

Abstract
Degeneration of the CA3 pyramidal and dentate hilar neurons in the adult rat hippocampus after an intracerebroventricular kainic acid (KA) administration, a model of temporal lobe epilepsy, leads to permanent loss of the calcium binding protein calbindin in major fractions of dentate granule cells and CA1 pyramidal neurons. We hypothesize that the enduring loss of calbindin in the dentate gyrus and the CA1 subfield after CA3-lesion is due to disruption of the hippocampal circuitry leading to hyperexcitability in these regions; therefore, specific cell grafts that are capable of both reconstructing the disrupted circuitry and suppressing hyper-excitability in the injured hippocampus can restore calbindin. We compared the effects of fetal CA3 or CA1 cell grafting into the injured CA3 region of adult rats at 45 days after KA-induced injury on the hippocampal calbindin. The calbindin immunoreactivity in the dentate granule cells and the CA1 pyramidal neurons of grafted animals was evaluated at 6 months after injury (i.e. at 4.5 months post-grafting). Compared with the intact hippocampus, the calbindin in “lesion-only” hippocampus was dramatically reduced at 6 months post-lesion. However, calbindin expression was restored in the lesioned hippocampus receiving CA3 cell grafts. In contrast, in the lesioned hippocampus receiving CA1 cell grafts, calbindin expression remained less than the intact hippocampus. Thus, specific cell grafting restores the injury-induced loss of calbindin in the adult hippocampus, likely via restitution of the disrupted circuitry. Since loss of calbindin after hippocampal injury is linked to hyperexcitability, re-expression of calbindin in both dentate gyrus and CA1 subfield following CA3 cell grafting may suggest that specific cell grafting is efficacious for ameliorating injury-induced hyperexcitability in the adult hippocampus. However, electrophysiological studies of KA-lesioned hippocampus receiving CA3 cell grafts are required in future to validate this possibility.
Keywords: calcium binding proteins, chronic epilepsy, dentate gyrus, graft-host connectivity, hippocampal CA3 cells, hippocampal injury, kainic acid, neural grafting, neural transplant integration, seizures
INTRODUCTION
Temporal lobe epilepsy (TLE) is one of the most prevalent neurodegenerative diseases, and seizures in a significant fraction of people with TLE are resistant to antiepileptic drugs (Engel, 1999). Because of the involvement of the hippocampus in the generation and maintenance of seizures in TLE, the conventional treatment of patients with medically intractable TLE has been neurosurgical excision of the hippocampus or both hippocampus and amygdala. This procedure, though greatly reduces seizure frequency (Clusmann et al., 2002; Engel, 2003), may lead to impairments in learning and memory functions (Nunn et al., 1998; Martin et al., 2002; Richardson et al., 2004). Therefore, alternative therapeutic options that not only suppress seizures but also preserve hippocampal-dependent learning and memory functions are critical. Multiple studies have shown that TLE is associated with profound hyperexcitability in the dentate gyrus of the hippocampal formation (Sutula et al., 1989; Williamson and Spencer, 1995; Isokawa, 1996; Isokawa et al., 1997; Nagerl et al., 2000). Hence, testing promising new therapies in animal models that exhibit hyperexcitability in the dentate gyrus have considerable importance.
Intracerebroventricular (ICV) kainic acid (KA) administration in rat is widely used as a model for studying hippocampal injury, hyperexcitability, and TLE. An ICV KA administration results in the degeneration of CA3 pyramidal and dentate hilar neurons (Shetty and Turner, 1999a; Nadler, 2003), which leads to reorganization of the circuitry, reductions in the number of GABA-ergic interneurons, loss of functional inhibition, and hyperexcitability in the hippocampus (Okazaki et al., 1995; Shetty and Turner, 1997, 1999b, 2000; Nadler, 2003). These changes are also associated with sustained loss of the calcium binding protein calbindin in major fractions of dentate granule and CA1 pyramidal cells (Shetty and Turner, 1995b), which is consistent with the loss of calbindin observed in the surviving dentate granule cells of TLE patients (Magloczky et al., 1997). Although the precise function of calbindin in the nervous system remains largely unclear, the binding of calcium by calbindin is believed to maintain homeostasis of intracellular calcium and to protect neurons against excessive intracellular calcium (Baimbridge and Miller, 1982, 1984; Miller and Baimbridge, 1983; Baimbridge et al., 1985, 1992; Sloviter, 1989; Cheng et al., 1994). From this perspective, it appears that the loss of calbindin in the surviving dentate granule and CA1 pyramidal cells after hippocampal injury and in TLE implies an elevated intracellular calcium concentration. However, physiological studies using hippocampal tissues from TLE patients imply that the loss of calbindin from granule cells increases the calcium-dependent inactivation of voltage-dependent calcium currents, and thereby diminishes calcium influx during repetitive neuronal firing, suggesting that the loss of calbindin contributes to the resistance of neurons to calcium-dependent neuronal damage related to hyperexcitability (Nagerl et al., 2000). Thus, though the precise reason for calbindin loss following hippocampal injury or during epilepsy is still unclear, the loss of calbindin in dentate granule cells and CA1 pyramidal cells after the KA-induced CA3-region injury implies the existence of hyperexcitability in both dentate gyrus and CA1 subfield. Indeed, physiological studies report the presence of hyperexcitability in both of these regions following CA3 region injury (Tauck and Nadler, 1985; Turner and Wheal, 1991; Perez et al., 1996). The mechanisms responsible for persistent hyperexcitability in the dentate gyrus and the CA1 subfield of the CA3-lesioned hippocampus likely include an aberrant reorganization of the disrupted circuitry, decreased afferent circuitry leading to interneurons, loss of the physiological efficacy of interneurons, and reductions in the number of GABA synthesizing interneurons (Mathern et al., 1993; Mello et al., 1993; Dudek et al., 1994; Okazaki et al., 1995; Shetty and Turner, 2000, 2001; Wuarin and Dudek, 2001; Buckmaster et al., 2002; Scharfman et al., 2003; Shetty et al., 2005) Therefore, strategies that facilitate appropriate reorganization of the disrupted circuitry may be critical for easing hyperexcitability in the injured hippocampus. Replacing the degenerated host neurons with grafting of specific fetal neural cells may be particularly beneficial in this regard. It is plausible that circuitry restoration within the injured hippocampus by appropriate cell grafts leads to afferent control over autonomous regions to reduce their seizure-generating output into other regions of the brain. Our previous studies using the ICV KA injury model of rat TLE have suggested that grafting of specific fetal cells facilitates appropriate reorganization of the damaged hippocampal circuitry (Shetty and Turner, 1997, 2000; Shetty et al., 2000, 2005). However, it is unknown whether specific cell grafting into the injured hippocampus is capable of normalizing the altered calcium homeostatic mechanisms in dentate granule and CA1 pyramidal cells.
We evaluated the efficacy of specific and nonspecific fetal hippocampal cell grafts placed into the lesioned CA3 region of KA-treated rats for restoring the expression of calbindin in dentate granule and CA1 pyramidal cells. We hypothesize that the enduring loss of calbindin in the dentate gyrus and the CA1 subfield after CA3-lesion is due to disruption of the hippocampal circuitry leading to hyperexcitability in these regions; therefore, specific cell grafts that are capable of both reconstructing the disrupted circuitry and suppressing hyperexcitability in the injured hippocampus can restore calbindin. The effects of two different fetal cell grafts (CA3 and CA1 cell grafts) were evaluated on the expression of calbindin in adult dentate granule cells and CA1 pyramidal cells of the adult hippocampus after an ICV KA-induced CA3 region injury, in comparison to the intact hippocampus and the lesion-only hippocampus. Grafts were placed at 45 days after KA administration, and calbindin expression in the host hippocampus was evaluated at 6 months post-lesion.
MATERIALS AND METHODS
Animals and Experimental Design
Young adult (4-month-old) male Fischer 344 rats purchased from Harlan Sprague-Dawley (Indianapolis, IN) were used in this study. Figure 1 illustrates various experiments and procedures employed in this study. Duke University Institutional Animal Care and Use Committee and the Animal Studies Subcommittee of the Durham Veterans Affairs Medical Center have approved all experiments performed in this study.
FIGURE 1.

Schematic of major experiments performed in this study. The experiments mainly include kainic acid (KA) lesions of host animals (illustrated in the center), the dissection and dissociation of fetal hippocampal CA3 and CA1 tissues from donor fetuses into single cell suspensions, grafting of donor cells into the injured CA3 region at 45 days post-KA administration, perfusion of animals, and processing of tissues for Nissl staining and calbindin immunostaining, and measurement of calbindin immunopositive neurons in the dentate granule cell layer and the CA1 pyramidal cell layer using stereology. F, Fimbria; ICV KA, intracerebroventricular kainic acid; S, Subiculum.
ICV KA Injections
KA was administered unilaterally into the posterior lateral ventricle of young adult rats, using established protocols (Shetty and Turner, 1995b, 1997, 2000) (Fig. 1). Each rat was anesthetized with an anesthetic mixture comprising ketamine (50 mg/ml), xylazine (6 mg/ml), and acepromazine (0.5 mg/ml) at a dose of 1.25 ml/kg body weight and fixed into a stereotaxic apparatus. Incisor bars in the stereotaxic apparatus were set at 3.7 mm below the interaural line, the dorsal surface of the skull was uncovered through an incision in the skin, and a burr hole was drilled in the skull at a spot, which is 3.7 mm caudal to bregma and 4.1 mm right lateral to the midline. Tenmicroliter Hamilton syringe fitted with 25G needle and filled with KA solution in saline was inserted through the burr hole, the cerebral cortex, and the corpus callosum into the posterior lateral ventricle. At 4.5 mm ventral to the surface of the brain, 1 μl of KA (0.5 μg) was injected at a rate of 0.2 μl/min. The needle was withdrawn at 15 min after the injection.
Preparation of CA3 and CA1 Cell Suspension From Embryonic Day (E) 19 Rat Hippocampus
Timed pregnant rats were deeply anesthetized on gestation day 19, fetuses were removed by cesarean section, and decapitated. The heads of fetuses were collected in a petri plate containing calcium- and magnesium-free Hank’s Balanced Salt Solution (HBSS; Sigma) with 0.6% glucose, 10 mM HEPES, and 1% penicillin–streptomycin. Using a dissection microscope, the two cerebral hemispheres were separated from the rest of the brain by cutting through cerebral peduncles. Each cerebral hemisphere was then cut coronally into four slices of equal size and slices comprising hippocampal tissue were identified. Middle two slices from each hemisphere were consistently found to contain hippocampal tissues. Hippocampal tissue was unfolded from each of these pieces, and subfields CA3 (lateral most part of hippocampus with attached choroid plexus) and CA1 (medial part of hippocampus adjoining subiculum) were separated by sharp cuts using scalpel blade and collected separately in fresh HBSS (Shetty and Turner, 2000; Zaman et al., 2000) (Fig. 1). The tiny primordial dentate gyrus was included in the CA1 tissue with this procedure. The hippocampal CA3 and CA1 tissues were processed separately for dissociation and preparation of single cell suspension using mechanical trituration. Tissue pieces were triturated 30–40 times in 2 ml of HBSS using a fire polished Pasteur pipette, and the resulting cell suspension was diluted with 10 ml of fresh HBSS. The diluted cell suspension was then sieved through a tissue strainer (pore size = 75 μm), centrifuged at 800 rpm for 10 min, and the pellet was resuspended in HBSS. Cells were washed twice by resuspension in HBSS and centrifugation. The final pellet was resuspended in 30 μl of HBSS and viability assessed using trypan blue exclusion method. The concentration of cells was then adjusted to 1 × 105 viable cells/μl and placed on ice during the grafting procedure.
Grafting of CA3 and CA1 Cells Into the CA3-Injured Hippocampus
Each host rat was anesthetized (using the mixture described earlier) at 45 days following the unilateral ICV KA injection and fixed into a stereotaxic apparatus with the plane of the incisor bar set at 3.3 ± 0.3 mm below the interaural line. The detailed transplantation procedure is described in our earlier reports (Shetty and Turner, 1995a, 1997). One microliter of the cell suspension containing 100,000 live cells was injected into each of the following three locations in the hippocampus ipsilateral to the KA lesion: (1) anterior-posterior (AP) = 3.0 mm posterior to bregma, lateral (L) = 2.2 mm right lateral to midline, and ventral (V) = 3.5 mm from the surface of brain; (2) AP = 3.8 mm, L = 3.0 mm right lateral, and V = 3.5 mm; and (3) AP = 4.6 mm, L = 4.0 mm right lateral, and V = 3.5 mm. Our earlier studies have shown that these locations are particularly useful for placing grafts in close vicinity of the degenerated CA3 pyramidal cell layer (Shetty and Turner, 2000; Shetty et al., 2005) (Fig. 1). Grafting was performed at a delayed time-point (i.e. at 45 days) after KA injections, as our earlier studies have shown that calbindin expression in both dentate granule cells and CA1 pyramidal neurons is considerably diminished during this period (Shetty and Turner, 1995b).
Analyses of KA Lesions and Selection of Section for Immunohistochemistry
The calbindin immunoreactivity was analyzed in the dentate granule cells and the CA1 pyramidal cells of intact control animals (naive controls n = 5), rats treated with ICV KA alone (at 6 months post-KA, n = 5), and ICV KA-treated rats receiving CA3 or CA1 cell grafts at 45 days post-KA and killed at 6 months post-KA (n = 5/transplant group). All animals were deeply anesthetized with halothane and perfused transcardially with 4% paraformaldehyde solution in phosphate buffer (PB). The brains were removed, post-fixed in 4% paraformaldehyde overnight at 4°C, cryoprotected in 30% sucrose solution in PB, and 20-μm-thick cryostat sections were cut coronally through the hippocampus. The sections were collected serially in PB and every 12th section through the dorsal hippocampus was processed for Nissl staining (Fig. 1). In brain sections of animals receiving ICV KA alone (lesion-only animals), Nissl staining demonstrated the extent and size of KA-induced injury.
Whereas, in brain sections of animals receiving ICV KA and grafts, Nissl staining revealed both the size of KA-induced lesion and the location and cytoarchitecture of transplants. In all intact and lesion-only animals, sections through the septal hippocampus at levels corresponding to 2.8–4.0 mm posterior to bregma (Paxinos et al., 1985) with a distance of 120 μm between them were selected for calbindin immunostaining (Fig. 1). This selection ensured that selected sections were independent from one another to clearly avoid analysis of calbindin immunoreactivity from adjacent sections, and hence, replication of the findings of the first section. All animals that received grafts at 45 days post-KA lesion exhibited surviving grafts inside the brain when examined at 6 months post-KA lesion; however, only the brains with at least one intrahippocampal graft were selected for calbindin staining; over 80% of grafted brains reached this threshold. Every sixth section through the grafted region from these brains was processed for calbindin immunostaining. In “lesioned, grafted” animals, it was also ensured that the selected sections contained a transplant in the close vicinity of the degenerated CA3 cell layer. Coronal sections through the dorsal hippocampus, at levels specified above, were preferred over those from ventral hippocampus because the injected transplants were clearly located in the dorsal hippocampus.
Calbindin Immunostaining
Free-floating sections were washed in 0.1 M phosphate-buffered saline (PBS) and treated with 1% sodium borohydride solution in distilled water for 15 min. The sodium borohydride treatment reduces free aldehyde groups and double bonds, and thereby improves immunoreactivity of calbindin (Toth and Freund, 1992). The sections were then washed in PBS, treated with 10% normal horse serum (NHS, for 30 min), and incubated in the primary antiserum (1:1,000) raised against Calbindin-D28k purified from chicken gut (Sigma Chemicals, St. Louis, MO) for 2 days at 4°C. This is a well-characterized antibody and has been shown to react specifically with calbindin-D28k in brain tissues from several species including rat (Celio, 1990; Bazzett et al., 1994; Gutierrez and Cusick, 1994; Ren and Ruda, 1994). The primary antibody solution was prepared in PBS and contained 3% NHS and 0.1% Triton X-100. The sections were washed three times in PBS, incubated in biotinylated horse anti-mouse IgG (1:200; Vector Laboratories, Burlingame, CA) for 45 min, washed in PBS, and then treated with avidin biotin complex (ABC) reagent (1:25; Vector) for 45 min. The sections were rinsed in PBS, and the peroxidase reaction was developed using diaminobenzidine (DAB) as chromogen (DAB kit; Vector). The sections from all KA-treated groups were processed and immunostained in parallel with some sections from control animals using the same dilutions of primary and secondary antibody and ABC solutions, the same number of washes, and the same concentrations of DAB and hydrogen peroxide. Under these conditions, the sections from control animals consistently demonstrated normal staining pattern. Negative control sections were processed in the same manner except that the primary antibody incubation step was replaced by continued incubation in normal horse serum. Neither immunostaining nor any recognizable background staining was observed under these conditions in the negative control sections. Sections were mounted on gelatinized slides, air dried, dehydrated, cleared, and permanently mounted using Permount.
Quantification of Calbindin Immunoreactive Cells in the Granule Cell Layer and the CA1 Pyramidal Cell Layer
The number of calbindin immunoreactive cells per unit volume of tissue in the dentate granule cell layer and the CA1 pyramidal cell layer was quantified using the optical fractionator method. In each animal belonging to different groups, four sections (each separated by a distance of 120 μm) through the dorsal hippocampus were quantified. In each section, calbindin-positive cells were counted from 42 randomly and systematically selected frames (22 on the granule cell layer and 22 on the CA1 pyramidal cell layer, each measuring 20 × 20 μm2, 0.0004 mm2 area) using the microscope eyepiece grid and 100× oil immersion objective lens (final magnification 1,000×). For these studies, we cut 20-μm-thick sections through the hippocampus using a cryostat that has been calibrated. Measurement of the thickness of sections immediately following sectioning has revealed that the variability between sections is minimal (i.e. ±1 μm). With calbindin immunostaining, sections showed significant shrinkage along the Z-axis. However, the extent of shrinkage between sections from control and KA lesioned groups was similar. Measurement of the thickness of sections in different groups revealed that the average thickness of sections was reduced to ~60% of the initial section thickness, and hence, at the time of data collection, the thickness of sections was about 12 μm in all groups. Every counting frame region was focused through the whole thickness of the section. The surface of the section was initially brought into focus, after which the plane of the focus was moved ~2 μm deeper through the section to get rid of the problem of uneven section surface. This plane served as the first point of the counting process, but calbindin-positive cells that were completely in focus at this level were not counted. Continuing to focus down, any additional calbindin-positive cells that came into focus were counted if they were entirely within the counting frame or touching the upper or right side of the counting frame. The calbindin-positive cells were counted through the total depth of the section and this thickness (i.e. 10 μm) was used as the height for calculation of the total number of cells per unit volume (Nv). The average value for every animal (i.e. average of 84 independent unit volumes; 21 unit volumes/section, 4 sections/animal) was calculated separately for each of the two cell layers (dentate granule cell layer and CA1 pyramidal cell layer) and expressed as counts per 0.1 mm3.
RESULTS
Hippocampal Cytoarchitecture After Unilateral ICV KA Administration
Unilateral ICV KA administration caused degeneration of pyramidal neurons in subfields CA3a–c and a fraction of neurons in the dentate hilus, in hippocampus ipsilateral to the KA administration. When compared with the cytoarchitecture of the intact control hippocampus (Figs. 2A1,A2), degeneration of neurons in the CA3 pyramidal cell layer on KA-treated side was striking at 45 days post-lesion (Figs. 2B1,B2). The loss of pyramidal neurons was virtually complete in CA3b and CA3c subregions but partial in CA3a subregion. Nissl staining also displayed gliosis in this region at this time-point. These features are consistent with our earlier reports on ICV KA model in rats (Shetty and Turner, 1995a,b, 1997, 2000; Shetty et al., 2003). The overall pattern of CA3 cell layer injury was consistent throughout the anteroposterior axis of the hippocampus. However, dentate granule cell layer was intact throughout the anteroposterior axis of the hippocampus. The CA1 pyramidal cell layer, on the other hand, remained intact in the anterior half but showed some cell loss in the posterior half of the hippocampus. When the hippocampus ipsilateral to KA was examined at 6 months post-KA, similar pattern of lesion was observed (Figs. 2C1,C2), but the amount of gliosis in the injured CA3 cell layer appeared reduced. Thus, unilateral ICV KA administration in rat leads to loss of CA3 pyramidal neurons in hippocampus ipsilateral to KA administration. Because the degenerated neurons are not replaced spontaneously, similar lesion persists at 6 months post-KA. However, in hippocampus contralateral to the KA administration, all principal cell layers remain intact even at 6 months post-KA (Figs. 2D1,D2).
FIGURE 2.

Extent of hippocampal injury after unilateral intracerebroventricular kainic acid (ICV KA) injection in young adult F344 rats. Figure compares the cytoarchitecture of an intact hippocampus (A1, A2), hippocampus ipsilateral to KA at 45 days post-administration (B1, B2), hippocampus ipsilateral to KA at 6 months post-administration (C1, C2), and hippocampus contralateral to KA at 6 months post-administration (D1, D2). Note the persistent loss of neurons in the CA3 pyramidal cell layer (denoted by asterisks) and the dentate hilus of hippocampi ipsilateral to KA administration at both 45 days and 6 months after KA administration. However, all hippocampal cell layers appear intact in hippocampus contralateral to KA administration. DG, dentate gyrus. Scale bar: A1, B1, C1, D1 = 500 μm; A2, B2, C2, D2 = 200 μm.
Calbindin Immunoreactivity in the Hippocampus of Naive Rats
In the hippocampus of naive rats, calbindin immunoreactivity was conspicuous in both the dentate gyrus and the CA1 subfield (Fig. 3). Calbindin immunoreactivity in the dentate gyrus was characterized by its presence in the granule cell layer, molecular layer, and the entire mossy fiber bundle located in the dentate hilus (Figs. 3A1–A3). Virtually all granule cells appeared robustly immunopositive for calbindin in the granule cell layer (Fig. 3A3). Thus, there is robust presence of calbindin in all compartments of dentate granule cells (cell bodies, dendrites, and axons). Calbindin immunoreactivity in the CA1 subfield was typified by its occurrence in the superficial pyramidal neurons of the stratum pyramidale and some scattered non-pyramidal neurons of strata radiatum, lacunosum moleculare, and oriens (Fig. 3A4). In the CA3 subfield, the principal mossy fiber bundle (i.e. in the strata lucidum) and some nonpyramidal neurons in the strata radiatum and oriens exhibited calbindin immunoreactivity. However, the CA3 pyramidal neurons lacked calbindin expression (Fig. 3A2). The above pattern of calbindin immunoreactivity in the intact adult hippocampus is consistent with multiple earlier reports (Baimbridge and Miller, 1982; Sloviter, 1989; Tonder et al., 1994; Shetty and Turner, 1995b).
FIGURE 3.

Calbindin immunoreactivity in the hippocampus of naive rats (A1–A4). Note dense calbindin immunoreactivity in the principal mossy fiber bundle (A2), dentate granule cells (A3), and in the superficial CA1 pyramidal neurons (A4). DG, dentate gyrus; DH, dentate hilus; pMFB, principal mossy fiber bundle; SO, stratum oriens; SP, stratum pyramidale; SR, stratum radiatum. Scale bar: A1 = 500 μm; A2, A3 = 200 μm; A4 = 100 μm.
Changes in Hippocampal Calbindin Immunoreactivity After ICV KA-Induced Injury
In hippocampus ipsilateral to KA administration (i.e. the hippocampus with CA3 region injury), calbindin immunoreactivity diminishes considerably in both the dentate gyrus and the CA1 subfield. Our earlier study has shown that reductions in calbindin immunoreactivity within the dentate gyrus and CA1 subfield of hippocampus ipsilateral to KA is apparent as early as 21 days post-KA and persists at least until 60 days post-KA (Shetty and Turner, 1995b). Examination of calbindin immunoreactivity at 6 months post-KA in this study demonstrated persistence of calbindin loss within these regions of the hippocampus ipsilateral to KA administration (Figs. 4A1–A5). In the dentate gyrus of hippocampus ipsilateral to KA administration, only fractions of granule cells expressed calbindin immunoreactivity in both upper and lower blades of the granule cell layer (Fig. 4A1). Because smaller groups of granule cells positive for calbindin were separated by granule cells that lacked calbindin, the overall pattern of calbindin immunoreactivity appeared patchy in the granule cell layer (Fig. 4A1). The appearance of intact granule cell layer in Nissl-stained adjacent section demonstrated that the patchy appearance of calbindin immunoreactivity is not due to degeneration of granule cells (Fig. 4A2). Calbindin immunoreactivity was also reduced in the principal mossy fiber bundle located in the CA3 region (Fig. 4A3). Interestingly, the intensity/amount of calbindin in granule cells that are positive was comparable to the intensity/amount typically observed in granule cells of the intact control hippocampus (Fig. 4A4), suggesting that some granule cells retain normal levels of calbindin despite complete loss in a vast of majority of granule cells. In the CA1 subfield of hippocampus ipsilateral to KA administration, calbindin immunoreactivity was absent in virtually all pyramidal cells (Fig. 4A5). Similar loss of calbindin immunoreactivity also occurred in the dentate gyrus and CA1 subfield of hippocampus contralateral to KA administration (Figs. 4B1–B3). Thus, unilateral ICV KA administration leads to persistent loss of calbindin in virtually all pyramidal cells of the CA1 subfield and a vast majority of neurons in the granule cell layer. Furthermore, the loss of calbindin immunoreactivity was bilateral even though the neurodegeneration inflicted by KA appeared unilateral in nature. Because loss of calbindin in dentate gyrus and CA1 subfield is suggestive of hyperexcitability in these regions, the observations suggest the presence of persistent hyperexcitability in hippocampi on both ipsi- and contra-lateral sides at 6 months following KA administration.
FIGURE 4.

Bilateral loss of hippocampal calbindin immunoreactivity following unilateral intracerebroventricular kainic acid (ICV KA), visualized at 6 months post-administration (A1–B3). A1 is an example of hippocampus ipsilateral to ICV KA demonstrating severe loss of calbindin in dentate granule cells and CA1 pyramidal neurons. Nissl-stained adjacent section however shows intact cell layers in the dentate gyrus and the CA1 subfield (A2). A3–A5 are magnified views from A1, illustrating diminished calbindin immunoreactivity in the principal mossy fiber bundle (pMFB) located in the CA3 region (A3), loss of calbindin in a major fraction of dentate granules (A4), and virtually all CA1 pyramidal neurons (A5) following KA administration. B1 is an example of hippocampus contralateral to ICV KA demonstrating loss of calbindin in dentate granule cells and CA1 pyramidal neurons. B2 and B3 are magnified views of regions from B1 illustrating loss of calbindin immunoreactivity in the dentate gyrus (B2) and CA1 subfield (B3). DG, dentate gyrus; DH, dentate hilus; SO, pMFB, principal mossy fiber bundle; stratum oriens; SP, stratum pyramidale; SR, stratum radiatum. Scale bar: A1, A2, B1 = 500 μm; A3 = 200 μm; A4, A5, B2, B3 = 100 μm.
Effects of Hippocampal CA3 Cell Grafting on Calbindin Expression
We transplanted fetal hippocampal CA3 cells into the injured CA3 region at 45 days post-KA administration in this study, with the aim of replacing the lost CA3 pyramidal neurons and repairing the hippocampal circuitry disrupted by KA-induced neurodegeneration. Based on our earlier study, loss of calbindin immunoreactivity is apparent at 45 days post-KA in both dentate gyrus and the CA1 subfield (Shetty and Turner, 1995b). Thus, at the time of transplantation, hippocampi on both ipsi-and contra-lateral sides of KA administration exhibit loss of calbindin immunoreactivity, which allowed analyses of the effects of grafting on hippocampal calbindin immunoreactivity. Examination of sections through the hippocampus using both calbindin immunostaining and Nissl staining revealed the survival of transplants at 6 months post-KA (~4.5 months post-grafting). In majority of animals, transplants were located just below the degenerated CA3 pyramidal cell layer (Figs. 5A1,A2). In addition, Nissl staining demonstrated a large number of surviving neurons within CA3 cell transplants (Figs. 5A2,A3), suggesting an excellent survival of grafted CA3 pyramidal neurons in the adult hippocampus with CA3-region injury. This finding is consistent with our earlier quantitative studies on both short-term and long-term survival of grafted cells in this injury model (Shetty and Turner, 1995b; Zaman et al., 2000; Zaman and Shetty, 2001). Larger CA3 pyramidal neurons within these grafts appeared to be organized in clusters (Fig. 5A3). The types of neurons encountered within CA3 cell transplants have been characterized previously and described in our earlier report (Zaman et al., 2000). These results have shown that the morphology and size of neurons encountered within CA3 cell transplants are comparable to hippocampal CA3 neurons developed in situ (Zaman et al., 2000). Furthermore, it was observed that a large number of neurons within CA3 cell grafts express the CA3 pyramidal cell specific markers, such as the nonphosphorylated neurofilament proteins (Zaman et al., 2000). Moreover, it was seen that neurons from CA3 cell grafts send robust efferent projections into the dentate inner molecular layer and the CA1 strata oriens and radiatum (Shetty et al., 2005) and the contralateral hippocampus (commissural projections) and the septum (Shetty et al., 2000), which are consistent with the efferent pathways observed for the CA3 pyramidal neurons developed in situ. Thus, hippocampal CA3 cell transplants contain a large number of CA3 pyramidal neurons.
FIGURE 5.

Calbindin immunoreactivity, and transplant location and morphology in a rat receiving CA3 cell transplants at 45 days post-KA and analyzed at 6 months post-KA. A1 shows the pattern of calbindin immunoreactivity, whereas A2 and A3 demonstrate transplant location and morphology in this rat. A4–A6 are magnified views of regions from A1 demonstrating the robust expression of calbindin in the principal mossy fiber bundle located in the CA3 region (A4), dentate granule cells (A5), and CA1 pyramidal neurons (A6). Similar calbindin immunoreactivity is also seen in the hippocampus contralateral to KA administration and CA3 cell grafting (B1). B2 and B3 are magnified views of regions from B1 demonstrating robust calbindin immunoreactivity in dentate granule cells (B2) and CA1 pyramidal neurons (B3). Note that calbindin expression in this KA treated rat receiving CA3 cell grafts after KA administration is equivalent to calbindin expression observed in an intact control rat (Fig. 3), and much greater than that observed in a rat receiving KA alone (Fig. 4) or a rat receiving KA and CA1 cell grafts (Fig. 6). Asterisks in A2 denote the degenerated CA3 cell layer. DG, dentate gyrus; DH, dentate hilus; ML, molecular layer; SO, stratum oriens; SP, stratum pyramidale; SR, stratum radiatum. Scale bar: A1, A2, B1 = 500 μm; A4 = 200 μm; A3, A5, A6, B2, B3 = 100 μm.
Interestingly, in rats receiving CA3 cell grafts at 45 days post-KA and analyzed at 6 months post-KA, calbindin immunoreactivity was restored in dentate gyrus and the CA1 subfield on both ipsi- and contra-lateral sides of KA administration and grafting (Fig. 5A1,B1). In addition, the CA3 cell transplants comprised some calbindin-positive neurons (presumably inter-neurons derived from grafts). The restoration of calbindin in hippocampi of grafted animals appeared striking when compared with age-matched hippocampi from rats receiving KA treatment alone (Fig. 4A1). The overall pattern of hippocampal calbindin immunoreactivity in rats receiving CA3 cell grafts resembled that of the intact control hippocampus. This was exemplified by the presence of robust calbindin immunoreactivity in virtually all granule cells of the dentate granule cell layer (Figs. 5A1,A5), the principal mossy fiber bundle located in the CA3 region (Figs. 5A1,A4), and a large number of pyramidal neurons in the CA1 subfield (Figs. 5A1,A6). In the dentate gyrus, the molecular layer comprising dendrites of granule cells also exhibited robust calbindin immunoreactivity. Similar restoration of calbindin immunoreactivity was observed in hippocampus contralateral to KA administration and CA3 cell grafting (Figs. 5B1–B3). Thus, just as the bilateral loss of calbindin following unilateral CA3-region injury, the restoration of calbindin was bilateral following unilateral cell grafting into the injured CA3 region, suggesting that the restoration of calbindin is linked to reconstruction of hippocampal circuitry. Restoration of calbindin may also be due to suppression of hyperexcitability mediated by CA3 cell grafts. However, EEG recordings of KA-treated animals receiving CA3 cell grafts are needed in future to validate this possibility.
Effects of Hippocampal CA1 Cell Grafting on Calbindin Expression
We transplanted fetal hippocampal CA1 cells into the injured CA3 region at 45 days post-KA administration in another group of rats with the aim of ascertaining the effects of cells that are nonspecific to the injured region on calbindin immunoreactivity. Because CA1 pyramidal neurons are not expected to repair the hippocampal circuitry, this approach helped in determining whether the effects of CA3 cell grafts observed above was specific or just a nonspecific effect of grafting itself. Examination of sections through the hippocampus using both calbindin immunostaining and Nissl staining revealed the survival of CA1 cell transplants at 6 months post-KA (~4.5 months post-grafting). In majority of animals, CA1 cell transplants were located close to the degenerated CA3 pyramidal cell layer (Figs. 6A1,A2). Nissl staining demonstrated surviving neurons within CA1 cell transplants (Figs. 6A2,A3). However, in comparison to CA3 cell transplants, the overall size and survival of grafted cells appeared modest, which is consistent with our earlier quantitative comparison of cell survival between CA3 and CA1 transplants in this injury model (Zaman et al., 2000). The neurons were smaller in size and were dispersed within CA1 cell grafts (Fig. 6A3). The types of neurons encountered within CA1 cell transplants have been characterized previously. These studies have demonstrated that the morphology and size of neurons encountered within CA1 cell transplants are comparable to hippocampal CA1 neurons developed in situ and neurons within CA1 cell grafts do not express CA3 cell markers such as nonphosphorylated neurofilament proteins (Zaman et al., 2000). Furthermore, it was observed that neurons from CA1 cell grafts placed into the injured CA3 region send local axonal projections mostly into to the CA1 subfield and only a few into the dentate inner molecular layer (Shetty et al., 2005), and send long-distance efferent projections into the septum but fail to establish commissural projections (Shetty et al., 2000). Additionally, the CA1 cell transplants in this study exhibited some calbindin-positive neurons (presumably graft-derived interneurons or a type of CA1 pyramidal neurons that express calbindin). The above observations suggest that hippocampal CA1 cell transplants appear to contain mostly neurons having characteristics of CA1 pyramidal neurons. Despite grafting into the CA3 region, they do not seem to adopt characteristics of CA3 pyramidal neurons. However, fetal CA1 cells transplanted into the ischemia-damaged CA1 region develop cytological features that are indistinguishable from their normal CA1 counterparts, express proteins that are specific to CA1 pyramidal neurons, and reconstruct damaged circuitry in the CA1 region (Mudrick et al., 1989; Mudrick and Baimbridge, 1991).
FIGURE 6.
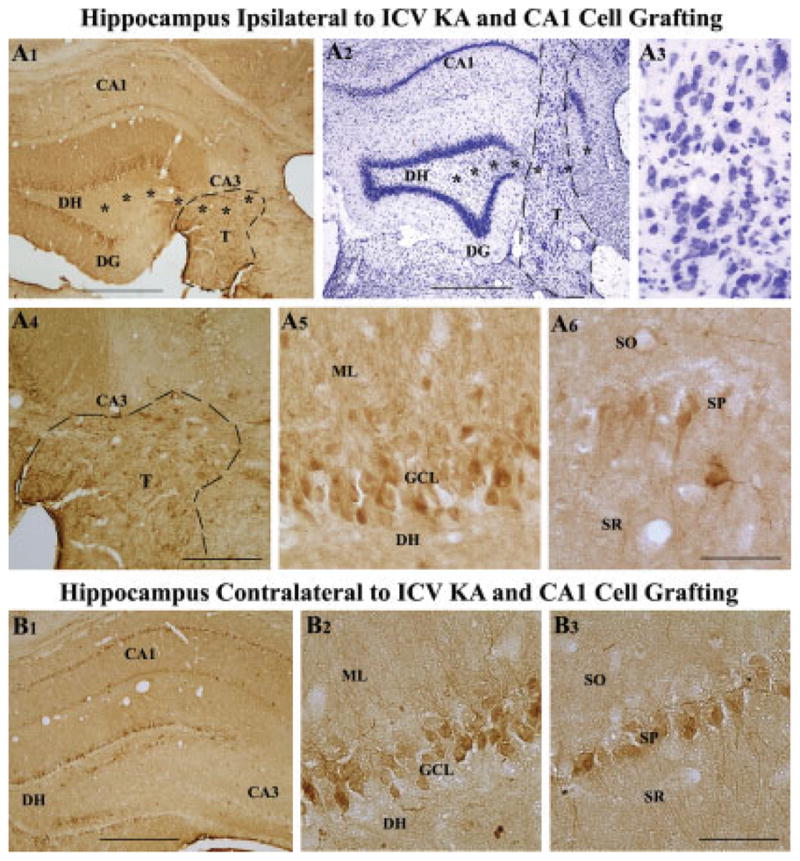
Calbindin immunoreactivity, and transplant location and morphology in a rat receiving CA1 cell transplants at 45 days post-KA and analyzed at 6 months post-KA. A1 shows the pattern of calbindin immunoreactivity, whereas A2 and A3 demonstrate transplant location and morphology in this rat. A4–A6 are magnified views of regions from A1 demonstrating the absence of calbindin in the principal mossy fiber bundle located in the CA3 region (A4), expression of calbindin in a fraction of dentate granule cells (A5), and a few CA1 pyramidal neurons (A6). B1 shows the pattern of calbindin immunoreactivity in the hippocampus contralateral to KA administration and CA1 cell grafting (B1). B2 and B3 are magnified views of regions from B1 demonstrating calbindin immunoreactivity in a fraction of dentate granule cells (B2) and CA1 pyramidal neurons (B3). Note that calbindin expression in this rat receiving CA1 cell grafts after KA administration is considerably less than calbindin expression observed in an intact control rat (Fig. 3) and a rat receiving KA and CA3 cell grafts (Fig. 5) but better than calbindin immunoreactivity seen in a rat receiving KA alone (Fig. 4). Asterisks in A1 and A2 denote the degenerated CA3 cell layer. DG, dentate gyrus; DH, dentate hilus; DG, dentate gyrus; ML, molecular layer; SO, stratum oriens; SP, stratum pyramidale; SR, stratum radiatum. Scale bar: A1, A2, B1 = 500 μm; A4 = 200 μm; A3, A5, A6, B2, B3 = 100 μm.
Hippocampal calbindin immunoreactivity in rats receiving CA1 cell grafts at 45 days post-KA and analyzed at 6 months post-KA (Fig. 6A1) appeared clearly less than the intact control hippocampus (Fig. 3A1). However, in comparison to rats receiving KA treatment alone (Fig. 4A1), the hippocampus ipsilateral to KA administration and CA1 cell grafting exhibited calbindin immunoreactivity in a greater fraction of granule cells in the dentate granule cell layer (Figs. 6A1,A5) and in a few neurons in the CA1 cell layer (Figs. 6A1,A6). The principal mossy fiber bundle was not calbindin immunoreactive in both dentate hilus and CA3 stratum lucidum (Figs. 6A1,A4). Similar pattern of changes in calbindin immunoreactivity were observed in hippocampus contralateral to KA administration and CA1 cell grafting (Figs. 6B1–B3). However, calbindin immunoreactivity in CA1 pyramidal cell layer appeared relatively greater in some rats, in comparison to CA1 pyramidal cell layer of hippocampus ipsilateral to KA administration. Thus, though the calbindin immunoreactivity following unilateral CA1 cell grafting into the injured CA3 region remained less than the intact control hippocampus, there was some recovery in calbindin immunoreactivity bilaterally in comparison to rats receiving KA treatment alone. The improvements were mainly in soma of a fraction of granule cells and a few CA1 pyramidal neurons. However, there was no perceivable restoration in dendrites and axons of granule cells.
Extent of Restoration in Hippocampal Calbindin Following Grafting of Fetal CA3 or CA1 Cells
To ascertain the degree of restoration in hippocampal calbindin following grafting of fetal CA3 or CA1 cells, we compared the total number of calbindin immunoreactive cells per unit volume in the dentate granule cell layer and the CA1 pyramidal cell layer between different groups (n = 5/group). The groups included hippocampi of intact control rats, hippocampi ipsilateral to KA administration (lesion-only hippocampi), hippocampi ipsilateral to KA administration and CA3 cell grafting, and hippocampi ipsilateral to KA and CA1 cell grafting (Fig. 7). These comparisons utilized one-way analysis of variance (ANOVA) with Student-Newman-Keuls multiple comparisons post hoc test. In the dentate granule cell layer of the intact hippocampus, 36,200 ± 1,063 (mean ± SEM) cells were immunoreactive in every 0.1 mm3 of tissue (Fig. 7A). Following KA-induced CA3 region injury, this number decreased to 13,050 ± 554 cells at 6 months post-KA (64% reduction, P < 0.001). However, when fetal CA3 cells were transplanted at 45 days post-KA and hippocampi examined at 6 months post-KA, the numbers of calbindin immunoreactive granule cells (32,650 ± 1,484) were highly similar to that observed in the intact control hippocampus (Fig. 7A). In contrast, when fetal CA1 cells were transplanted at 45 days post-KA and hippocampi examined at 6 months post-KA, the numbers of calbindin immunoreactive cells (21,145 ± 1,687) were improved in comparison to lesion-only hippocampus (62% increase, P < 0.001; Fig. 7A). However, the numbers of immunoreactive cells remained 42% less than that in the intact control hippocampus (P < 0.001) and 35% less than the injured hippocampi receiving CA3 cell grafts (P < 0.001; Fig. 7A). Thus, restoration of calbindin in granule cell layer is complete following CA3 cell grafting but only partial following CA1 cell grafting.
FIGURE 7.

Comparison of the total number of calbindin immunoreactive neurons per 0.1 mm3 of tissue in the dentate granule cell layer (A) and the CA1 pyramidal cell layer (B) between hippocampi from intact rats, rats treated with KA alone (lesion only rats), KA treated rats receiving CA3 cell grafts, and KA treated rats receiving CA1 cell grafts. Comparisons utilized one-way analysis of variance (ANOVA) with Student-Newman-Keuls multiple comparisons post hoc test. All data are presented as means ± standard errors (SEM). Note the dramatic loss of calbindin in both dentate granule cells and CA1 pyramidal neurons in rats receiving KA alone and complete restoration of calbindin in KA treated rats receiving CA3 cell grafts. Also, note that CA1 cell grafting in KA treated rats leads to only a partial enhancement of calbindin in the granule cell layer but no increase in CA1 pyramidal cell layer. ***P < 0.001.
In the CA1 pyramidal cell layer of the intact hippocampus, 14,450 ± 519 (mean ± SEM) cells were immunoreactive in every 0.1 mm3 of tissue (Fig. 7B). After KA-induced CA3 region injury, this number diminished drastically to 1,225 ± 195 cells at 6 months post-KA (91% reduction, P < 0.001). However, when fetal CA3 cells were transplanted at 45 days post-KA and hippocampi examined at 6 months post-KA, the numbers of calbindin immunoreactive CA1 pyramidal neurons (13,875 ± 2,227) were similar to that in the intact control hippocampus (Fig. 7B). On the contrary, when fetal CA1 cells were grafted at 45 days post-KA and hippocampi examined at 6 months post-KA, the numbers of calbindin immunoreactive cells (3,425 ± 1,016) remained statistically comparable to lesion-only hippocampi (P > 0.05; Fig. 7B). The overall numbers were 76% less than the intact control hippocampus (P < 0.001) and 75% less than the injured hippocampi receiving CA3 cell grafts (P < 0.001; Fig. 7B). Thus, restoration of calbindin in CA1 pyramidal cell layer is complete following CA3 cell grafting. In contrast, CA1 cell grafting did not induce significant improvement.
DISCUSSION
This study provides the first evidence for the efficacy of fetal hippocampal CA3 cell grafting for restoring the expression of calbindin in both dentate granule cells and CA1 pyramidal neurons of the injured adult hippocampus in a rat model of TLE. Restoration of calbindin expression following grafting likely indicates amelioration of hyperexcitability in these neurons, because loss of calbindin in these cells following CA3 region injury is thought to reflect the persistence of hyperexcitability in the injured hippocampus. However, EEG recordings of KA-treated animals receiving CA3 cell grafts are needed in future to validate this possibility. As grafts were placed at 45 days post-injury (i.e. after the occurrence of calbindin loss), the results also imply the presence of a larger window after injury for application of intervention strategies that have the ability for both reconstructing the disrupted circuitry and suppressing epileptogenesis in the injured hippocampus. The study also demonstrates that fetal CA1 cell grafting is not efficacious for restoring normal expression of calbindin in adult hippocampus with CA3 region injury. Thus, fetal CA3 cell grafting is a useful approach for restoring calbindin expression in the adult hippocampus after injury.
Significance of Calbindin Loss Following Hippocampal Injury or Seizures
Calbindin is present in the entire cell volume of all dentate granule cells (Baimbridge, 1992) and a significant fraction of CA1 pyramidal neurons in the hippocampus. Because of the robust expression of the calbindin, these neurons are believed to have a large calcium buffer capacity (Baimbridge and Miller, 1982; Sloviter et al., 1991; Muller et al., 2005). Indeed, it has been observed that calbindin plays an important role in controlling abnormal discharges in hippocampal neurons via its function as an intracellular facilitator of calcium diffusion with a high affinity and selectivity for calcium in the micromolar range (Baimbridge et al., 1992). A recent study suggests that reduced expression of calcium binding proteins induced by seizure activity is indicative of hyperactivity in calcium binding protein positive neurons (Kim et al., 2006). Therefore, loss of calbindin in neurons following seizure activity has been considered as a sign of neuronal malfunction that likely result in hyperexcitability of neurons (Kim et al., 2006). Furthermore, it is well known that a significant fraction of dentate granule cells in both human and rodent hippocampus lose their cellular calbindin content during the course of TLE (Sloviter et al., 1991; Shetty and Turner, 1995b; Magloczky et al., 1997; Nagerl et al., 2000; Selke et al., 2006). Considering the functions, the deficiency of calbindin in the existing dentate granule and CA1 pyramidal cells after hippocampal injury and in TLE suggests an elevated intracellular calcium concentration. However, electrophysiological studies in excised hippocampal tissues from TLE patients reveal that calbindin loss in granule cells enhances the calcium-dependent inactivation of voltage-dependent calcium currents, which likely weakens the invasion of calcium during rhythmic neuronal firing (Nagerl et al., 2000). Thus, it appears that the loss of calbindin contributes to the defiance of dentate granule cells to possible calcium-related neuronal damage or death during seizures/hyperexcitability (Nagerl et al., 2000). A recent study showing that ~50% of granule cells in sclerotic hippocampi exhibit both hyperexcitability and a severe loss of calbindin supports the above plasticity in granule cells (Selke et al., 2006). Taken together, the above observations suggest a link between hyperexcitability and calbindin loss in hippocampal neurons, though it is currently unclear whether hyperexcitability precedes or succeeds calbindin loss in these neurons. Nevertheless, persistent loss of calbindin in these neurons indicates hyperexcitability.
Decreased calbindin content in dentate granule cells and CA1 pyramidal neurons may also induce impairments in both spatial learning and long-term potentiation because of the following. First, learning deficits in mice expressing Alzheimer’s disease-mutant human amyloid precursor protein correlates strongly with decreased levels of calbindin in granule cells of the dentate gyrus (Palop et al., 2003). This observation is consistent with the association of reduced hippocampal calbindin and hippocampal-dependent learning and memory deficits observed during normal aging (Shetty and Turner, 1998; Wilson et al., 2006). Second, transgenic mice deficient in calbindin exhibit selective impairments in spatial learning tasks that are dependent on normal hippocampal function (Molinari et al., 1996). Mice deficient in calbindin also fail to maintain long-term potentiation though short-lasting enhancement of the synaptic response persists (Molinari et al., 1996; Jouvenceau et al., 1999, 2002). Although the exact link between calbindin and long-term potentiation is unclear, several hypotheses have been proposed. One of the propositions is that calbindin plays an important role in spatially transferring and/or releasing calcium within the neuronal cytoplasm, which in turn prolongs an intracellular calcium signal (Lledo et al., 1992). This “slow release” calcium buffer is considered important for activation of calcium-activated kinases such as protein kinase C or calcium CaM kinase II, proteins required for the maintenance of long-term potentiation (Malinow et al., 1988; Malenka et al., 1989; Molinari et al., 1996). The second suggestion is that neurons depleted of their calbindin content exhibit a markedly increased calcium-dependent inactivation of calcium currents, which leads to only a weak rise in intracellular calcium that falls short of reaching the threshold level required for the maintenance of LTP (Kohr et al., 1991; Molinari et al., 1996). However, a recent study suggests that long-term potentiation impairment in calbindin-deficient mice is due to a disturbed control of intracellular calcium elevation (Jouvenceau et al., 2002). Considering the above, it is likely that decreased calbindin content in dentate granule cells and CA1 pyramidal neurons in the chronically injured/epileptic hippocampus contributes to impairments in both spatial learning and long-term potentiation observed during chronic epilepsy. These include reduced capacity for long-term potentiation and impaired spatial learning observed in adult rats after KA-induced seizures evoked during early postnatal development (Lynch et al., 2000), and failure to induce long-term potentiation in 50% of epileptic rats using high-frequency tetanic stimulation (Bragin et al., 2002).
Potential Mechanisms of Calbindin Restoration in the Injured Hippocampus After Fetal CA3 Cell Grafting
Following ICV KA-induced hippocampal injury, numbers of calbindin-positive neurons were reduced by 64% in the dentate granule cell layer and 91% in the CA1 pyramidal cell layer at 6 months post-KA. In spite of this, with CA3 cell grafting at 45 days post-KA, the numbers were restored to 90–96% of control levels at 6 months post-KA. Thus, the overall effect of CA3 cell grafting is very robust in terms of restoring calbindin expression in both dentate granule cells and CA1 pyramidal neurons. Fitting repair of the injured hippocampus by fetal CA3 cell grafts leading to amelioration of hyperexcitability likely underlies calbindin reinstatement. Hyperexcitability (and the associated calbindin loss) in the ICV KA model of TLE is thought to be caused by the disruption and abnormal reorganization of the hippocampal circuitry, and reductions/alterations in hippocampal GABA-ergic interneurons after degeneration of sizeable fractions of CA3 pyramidal and dentate hilar neurons (Nadler et al., 1980a,b; Shetty, 2002; Shetty et al., 2005). Disruptions in circuitry due to degeneration of CA3 pyramidal and hilar neurons comprise loss of synapses at: dentate granule cell dendrites in the inner molecular layer, terminals of dentate mossy fibers (axons of granule cells) in the dentate hilus and CA3 stratum lucidum, Schaeffer collateral terminals in CA1 strata oriens and radiatum, and commissural pathway terminals in multiple sites of the contralateral hippocampus.
The synaptic reorganization following disruption of hippocampal circuitry mainly comprises abnormal sprouting of dentate mossy fibers into the dentate inner molecular layer (Shetty and Turner, 1995a,b, 1997, 1999b; Shetty et al., 2003, 2005), sprouting of entorhinal axons in the CA1 strata radiatum and lacunosum (Shetty, 2002), and sprouting of axons of CA1 pyramidal neurons (Perez et al., 1996). The reductions in GABA-ergic interneuron numbers following ICV KA-induced injury have been observed in both dentate gyrus and CA1 and CA3 subfields of the hippocampus (Shetty and Turner, 2001), which is consistent with loss of functional inhibition observed in this model (Wheal, 1989; Turner and Wheal, 1991; Chen et al., 1999). However, these reductions appeared to be due to loss of glutamate decarboxylase (synthesizing enzyme of GABA) rather than interneuron degeneration per se (Shetty and Turner, 2001). It is believed that dysfunction of hippocampal inter-neurons is linked to loss or reductions in afferent input and trophic support due to degeneration of CA3 pyramidal neurons, as most of the GABA-ergic interneurons in the hippocampus are innervated by axons of CA3 pyramidal neurons (Shetty and Turner, 2000; Wierenga and Wadman, 2003).
However, many of the above alterations following CA3 region injury can be modulated considerably with transplantation of fetal CA3 cells. First, with CA3 cell transplantation, host mossy fibers robustly grow into the transplant where they surround the clusters of CA3 pyramidal neurons (Shetty et al., 2005). The CA3 transplant-mediated restitution of the disrupted circuitry is also associated with a dramatic inhibition of the aberrant mossy fiber sprouting into the dentate inner molecular layer (Shetty et al., 2005). Because sprouted mossy fibers make excitatory connections with the dendrites of granule cells and dentate gyrus with aberrant mossy fiber sprouting has increased seizure susceptibility (Mathern et al., 1996, 1998; Sutula, 2002; Nadler, 2003; Santhakumar et al., 2005), it is likely that suppression of the aberrant mossy fiber sprouting mediated by fetal CA3 cell grafts contributes to decreased hyperexcitability in the dentate gyrus of the injured hippocampus. Second, studies have suggested that neural transplant mediated functional recovery after injury to discrete brain regions requires specific axon growth from transplanted neurons into both local and distant host neurons leading to at least partial restitution of the damaged circuitry (Wictorin, 1992; Dunnett, 1995; Dunnett et al., 1997; Isacson and Deacon, 1997; Isacson, 2003; Turner and Shetty, 2003; Ferrari et al., 2006). Indeed, specific and vigorous axon growth occurs from CA3 cell grafts into the deafferented inner molecular layer of the dentate gyrus and strata oriens and radiatum of the CA1 sub-field in the injured hippocampus (Shetty et al., 2005), and into distant host sites such as the septal nuclei and the contralateral hippocampus (Shetty et al., 2000). Third, placement of grafts containing CA3 pyramidal neurons into the injured hippocampus normalizes the depletions observed in GABA-ergic inter-neuron numbers in the dentate gyrus and the CA1 subfield (Shetty and Turner, 2000). Thus, with CA3 cell grafting, it is possible to minimize the evolution of the abnormal mossy fiber circuitry, reconstruct the disrupted circuitry, and restore depletions in hippocampal GABA-ergic interneuron numbers in the injured hippocampus. As all of these changes are beneficial for preventing/suppressing hyperexcitability of neurons in the injured hippocampus, restoration of hippocampal calbindin following CA3 cell grafting observed in this study is likely a manifestation of these changes leading to normalization in the excitatory and inhibitory connectivity of dentate granule cells and CA1 pyramidal neurons. Additionally, as decreased hippocampal calbindin content likely contributes to impairments in both spatial learning and long-term potentiation, restoration of calbindin mediated by fetal CA3 cell grafts may also be useful for ameliorating learning and memory deficits observed during chronic epilepsy. However, to confirm the above functional effects, rigorous studies that specifically test hippocampal hyperexcitability, hippocampal-dependent learning and memory function following KA-induced injury, and CA3 cell grafting are needed in future.
Probable Reasons for Lack of Improvement in Calbindin After Fetal CA1 Cell Grafting
With CA1 cell grafting at 45 days post-KA, numbers of calbindin-positive neurons remained 42% below control levels in the granule cell layer and 76% below control levels in the CA1 pyramidal cell layer at 6 months post-KA. Thus, in divergence to CA3 cell grafting, CA1 cell grafting was largely ineffective for restoring calbindin in the injured hippocampus. This failure reveals inability of fetal CA1 cell grafts to suitably repair and to prevent/suppress epileptogenic changes in the injured hippocampus, which likely leads to persistence of hyperexcitability and calbindin loss. This supposition is based on the following observations in our prior grafting studies utilizing this model. To begin with, fetal CA1 cell grafts fail to curb aberrant mossy fiber sprouting into the dentate inner molecular layer of the injured hippocampus. This is owing to their incapability to draw host mossy fibers and to send out axons into the deafferented inner molecular layer of the dentate gyrus (Koyama and Ikegaya, 2004; Shetty et al., 2005). Given that CA1 cell grafting does not offer target neurons to the sprouting mossy fibers and does not fill up vacant synaptic sites in the dentate inner molecular layer, the aberrant mossy fiber growth proceeds unhindered into the dentate inner molecular layer (Shetty et al., 2005). Second, fetal CA1 cell grafts placed into the injured CA3 region fail to appropriately rebuild the disrupted circuitry in the CA3-lesioned hippocampus (Shetty et al., 2000). Third, fetal CA1 cell grafting does not normalize GABA-ergic interneuron numbers in the dentate gyrus or the CA1 subfield, likely due to their inability to innervate GABA-deficient interneurons in the injured hippocampus (Shetty and Turner, 2000). Because CA1 cell grafting does not lead to suppression of abnormal mossy fiber circuitry, appropriate reconstruction of the disrupted circuitry, and normalization of GABA-ergic interneuron numbers in the injured hippocampus, hyperexcitability in the injured hippocampus is likely not altered with CA1 cell grafting. Thus, it is plausible that lack of restoration of hippocampal calbindin following CA1 cell grafting reflects persistence of hyperexcitability. However, EEG recordings of KA-treated animals receiving CA1 cell grafts are needed in future to confirm this probability.
CONCLUSIONS
Our results indicate that specific cell grafting is effective for reinstating the injury-induced calbindin loss in the adult hippocampus. When taken in concert with findings of our previous studies, it emerges that proper restitution of the disrupted circuitry by fetal CA3 cell grafts underlies this repair. Because enduring loss of calbindin in a large fraction of dentate granule cells in both human TLE and animal models of TLE is tied to the persistence of hyperexcitability in these neurons, re-expression of calbindin in both dentate gyrus and CA1 subfield after CA3 cell grafting in this study suggests that specific cell grafting approach has the promise for restraining hyperexcitability and spontaneous recurrent seizures in the injured/epileptic adult hippocampus. However, EEG recordings of KA-treated animals receiving CA3 cell grafts are needed in future to corroborate these possibilities. Furthermore, as decreased hippocampal calbindin contributes to impairments in both spatial learning and long-term potentiation, restoration of calbindin mediated by fetal CA3 cell grafts may also ameliorate learning and memory deficits observed during chronic epilepsy. Careful long-term studies on graft function and behavior in animal models that exhibit spontaneous seizures after hippocampal injury are however required in future for promoting this intervention strategy as preventive treatment that blocks evolution of the initial hippocampal injury into chronic epilepsy and learning and memory deficits.
Acknowledgments
Grant sponsor: National Institute of Neurological Disorders and Stroke; Grant numbers: R01 NS 054780, R01 NS 043507; Grant sponsor: Department of Veterans Affairs.
References
- Baimbridge KG. Calcium-binding proteins in the dentate gyrus. Epilepsy Res Suppl. 1992;7:211–220. [PubMed] [Google Scholar]
- Baimbridge KG, Miller JJ. Immunohistochemical localization of calcium-binding protein in the cerebellum, hippocampal formation and olfactory bulb of the rat. Brain Res. 1982;245:223–229. doi: 10.1016/0006-8993(82)90804-6. [DOI] [PubMed] [Google Scholar]
- Baimbridge KG, Miller JJ. Hippocampal calcium-binding protein during commissural kindling-induced epileptogenesis: Progressive decline and effects of anticonvulsants. Brain Res. 1984;324:85–90. doi: 10.1016/0006-8993(84)90624-3. [DOI] [PubMed] [Google Scholar]
- Baimbridge KG, Mody I, Miller JJ. Reduction of rat hippocampal calcium-binding protein following commissural, amygdala, septal, perforant path, and olfactory bulb kindling. Epilepsia. 1985;26:460–465. doi: 10.1111/j.1528-1157.1985.tb05681.x. [DOI] [PubMed] [Google Scholar]
- Baimbridge KG, Celio MR, Rogers JH. Calcium-binding proteins in the nervous system. Trends Neurosci. 1992;15:303–308. doi: 10.1016/0166-2236(92)90081-i. [DOI] [PubMed] [Google Scholar]
- Bazzett TJ, Becker JB, Falik RC, Albin RL. Chronic intrastriatal quinolinic acid produces reversible changes in perikaryal calbindin and parvalbumin immunoreactivity. Neuroscience. 1994;60:837–841. doi: 10.1016/0306-4522(94)90266-6. [DOI] [PubMed] [Google Scholar]
- Bragin A, Wilson CL, Engel J., Jr Rate of interictal events and spontaneous seizures in epileptic rats after electrical stimulation of hippocampus and its afferents. Epilepsia. 2002;43 (Suppl 5):81–85. doi: 10.1046/j.1528-1157.43.s.5.22.x. [DOI] [PubMed] [Google Scholar]
- Buckmaster PS, Zhang GF, Yamawaki R. Axon sprouting in a model of temporal lobe epilepsy creates a predominantly excitatory feedback circuit. J Neurosci. 2002;22:6650–6658. doi: 10.1523/JNEUROSCI.22-15-06650.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celio MR. Calbindin D-28k and parvalbumin in the rat nervous system. Neuroscience. 1990;35:375–475. doi: 10.1016/0306-4522(90)90091-h. [DOI] [PubMed] [Google Scholar]
- Chen Y, Chad JE, Cannon RC, Wheal HV. Reduced Mg2+ blockade of synaptically activated N-methyl-D-aspartate receptor-channels in CA1 pyramidal neurons in kainic acid-lesioned rat hippocampus. Neuroscience. 1999;88:727–739. doi: 10.1016/s0306-4522(98)00253-x. [DOI] [PubMed] [Google Scholar]
- Cheng B, Goodman Y, Begley JG, Mattson MP. Neurotrophin-4/5 protects hippocampal and cortical neurons against energy deprivation- and excitatory amino acid-induced injury. Brain Res. 1994;650:331–335. doi: 10.1016/0006-8993(94)91801-5. [DOI] [PubMed] [Google Scholar]
- Clusmann H, Schramm J, Kral T, Helmstaedter C, Ostertun B, Fimmers R, Haun D, Elger CE. Prognostic factors and outcome after different types of resection for temporal lobe epilepsy. J Neurosurg. 2002;97:1131–1141. doi: 10.3171/jns.2002.97.5.1131. [DOI] [PubMed] [Google Scholar]
- Dudek FE, Obenaus A, Schweitzer JS, Wuarin JP. Functional significance of hippocampal plasticity in epileptic brain: Electrophysiological changes of the dentate granule cells associated with mossy fiber sprouting. Hippocampus. 1994;4:259–265. doi: 10.1002/hipo.450040306. [DOI] [PubMed] [Google Scholar]
- Dunnett SB. Functional repair of striatal systems by neural transplants: Evidence for circuit reconstruction. Behav Brain Res. 1995;66:133–142. doi: 10.1016/0166-4328(94)00134-2. [DOI] [PubMed] [Google Scholar]
- Dunnett SB, Kendall AL, Watts C, Torres EM. Neuronal cell transplantation for Parkinson’s and Huntington’s diseases. Br Med Bull. 1997;53:757–776. doi: 10.1093/oxfordjournals.bmb.a011646. [DOI] [PubMed] [Google Scholar]
- Engel J., Jr The timing of surgical intervention for mesial temporal lobe epilepsy: A plan for a randomized clinical trial. Arch Neurol. 1999;56:1338–1341. doi: 10.1001/archneur.56.11.1338. [DOI] [PubMed] [Google Scholar]
- Engel J., Jr A Greater Role for Surgical Treatment of Epilepsy: Why and When? Epilepsy Curr. 2003;3:37–40. doi: 10.1046/j.1535-7597.2003.03201.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari D, Sanchez-Pernaute R, Lee H, Studer L, Isacson O. Transplanted dopamine neurons derived from primate ES cells preferentially innervate DARPP-32 striatal progenitors within the graft. Eur J Neurosci. 2006;24:1885–1896. doi: 10.1111/j.1460-9568.2006.05093.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutierrez C, Cusick CG. Effects of chronic monocular enucleation on calcium binding proteins calbindin-D28k and parvalbumin in the lateral geniculate nucleus of adult rhesus monkeys. Brain Res. 1994;651:300–310. doi: 10.1016/0006-8993(94)90710-2. [DOI] [PubMed] [Google Scholar]
- Isacson O. The production and use of cells as therapeutic agents in neurodegenerative diseases. Lancet Neurol. 2003;2:417–424. doi: 10.1016/s1474-4422(03)00437-x. [DOI] [PubMed] [Google Scholar]
- Isacson O, Deacon T. Neural transplantation studies reveal the brain’s capacity for continuous reconstruction. Trends Neurosci. 1997;20:477–482. doi: 10.1016/s0166-2236(97)01081-3. [DOI] [PubMed] [Google Scholar]
- Isokawa M. Decrement of GABAA receptor-mediated inhibitory postsynaptic currents in dentate granule cells in epileptic hippocampus. J Neurophysiol. 1996;75:1901–1908. doi: 10.1152/jn.1996.75.5.1901. [DOI] [PubMed] [Google Scholar]
- Isokawa M, Levesque M, Fried I, Engel J., Jr Glutamate currents in morphologically identified human dentate granule cells in temporal lobe epilepsy. J Neurophysiol. 1997;77:3355–3369. doi: 10.1152/jn.1997.77.6.3355. [DOI] [PubMed] [Google Scholar]
- Jouvenceau A, Potier B, Battini R, Ferrari S, Dutar P, Billard JM. Glutamatergic synaptic responses and long-term potentiation are impaired in the CA1 hippocampal area of calbindin D(28k)-deficient mice. Synapse. 1999;33:172–180. doi: 10.1002/(SICI)1098-2396(19990901)33:3<172::AID-SYN2>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- Jouvenceau A, Potier B, Poindessous-Jazat F, Dutar P, Slama A, Epelbaum J, Billard JM. Decrease in calbindin content significantly alters LTP but not NMDA receptor and calcium channel properties. Neuropharmacology. 2002;42:444–458. doi: 10.1016/s0028-3908(01)00202-7. [DOI] [PubMed] [Google Scholar]
- Kim JE, Kwak SE, Kim DS, Won MH, Kwon OS, Choi SY, Kang TC. Reduced calcium binding protein immunoreactivity induced by electroconvulsive shock indicates neuronal hyperactivity, not neuronal death or deactivation. Neuroscience. 2006;137:317–326. doi: 10.1016/j.neuroscience.2005.08.052. [DOI] [PubMed] [Google Scholar]
- Kohr G, Lambert CE, Mody I. Calbindin-D28K (CaBP) levels and calcium currents in acutely dissociated epileptic neurons. Exp Brain Res. 1991;85:543–551. doi: 10.1007/BF00231738. [DOI] [PubMed] [Google Scholar]
- Koyama R, Ikegaya Y. Mossy fiber sprouting as a potential therapeutic target for epilepsy. Curr Neurovasc Res. 2004;1:3–10. doi: 10.2174/1567202043480242. [DOI] [PubMed] [Google Scholar]
- Lledo PM, Somasundaram B, Morton AJ, Emson PC, Mason WT. Stable transfection of calbindin-D28k into the GH3 cell line alters calcium currents and intracellular calcium homeostasis. Neuron. 1992;9:943–954. doi: 10.1016/0896-6273(92)90246-a. [DOI] [PubMed] [Google Scholar]
- Lynch M, Sayin U, Bownds J, Janumpalli S, Sutula T. Long-term consequences of early postnatal seizures on hippocampal learning and plasticity. Eur J Neurosci. 2000;12:2252–2264. doi: 10.1046/j.1460-9568.2000.00117.x. [DOI] [PubMed] [Google Scholar]
- Magloczky Z, Halasz P, Vajda J, Czirjak S, Freund TF. Loss of Calbindin-D28K immunoreactivity from dentate granule cells in human temporal lobe epilepsy. Neuroscience. 1997;76:377–385. doi: 10.1016/s0306-4522(96)00440-x. [DOI] [PubMed] [Google Scholar]
- Malenka RC, Kauer JA, Perkel DJ, Mauk MD, Kelly PT, Nicoll RA, Waxham MN. An essential role for postsynaptic calmodulin and protein kinase activity in long-term potentiation. Nature. 1989;340:554–557. doi: 10.1038/340554a0. [DOI] [PubMed] [Google Scholar]
- Malinow R, Madison DV, Tsien RW. Persistent protein kinase activity underlying long-term potentiation. Nature. 1988;335:820–824. doi: 10.1038/335820a0. [DOI] [PubMed] [Google Scholar]
- Martin RC, Kretzmer T, Palmer C, Sawrie S, Knowlton R, Faught E, Morawetz R, Kuzniecky R. Risk to verbal memory following anterior temporal lobectomy in patients with severe left-sided hippocampal sclerosis. Arch Neurol. 2002;59:1895–1901. doi: 10.1001/archneur.59.12.1895. [DOI] [PubMed] [Google Scholar]
- Mathern GW, Cifuentes F, Leite JP, Pretorius JK, Babb TL. Hippocampal EEG excitability and chronic spontaneous seizures are associated with aberrant synaptic reorganization in the rat intra-hippocampal kainate model. Electroencephalogr Clin Neurophysiol. 1993;87:326–339. doi: 10.1016/0013-4694(93)90186-y. [DOI] [PubMed] [Google Scholar]
- Mathern GW, Leite JP, Pretorius JK, Quinn B, Peacock WJ, Babb TL. Severe seizures in young children are associated with hippocampal neuron losses and aberrant mossy fiber sprouting during fascia dentata postnatal development. Epilepsy Res Suppl. 1996;12:33–43. [PubMed] [Google Scholar]
- Mathern GW, Price G, Rosales C, Pretorius JK, Lozada A, Mendoza D. Anoxia during kainate status epilepticus shortens behavioral convulsions but generates hippocampal neuron loss and supra-granular mossy fiber sprouting. Epilepsy Res. 1998;30:133–151. doi: 10.1016/s0920-1211(97)00103-4. [DOI] [PubMed] [Google Scholar]
- Mello LE, Cavalheiro EA, Tan AM, Kupfer WR, Pretorius JK, Babb TL, Finch DM. Circuit mechanisms of seizures in the pilocarpine model of chronic epilepsy: Cell loss and mossy fiber sprouting. Epilepsia. 1993;34:985–995. doi: 10.1111/j.1528-1157.1993.tb02123.x. [DOI] [PubMed] [Google Scholar]
- Miller JJ, Baimbridge KG. Biochemical and immunohistochemical correlates of kindling-induced epilepsy: Role of calcium binding protein. Brain Res. 1983;278:322–326. doi: 10.1016/0006-8993(83)90264-0. [DOI] [PubMed] [Google Scholar]
- Molinari S, Battini R, Ferrari S, Pozzi L, Killcross AS, Robbins TW, Jouvenceau A, Billard JM, Dutar P, Lamour Y, Baker WA, Cox H, Emson PC. Deficits in memory and hippocampal long-term potentiation in mice with reduced calbindin D28K expression. Proc Natl Acad Sci USA. 1996;93:8028–8033. doi: 10.1073/pnas.93.15.8028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mudrick LA, Baimbridge KG. Hippocampal neurons transplanted into ischemically lesioned hippocampus: Anatomical assessment of survival, maturation and integration. Exp Brain Res. 1991;86:233–247. doi: 10.1007/BF00228948. [DOI] [PubMed] [Google Scholar]
- Mudrick LA, Baimbridge KG, Peet MJ. Hippocampal neurons transplanted into ischemically lesioned hippocampus: Electroresponsiveness and reestablishment of circuitries. Exp Brain Res. 1989;76:333–342. doi: 10.1007/BF00247893. [DOI] [PubMed] [Google Scholar]
- Muller A, Kukley M, Stausberg P, Beck H, Muller W, Dietrich D. Endogenous Ca2+ buffer concentration and Ca2+ microdomains in hippocampal neurons. J Neurosci. 2005;25:558–565. doi: 10.1523/JNEUROSCI.3799-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadler JV. The recurrent mossy fiber pathway of the epileptic brain. Neurochem Res. 2003;28:1649–1658. doi: 10.1023/a:1026004904199. [DOI] [PubMed] [Google Scholar]
- Nadler JV, Perry BW, Cotman CW. Selective reinnervation of hippocampal area CA1 and the fascia dentata after destruction of CA3–CA4 afferents with kainic acid. Brain Res. 1980a;182:1–9. doi: 10.1016/0006-8993(80)90825-2. [DOI] [PubMed] [Google Scholar]
- Nadler JV, Perry BW, Gentry C, Cotman CW. Loss and reacquisition of hippocampal synapses after selective destruction of CA3–CA4 afferents with kainic acid. Brain Res. 1980b;191:387–403. doi: 10.1016/0006-8993(80)91289-5. [DOI] [PubMed] [Google Scholar]
- Nagerl UV, Mody I, Jeub M, Lie AA, Elger CE, Beck H. Surviving granule cells of the sclerotic human hippocampus have reduced Ca(2+) influx because of a loss of calbindin-D(28k) in temporal lobe epilepsy. J Neurosci. 2000;20:1831–1836. doi: 10.1523/JNEUROSCI.20-05-01831.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunn JA, Polkey CE, Morris RG. Selective spatial memory impairment after right unilateral temporal lobectomy. Neuropsychologia. 1998;36:837–848. doi: 10.1016/s0028-3932(98)00030-x. [DOI] [PubMed] [Google Scholar]
- Okazaki MM, Evenson DA, Nadler JV. Hippocampal mossy fiber sprouting and synapse formation after status epilepticus in rats: Visualization after retrograde transport of biocytin. J Comp Neurol. 1995;352:515–534. doi: 10.1002/cne.903520404. [DOI] [PubMed] [Google Scholar]
- Palop JJ, Jones B, Kekonius L, Chin J, Yu GQ, Raber J, Masliah E, Mucke L. Neuronal depletion of calcium-dependent proteins in the dentate gyrus is tightly linked to Alzheimer’s disease-related cognitive deficits. Proc Natl Acad Sci USA. 2003;100:9572–9577. doi: 10.1073/pnas.1133381100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Watson C, Pennisi M, Topple A. Bregma, lambda and the interaural midpoint in stereotaxic surgery with rats of different sex, strain and weight. J Neurosci Methods. 1985;13:139–143. doi: 10.1016/0165-0270(85)90026-3. [DOI] [PubMed] [Google Scholar]
- Perez Y, Morin F, Beaulieu C, Lacaille JC. Axonal sprouting of CA1 pyramidal cells in hyperexcitable hippocampal slices of kainate-treated rats. Eur J Neurosci. 1996;8:736–748. doi: 10.1111/j.1460-9568.1996.tb01259.x. [DOI] [PubMed] [Google Scholar]
- Ren K, Ruda MA. A comparative study of the calcium-binding proteins calbindin-D28K, calretinin, calmodulin and parvalbumin in the rat spinal cord. Brain Res Brain Res Rev. 1994;19:163–179. doi: 10.1016/0165-0173(94)90010-8. [DOI] [PubMed] [Google Scholar]
- Richardson MP, Strange BA, Thompson PJ, Baxendale SA, Duncan JS, Dolan RJ. Pre-operative verbal memory fMRI predicts post-operative memory decline after left temporal lobe resection. Brain. 2004;127:2419–2426. doi: 10.1093/brain/awh293. [DOI] [PubMed] [Google Scholar]
- Santhakumar V, Aradi I, Soltesz I. Role of mossy fiber sprouting and mossy cell loss in hyperexcitability: A network model of the dentate gyrus incorporating cell types and axonal topography. J Neurophysiol. 2005;93:437–453. doi: 10.1152/jn.00777.2004. [DOI] [PubMed] [Google Scholar]
- Scharfman HE, Sollas AE, Berger RE, Goodman JH, Pierce JP. Perforant path activation of ectopic granule cells that are born after pilocarpine-induced seizures. Neuroscience. 2003;121:1017–1029. doi: 10.1016/s0306-4522(03)00481-0. [DOI] [PubMed] [Google Scholar]
- Selke K, Muller A, Kukley M, Schramm J, Dietrich D. Firing pattern and calbindin-D28k content of human epileptic granule cells. Brain Res. 2006;1120:191–201. doi: 10.1016/j.brainres.2006.08.072. [DOI] [PubMed] [Google Scholar]
- Shetty AK. Entorhinal axons exhibit sprouting in CA1 subfield of the adult hippocampus in a rat model of temporal lobe epilepsy. Hippocampus. 2002;12:534–542. doi: 10.1002/hipo.10031. [DOI] [PubMed] [Google Scholar]
- Shetty AK, Turner DA. Enhanced cell survival in fetal hippocampal suspension transplants grafted to adult rat hippocampus following kainate lesions: A three-dimensional graft reconstruction study. Neuroscience. 1995a;67:561–582. doi: 10.1016/0306-4522(95)00025-e. [DOI] [PubMed] [Google Scholar]
- Shetty AK, Turner DA. Intracerebroventricular kainic acid administration in adult rat alters hippocampal calbindin and non-phosphorylated neurofilament expression. J Comp Neurol. 1995b;363:581–599. doi: 10.1002/cne.903630406. [DOI] [PubMed] [Google Scholar]
- Shetty AK, Turner DA. Development of long-distance efferent projections from fetal hippocampal grafts depends upon pathway specificity and graft location in kainate-lesioned adult hippocampus. Neuroscience. 1997;76:1205–1219. doi: 10.1016/s0306-4522(96)00413-7. [DOI] [PubMed] [Google Scholar]
- Shetty AK, Turner DA. Hippocampal interneurons expressing glutamic acid decarboxylase and calcium-binding proteins decrease with aging in Fischer 344 rats. J Comp Neurol. 1998;394:252–269. [PubMed] [Google Scholar]
- Shetty AK, Turner DA. Vulnerability of the dentate gyrus to aging and intracerebroventricular administration of kainic acid. Exp Neurol. 1999a;158:491–503. doi: 10.1006/exnr.1999.7107. [DOI] [PubMed] [Google Scholar]
- Shetty AK, Turner DA. Aging impairs axonal sprouting response of dentate granule cells following target loss and partial deafferentation. J Comp Neurol. 1999b;414:238–254. doi: 10.1002/(sici)1096-9861(19991115)414:2<238::aid-cne7>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- Shetty AK, Turner DA. Fetal hippocampal grafts containing CA3 cells restore host hippocampal glutamate decarboxylase-positive interneuron numbers in a rat model of temporal lobe epilepsy. J Neurosci. 2000;20:8788–8801. doi: 10.1523/JNEUROSCI.20-23-08788.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shetty AK, Turner DA. Glutamic acid decarboxylase-67-positive hippocampal interneurons undergo a permanent reduction in number following kainic acid-induced degeneration of ca3 pyramidal neurons. Exp Neurol. 2001;169:276–297. doi: 10.1006/exnr.2001.7668. [DOI] [PubMed] [Google Scholar]
- Shetty AK, Zaman V, Turner DA. Pattern of long-distance projections from fetal hippocampal field CA3 and CA1 cell grafts in lesioned CA3 of adult hippocampus follows intrinsic character of respective donor cells. Neuroscience. 2000;99:243–255. doi: 10.1016/s0306-4522(00)00178-0. [DOI] [PubMed] [Google Scholar]
- Shetty AK, Zaman V, Shetty GA. Hippocampal neurotrophin levels in a kainate model of temporal lobe epilepsy: A lack of correlation between brain-derived neurotrophic factor content and progression of aberrant dentate mossy fiber sprouting. J Neurochem. 2003;87:147–159. doi: 10.1046/j.1471-4159.2003.01979.x. [DOI] [PubMed] [Google Scholar]
- Shetty AK, Zaman V, Hattiangady B. Repair of the injured adult hippocampus through graft-mediated modulation of the plasticity of the dentate gyrus in a rat model of temporal lobe epilepsy. J Neurosci. 2005;25:8391–8401. doi: 10.1523/JNEUROSCI.1538-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloviter RS. Calcium-binding protein (calbindin-D28k) and parvalbumin immunocytochemistry: Localization in the rat hippocampus with specific reference to the selective vulnerability of hippocampal neurons to seizure activity. J Comp Neurol. 1989;280:183–196. doi: 10.1002/cne.902800203. [DOI] [PubMed] [Google Scholar]
- Sloviter RS, Sollas AL, Barbaro NM, Laxer KD. Calcium-binding protein (calbindin-D28K) and parvalbumin immunocytochemistry in the normal and epileptic human hippocampus. J Comp Neurol. 1991;308:381–396. doi: 10.1002/cne.903080306. [DOI] [PubMed] [Google Scholar]
- Sutula T. Seizure-Induced Axonal Sprouting: Assessing Connections Between Injury, Local Circuits, and Epileptogenesis. Epilepsy Curr. 2002;2:86–91. doi: 10.1046/j.1535-7597.2002.00032.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutula T, Cascino G, Cavazos J, Parada I, Ramirez L. Mossy fiber synaptic reorganization in the epileptic human temporal lobe. Ann Neurol. 1989;26:321–330. doi: 10.1002/ana.410260303. [DOI] [PubMed] [Google Scholar]
- Tauck DL, Nadler JV. Evidence of functional mossy fiber sprouting in hippocampal formation of kainic acid-treated rats. J Neurosci. 1985;5:1016–1022. doi: 10.1523/JNEUROSCI.05-04-01016.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tonder N, Kragh J, Bolwig T, Zimmer J. Transient decrease in calbindin immunoreactivity of the rat fascia dentata granule cells after repeated electroconvulsive shocks. Hippocampus. 1994;4:79–83. doi: 10.1002/hipo.450040110. [DOI] [PubMed] [Google Scholar]
- Toth K, Freund TF. Calbindin D28k-containing nonpyramidal cells in the rat hippocampus: Their immunoreactivity for GABA and projection to the medial septum. Neuroscience. 1992;49:793–805. doi: 10.1016/0306-4522(92)90357-8. [DOI] [PubMed] [Google Scholar]
- Turner DA, Shetty AK. Clinical prospects for neural grafting therapy for hippocampal lesions and epilepsy. Neurosurgery. 2003;52:632–644. doi: 10.1227/01.neu.0000047825.91205.e6. discussion 641–634. [DOI] [PubMed] [Google Scholar]
- Turner DA, Wheal HV. Excitatory synaptic potentials in kainic acid-denervated rat CA1 pyramidal neurons. J Neurosci. 1991;11:2786–2794. doi: 10.1523/JNEUROSCI.11-09-02786.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheal HV. Function of synapses in the CA1 region of the hippocampus: Their contribution to the generation or control of epileptiform activity. Comp Biochem Physiol A. 1989;93:211–220. doi: 10.1016/0300-9629(89)90209-0. [DOI] [PubMed] [Google Scholar]
- Wictorin K. Anatomy and connectivity of intrastriatal striatal transplants. Prog Neurobiol. 1992;38:611–639. doi: 10.1016/0301-0082(92)90044-f. [DOI] [PubMed] [Google Scholar]
- Wierenga CJ, Wadman WJ. Functional relation between inter-neuron input and population activity in the rat hippocampal cornu ammonis 1 area. Neuroscience. 2003;118:1129–1139. doi: 10.1016/s0306-4522(03)00060-5. [DOI] [PubMed] [Google Scholar]
- Williamson A, Spencer D. Zinc reduces dentate granule cell hyperexcitability in epileptic humans. Neuroreport. 1995;6:1562–1564. doi: 10.1097/00001756-199507310-00024. [DOI] [PubMed] [Google Scholar]
- Wilson IA, Gallagher M, Eichenbaum H, Tanila H. Neurocognitive aging: Prior memories hinder new hippocampal encoding. Trends Neurosci. 2006;29:662–670. doi: 10.1016/j.tins.2006.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wuarin JP, Dudek FE. Excitatory synaptic input to granule cells increases with time after kainate treatment. J Neurophysiol. 2001;85:1067–1077. doi: 10.1152/jn.2001.85.3.1067. [DOI] [PubMed] [Google Scholar]
- Zaman V, Shetty AK. Fetal hippocampal CA3 cell grafts transplanted to lesioned CA3 region of the adult hippocampus exhibit long-term survival in a rat model of temporal lobe epilepsy. Neurobiol Dis. 2001;8:942–952. doi: 10.1006/nbdi.2001.0440. [DOI] [PubMed] [Google Scholar]
- Zaman V, Turner DA, Shetty AK. Survival of grafted fetal neural cells in kainic acid lesioned CA3 region of adult hippocampus depends upon cell specificity. Exp Neurol. 2000;161:535–561. doi: 10.1006/exnr.1999.7304. [DOI] [PubMed] [Google Scholar]