neutrophils, Tregs, and MDSCs pro-tumoral populations can be inactivated or targeted for depletion with specific antibodies. Ipilimumab antagonizes the T cell inhibitor CTLA4 and increases survival of patients with melanoma; antagonists of CXCR4-SDF1 interactions or antagonist of other chemotactic factors (CCL2, CXXL12) are showing promising results inhibiting the arrival of helping populations to primary and secondary tumor sites, thus inhibiting tumor growth and metastasis. A SDF-1 peptide analog has been approved by the FDA for treatment of osteosarcoma. The antiangiogenic drug sunitinib and antibody bevacizumab reduce the numbers of MDSCs in tumors and have also shown anti-tumor activity in preclinical studies. Taking together all these observations, the incorporation of a measure of tumor immunological activity, or an immunoscore, has been proposed to be added into the traditional classification schemes of tumor prognosis.
In conclusion, because of the multiple genetic and epigenetic changes that lead a cell to undergo oncogenic transformation, the lesser factors involved in tumor-supportive mechanisms could be more effective targets for therapy, among many types of cancer and even for advanced stages. Still, it is highly unlikely to find a sole mechanism to enhance immunosurveillance while antagonizing oncotraining and oncopromotion mechanisms, but perhaps the combined action of targeted therapy against these mechanisms together with more traditional chemotherapy and radiotherapy will result in more efficient and less toxic cancer treatments.
Authors' Contribution
G. K. Chimal-Ramírez and N. A. Espinoza-Sánchez contributed equally to this work.
Acknowledgments
This work was supported by Fondo de Investigación en Salud (FIS/IMSS/PROT/G11/969). This work constitutes a partial fulfillment of the Graduate Program of Doctor Degree in Chemical and Biological Sciences of the Instituto Politecnico Nacional, IPN (GKC-R), and Doctor Degree in Biomedical Science of the Universidad Nacional Autonoma de Mexico, UNAM (NAE-S). G. K. Chimal-Ramírez and N. A. Espinoza-Sanchez acknowledge the scholarship and financial support provided by the National Council of Science and Technology (CONACyT) and the Mexican Institute for Social Security (IMSS). The authors kindly acknowledge the critical review to this paper provided by Dr. Jack F. Treml.
Abbreviations
Cell Populations
- APCs:
Antigen-presenting cells
- CTLs:
Cytotoxic T cells
- DCs:
Dendritic cells
- MCs:
Mast cells
- MDSCs:
Myeloid-derived suppressor cells
- NKs:
Natural killer cells
- PMN:
Polymorphonuclear cells
- TAMs:
Tumor-associated macrophages
- Th:
T helper cells
- Tregs:
Regulatory T cells
- EMT:
Epithelial-mesenchymal transition.
Molecules
- ADAM:
Metalloproteinase disintegrin
- Akt:
Protein kinase B
- BAD:
Bcl-2-associated death promoter
- BCL-2:
B cell lymphoma 2
- Bcl-xL:
B-cell lymphomaextra large protein
- CagA:
Cytotoxin-associated gene A
- CAR:
Coxsackievirus and adenovirus receptor
- CCL:
Chemokines with two adjacent cysteines
- COX-2:
Cyclooxygenase 2
- CSF1:
Colony-stimulating factor 1
- CXCL:
Chemokines with N-terminal cysteines separated by one amino acid
- CXCR:
CXCL chemokine receptor
- EGF:
Epithelial growth factor
- EGFR:
Epithelial growth factor receptor
- ECM:
Extracellular matrix
- FGF:
Fibroblast growth factor
- FOXP3:
Forkhead box P3
- G-CSF:
Granulocyte colony-stimulating factor
- GM-CSF:
Granulocyte-macrophage colony stimulating factor
- HBV:
Hepatitis B virus
- HCV:
Hepatitis C virus
- HGF:
Hepatocyte growth factor
- HIF-1α:
Hypoxia inducible factor alpha
- HLA:
Human Leukocyte antigen
- HPV16:
Human papilloma virus 16
- ICAM-1:
Intercellular adhesion molecule 1
- IDO:
Indoleamine 2,3-dioxygenase
- IFN-γ:
Interferon gamma
- iNOS:
Inducible nitric oxide synthase
- KIR:
Killer cell immunoglobulin-like receptor
- M-CSF:
Macrophage colony-stimulating factor
- MHC:
Major histocompatibility complex
- MIP-1α:
Macrophage inflammatory protein 1 alpha
- MMP:
Matrix metalloprotease
- NAIP5:
Neuronal apoptosis inhibitory protein
- NFκB:
Nuclear factor kappa-light-chain enhancer of activated B cells
- NKG2D:
Natural killer group 2, member D receptor
- PAMP:
Pathogen-associated molecular patterns
- PDGF:
Platelet-derived growth factor
- PDL1:
Programmed cell death 1 ligand 1
- PGE2:
Prostaglandin 2
- PI3K:
Phosphatidylinositide 3-kinases
- PIP2:
Phosphatidylinositol (4,5) bisphosphate
- PIP3:
Phosphatidylinositol (3,4,5) triphosphate
- RAS:
Rat sarcoma proteins
- RNS:
Reactive nitrogen Intermediates
- ROS:
Reactive oxygen species
- RNI:
Reactive nitrogen species
- TGF-β:
Transforming growth factor beta
- TNF-α:
Tumor necrosis factor alpha
- TLR:
Toll-like receptor
- uPA:
Urokinase-like plasminogen activator
- VLA4:
Integrin alpha4beta1(very late antigen-4)
- VEGF:
Vascular endothelial growth factor
- VEGFR:
Vascular endothelial growth factor receptor
- RANKL:
Receptor activator of nuclear factor kappa-B ligand
- SCF:
Stem cell factor
- TCR:
T Cell antigen receptor.
References
- 1.Burnet FM. The concept of immunological surveillance. Progress in Experimental Tumor Research. 1970;13:1–27. doi: 10.1159/000386035. [DOI] [PubMed] [Google Scholar]
- 2.Thomas L. On immunosurveillance in human cancer. The Yale Journal of Biology and Medicine. 1982;55(3-4):329–333. [PMC free article] [PubMed] [Google Scholar]
- 3.Balkwill FR, Mantovani A. Cancer-related inflammation: common themes and therapeutic opportunities. Seminars in Cancer Biology. 2012;22(1):33–40. doi: 10.1016/j.semcancer.2011.12.005. [DOI] [PubMed] [Google Scholar]
- 4.DeNardo DG, Andreu P, Coussens LM. Interactions between lymphocytes and myeloid cells regulate pro-versus anti-tumor immunity. Cancer and Metastasis Reviews. 2010;29(2):309–316. doi: 10.1007/s10555-010-9223-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shankaran V, Ikeda H, Bruce AT, et al. IFNγ, and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature. 2001;410(6832):1107–1111. doi: 10.1038/35074122. [DOI] [PubMed] [Google Scholar]
- 6.Ribatti D, Crivellato E. Mast cells, angiogenesis and cancer. Advances in Experimental Medicine and Biology. 2011;716:270–288. doi: 10.1007/978-1-4419-9533-9_14. [DOI] [PubMed] [Google Scholar]
- 7.Kokenyesi R. Ovarian carcinoma cells synthesize both chondroitin sulfate and heparan sulfate cell surface proteoglycans that mediate cell adhesion to interstitial matrix. Journal of Cellular Biochemistry. 2001;83(2):259–270. doi: 10.1002/jcb.1230. [DOI] [PubMed] [Google Scholar]
- 8.Oldford SA, Haidl ID, Howatt MA, Leiva CA, Johnston B, Marshall JS. A critical role for mast cells and mast cell-derived IL-6 in TLR2-mediated inhibition of tumor growth. Journal of Immunology. 2010;185(11):7067–7076. doi: 10.4049/jimmunol.1001137. [DOI] [PubMed] [Google Scholar]
- 9.Ch’ng S, Wallis RA, Yuan L, Davis PF, Tan ST. Mast cells and cutaneous malignancies. Modern Pathology. 2006;19(1):149–159. doi: 10.1038/modpathol.3800474. [DOI] [PubMed] [Google Scholar]
- 10.Maltby S, Khazaie K, McNagny KM. Mast cells in tumor growth: angiogenesis, tissue remodelling and immune-modulation. Biochimica et Biophysica Acta. 2009;1796(1):19–26. doi: 10.1016/j.bbcan.2009.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Della Rovere F, Granata A, Monaco M, Basile G. Phagocytosis of cancer cells by mast cells in breast cancer. Anticancer Research. 2009;29(8):3157–3161. [PubMed] [Google Scholar]
- 12.Pham CTN. Neutrophil serine proteases: specific regulators of inflammation. Nature Reviews Immunology. 2006;6(7):541–550. doi: 10.1038/nri1841. [DOI] [PubMed] [Google Scholar]
- 13.Scapini P, Lapinet-Vera JA, Gasperini S, Calzetti F, Bazzoni F, Cassatella MA. The neutrophil as a cellular source of chemokines. Immunological Reviews. 2000;177:195–203. doi: 10.1034/j.1600-065x.2000.17706.x. [DOI] [PubMed] [Google Scholar]
- 14.Colombo MP, Lombardi L, Stoppacciaro A, et al. Granulocyte colony-stimulating factor (G-CSF) gene transduction in murine adenocarcinoma drives neutrophil-mediated tumor inhibition in vivo: neutrophils discriminate between G-CSF-producing and G-CSF-nonproducing tumor cells. Journal of Immunology. 1992;149(1):113–119. [PubMed] [Google Scholar]
- 15.Di Carlo E, Forni G, Lollini P, Colombo MP, Modesti A, Musiani P. The intriguing role of polymorphonuclear neutrophils in antitumor reactions. Blood. 2001;97(2):339–345. doi: 10.1182/blood.v97.2.339. [DOI] [PubMed] [Google Scholar]
- 16.Welch DR, Schissel DJ, Howrey RP, Aeed PA. Tumor-elicited polymorphonuclear cells, in contrast to ’normal’ circulating polymorphonuclear cells, stimulate invasive and metastatic potentials of rat mammary adenocarcinoma cells. Proceedings of the National Academy of Sciences of the United States of America. 1989;86(15):5859–5863. doi: 10.1073/pnas.86.15.5859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fridlender ZG, Sun J, Kim S, et al. Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” versus “N2” TAN. Cancer Cell. 2009;16:183–194. doi: 10.1016/j.ccr.2009.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gregory AD, Houghton AM. Tumor-associated neutrophils: new targets for cancer therapy. Cancer Research. 2011;71(7):2411–2416. doi: 10.1158/0008-5472.CAN-10-2583. [DOI] [PubMed] [Google Scholar]
- 19.Rittmeyer D, Lorentz A. Relationship between allergy and cancer: an overview. International Archives of Allergy and Immunology. 2012;159:216–225. doi: 10.1159/000338994. [DOI] [PubMed] [Google Scholar]
- 20.Turner MC. Epidemiology: allergy history, IgE, and cancer. Cancer Immunology, Immunotherapy. 2011;61(9):1493–1510. doi: 10.1007/s00262-011-1180-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Geissmann F, Manz MG, Jung S, Sieweke MH, Merad M, Ley K. Development of monocytes, macrophages, and dendritic cells. Science. 2010;327(5966):656–661. doi: 10.1126/science.1178331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Karre K, Ljunggren HG, Piontek G, Kiessling R. Selective rejection of H-2-deficient lymphoma variants suggests alternative immune defence strategy. Nature. 1986;319(6055):675–678. doi: 10.1038/319675a0. [DOI] [PubMed] [Google Scholar]
- 23.Raulet DH, Vance RE, McMahon CW. Regulation of the natural killer cell receptor repertoire. Annual Review of Immunology. 2001;19:291–330. doi: 10.1146/annurev.immunol.19.1.291. [DOI] [PubMed] [Google Scholar]
- 24.Langers I, Renoux VM, Thiry M, Delvenne P, Jacobs N. Natural killer cells: role in local tumor growth and metastasis. Biologics: Targets & Therapy. 2012;6:73–82. doi: 10.2147/BTT.S23976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.O’Connor RA, Taams LS, Anderton SM. Translational mini-review series on Th17 cells: CD4+ T helper cells: functional plasticity and differential sensitivity to regulatory T cell-mediated regulation. Clinical and Experimental Immunology. 2010;159(2):137–147. doi: 10.1111/j.1365-2249.2009.04040.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wing JB, Sakaguchi S. Multiple treg suppressive modules and their adaptability. Frontiers in Immunology. 2012;3:p. 178. doi: 10.3389/fimmu.2012.00178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Weaver CT, Harrington LE, Mangan PR, Gavrieli M, Murphy KM. Th17: an effector CD4 T cell lineage with regulatory T cell ties. Immunity. 2006;24(6):677–688. doi: 10.1016/j.immuni.2006.06.002. [DOI] [PubMed] [Google Scholar]
- 28.Whitfield JR, Soucek L. Tumor microenvironment: becoming sick of Myc. Cellular and Molecular Life Sciences. 2012;69:931–934. doi: 10.1007/s00018-011-0860-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fuentes-Pananá E, Camorlinga-Ponce M, Maldonado-Bernal C. Infection, inflammation and gastric cancer. Salud Publica de Mexico. 2009;51(5):427–433. doi: 10.1590/s0036-36342009000500010. [DOI] [PubMed] [Google Scholar]
- 30.Colotta F, Allavena P, Sica A, Garlanda C, Mantovani A. Cancer-related inflammation, the seventh hallmark of cancer: links to genetic instability. Carcinogenesis. 2009;30(7):1073–1081. doi: 10.1093/carcin/bgp127. [DOI] [PubMed] [Google Scholar]
- 31.Fiaschi T, Chiarugi P. Oxidative stress, tumor microenvironment, and metabolic reprogramming: a diabolic liaison. International Journal of Cell Biology. 2012;2012:8 pages. doi: 10.1155/2012/762825.762825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Harada A, Sekido N, Akahoshi T, Wada T, Mukaida N, Matsushima K. Essential involvement of interleukin-8 (IL-8) in acute inflammation. Journal of Leukocyte Biology. 1994;56(5):559–564. [PubMed] [Google Scholar]
- 33.Johansson M, Denardo DG, Coussens LM. Polarized immune responses differentially regulate cancer development. Immunological Reviews. 2008;222(1):145–154. doi: 10.1111/j.1600-065X.2008.00600.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010;140(6):883–899. doi: 10.1016/j.cell.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xu Q, Wang L, Li H, et al. Mesenchymal stem cells play a potential role in regulating the establishment and maintenance of epithelial-mesenchymal transition in MCF7 human breast cancer cells by paracrine and induced autocrine TGF-beta. International Journal of Oncology. 2012;41(3) doi: 10.3892/ijo.2012.1541. [DOI] [PubMed] [Google Scholar]
- 36.Wang H, Wu J, Zhang Y, et al. Transforming growth factor beta-induced epithelial-mesenchymal transition increases cancer stem-like cells in the PANC-1 cell line. Oncology Letters. 2012;3:229–233. doi: 10.3892/ol.2011.448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Malinowsky K, Raychaudhuri M, Buchner T, et al. Common protein biomarkers assessed by reverse phase protein arrays show considerable intratumoral heterogeneity in breast cancer tissues. PloS One. 2012;7 doi: 10.1371/journal.pone.0040285.e40285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schillaci R, Guzman P, Cayrol F, et al. Clinical relevance of ErbB-2/HER2 nuclear expression in breast cancer. BMC Cancer. 2012;12(article 74) doi: 10.1186/1471-2407-12-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Giannios J, Ioannidou-Mouzaka L. Molecular aspects of breast and ovarian cancer. European Journal of Gynaecological Oncology. 1997;18(5):387–393. [PubMed] [Google Scholar]
- 40.Tindall EA, Severi G, Hoang HN, et al. Interleukin-6 promoter variants, prostate cancer risk, and survival. The Prostate. 2012;72(16):1701–1707. doi: 10.1002/pros.22557. [DOI] [PubMed] [Google Scholar]
- 41.Wang Y, Li L, Guo X, et al. Interleukin-6 signaling regulates anchorage-independent growth, proliferation, adhesion and invasion in human ovarian cancer cells. Cytokine. 2012;59:228–236. doi: 10.1016/j.cyto.2012.04.020. [DOI] [PubMed] [Google Scholar]
- 42.Mantovani A, Sica A. Macrophages, innate immunity and cancer: balance, tolerance, and diversity. Current Opinion in Immunology. 2010;22(2):231–237. doi: 10.1016/j.coi.2010.01.009. [DOI] [PubMed] [Google Scholar]
- 43.Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nature Reviews Immunology. 2012;12:253–268. doi: 10.1038/nri3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ben-Neriah Y, Karin M. Inflammation meets cancer, with NF-kappaB as the matchmaker. Nature Immunology. 2011;12:715–723. doi: 10.1038/ni.2060. [DOI] [PubMed] [Google Scholar]
- 45.Greten FR, Eckmann L, Greten TF, et al. IKKβ links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell. 2004;118(3):285–296. doi: 10.1016/j.cell.2004.07.013. [DOI] [PubMed] [Google Scholar]
- 46.Nehra R, Riggins RB, Shajahan AN, Zwart A, Crawford AC, Clarke R. BCL2 and CASP8 regulation by NF-κB differentially affect mitochondrial function and cell fate in antiestrogen-sensitive and -resistant breast cancer cells. The FASEB Journal. 2010;24(6):2040–2055. doi: 10.1096/fj.09-138305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Baldwin AS. Control of oncogenesis and cancer therapy resistance by the transcription factor NF-κB. Journal of Clinical Investigation. 2001;107(3):241–246. doi: 10.1172/JCI11991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sheppard K, Kinross KM, Solomon B, Pearson RB, Phillips WA. Targeting PI3 kinase/AKT/mTOR signaling in cancer. Critical Reviews in Oncogenesis. 2012;17:69–95. doi: 10.1615/critrevoncog.v17.i1.60. [DOI] [PubMed] [Google Scholar]
- 49.Trojan L, Thomas D, Knoll T, Grobholz R, Alken P, Michel MS. Expression of pro-angiogenic growth factors VEGF, EGF and bFGF and their topographical relation to neovascularisation in prostate cancer. Urological Research. 2004;32(2):97–103. doi: 10.1007/s00240-003-0383-5. [DOI] [PubMed] [Google Scholar]
- 50.Pugh CW, Ratcliffe PJ. Regulation of angiogenesis by hypoxia: role of the HIF system. Nature Medicine. 2003;9(6):677–684. doi: 10.1038/nm0603-677. [DOI] [PubMed] [Google Scholar]
- 51.Shen K, Ji L, Gong C, et al. Notoginsenoside Ft1 promotes angiogenesis via HIF-1α mediated VEGF secretion and the regulation of PI3K/AKT and Raf/MEK/ERK signaling pathways. Biochemical Pharmacology. 2012;84(6):784–792. doi: 10.1016/j.bcp.2012.05.024. [DOI] [PubMed] [Google Scholar]
- 52.Hicklin DJ, Ellis LM. Role of the vascular endothelial growth factor pathway in tumor growth and angiogenesis. Journal of Clinical Oncology. 2005;23(5):1011–1027. doi: 10.1200/JCO.2005.06.081. [DOI] [PubMed] [Google Scholar]
- 53.Wels J, Kaplan RN, Rafii S, Lyden D. Migratory neighbors and distant invaders: tumor-associated niche cells. Genes and Development. 2008;22(5):559–574. doi: 10.1101/gad.1636908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang TB, Chen ZG, Wei XQ, Wei B, Dong WG. Serum vascular endothelial growth factor-C and lymphoangiogenesis are associated with the lymph node metastasis and prognosis of patients with colorectal cancer. ANZ Journal of Surgery. 2011;81:694–699. doi: 10.1111/j.1445-2197.2010.05539.x. [DOI] [PubMed] [Google Scholar]
- 55.Talbot LJ, Bhattacharya SD, Kuo PC. Epithelial-mesenchymal transition, the tumor microenvironment, and metastatic behavior of epithelial malignancies. International Journal of Biochemistry and Molecular Biology. 2012;3:117–136. [PMC free article] [PubMed] [Google Scholar]
- 56.Gunasinghe NP, Wells A, Thompson EW, Hugo HJ. Mesenchymal-epithelial transition (MET) as a mechanism for metastatic colonisation in breast cancer. Cancer Metastasis Reviews. 2012;31(3-4):469–478. doi: 10.1007/s10555-012-9377-5. [DOI] [PubMed] [Google Scholar]
- 57.Onitilo AA, Aryal G, Engel JM. Hereditary diffuse Gastric cancer: a family diagnosis and treatment. Clinical Medicine & Research. 2012 doi: 10.3121/cmr.2012.1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Masciari S, Larsson N, Senz J, et al. Germline E-cadherin mutations in familial lobular breast cancer. Journal of Medical Genetics. 2007;44(11):726–731. doi: 10.1136/jmg.2007.051268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Huang FY, Chan AO, Rashid A, Wong DK, Cho CH, Yuen MF. Helicobacter pylori induces promoter methylation of E-cadherin via interleukin-1β activation of nitric oxide production in gastric cancer cells. Cancer. 2012;118(20):4969–4980. doi: 10.1002/cncr.27519. [DOI] [PubMed] [Google Scholar]
- 60.Hoy B, Geppert T, Boehm M, et al. Distinct roles of secreted HtrA proteases from gram-negative pathogens in cleaving the junctional protein and tumor suppressor E-cadherin. The Journal of Biological Chemistry. 2012;287:10115–10120. doi: 10.1074/jbc.C111.333419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Murata-Kamiya N, Kurashima Y, Teishikata Y, et al. Helicobacter pylori CagA interacts with E-cadherin and deregulates the β-catenin signal that promotes intestinal transdifferentiation in gastric epithelial cells. Oncogene. 2007;26(32):4617–4626. doi: 10.1038/sj.onc.1210251. [DOI] [PubMed] [Google Scholar]
- 62.Mutoh H, Sakurai S, Satoh K, et al. Cdx1 induced intestinal metaplasia in the transgenic mouse stomach: comparative study with Cdx2 transgenic mice. Gut. 2004;53(10):1416–1423. doi: 10.1136/gut.2003.032482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Saito Y, Murata-Kamiya N, Hirayama T, Ohba Y, Hatakeyama M. Conversion of Helicobacter pylori CagA from senescence inducer to oncogenic driver through polarity-dependent regulation of p21. The Journal of Experimental Medicine. 2010;207(10):2157–2174. doi: 10.1084/jem.20100602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hussain F, Morton PE, Snippe M, et al. CAR modulates E-cadherin dynamics in the presence of adenovirus type 5. PLoS One. 2011;6(8) doi: 10.1371/journal.pone.0023056.e23056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Matthews K, Leong CM, Baxter L, et al. Depletion of Langerhans cells in human papillomavirus type 16-infected skin is associated with E6-mediated down regulation of E-cadherin. Journal of Virology. 2003;77(15):8378–8385. doi: 10.1128/JVI.77.15.8378-8385.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lee JO, Kwun HJ, Jung JK, Choi KH, Min DS, Jang KL. Hepatitis B virus X protein represses E-cadherin expression via activation of DNA methyltransferase 1. Oncogene. 2005;24(44):6617–6625. doi: 10.1038/sj.onc.1208827. [DOI] [PubMed] [Google Scholar]
- 67.Lee JM, Dedhar S, Kalluri R, Thompson EW. The epithelial-mesenchymal transition: new insights in signaling, development, and disease. Journal of Cell Biology. 2006;172(7):973–981. doi: 10.1083/jcb.200601018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Jing Y, Han Z, Zhang S, Liu Y, Wei L. Epithelial-Mesenchymal transition in tumor microenvironment. Cell & Bioscience. 2011;1(article 29) doi: 10.1186/2045-3701-1-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.López-Otín C, Hunter T. The regulatory crosstalk between kinases and proteases in cancer. Nature Reviews Cancer. 2010;10(4):278–292. doi: 10.1038/nrc2823. [DOI] [PubMed] [Google Scholar]
- 70.Mason SD, Joyce JA. Proteolytic networks in cancer. Trends in Cell Biology. 2011;21(4):228–237. doi: 10.1016/j.tcb.2010.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bergers G, Brekken R, McMahon G, et al. Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nature Cell Biology. 2000;2(10):737–744. doi: 10.1038/35036374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Victor BC, Anbalagan A, Mohamed MM, Sloane BF, Cavallo-Medved D. Inhibition of cathepsin B activity attenuates extracellular matrix degradation and inflammatory breast cancer invasion. Breast Cancer Research. 2011;13(article R115) doi: 10.1186/bcr3058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Duffy MJ, Maguire TM, McDermott EW, O'Higgins N. Urokinase plasminogen activator: a prognostic marker in multiple types of cancer. Journal of Surgical Oncology. 1999;71:130–135. doi: 10.1002/(sici)1096-9098(199906)71:2<130::aid-jso14>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 74.Duffy MJ. Urokinase-type plasminogen activator: a potent marker of metastatic potential in human cancers. Biochemical Society Transactions. 2002;30(2):207–210. doi: 10.1042/. [DOI] [PubMed] [Google Scholar]
- 75.Jelisavac-Cosic S, Sirotkovic-Skerlev M, Kulic A, Jakic-Razumovic J, Kovac Z, Vrbanec D. Prognostic significance of urokinase-type plasminogen activator (uPA) and plasminogen activator inhibitor (PAI-1) in patients with primary invasive ductal breast carcinoma—a 7.5-year follow-up study. Tumori. 2011;97:532–539. doi: 10.1177/030089161109700419. [DOI] [PubMed] [Google Scholar]
- 76.Whitley BR, Palmieri D, Twerdi CD, Church FC. Expression of active plasminogen activator inhibitor-1 reduces cell migration and invasion in breast and gynecological cancer cells. Experimental Cell Research. 2004;296(2):151–162. doi: 10.1016/j.yexcr.2004.02.022. [DOI] [PubMed] [Google Scholar]
- 77.Magdolen V, Krüger A, Sato S, et al. Inhibition of the tumor-associated urokinase-type plasminogen activation system: effects of high-level synthesis of soluble urokinase receptor in ovarian and breast cancer cells in vitro and in vivo. Recent Results in Cancer Research. 2003;162:43–63. doi: 10.1007/978-3-642-59349-9_4. [DOI] [PubMed] [Google Scholar]
- 78.Giraudo E, Inoue M, Hanahan D. An amino-bisphosphonate targets MMP-9—expressing macrophages and angiogenesis to impair cervical carcinogenesis. Journal of Clinical Investigation. 2004;114(5):623–633. doi: 10.1172/JCI22087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Coussens LM, Tinkle CL, Hanahan D, Werb Z. MMP-9 supplied by bone marrow-derived cells contributes to skin carcinogenesis. Cell. 2000;103(3):481–490. doi: 10.1016/s0092-8674(00)00139-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Coleman RE. Bisphosphonates in breast cancer. Annals of Oncology. 2005;16(5):687–695. doi: 10.1093/annonc/mdi162. [DOI] [PubMed] [Google Scholar]
- 81.Tan W, Zhang W, Strasner A, et al. Tumour-infiltrating regulatory T cells stimulate mammary cancermetastasis through RANKL-RANK signalling. Nature. 2011;470(7335):548–553. doi: 10.1038/nature09707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kaplan RN, Riba RD, Zacharoulis S, et al. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature. 2005;438(7069):820–827. doi: 10.1038/nature04186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Shokeen M, Zheleznyak A, Wilson JM, et al. Molecular imaging of very late antigen-4 (alpha4beta1 integrin) in the premetastatic niche. Journal of Nuclear Medicine. 2012;53:779–786. doi: 10.2967/jnumed.111.100073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bolat F, Kayaselcuk F, Nursal TZ, Yagmurdur MC, Bal N, Demirhan B. Microvessel density, VEGF expression, and tumor-associated macrophages in breast tumors: correlations with prognostic parameters. Journal of Experimental and Clinical Cancer Research. 2006;25(3):365–372. [PubMed] [Google Scholar]
- 85.Osinsky S, Bubnovskaya L, Ganusevich I, et al. Hypoxia, tumour-associated macrophages, microvessel density, VEGF and matrix metalloproteinases in human gastric cancer: interaction and impact on survival. Clinical and Translational Oncology. 2011;13(2):133–138. doi: 10.1007/s12094-011-0630-0. [DOI] [PubMed] [Google Scholar]
- 86.de Visser KE, Eichten A, Coussens LM. Paradoxical roles of the immune system during cancer development. Nature Reviews Cancer. 2006;6(1):24–37. doi: 10.1038/nrc1782. [DOI] [PubMed] [Google Scholar]
- 87.Rabenhorst A, Schlaak M, Heukamp LC, et al. Mast cells play a protumorigenic role in primary cutaneous lymphoma. Blood. 2012;120(10):2042–2054. doi: 10.1182/blood-2012-03-415638. [DOI] [PubMed] [Google Scholar]
- 88.Priceman SJ, Sung JL, Shaposhnik Z, et al. Targeting distinct tumor-infiltrating myeloid cells by inhibiting CSF-1 receptor: combating tumor evasion of antiangiogenic therapy. Blood. 2010;115(7):1461–1471. doi: 10.1182/blood-2009-08-237412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lin EY, Nguyen AV, Russell RG, Pollard JW. Colony-stimulating factor 1 promotes progression of mammary tumors to malignancy. The Journal of Experimental Medicine. 2001;193(6):727–740. doi: 10.1084/jem.193.6.727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Patsialou A, Wyckoff J, Wang Y, Goswami S, Stanley ER, Condeelis JS. Invasion of human breast cancer cells in vivo requires both paracrine and autocrine loops involving the colony-stimulating factor-1 receptor. Cancer Research. 2009;69(24):9498–9506. doi: 10.1158/0008-5472.CAN-09-1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Agarwal S, Reddy GV, Reddanna P. Eicosanoids in inflammation and cancer: the role of COX-2. Expert Review of Clinical Immunology. 2009;5(2):145–165. doi: 10.1586/1744666X.5.2.145. [DOI] [PubMed] [Google Scholar]
- 92.Wang D, Dubois RN. Eicosanoids and cancer. Nature Reviews Cancer. 2010;10(3):181–193. doi: 10.1038/nrc2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Cuzick J, Otto F, Baron JA, et al. Aspirin and non-steroidal anti-inflammatory drugs for cancer prevention: an international consensus statement. The Lancet Oncology. 2009;10(5):501–507. doi: 10.1016/S1470-2045(09)70035-X. [DOI] [PubMed] [Google Scholar]
- 94.Ferrandez A, Piazuelo E, Castells A. Aspirin and the prevention of colorectal cancer. Best Practice & Research. Clinical Gastroenterology. 2012;26(2):185–195. doi: 10.1016/j.bpg.2012.01.009. [DOI] [PubMed] [Google Scholar]
- 95.Garaude J, Kent A, van Rooijen N, Blander JM. Simultaneous targeting of toll- and nod-like receptors induces effective tumor-specific immune responses. Science Translational Medicine. 2012;4 doi: 10.1126/scitranslmed.3002868.120ra116 [DOI] [PubMed] [Google Scholar]
- 96.Pollard JW. Trophic macrophages in development and disease. Nature Reviews Immunology. 2009;9(4):259–270. doi: 10.1038/nri2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Steidl C, Lee T, Shah SP, et al. Tumor-associated macrophages and survival in classic Hodgkin’s lymphoma. The New England Journal of Medicine. 2010;362(10):875–885. doi: 10.1056/NEJMoa0905680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Subimerb C, Pinlaor S, Khuntikeo N, et al. Tissue invasive macrophage density is correlated with prognosis in cholangiocarcinoma. Molecular Medicine Reports. 2010;3(4):597–605. doi: 10.3892/mmr_00000303. [DOI] [PubMed] [Google Scholar]
- 99.Kurahara H, Takao S, Maemura K, et al. M2-polarized tumor-associated macrophage infiltration of regional lymph nodes is associated with nodal lymphangiogenesis and occult nodal involvement in pN0 pancreatic cancer. Pancreas. 2013;42(1):155–159. doi: 10.1097/MPA.0b013e318254f2d1. [DOI] [PubMed] [Google Scholar]
- 100.Mahmoud SM, Lee AH, Paish EC, Macmillan RD, Ellis IO, Green AR. Tumour-infiltrating macrophages and clinical outcome in breast cancer. Journal of Clinical Pathology. 2012;65(2):159–163. doi: 10.1136/jclinpath-2011-200355. [DOI] [PubMed] [Google Scholar]
- 101.Wei Q, Fang W, Ye L, et al. Density of tumor-associated macrophages correlates with lymph node metastasis in papillary thyroid carcinoma. Thyroid. 2012;22(9):905–910. doi: 10.1089/thy.2011.0452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell. 2010;141(1):39–51. doi: 10.1016/j.cell.2010.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Morandi A, Barbetti V, Riverso M, Dello Sbarba P, Rovida E. The colony-stimulating factor-1 (CSF-1) receptor sustains ERK1/2 activation and proliferation in breast cancer cell lines. PloS One. 2011;6(11) doi: 10.1371/journal.pone.0027450.e27450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Lee Y, Chittezhath M, Andre V, et al. Protumoral role of monocytes in human B-cell precursor acute lymphoblastic leukemia: involvement of the chemokine CXCL10. Blood. 2012;119(1):227–237. doi: 10.1182/blood-2011-06-357442. [DOI] [PubMed] [Google Scholar]
- 105.Hao NB, Lu MH, Fan YH, Cao YL, Zhang ZR, Yang SM. Macrophages in tumor microenvironments and the progression of tumors. Clinical & Developmental Immunology. 2012;2012:11 pages. doi: 10.1155/2012/948098.948098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Smith PD, Smythies LE, Shen R, Greenwell-Wild T, Gliozzi M, Wahl SM. Intestinal macrophages and response to microbial encroachment. Mucosal Immunology. 2011;4(1):31–42. doi: 10.1038/mi.2010.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Weisser SB, Brugger HK, Voglmaier NS, McLarren KW, van Rooijen N, Sly LM. SHIP-deficient, alternatively activated macrophages protect mice during DSS-induced colitis. Journal of Leukocyte Biology. 2011;90(3):483–492. doi: 10.1189/jlb.0311124. [DOI] [PubMed] [Google Scholar]
- 108.Wahl SM, Chen W. Transforming growth factor β-induced regulatory T cells referee inflammatory and autoimmune diseases. Arthritis Research and Therapy. 2005;7(2):62–68. doi: 10.1186/ar1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Pandiyan P, Zheng L, Ishihara S, Reed J, Lenardo MJ. CD4+CD25+Foxp3+ regulatory T cells induce cytokine deprivation-mediated apoptosis of effector CD4+ T cells. Nature Immunology. 2007;8(12):1353–1362. doi: 10.1038/ni1536. [DOI] [PubMed] [Google Scholar]
- 110.Baratelli F, Lin Y, Zhu L, et al. Prostaglandin E2 induces FOXP3 gene expression and T regulatory cell function in human CD4+ T cells. Journal of Immunology. 2005;175(3):1483–1490. doi: 10.4049/jimmunol.175.3.1483. [DOI] [PubMed] [Google Scholar]
- 111.Condeelis J, Pollard JW. Macrophages: obligate partners for tumor cell migration, invasion, and metastasis. Cell. 2006;124(2):263–266. doi: 10.1016/j.cell.2006.01.007. [DOI] [PubMed] [Google Scholar]
- 112.Rogers TL, Holen I. Tumour macrophages as potential targets of bisphosphonates. Journal of Translational Medicine. 2011;9(article 177) doi: 10.1186/1479-5876-9-177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Bellocq A, Antoine M, Flahault A, et al. Neutrophil alveolitis in bronchioloalveolar carcinoma: induction by tumor-derived interleukin-8 and relation to clinical outcome. American Journal of Pathology. 1998;152(1):83–92. [PMC free article] [PubMed] [Google Scholar]
- 114.Pekarek LA, Starr BA, Toledano AY, Schreiber H. Inhibition of tumor growth by elimination of granulocytes. The Journal of Experimental Medicine. 1995;181(1):435–440. doi: 10.1084/jem.181.1.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Jensen HK, Donskov F, Marcussen N, Nordsmark M, Lundbeck F, von der Maase H. Presence of intratumoral neutrophils is an independent prognostic factor in localized renal cell carcinoma. Journal of Clinical Oncology. 2009;27(28):4709–4717. doi: 10.1200/JCO.2008.18.9498. [DOI] [PubMed] [Google Scholar]
- 116.Wislez M, Rabbe N, Marchal J, et al. Hepatocyte growth factor production by neutrophils infiltrating bronchioloalveolar subtype pulmonary adenocarcinoma: role in tumor progression and death. Cancer Research. 2003;63(6):1405–1412. [PubMed] [Google Scholar]
- 117.Kowanetz M, Wu X, Lee J, et al. Granulocyte-colony stimulating factor promotes lung metastasis through mobilization of Ly6G+Ly6C+ granulocytes. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(50):21248–21255. doi: 10.1073/pnas.1015855107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Houghton AM, Rzymkiewicz DM, Ji H, et al. Neutrophil elastase-mediated degradation of IRS-1 accelerates lung tumor growth. Nature Medicine. 2010;16(2):219–223. doi: 10.1038/nm.2084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Conti P, Castellani ML, Kempuraj D, et al. Role of mast cells in tumor growth. Annals of Clinical and Laboratory Science. 2007;37(4):315–322. [PubMed] [Google Scholar]
- 120.Huang B, Lei Z, Zhang GM, et al. SCF-mediated mast cell infiltration and activation exacerbate the inflammation and immunosuppression in tumor microenvironment. Blood. 2008;112(4):1269–1279. doi: 10.1182/blood-2008-03-147033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Norrby K. Mast cells and angiogenesis. APMIS. 2002;110(5):355–371. doi: 10.1034/j.1600-0463.2002.100501.x. [DOI] [PubMed] [Google Scholar]
- 122.Coussens LM, Raymond WW, Bergers G, et al. Inflammatory mast cells up-regulate angiogenesis during squamous epithelial carcinogenesis. Genes and Development. 1999;13(11):1382–1397. doi: 10.1101/gad.13.11.1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Ribatti D, Crivellato E. The controversial role of mast cells in tumor growth. International Review of Cell and Molecular Biology. 2009;275:89–131. doi: 10.1016/S1937-6448(09)75004-X. [DOI] [PubMed] [Google Scholar]
- 124.Wasiuk A, de Vries VC, Hartmann K, Roers A, Noelle RJ. Mast cells as regulators of adaptive immunity to tumours. Clinical and Experimental Immunology. 2009;155(2):140–146. doi: 10.1111/j.1365-2249.2008.03840.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Mancino A, Schioppa T, Larghi P, et al. Divergent effects of hypoxia on dendritic cell functions. Blood. 2008;112(9):3723–3734. doi: 10.1182/blood-2008-02-142091. [DOI] [PubMed] [Google Scholar]
- 126.Elia AR, Cappello P, Puppo M, et al. Human dendritic cells differentiated in hypoxia down-modulate antigen uptake and change their chemokine expression profile. Journal of Leukocyte Biology. 2008;84(6):1472–1482. doi: 10.1189/jlb.0208082. [DOI] [PubMed] [Google Scholar]
- 127.Yang M, Ma C, Liu S, et al. HIF-dependent induction of adenosine receptor A2b skews human dendritic cells to a Th2-stimulating phenotype under hypoxia. Immunology and Cell Biology. 2010;88(2):165–171. doi: 10.1038/icb.2009.77. [DOI] [PubMed] [Google Scholar]
- 128.Scarlett UK, Rutkowski MR, Rauwerdink AM, et al. Ovarian cancer progression is controlled by phenotypic changes in dendritic cells. The Journal of Experimental Medicine. 2012;209(3):495–506. doi: 10.1084/jem.20111413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Novitskiy SV, Ryzhov S, Zaynagetdinov R, et al. Adenosine receptors in regulation of dendritic cell differentiation and function. Blood. 2008;112(5):1822–1831. doi: 10.1182/blood-2008-02-136325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Tan TT, Coussens LM. Humoral immunity, inflammation and cancer. Current Opinion in Immunology. 2007;19(2):209–216. doi: 10.1016/j.coi.2007.01.001. [DOI] [PubMed] [Google Scholar]
- 131.DeNardo DG, Coussens LM. Inflammation and breast cancer. Balancing immune response: crosstalk between adaptive and innate immune cells during breast cancer progression. Breast Cancer Research. 2007;9(4):p. 212. doi: 10.1186/bcr1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Zapata JM, Llobet D, Krajewska M, Lefebvre S, Kress CL, Reed JC. Lymphocyte-specific TRAF3 transgenic mice have enhanced humoral responses and develop plasmacytosis, autoimmunity, inflammation, and cancer. Blood. 2009;113(19):4595–4603. doi: 10.1182/blood-2008-07-165456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.de Visser KE, Korets LV, Coussens LM. De novo carcinogenesis promoted by chronic inflammation is B lymphocyte dependent. Cancer Cell. 2005;7(5):411–423. doi: 10.1016/j.ccr.2005.04.014. [DOI] [PubMed] [Google Scholar]
- 134.Talmadge JE, Donkor M, Scholar E. Inflammatory cell infiltration of tumors: Jekyll or Hyde. Cancer and Metastasis Reviews. 2007;26(3-4):373–400. doi: 10.1007/s10555-007-9072-0. [DOI] [PubMed] [Google Scholar]
- 135.Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science. 2011;331(6024):1565–1570. doi: 10.1126/science.1203486. [DOI] [PubMed] [Google Scholar]
- 136.Vesely MD, Kershaw MH, Schreiber RD, Smyth MJ. Natural innate and adaptive immunity to cancer. Annual Review of Immunology. 2011;29:235–271. doi: 10.1146/annurev-immunol-031210-101324. [DOI] [PubMed] [Google Scholar]
- 137.Josefowicz SZ, Lu LF, Rudensky AY. Regulatory T cells: mechanisms of differentiation and function. Annual Review of Immunology. 2012;30:531–564. doi: 10.1146/annurev.immunol.25.022106.141623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Onizuka S, Tawara I, Shimizu J, Sakaguchi S, Fujita T, Nakayama E. Tumor rejection by in vivo administration of anti-CD25 (interleukin-2 receptor α) monoclonal antibody. Cancer Research. 1999;59(13):3128–3133. [PubMed] [Google Scholar]
- 139.Driessler F, Venstrom K, Sabat R, Asadullah K, Schottelius AJ. Molecular mechanisms of interleukin-10-mediated inhibition of NF-κB activity: a role for p50. Clinical and Experimental Immunology. 2004;135(1):64–73. doi: 10.1111/j.1365-2249.2004.02342.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Ouyang W, Rutz S, Crellin NK, Valdez PA, Hymowitz SG. Regulation and functions of the IL-10 family of cytokines in inflammation and disease. Annual Review of Immunology. 2011;29:71–109. doi: 10.1146/annurev-immunol-031210-101312. [DOI] [PubMed] [Google Scholar]
- 141.Prieto PA, Yang JC, Sherry RM, et al. CTLA-4 blockade with ipilimumab: long-term follow-up of 177 patients with metastatic melanoma. Clinical Cancer Research. 2012;18:2039–2047. doi: 10.1158/1078-0432.CCR-11-1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Zhou G, Levitsky H. Towards curative cancer immunotherapy: overcoming posttherapy tumor escape. Clinical and Developmental Immunology. 2012;2012:12 pages. doi: 10.1155/2012/124187.124187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nature Reviews Immunology. 2009;9(3):162–174. doi: 10.1038/nri2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Pak AS, Wright MA, Matthews JP, Collins SL, Petruzzelli GJ, Young MRI. Mechanisms of immune suppression in patients with head and neck cancer: presence of CD34+ cells which suppress immune functions within cancers that secrete granulocyte-macrophage colony-stimulating factor. Clinical Cancer Research. 1995;1(1):95–103. [PubMed] [Google Scholar]
- 145.Young MRI, Kolesiak K, Wright MA, Gabrilovich DI. Chemoattraction of femoral CD34+ progenitor cells by tumor-derived vascular endothelial cell growth factor. Clinical and Experimental Metastasis. 1999;17(10):881–888. doi: 10.1023/a:1006708607666. [DOI] [PubMed] [Google Scholar]
- 146.Young MRI, Wright MA. Myelopoiesis-associated immune suppressor cells in mice bearing metastatic Lewis lung carcinoma tumors: γ interferon plus tumor necrosis factor α synergistically reduces immune suppressor and tumor growth-promoting activities of bone marrow cells and diminishes tumor recurrence and metastasis. Cancer Research. 1992;52(22):6335–6340. [PubMed] [Google Scholar]
- 147.Peranzoni E, Zilio S, Marigo I, et al. Myeloid-derived suppressor cell heterogeneity and subset definition. Current Opinion in Immunology. 2010;22(2):238–244. doi: 10.1016/j.coi.2010.01.021. [DOI] [PubMed] [Google Scholar]
- 148.Greten TF, Manns MP, Korangy F. Myeloid derived suppressor cells in human diseases. International Immunopharmacology. 2011;11(7):802–806. doi: 10.1016/j.intimp.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Filipazzi P, Valenti R, Huber V, et al. Identification of a new subset of myeloid suppressor cells in peripheral blood of melanoma patients with modulation by a granulocyte-macrophage colony-stimulation factor-based antitumor vaccine. Journal of Clinical Oncology. 2007;25(18):2546–2553. doi: 10.1200/JCO.2006.08.5829. [DOI] [PubMed] [Google Scholar]
- 150.Waight JD, Hu Q, Miller A, Liu S, Abrams SI. Tumor-derived G-CSF facilitates neoplastic growth through a granulocytic myeloid-derived suppressor cell-dependent mechanism. PloS One. 2011;6 doi: 10.1371/journal.pone.0027690.e27690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Stairs DB, Bayne LJ, Rhoades B, et al. Deletion of p120-catenin results in a tumor microenvironment with inflammation and cancer that establishes it as a tumor suppressor gene. Cancer Cell. 2011;19(4):470–483. doi: 10.1016/j.ccr.2011.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Dilek N, Vuillefroy de Silly R, Blancho G, Vanhove B. Myeloid-derived suppressor cells: mechanisms of action and recent advances in their role in transplant tolerance. Frontiers in Immunology. 2012;3:p. 208. doi: 10.3389/fimmu.2012.00208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Nagaraj S, Gabrilovich DI. Tumor escape mechanism governed by myeloid-derived suppressor cells. Cancer Research. 2008;68(8):2561–2563. doi: 10.1158/0008-5472.CAN-07-6229. [DOI] [PubMed] [Google Scholar]
- 154.Ostrand-Rosenberg S, Sinha P. Myeloid-derived suppressor cells: linking inflammation and cancer. Journal of Immunology. 2009;182(8):4499–4506. doi: 10.4049/jimmunol.0802740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Sica A, Bronte V. Altered macrophage differentiation and immune dysfunction in tumor development. Journal of Clinical Investigation. 2007;117(5):1155–1166. doi: 10.1172/JCI31422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Hanson EM, Clements VK, Sinha P, Ilkovitch D, Ostrand-Rosenberg S. Myeloid-derived suppressor cells down-regulate L-selectin expression on CD4+ and CD8+ T cells. Journal of Immunology. 2009;183(2):937–944. doi: 10.4049/jimmunol.0804253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Molon B, Ugel S, Del Pozzo F, et al. Chemokine nitration prevents intratumoral infiltration of antigen-specific T cells. The Journal of Experimental Medicine. 2011;208(10):1949–1962. doi: 10.1084/jem.20101956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Pan PY, Ma G, Weber KJ, et al. Immune stimulatory receptor CD40 is required for T-cell suppression and T regulatory cell activation mediated by myeloid-derived suppressor cells in cancer. Cancer Research. 2010;70(1):99–108. doi: 10.1158/0008-5472.CAN-09-1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Huang B, Pan PY, Li Q, et al. Gr-1+CD115+ immature myeloid suppressor cells mediate the development of tumor-induced T regulatory cells and T-cell anergy in tumor-bearing host. Cancer Research. 2006;66(2):1123–1131. doi: 10.1158/0008-5472.CAN-05-1299. [DOI] [PubMed] [Google Scholar]
- 160.Centuori SM, Trad M, Lacasse CJ, et al. Myeloid-derived suppressor cells from tumor-bearing mice impair TGF-β-induced differentiation of CD4+CD25+Foxp3+ Tregs from CD4+CD25−FoxP3− T cells. Journal of Leukocyte Biology. 2012;92(5):987–997. doi: 10.1189/jlb.0911465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Li H, Han Y, Guo Q, Zhang M, Cao X. Cancer-expanded myeloid-derived suppressor cells induce anergy of NK cells through membrane-bound TGF-β1. Journal of Immunology. 2009;182(1):240–249. doi: 10.4049/jimmunol.182.1.240. [DOI] [PubMed] [Google Scholar]
- 162.Suzuki E, Kapoor V, Jassar AS, Kaiser LR, Albelda SM. Gemcitabine selectively eliminates splenic Gr-1+/CD11b + myeloid suppressor cells in tumor-bearing animals and enhances antitumor immune activity. Clinical Cancer Research. 2005;11(18):6713–6721. doi: 10.1158/1078-0432.CCR-05-0883. [DOI] [PubMed] [Google Scholar]
- 163.Narita Y, Wakita D, Ohkuri T, Chamoto K, Nishimura T. Potential differentiation of tumor bearing mouse CD11b+Gr-1+ immature myeloid cells into both suppressor macrophages and immunostimulatory dendritic cells. Biomedical Research. 2009;30(1):7–15. doi: 10.2220/biomedres.30.7. [DOI] [PubMed] [Google Scholar]
- 164.Corzo CA, Condamine T, Lu L, et al. HIF-1α regulates function and differentiation of myeloid-derived suppressor cells in the tumor microenvironment. The Journal of Experimental Medicine. 2010;207(11):2439–2453. doi: 10.1084/jem.20100587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Yang L, DeBusk LM, Fukuda K, et al. Expansion of myeloid immune suppressor Gr+CD11b+ cells in tumor-bearing host directly promotes tumor angiogenesis. Cancer Cell. 2004;6(4):409–421. doi: 10.1016/j.ccr.2004.08.031. [DOI] [PubMed] [Google Scholar]
- 166.Toh B, Wang X, Keeble J, et al. Mesenchymal transition and dissemination of cancer cells is driven by myeloid-derived suppressor cells infiltrating the primary tumor. PLoS Biology. 2011;9 doi: 10.1371/journal.pbio.1001162.e1001162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Grivennikov S, Karin E, Terzic J, et al. IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell. 2009;15(2):103–113. doi: 10.1016/j.ccr.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Lesina M, Kurkowski MU, Ludes K, et al. Stat3/Socs3 activation by IL-6 transsignaling promotes progression of pancreatic intraepithelial neoplasia and development of pancreatic cancer. Cancer Cell. 2011;19(4):456–469. doi: 10.1016/j.ccr.2011.03.009. [DOI] [PubMed] [Google Scholar]
- 169.Abe F, Dafferner AJ, Donkor M, et al. Myeloid-derived suppressor cells in mammary tumor progression in FVB Neu transgenic mice. Cancer Immunology, Immunotherapy. 2010;59(1):47–62. doi: 10.1007/s00262-009-0719-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Diaz-Montero CM, Salem ML, Nishimura MI, Garrett-Mayer E, Cole DJ, Montero AJ. Increased circulating myeloid-derived suppressor cells correlate with clinical cancer stage, metastatic tumor burden, and doxorubicin-cyclophosphamide chemotherapy. Cancer Immunology, Immunotherapy. 2009;58(1):49–59. doi: 10.1007/s00262-008-0523-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Solito S, Falisi E, Diaz-Montero CM, et al. A human promyelocytic-like population is responsible for the immune suppression mediated by myeloid-derived suppressor cells. Blood. 2011;118:2254–2265. doi: 10.1182/blood-2010-12-325753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Raychaudhuri B, Rayman P, Ireland J, et al. Myeloid-derived suppressor cell accumulation and function in patients with newly diagnosed glioblastoma. Neuro-Oncology. 2011;13(6):591–599. doi: 10.1093/neuonc/nor042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Ostrand-Rosenberg S, Sinha P, Beury DW, Clements VK. Cross-talk between myeloid-derived suppressor cells (MDSC), macrophages, and dendritic cells enhances tumor-induced immune suppression. Seminars in Cancer Biology. 2012;22(4):275–281. doi: 10.1016/j.semcancer.2012.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Ochando JC, Chen SH. Myeloid-derived suppressor cells in transplantation and cancer. Immunologic Research. 2012;54(1–3):275–285. doi: 10.1007/s12026-012-8335-1. [DOI] [PMC free article] [PubMed] [Google Scholar]