

Abstract
Many previous studies demonstrate that hepatocytes can be reprogrammed into insulin-producing cells (IPCs) utilizing viral vector-mediated delivery of pancreatic transcription factors (PTFs). However, whether these liver-derived IPCs are susceptible to autoimmune attack in animal models of type 1 diabetes remains unclear, in part due to the immunogenicity of the viral vectors used to introduce PTF genes. Adeno-associated virus serotype 2 vector-expressing Pdx1-VP16 (Pdx1) and Ngn3 were prepared and injected into the portal vein of streptozotocin (Stz)/diabetic NOD/SCID mice. The presence of glucose-responsive liver-IPCs and their susceptibility to anti-beta cell autoimmunity were assessed by blood glucose levels, insulin content, IPC cell distribution, and intraperitoneal glucose tolerance test following subtotal pancreatectomy (Px) and passive transfer of diabetogenic splenocytes isolated from diabetic female NOD mice. A combination of two PTF genes (Pdx1/Ngn3) effectively reprogrammed liver cells into glucose-responsive IPCs. These IPCs corrected hyperglycemia in Stz/diabetic NOD/SCID mice and maintained normoglycemia following subtotal Px, indicating that liver-derived IPCs could maintain glucose homeostasis. Importantly, we also demonstrated that the glucose-responsive liver–derived IPCs were susceptible to autoimmune destruction by diabetogenic splenocytes, as indicated by progressive elevation in blood glucose levels as well as mixed T-, and B-lymphocytic infiltrates surrounding liver-IPCs 2~3 weeks following transferring of diabetogenic splenocytes into NOD/SCID mice, and confirmed by immunohistochemical studies. In conclusion, genetically reprogrammed liver-IPCs, like pancreatic islet beta-cells, are susceptible to autoimmune attack, suggesting that for cell-replacement therapy of treating type 1 diabetes, beta-cell surrogates may require concomitant immunotherapy to avoid autoimmune destruction.
Keywords: Reprogram, hepatocytes, insulin-producing beta cells, autoimmunity, type 1 diabetes, gene therapy
Introduction
Because current methods for treating type 1 diabetes (T1D) are ineffective in preventing long-term complications, researchers have sought to identify alternative therapies that regenerate pancreatic beta cells, differentiate pluripotent/progenitor stem cells, and/or reprogram autologous non-pancreatic somatic cells, such as liver cells, into insulin-producing cells (IPCs) for restoring glucose homeostasis. Growing evidence, both in vitro [1-6] and in vivo, [7-11] indicates that the introduction of key pancreatic transcription factor (PTF) gene(s) into liver cells using viral vectors can reprogram hepatocytes into IPCs that produce insulin at levels sufficient for maintaining glucose homeostasis in diabetic animals. Even so, in vivo studies using adenovirus to deliver PTF genes such as Pdx1 [7,8], Pdx1/NeuroD [9,11], Pdx1/neurogenin 3 (Ngn3) [10-12], have shown variable degrees of success in terms of reprogramming liver cells into functional IPCs. While these studies have provided proof-of-principle evidence that liver cells can be genetically reprogrammed into glucose-sensitive IPCs, they do not address the critical question of whether liver-derived IPCs are susceptible to an autoimmune attack observed in settings of T1D therapy. Most in vivo liver-reprogramming studies have employed adenovirus (AD) to introduce PTF genes into the liver cells. Adenoviral vectors can potently transduce liver cells, but AD infection can stimulate a strong immune response [13,14] and these AD-reprogrammed liver-IPCs are often short-lived, being quickly eliminated by the immune response [11]. Therefore it is impossible to determine whether these AD-treated liver-reprogrammed IPCs can resist autoimmune attack mounted by diabetogenic immune cells in T1D animal models.
Recombinant adeno-associated virus (AAV) is a more promising vector for test autoimmunity of the liver-IPCs, given its ability to mediate efficient gene transfer with stable gene expression and low immunogenicity [15]. Derived from a nonpathogenic human parvovirus, AAV vectors have gained popularity during the last decade in the treatment of a variety of genetic and inherited diseases [16]. AAV vectors can transduce both dividing and nondividing cells, allowing differentiated tissues to be used as targets and permitting long-term expression of the therapeutic gene product in animal models [15,17]. In this study, we used Stz-induced diabetic mice and achieved AAV-mediated transgene expression of Pdx1-VP16 (a super active form of Pdx1, hereafter abbreviated as Pdx1) and Ngn3 in the NOD/SCID mouse livers by the onetime portal vein injection to produce liver IPCs. We addressed two important questions: 1) Can the genetically reprogrammed liver-IPCs alone restore and maintain euglycemia in Stz/diabetic mice? And 2) can the liver-IPCs escape autoimmune-mediated destruction as occurs in pancreatic islets of mice with T1D? We demonstrate that, while genetically-reprogrammed liver-IPCs alone can maintain normoglycemia following subtotal (~90%) pancreatectomy (Px), these IPCs are susceptible to attack and destruction by diabetogenic immune cells. Our results suggest that non-pancreatic IPCs may share a similar surface autoantigen profile with pancreatic beta-cells. Thus, beta-cell replacement and regeneration therapies via a genetic reprogramming may require concomitantly an immunologic approach to suppress the elimination of reprogrammed IPCs by pre-existing autoreactive lymphocytes.
Methods
Construction and generation of AAV serotype 2 vectors
A fused CMV-chicken beta-actin (CB) promoter was used to drive the expression of GFP (Invitrogen), mouse Pdx1-VP16 (kindly provided by Marko Horb), and mouse Ngn3 (kindly provided by Michael German) by cloning them into recombinant AAV2 vector (UF11), hereafter called AAV. In brief, PCR-amplified cDNAs of Pdx1-VP16, Ngn3 and GFP were inserted into AAVs. Pdx1-VP16 was constructed by fusing the activation domain of VP16 (80 amino acids) to the mouse COOH-terminus of Pdx1 as previously described [18]. AAV2 viruses were made as previously described [19,20]. Briefly, HEK 293 cells were co-transfected by the AAV plasmid and the helper plasmid pDG for 48-60 h. Cells were harvested, and the crude lysate was purified through an iodixanol step gradient followed by heparin affinity chromatography. Titers of total AAV viral particles were determined by Quantitative competitive polymerase chain reaction (QC-PCR) and AAV2 Titration ELISA (American Research Products, Inc. MA USA), respectively. The vectors were 99% pure as illustrated by silver-stained SDS-PAGE. Two AAV products contained viral particles of 3.11x1012 (Pdx1-VP16, hereafter abbreviated as Pdx1) and 6.74 x1012 (Ngn3) in a volume of ~300 μl.
Hyperglycemic animal model
All mice were housed in an SPF environment and handled according to institutional guidelines of the University of Florida. Six to eight-week-old NOD/SCID mice (NOD.CB17-Prkdcscid/J) were purchased from the Jackson Laboratory and injected with Stz at 50 mg/g body weight i.p. daily for five consecutive days to induce diabetes according to our previously published procedures [5,18,21]. Fasting blood glucose levels were measured using an AccuChek Advantage glucose detector (TYP 033304510, Roche Diagnostics, IN) after the mice were fasted for 6 h. The upper limit of this glucose meter was 600 mg/dL, and values above that gave a reading of “high”. Animals with fasting blood glucose levels of ≥ 250 mg/dL for two consecutive readings within a period of 24 hours were defined as diabetic and received a portal vein injection of AAVs.
Portal vein injection of AAV vectors
Eight to ten-week-old adult male or female mice were anesthetized with isoflurane. Mice were injected with 106, 108, or 1010 AAV viral particles in PBS via the portal vein using a 0.5 ml syringe with 30-gauge needle. Liver cells expressing GFP protein were evaluated at day 10 post-injection. All procedures were performed under anesthesia and all experiments were carried out in compliance with the guidelines on the IACUC, University of Florida. Blood glucose levels were measured from a tail vein snipping using a glucose meter every three days.
Subtotal pancreatectomy (Px)
Mice were anesthetized by administration of isoflurane. The abdomen was opened through a left lateral incision. The entire splenic portion and most of the duodenum portion of the pancreas was surgically removed. The removed pancreas was weighed and subjected to immunohistochemistry (IHC), RT-PCR, and ELISA for tissue insulin content measurement. A pilot study was carried out in five mice undergoing subtotal Px (~90%), revealing that there was no spontaneous recovery of normoglycemia due to insufficient pancreatic beta cell regeneration. All five mice had persistent hyperglycemia (glucose >450 mg/dL) for 3 weeks before they were sacrificed. Intraperitoneal glucose tolerance test (IPGTT) was performed three days after Px to allow animals to recover from surgery.
IHC and immunofluorescence (IF)
The harvested tissues were fixed in 10% formalin/PBS and embedded in paraffin. The 5-μm paraffin sections from the liver and pancreas tissues were double immunostained with polyclonal antibodies against Pdx1 generated in our laboratory (1:500 dilution) [21] and insulin (Dako, 1:1000); or insulin and glucagon (Dako, 1:2000) according to our previously published procedures [21]. Immunihistochemistry (IHC) for CD3 (SeriTec, 1:1000), B220 (BD, 1:500), anti-F4/80 (CAT-TAT, 1:50) and Ki67 (Dako, 1:50) was performed by Pathology Core Laboratory, University of Florida. Staining of antibody-specific cells was photographed using Zeiss Axioskop 2 Plus camera and visualized either in brown (DAB) or Liquid Permanent Red (LPR) chromagens. For detection of GFP expression, the liver tissues were fixed in 4% paraformaldehyde for 12 hours, then transferred into 30% sucrose solution for overnight, embedded in TissueTek OCT compound, quickly frozen in liquid nitrogen, and cut into 6-μm sections with the Microtome HM505E cryostat (Carl Zeiss Co, Jena, Germany). The frozen sections were covered with coverslips using mounting medium containing DAPI (Vector Lab.) to highlight cell nuclei. Three sections at different levels were used for morphologic quantification of the percentage of fluorescent cells (GFP) or immunoreactive cells by counting 10 fields under the 40X lens in representative areas. In all cases, over 500 positive cells were counted. Pancreatic and liver sections were incubated with guinea pig anti-insulin (Dako, 1:500) and rabbit anti-glucagon (Dako, 1:500) antibodies for 10 hrs at 4°C, followed by washing five times. Staining was visualized using anti-guinea pig-AF488 (Green, Invitrogen) and goat-anti-rabbit-AF555 (Red, Invitrogen) for 2 hours at room temperature under a fluorescence microscope and photographed using Zeiss Axioskop 2 Plus camera.
Intraperitoneal glucose tolerance test (IPGTT)
Following a 6h fast, the mice were injected with glucose (2 g/kg body weight i.p.). Blood glucose levels were determined at 0, 5, 15, 30, 60, and 120 min post-injection via the tail vein snipping [18,21].
Tissue insulin measurement by ELISA
For tissue insulin, whole livers and whole pancreas from at least three mice per group were harvested, weighed, and immediately placed in acid-ethanol solution (180mM HCl in 70% ethanol) on ice along with 1 ml buffer/0.1g liver or 1 ml/0.05 g pancreas as described previously [21]. Insulin levels were measured using an ultrasensitive mouse insulin ELISA kit (ALPCO, Salem, NH). Absorbance was measured immediately by a BIO-RAD 3550-UV microplate reader, with final results converted to ng-insulin/g-liver or ng-insulin/mg-pancreas tissue.
RT-PCR
Total tissue RNA was prepared from the liver and pancreas using TRIZOL Reagent, complementary DNA (cDNA) was prepared, and insulin and glucagon gene expression levels were determined by RT-PCR and the forward and reverse PCR primers were designed to be located in different exon(s) as previously described [6,18].
Splenocyte adoptive transfer (SAT)
Diabetogenic splenocytes were harvested from new-onset (within a week of hyperglycemia) diabetic female NOD mice according standard procedures [22-24]. The spleen was flushed with PBS and a cell suspension was treated with hypotonic buffer to lyse red blood cells. Total splenocytes were transferred without further fractionation. NOD/SCID mice were injected with diabetogenic splenocytes (2 x107 cells/mouse i.p., viability > 95%). Fasting blood glucose levels were monitored twice weekly via the tail vein snipping. Two consecutive measurements of blood glucose levels >250 mg/dL defined mice as diabetic. Our pilot study showed that more than 80% of mice receiving SAT became diabetic within 4 weeks and all mice were diabetic at 6 weeks post-SAT.
Statistical analysis
Statistical significance was analyzed using an independent sample t-test, requiring a P value of less than 0.05 for the data to be considered statistically significant.
Results
AAV2 mediates efficient gene expression in liver via portal vein injection
To optimize the AAV vector dose, we first examined gene transduction efficiency in normal livers of non-diabetic mice injected into the portal vein with an AAV2-GFP reporter gene. The result of injecting 106, 108, or 1010 AAV viral particles via the portal vein and the liver cells expressing GFP protein were evaluated at day-10 post-injection. Control mice were injected with saline via the portal vein. Liver sections were fixed and stained for fluorescence microscopy.
As shown in Figure 1A, hepatocyte GFP fluorescence was used to indicate the presence of GFP protein in livers at all doses, and exhibited a dose-dependent increase in both the intensity of staining and the proportion of hepatocytes transduced. But GFP fluorescence varied in different areas. By morphologic estimation, overall 5-20% of liver cells expressed GFP. No GFP fluorescence was detected in the control livers. These results indicate that AAV2 vector can mediate satisfactory liver gene transduction via portal vein injection. On the basis of the GFP-fluorescence data, we chose to use a dose of 108 viral particles per mouse for subsequent studies (~10-20% transduction efficiency).
Figure 1.
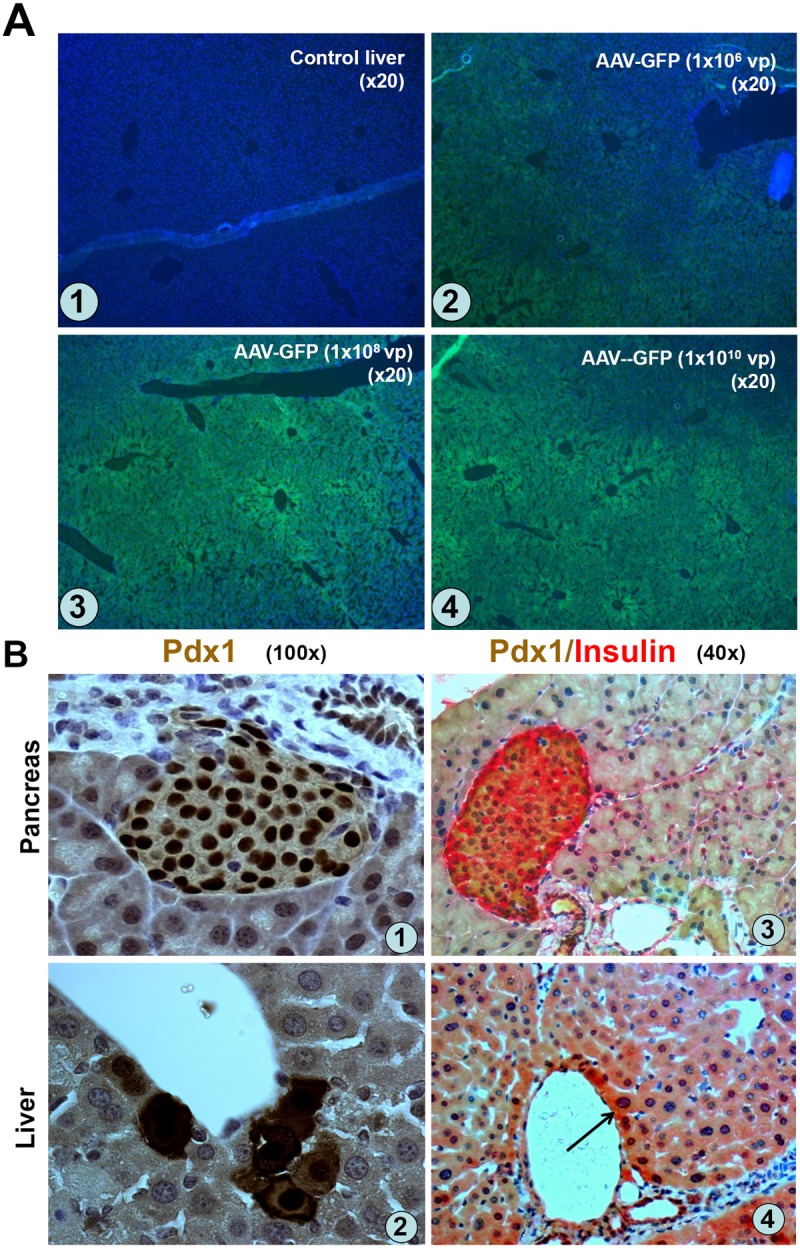
AAV mediated GFP and Pdx1 expression in liver following portal vein injection. A. Dose-dependent GFP expression in liver. Normal mice were injected with 106, 108, or 1010 viral particles (vp)/mouse, (n=2/dose) of AAV2-GFP via the portal vein. Expression of GFP protein in the liver cells was evaluated at day10 post-injection. Liver sections were photographed under a fluorescence microscope. A-1 (Control liver), A-2 (106 vp), A-3 (108 vp), and A-4 (1010 vp). B. Pdx1 and insulin expression in liver. Normal mice (n=3) were injected with AAV-Pdx1-VP16 (108 vp/mouse) and Pdx1 and insulin expression in the liver sections was detected with anti-Pdx1 antibody at day 12 post-injection. B-(1): Pancreatic islet as positive control for Pdx1. B-(2): high power views of Pdx1 protein located both in cytoplasm and nuclei of the liver cells. B-(3): Pancreatic islet as a positive control for double Pdx1 (brown) and insulin (red) IHC. B-(4): Pdx1 and insulin protein in cytoplasm and nuclei of the liver cells. Arrow indicates double Pdx1/insulin-positive cells.
We next determined in vivo AAV2-mediated liver transduction efficiency for Pdx1 expression by the portal vein injection into diabetic mice of 108 viral particles/mouse of AAV-Pdx1. The mice were killed at day-12 to evaluate Pdx1 expression by IHC using anti-Pdx1-antibody. Approximately 5% of liver cells strongly expressed Pdx1 protein in both nuclei and cytoplasm (Figure 1B-2) and occasional double Pdx1-/insulin-positive cells (Figure 1B-4, arrow); the Pdx1-expressing hepatocytes were located mainly along the edge of central veins, consistent with the direction of blood flow. Pdx1 or insulin expression could not be detected in control livers (data not shown). These results demonstrated that AAV-Pdx1 injection resulted in Pdx1 and insulin expression in the liver cells after 12 days. We also confirmed Ngn3 expression following AAV-Ngn3 transduction of cultured rat liver stem cells by IHC (data not shown), since antibodies against Ngn3 did not work on paraffin-sections.
AAV-PTF-reprogrammed liver-IPCs restore normoglycemia in Stz/diabetic mice
First, we obtained the baseline body weight, blood glucose levels, IPGTT, and pancreas and liver tissue insulin content from six Stz-induced diabetic mice at day 10 post-Stz injection, but before the AAV injection, as shown in Figure 2A. To determine whether ectopically expressed PTFs in the liver could control hyperglycemia, next, Stz-induced diabetic mice were injected with AAV-Pdx1, AAV-Ngn3, or both vectors (108 viral particles/mouse) via the portal vein. Blood glucose levels were monitored to observe the effect of transduced PTFs. As showed in Figure 2B, blood glucose levels gradually declined in the mice receiving AAV-Pdx1, from over 350 mg/dl to 200 mg/dl 2-3 weeks post-injection. The blood glucose levels decreased further in mice receiving AAV-Pdx1/Ngn3, reaching near-normal levels (120-150 mg/dL) within 2-3 weeks post-injection (normal range 80-120 mg/dL). However, the mice receiving only the AAV-Ngn3 vector remained hyperglycemic with glucose levels around 300 mg/dl. As expected, the mice receiving AAV-GFP remained hyperglycemic with glucose levels > 400 mg/dL (data not shown). After ~30 days, the blood glucose levels in the Pdx1 only group reached levels seem comparable to those in the AAV-Pdx1/Ngn3 group (~150 mg/dl).
Figure 2.

Reversal of Stz/diabetes by liver-IPCs reprogrammed by AAV-PTFs. A. Baselines of Stz/diabetic NOD/SCID mice. NOD/SCID mice (10-12 weeks, n=6) were treated daily with Stz for five days and blood glucose levels and body weight were monitored every other day until the onset of diabetes (two measurements 24 h apart). Mice were sacrificed at glucose levels between 250 to 350 mg/dL and IPGTT was performed before the scarification. Tissue insulin was extracted from the pancreata and livers and quantified by ELISA. 1. Body weights of normal and diabetic NOD-SCID mice; 2. Blood glucose levels; 3. IPGTT; 4. Tissue insulin content of the liver and pancreas. Baseline information (1-3) was obtained just before the mice were sacrificed. B. Effects of AAV-mediated expression of PTFs on blood glucose levels. Stz-induced diabetic (>250 mg/dL) mice were injected via the portal vein with various AAV viruses (108 vp/mouse) expressing GFP, Ngn3, Pdx1, or a combination of Pdx1/Ngn3. Blood glucose levels were measured. Triangles, mice receiving AAV-Ngn3 (n=4); diamonds, mice receiving AAV-Pdx1 (n=8); squares, mice receiving both vectors (n=8). At day 24 post-injection, at least one mouse from each group was sacrificed for IHC (black arrows). At day 30, three mice from the AAV-GFP group (data not shown here) and AAV-Pdx1/Ngn3 groups were killed for tissue insulin measurements (brown arrow). Around day 40, two mice from each group underwent subtotal Px (red arrows). C. Changes of blood glucose levels following subtotal Px. To investigate a role of pancreatic beta-cells in normalizing the blood glucose levels, subtotal Px was performed on two mice from each group and changes of blood glucose levels were recorded (P1 and P2). Red lines represent mice receiving Pdx1/Ngn3; Blue lines, mice receiving Pdx1; and green lines, mice receiving Ngn3. One mouse in Ngn3 group (P2) died five days post surgery. D. Insulin IHC in Stz/Diabetic Pancreas. Formalin-fixed and paraffin embedded pancreatic sections were incubated with anti-insulin antibodies and visualized by DA B . Control (day 10) tissue was obtained after the first dose of Stz. The area shown was the only region containing islets in the entire pancreas. The remaining photographs are representative sections from Stz-induced diabetic mice at day-24 post-treatment via the portal vein with AAV-GFP, AAV-Ngn3, AAV-Pdx1, and AAV-Pdx1/Ngn3.
To investigate the possible role of pancreatic beta-cell regeneration in normalizing the blood glucose levels, we performed subtotal Px by removing the entire splenic portion and most of the duodenal portion (the “head” of the pancreas) of the pancreas. Two mice from each group underwent subtotal Px and changes in the blood glucose levels were examined two-days post surgery (Figure 2C). Surprisingly, subtotal Px had little or no effect on blood glucose levels in mice receiving Pdx1/Ngn3 (solid lines), suggesting that non-pancreatic IPCs from the liver play a major role in maintaining the blood glucose levels. However, the mice receiving Ngn3 alone showed a sharp increase in blood glucose levels after Px (dotted line). Interestingly, mice receiving Pdx1(dotted lines) showed a small rebound of blood glucose levels in the following week, which then decreased again, suggesting the regenerated pancreatic beta-cells may play a minor role in maintaining blood glucose levels, but the liver-IPCs may assume major responsibility for maintaining blood glucose levels following subtotal Px. Insulin IHC performed on the pancreatic sections (Figure 2D) confirmed the presence of endogenous pancreatic beta cell regeneration in all AAV-PTF-treated groups, with various degrees of pancreatic islet abundance (Pdx1/Ngn3 > Pdx1 > Ngn3 > GFP). As expected, the diabetic mice treated with Pdx1 or Pdx1/Ngn3 returned to normal age-specific body weight, even after subtotal Px, as compared to the Ngn3-treated groups that continued to lose body weight (data not shown).
PTF-reprogrammed liver-IPCs are glucose-responsive
To determine whether the PTF-reprogrammed IPCs in the livers could respond to a glucose challenge, we first performed IPGTT 12~14 days post subtotal Px. As shown in Figure 3A, there was a marked difference in glucose tolerance tests between the groups treated with Ngn3 and treated with Pdx1 or Pdx1/Ngn3. The latter groups could respond to a glucose challenge with a clearance rate similar to that of healthy mice. However, mice treated with Ngn3 remained hyperglycemic throughout the test. These data indicate that the genetically reprogrammed liver-IPCs exhibited pancreatic beta-cell function, responding to the loaded glucose by secreting insulin, resulting in lower blood glucose levels in the Pdx1 or Pdx1/Ngn3-treated mice.
Figure 3.

Reprogrammed liver-IPCs are glucose responsive. A. IPGTT after subtotal Px. After recovery from surgery around 22-24 days post-Px, mice were injected with glucose (2 mg/g body weight i.p.) and the blood glucose levels were recorded at indicated times. B. Expression of glucagon and insulin genes in the liver. Expression of glucagon and insulin mRNAs was assayed in the livers of normal mice (L1), or Stz/diabetic mice treated with GFP (L2) or Pdx1/Ngn3 (L3) by RT-PCR. Pancreatic tissue (P) serves as positive control. C. & D. Immunohistochemical analysis of liver sections. Liver sections harvested from mice at day 24 post-AAV-treatment (as indicated) were immunostained with anti-insulin (C) or anti-insulin and glucagon (D) antibodies. Islet in the mouse pancreatic tissue serves as positive controls. Either insulin-positive (green) or glucagon-positive (red) or both-positive liver cells are indicated by arrows. Original magnification is 40x.
Insulin and glucagon expression in liver after AAV-PTF treatment
To confirm pancreatic hormones were indeed expressed and synthesized in the liver, we examined insulin and glucagon gene and protein expression after AAV-PTF treatment. Figure 3B shows that expression of insulin and glucagon genes was detectable only in the liver of the mice treated with Pdx1/Ngn3 (lane L3), whereas not detectable in the livers of normal (L1) or Stz/diabetic (L2) mice. Figure 3C shows that the Pdx1/Ngn3-treated liver cells along the central vein or in the portal tract areas at day-24 post-injection exhibit strong staining for insulin by IHC, whereas relatively weak-staining insulin-positive liver cells were seen in Pdx1-treated liver cells and very weak staining here but none in other sections in Ngn3-treated liver cells. Although insulin-staining intensity is not quantitative or representative of the tissue insulin content, the more intense insulin-staining cells in the liver section in the combined Pdx1/Ngn3 treatment is consistent with their ability to more efficiently reduce blood glucose levels.
We then examined whether there is co-expression of both insulin and glucagon positive cells in Pdx1/Ngn3-treated liver cells at day 24-post-portal vein injection. Following Pdx1/Ngn3 treatment the liver cells expressed either insulin or glucagon, or both. Scattered rare double insulin/glucagon-positive cells were detected in some areas and single positive cells in other areas in the liver as shown by double IF in Figure 3D. The insulin and glucagon positive cells were mainly in the proximity of central veins. Additionally, we examined if there were IPCs in the spleen and kidneys on the same slide by IHC. However, none were detected (data not shown).
Liver and pancreas insulin quantification
To confirm liver insulin production and evaluate the relative contribution of regenerating pancreatic beta-cells vs. the liver-IPCs to restoration of normoglycemia, the whole liver and pancreas insulin content was examined (ELISA). Three mice each from the AAV-GFP and AAV-Pdx1/Ngn3 groups were sacrificed at day 30 post-portal vein AAV injection of Stz/diabetic mice and the liver and pancreas tissues were collected for quantification of tissue insulin. As shown in Figure 4A (right panel), there was an 8.7-fold increase in insulin content from the livers of mice receiving Pdx1/Ngn3 in comparison to control mice, which quantitatively confirms the liver insulin production detected by other methods. The large fraction of insulin from the liver indicated that the liver-derived IPCs significantly contribute to restoring normoglycemia. In comparison, the pancreatic insulin content (Figure 4A, left panel) in diabetic mice treated with Pdx1/Ngn3 was ~ 50% of normal mice, but ~ 10-times higher than AAV-GFP-treated control mice, supporting a role of Pdx1/Ngn3-mediated beta-cell regeneration in restoring and maintaining of blood glucose homeostasis. Figure 4B shows a noticeable increase in the numbers of liver-IPCs and the formation of small residual pancreatic islets at day 30, whereas there were no liver-IPCs and only rare tiny islets in the control AAV-GFP mice (data not shown).
Figure 4.
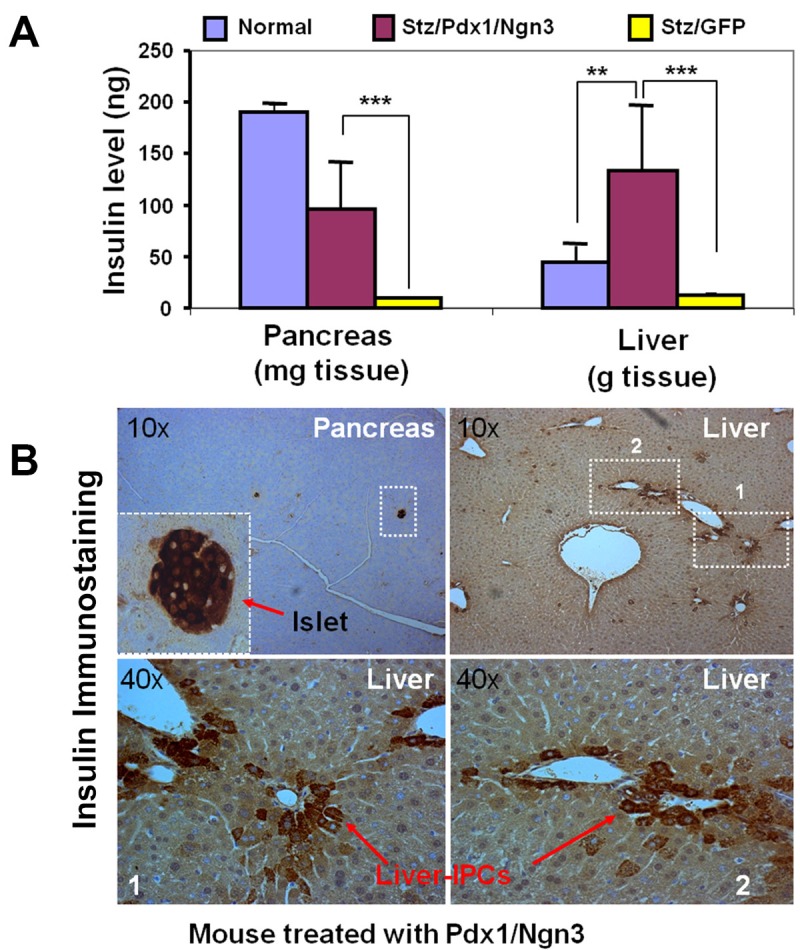
Tissue insulin in liver and pancreas. A. Insulin content in the livers and pancreata. Insulin was extracted from the livers and pancreata from normal (n=4) and AAV-treated Stz/diabetic mice either with GFP (n=4) or with Pdx1/Ngn3 (n=4) ~ 30 days post-treatment. Tissue insulin content was determined by ELISA using an ultrasensitive kit (Aplco). **p<0.01. ***p<0.001. B. IPCs in the pancreas and livers of AAV-Pdx1/Ngn3-treated mice. Pancreas and liver sections were incubated with anti-insulin antibody (1:500) overnight at 4°C. Small residual islets in the pancreas (left upper panel) and liver-derived IPCs are indicated by arrows. Lower panels represent enlarged views of areas 1 and 2.
PTF-reprogrammed liver-IPCs elicit autoimmune attack
We have demonstrated that PTF-reprogrammed mouse liver-IPCs can effectively reverse hyperglycemia in stz-induced diabetic mice and can maintain euglycemia after near total Px by releasing insulin in response to elevated blood glucose levels. Next, we addressed whether these liver-derived IPCs were susceptible to autoimmune attack following T1D passive transfer by diabetogenic splenocytes. Female NOD mice spontaneously develop autoimmunity toward their islets of Langerhans [25]. Such autoimmunity also destroys congenic and syngeneic transplanted islets. Adoptive transfer of splenocytes from prediabetic and diabetic NOD mice into recipient NOD/SCID mice shows that the recipient mice develop diabetes [23,24].
To examine whether autoimmunity toward the liver-IPCs developed, we employed a two-step approach using congenic NOD/SCID mice (Figure 5A). As shown in Figure 5B, NOD/SCID mice first were induced to become diabetic (250 ~300 mg/dL fasting glucose) by daily i.p. injections of Stz for 5 days, and then AAV-Pdx1/Ngn3 was delivered into the mouse livers via the portal vein. Following normalization of the blood glucose levels, three mice (#1, 2, and 6) underwent subtotal Px. Blood glucose levels remained normal suggesting that the liver-IPCs played a major role in maintaining glucose homeostasis. Mice then received non-fractionated splenocytes (2X107/mouse i.p.) harvested from new-onset diabetic NOD mice (14-20 weeks). Within 2-3 weeks, all mice became hyperglycemic (Figure 5B). At three time points, IPGTTs (Figure 5C) were performed before (1) and after (2) subtotal Px to confirm the vital role of the liver-IPCs in maintaining euglycemia. By the end of the experiment (3), all mice lost their ability to handle a glucose challenge by secreting inappropriate amount of insulin.
Figure 5.

Autoimmune attack of liver-derived IPCs. A. Experimental strategy. To test whether liver-derived IPCs can escape autoimmune attack, NOD/SCID mice were given diabetes by injecting Stz. The liver-derived IPCs were expected to be reprogrammed by AAV-Pdx1/Ngn3 and these liver-derived IPCs were expected to correct hyperglycemia, confirmed by performing subtotal Px. Once confirmed, diabetogenic SAT was performed to determine if the liver-IPCs resist autoimmune attack by monitoring changes of the blood glucose levels (resistant = normal glucose levels, susceptible = abnormally high glucose levels). B. Testing autoimmunity of the liver-IPCs. Stz-induced diabetic (>250 mg/dL) NOD/SCID mice (n=6) received AAV-Pdx1/Ngn3 viruses (108 vp/mouse). Blood glucose levels were monitored until the end of the experiment. Near normoglycemic mice #1, #2, and #6 underwent subtotal Px and remained normoglycemic. Diabetogenic NOD splenocytes (2X107 cells/mouse), were transferred i.p. to all Pdx1/Ngn3-treated mice including the three mice receiving Px at day 10 post-surgery. C. IPGTT. Mice were injected with glucose (2 mg/g body weight i.p.) and blood glucose levels were measured at indicated time points. Three IPGTTs were performed at various times (1. Before Px; 2. Day-8 post Px; and 3. Day 16 SAT). All mice were sacrificed after the last IPGTT. D. Autoimmune attack of the liver-IPCs. The livers and pancreata from the above mice were harvested for morphologic evaluation. Liver and pancreas sections were H&E stained (left panel) or immunostained with anti-insulin (middle panel) or anti-CD3 (right panel) antibodies. Arrows indicate infiltrating lymphocytes. E. Mixed cellular response of liver-derived IPCs. Mice were sacrificed 2-3 weeks post-SAT. Double IHC for identification of T-cells (CD3+), B-cells (B220+), and macrophages (F4/80+) was performed on AAV-Pdx1/Ngn3-treated mouse liver and pancreas sections following SAT. The liver sections were sequentially immunostained for CD3, B220, and anti-F4/80 for 1 hr, followed by appropriate secondary antibodies. Staining of T-cells, B-cells, and macrophages in the inflammatory infiltrate was visualized either in brown (DAB) or red (LPR) as indicated. F. Proliferative lymphoid cells attacking IPCs. Insulin (red) and Ki-67(brown) double IHC was performed on the pancreas and liver sections in the AAV-Pdx1/Ngn3-treated mice 2-3 weeks following SAT. Photographs 1-3 represent the total residual pancreas following subtotal Px. Black arrows indicate proliferating lymphocytes surrounding residual pancreatic insulin-producing beta cells (White arrows, panels 4-6) and liver-IPCs (white arrows, panels 7-9).
Control NOD/SCID mice of a similar age (n=10) were injected with diabetogenic splenocytes (2X107/mouse i.p.) and 100% of mice become diabetic within 6 weeks (80% diabetic within 4 weeks, data not shown), confirming that passively transferred splenocytes indeed mount a powerful autoimmune attack on pancreatic islet beta-cells. These results indicated that the PTF-reprogrammed liver-IPCs evoked an autoimmune response that is likely responsive for destroying the liver-IPCs.
To further investigate autoimmune attack on liver-IPCs, mice showing elevated blood glucose levels were sacrificed at day 55 post-AAV-PTF injection or 2.5 weeks post-SAT. Patchy lymphohistocytic infiltrates surrounding the liver-IPCs were apparent in H&E stained (Figure 5D, left panel), and in insulin and CD3 immunostained (Figure 5D, middle and right panels) liver sections. Insulin staining in the liver sections revealed a high background, likely due to dying or degenerating IPCs. However, CD3 immunostaining clearly demonstrated T-cells infiltrating this area. Double IHC on the liver sections confirmed a mixed infiltrate of CD3+ T cells, B220+ B cells, and F4/80+ macrophages (Figure 5E). Furthermore, insulin(red)/Ki-67(brown) double IHC of the pancreas and liver in the three mice (#1, 2, and 6) that underwent subtotal Px revealed only small fragments of residual pancreatic tissue with a total of 1-2 small islets (Figure 5F, 1-3). These were infiltrated by Ki-67-positive proliferating lymphocytes (Figure 5F, 4-6). Similarly, liver-IPCs around the central veins and portal tracts also were undergoing attack by Ki-67-positive proliferating lymphocytes (Figure 5F, 7-9).
To verify that the lymphocytic infiltrate in the liver was not caused by the AAV vector or foreign VP16 viral protein; we investigated three Stz/diabetic NOD/SCID mice treated with AAV-GFP via the portal vein, two weeks later followed by SAT. These control mice were sacrificed at week 2, 2.5, and 3 post-SAT and there was no noticeable lymphocytic infiltrate around central veins or portal tracts on histology examination (Data not shown), indicating that the autoimmunity to the liver-IPCs was not likely to have been caused by immunity to a foreign protein or the AAV vector. All serum samples for Pdx1 autoantibodies from these NOD/SCID mice were negative by ELISA for Pdx1 autoantibodies before and after SAT [26], suggesting that Pdx1 is not a major target for this autoimmune response to the liver-IPCs.
Discussion
Reprogramming of non-pancreatic somatic cells into insulin-secreting cells as pancreatic beta-cell surrogates is regarded as one of the most promising strategies to treat T1D. Since most in vivo reprogramming studies utilize the highly immunogenic adenovirus as a vehicle to transfer genes of PTFs into target cells, it is not feasible to ask whether these beta-cell surrogates are susceptible to autoimmune attack. The major challenge to address this key question in T1D mouse models is to be able to distinguish immunity against virally-encoded antigens from autoimmunity to the IPCs. Here, we provide direct evidence that liver-IPCs are susceptible to autoimmune attack. In the present study, the following objectives were achieved: 1) development of a strategy to minimize the immunogenicity of viral antigens in the host IPCs was developed by using recombinant AAV; 2) demonstration that liver-IPCs are glucose-responsive and can maintain glucose homeostasis in diabetic mice; and 3) direct observation that liver-IPCs are susceptible to autoimmune attack. To minimize the vector-specific immune response, we selected AAV serotype 2 vectors to deliver Pdx1 and Ngn3 genes into liver cells via a onetime portal vein injection of AAV-Pdx1/AAV-Ngn3 viral particles. Delivery of Pdx1/Ngn3 transgenes directly into the liver using AAV can help to minimize immunity to viral capsid (the protein shell of a virus) and efficiently introduces gene products [27,28]. AAV vectors have been widely adopted as gene delivery vehicles because of their ability to transduce a wide variety of tissues, mediate long-term expression of the transgene after a single in vivo administration [29-32] and minimize immunogenicity of host cells [32,33]. Compared to other target tissues, the liver has several advantages as a target cell for IPC reprogramming by gene transfer [4,34]. In addition to derivation from the endoderm and sharing a common progenitor and many transcription factors, hepatocytes and pancreatic beta cells have a built-in glucose sensing system (glucose kinase and glucose transporter-2) [35,36]. These biologic features make hepatocytes ideal target cells for IPC reprogramming. As a target organ, stable and sustained expression of a transgene in AAV-transduced hepatocytes is associated with the induction of tolerance rather than immunity [28], perhaps due to poor transduction efficiency of antigen presenting cells and up-regulation of T-regs [37]. This conclusion comes from detailed studies of the hepatic administration of an AAV vector expressing human coagulation factor Factor IX. (F.IX) [32,33]. In contrast to muscle-directed delivery of human F.IX using AAV, liver-directed delivery of the same vector results in antigen-specific immunological unresponsiveness to the transgene products in several strains of mice. Tolerance can be transferred to naïve recipients by adoptive T cell transfer after hepatic gene transfer with AAV and once established, it is not broken by immunological challenge with F.IX in adjuvant. Further studies have revealed that hepatic delivery of an AAV gene therapy vector encoding ovalbumin induces antigen-specific CD4+CD25+ regulatory T cells [37-39]. The advantages of AAV-mediated hepatic gene transfer have allowed us to address the issue of whether liver-IPCs are protected from autoimmunity.
To examine the glucose responsiveness of liver IPCs, we chose the AAV2 vector to deliver two key pancreatic transcription factors (mouse Pdx1 and Ngn3) via the portal vein. Consistent with other studies [7,8,10-12,40], we found that hepatic administration of AAV-Pdx1/Ngn3 can indeed reprogram the hepatocytes into glucose-responsible IPCs as evidenced by the restoration of normoglycemia in Stz/diabetic mice and the responsiveness to glucose challenge in IPGTT before and after nearly total Px. We further demonstrated that the liver-IPCs play a major role in maintaining glucose homeostasis by removing more than 90% of the pancreas. By IHC, we showed that the IPCs in treated liver are distributed mainly along the central veins, which is consistent with the path of the injected AAV- Pdx1/Ngn3 vectors. We found abundant hepatic insulin in the AAV-Pdx1/Ngn3-treated mice measured by several methods (insulin tissue content, insulin immunochemistry, and insulin gene expression). AAV-Pdx1/Ngn3 expression effectively reprogrammed the hepatocytes into functional IPCs and could maintain blood glucose homeostasis. Despite subtotal Px, Pdx1/Ngn3-treated mice remained normoglycemic for months, indicating that the liver-IPCs largely compensated the lack of pancreatic beta-cells by producing insulin. This effect persisted until the end of the observation period (2-4 months). Histological studies of the residual pancreatic tissues revealed very rare small regenerated islets, which were insufficient to maintain normoglycemia in mice as observed in control mice following subtotal Px. The lack of robust pancreatic islet beta-cell regeneration probably reflects the scarcity of residual pancreatic tissue following Px. Based on these data, we conclude that the AAV-Pdx1/Ngn3-reprogrammed liver-IPCs are functional and able to independently regulate and restore normal blood glucose levels in Stz/diabetic mice.
To directly demonstrate that liver-IPCs are glucose-sensitive, in situ liver perfusion of low and high glucose buffer via the inferior vena cava (inflow) and the portal vein (outflow) in a normoglycemic mouse that was treated with AAV-Pdx1/Ngn3 following subtotal Px can be attempted in future studies to thoroughly examine the pattern and intensity of the liver-IPCs in glucose-stimulated insulin release.
A three-step strategy was employed to address the question of whether liver IPCs are susceptible to autoimmunity; first, AAV-Pdx1/Ngn3 transgenes were expressed in hepatocytes. Next, the pancreas was removed to see if the mice remained normoglycemic (indicating the liver-IPCs are functional). Finally, diabetogenic splenocytes were adoptively transferred into the liver-IPC-mediated normoglycemic mice to see if they could induce autoimmune diabetes. Our data (Figure 5) clearly indicate that the liver-IPCs were functional and that they were fully susceptible to autoimmune attack, as 6/6 mice receiving diabetogenic splenocytes became hyperglycemic within 3 weeks. Thus, it is likely that after transdifferentiation, the liver-IPCs express at least some of the beta-cell autoantigenic targets of diabetogenic T cells. This interpretation is supported by the appearance of dense CD3+ T cell infiltrates surrounding the liver-IPCs along with antigen presenting B-cells and macrophages. Unfortunately, it was difficult to show clearly insulin-positive cells due to high background and it also was unclear whether the transfer of splenocytes from diabetic NOD mice led to the destruction of the transdifferentiated hepatic cells or just a loss of function. Additional future studies are needed to more carefully examine the time course of this destruction including the overall hepatic function in the mice after splenocyte transfer.
To exclude the possibility that the CD3+ T cells were reactive with the VP16 segment attached to the C-terminus of Pdx1, we performed similar experiments using AAV-GFP (a foreign gene to the mice), followed by splenocyte adoptive transfer. The absence of a T cell infiltrate in these mice suggests that there was not an immune response to the products of either transgenes or viral genes. Taken together with evidence that the AAV capsid is low immunogenic and that hepatic delivery of AAV promotes immune tolerance [33,39,41] and previous observations of hepatic delivery of Pdx1 modulates autoimmunity in NOD mice [26,42], the present data are consistent with the possibility that liver-derived (reprogrammed) insulin-producing/secreting cells share common antigens with pancreatic beta-cells and suffer the same fate as the beta-cells in the presence of diabetogenic (NOD-derived) splenocytes.
We have demonstrated that AAV-Pdx1/Ngn3-reprogrammed liver-IPCs can function to maintain blood glucose homeostasis. However, these liver-IPCs share immunologic markers with native pancreatic beta-cells that make them susceptible to the attack by the diabetogenic T cells from NOD mice. Therefore, cell regeneration/cell reprogramming will likely need to be coupled with immune modulation in order to achieve lasting control of T1D.
Acknowledgements
This work was supported in part by grants from the National Institutes of Health, NIDDK DK064054 and DK071831 (to LJ Yang) and HL077602 (to MI Phillips). We thank Professor Westley Reeves, University of Florida, College of Medicine, for the editorial assistance.
Author Contributions: DQ Tang, S Lu, V Koya, K Qian, Y Sun, QW Wang, H Wang, Y Sun, SW Li, and C Zhang generated researched data; LJ Yang analyzed researched data and wrote the manuscript, and DL Purich, M Atkinson, BC Hansen, and MI Phillips reviewed and edited the manuscript.
Abbreviations
- AAV
adeno-associated virus
- AD
adenovirus
- IPCs
insulin-producing cells
- IPGTT
Intraperitoneal glucose tolerance test
- PTFs
pancreatic transcription factors
- Px
pancreatectomy
- NOD/SCID
non-obese diabetic/severe combined immunodeficiency
References
- 1.Zalzman M, Gupta S, Giri RK, Berkovich I, Sappal BS, Karnieli O, Zern MA, Fleischer N, Efrat S. Reversal of hyperglycemia in mice by using human expandable insulin-producing cells differentiated from fetal liver progenitor cells. Proc Natl Acad Sci U S A. 2003;100:7253–7258. doi: 10.1073/pnas.1136854100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sapir T, Shternhall K, Meivar-Levy I, Blumenfeld T, Cohen H, Skutelsky E, Eventov-Friedman S, Barshack I, Goldberg I, Pri-Chen S, Ben Dor L, Polak-Charcon S, Karasik A, Shimon I, Mor E, Ferber S. Cell-replacement therapy for diabetes: Generating functional insulin-producing tissue from adult human liver cells. Proc Natl Acad Sci U S A. 2005;102:7964–7969. doi: 10.1073/pnas.0405277102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li WC, Horb ME, Tosh D, Slack JM. In vitro transdifferentiation of hepatoma cells into functional pancreatic cells. Mech Dev. 2005;122:835–847. doi: 10.1016/j.mod.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 4.Yang LJ, Li S, Hatch H, Ahrens K, Cornelius JG, Petersen BE, Peck AB. In vitro trans-differentiation of adult hepatic stem cells into pancreatic endocrine hormone-producing cells. Proc Natl Acad Sci U S A. 2002;99:8078–8083. doi: 10.1073/pnas.122210699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tang DQ, Lu S, Sun YP, Rodrigue E, Chou W, Yang C, Chang LJ, Yang LJ. Reprogramming Liver-Stem WB Cells into Functional Insulin-Producing Cells by Persistent Expression of Pdx1- and Pdx1-VP16 Mediated by Lentiviral Vectors. Lab Invest. 2003;86:83–93. doi: 10.1038/labinvest.3700368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tang DQ, Cao LZ, Chou W, Shun L, Farag C, Atkinson MA, Li SW, Chang LJ, Yang LJ. Role of Pax4 in Pdx1-VP16-mediated liver-to-endocrine pancreas transdifferentiation. Lab Invest. 2006;86:829–841. doi: 10.1038/labinvest.3700434. [DOI] [PubMed] [Google Scholar]
- 7.Ferber S, Halkin A, Cohen H, Ber I, Einav Y, Goldberg I, Barshack I, Seijffers R, Kopolovic J, Kaiser N, Karasik A. Pancreatic and duodenal homeobox gene 1 induces expression of insulin genes in liver and ameliorates streptozotocin-induced hyperglycemia. Nat Med. 2000;6:568–572. doi: 10.1038/75050. [DOI] [PubMed] [Google Scholar]
- 8.Ber I, Shternhall K, Perl S, Ohanuna Z, Goldberg I, Barshack I, Benvenisti-Zarum L, Meivar-Levy I, Ferber S. Functional, persistent, and extended liver to pancreas transdifferentiation. J Biol Chem. 2003;278:31950–31957. doi: 10.1074/jbc.M303127200. [DOI] [PubMed] [Google Scholar]
- 9.Kojima H, Fujimiya M, Matsumura K, Younan P, Imaeda H, Maeda M, Chan L. NeuroD-betacellulin gene therapy induces islet neogenesis in the liver and reverses diabetes in mice. Nat Med. 2003;9:596–603. doi: 10.1038/nm867. [DOI] [PubMed] [Google Scholar]
- 10.Yechoor V, Liu V, Espiritu C, Paul A, Oka K, Kojima H, Chan L. Neurogenin3 is sufficient for transdetermination of hepatic progenitor cells into neo-islets in vivo but not transdifferentiation of hepatocytes. Dev Cell. 2009;16:358–373. doi: 10.1016/j.devcel.2009.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kaneto H, Nakatani Y, Miyatsuka T, Matsuoka TA, Matsuhisa M, Hori M, Yamasaki Y. PDX-1/VP16 fusion protein, together with NeuroD or Ngn3, markedly induces insulin gene transcription and ameliorates glucose tolerance. Diabetes. 2005;54:1009–1022. doi: 10.2337/diabetes.54.4.1009. [DOI] [PubMed] [Google Scholar]
- 12.Wang AY, Ehrhardt A, Xu H, Kay MA. Adenovirus transduction is required for the correction of diabetes using Pdx-1 or Neurogenin-3 in the liver. Mol Ther. 2007;15:255–263. doi: 10.1038/sj.mt.6300032. [DOI] [PubMed] [Google Scholar]
- 13.Seiler MP, Cerullo V, Lee B. Immune response to helper dependent adenoviral mediated liver gene therapy: challenges and prospects. Curr Gene Ther. 2007;7:297–305. doi: 10.2174/156652307782151452. [DOI] [PubMed] [Google Scholar]
- 14.Campos SK, Barry MA. Current advances and future challenges in Adenoviral vector biology and targeting. Curr Gene Ther. 2007;7:189–204. doi: 10.2174/156652307780859062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kapturczak MH, Flotte T, Atkinson MA. Adenoassociated virus (AAV) as a vehicle for therapeutic gene delivery: improvements in vector design and viral production enhance potential to prolong graft survival in pancreatic islet cell transplantation for the reversal of type 1 diabetes. Curr Mol Med. 2001;1:245–258. doi: 10.2174/1566524013363979. [DOI] [PubMed] [Google Scholar]
- 16.Flotte TR. Recent developments in recombinant AAV-mediated gene therapy for lung diseases. Curr Gene Ther. 2005;5:361–366. doi: 10.2174/1566523054064986. [DOI] [PubMed] [Google Scholar]
- 17.Cotugno G, Annunziata P, Tessitore A, O’Malley T, Capalbo A, Faella A, Bartolomeo R, O’Donnell P, Wang P, Russo F, Sleeper MM, Knox VW, Fernandez S, Levanduski L, Hopwood J, De LE, Haskins M, Auricchio A. Long-term amelioration of feline Mucopolysaccharidosis VI after AAV-mediated liver gene transfer. Mol Ther. 2001;19:461–469. doi: 10.1038/mt.2010.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cao LZ, Tang DQ, Horb ME, Li SW, Yang LJ. High glucose is necessary for complete maturation of pdx-1-vp16-expressing hepatic cells into functional insulin-producing cells. Diabetes. 2004;53:3168–3178. doi: 10.2337/diabetes.53.12.3168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cheng H, Wolfe SH, Valencia V, Qian K, Shen L, Phillips MI, Chang LJ, Zhang YC. Efficient and persistent transduction of exocrine and endocrine pancreas by adeno-associated virus type 8. J Biomed Sci. 2007;14:585–594. doi: 10.1007/s11373-007-9159-1. [DOI] [PubMed] [Google Scholar]
- 20.Phillips MI, Mohuczy-Dominiak D, Coffey M, Galli SM, Kimura B, Wu P, Zelles T. Prolonged reduction of high blood pressure with an in vivo, nonpathogenic, adeno-associated viral vector delivery of AT1-R mRNA antisense. Hypertension. 1997;29:374–380. doi: 10.1161/01.hyp.29.1.374. [DOI] [PubMed] [Google Scholar]
- 21.Koya V, Lu S, Sun YP, Purich DL, Atkinson MA, Li SW, Yang LJ. Reversal of streptozotocin-induced diabetes in mice by cellular transduction with recombinant pancreatic transcription factor pancreatic duodenal homeobox-1: a novel protein transduction domain-based therapy. Diabetes. 2008;57:757–769. doi: 10.2337/db07-1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hutchings PR, Cooke A. The transfer of autoimmune diabetes in NOD mice can be inhibited or accelerated by distinct cell populations present in normal splenocytes taken from young males. J Autoimmun. 1990;3:175–185. doi: 10.1016/0896-8411(90)90139-j. [DOI] [PubMed] [Google Scholar]
- 23.Bendelac A, Carnaud C, Boitard C, Bach JF. Syngeneic transfer of autoimmune diabetes from diabetic NOD mice to healthy neonates. Requirement for both L3T4+ and Lyt-2+ T cells. J Exp Med. 1987;166:823–832. doi: 10.1084/jem.166.4.823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.O’Reilly LA, Hutchings PR, Crocker PR, Simpson E, Lund T, Kioussis D, Takei F, Baird J, Cooke A. Characterization of pancreatic islet cell infiltrates in NOD mice: effect of cell transfer and transgene expression. Eur J Immunol. 1991;21:1171–1180. doi: 10.1002/eji.1830210512. [DOI] [PubMed] [Google Scholar]
- 25.Giarratana N, Penna G, Adorini L. Animal models of spontaneous autoimmune disease: type 1 diabetes in the nonobese diabetic mouse. Methods Mol Biol. 2007;380:285–311. doi: 10.1007/978-1-59745-395-0_17. [DOI] [PubMed] [Google Scholar]
- 26.Li SW, Koya V, Li Y, Donelan W, Lin P, Reeves WH, Yang LJ. Pancreatic duodenal homeobox 1 protein is a novel beta-cell-specific autoantigen for type I diabetes. Lab Invest. 2010;90:31–39. doi: 10.1038/labinvest.2009.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Herzog RW. Hepatic AAV gene transfer and the immune system: friends or foes? Mol Ther. 2010;18:1063–1066. doi: 10.1038/mt.2010.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Loduca PA, Hoffman BE, Herzog RW. Hepatic gene transfer as a means of tolerance induction to transgene products. Curr Gene Ther. 2009;9:104–114. doi: 10.2174/156652309787909490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Arruda VR, Stedman HH, Nichols TC, Haskins ME, Nicholson M, Herzog RW, Couto LB, High KA. Regional intravascular delivery of AAV-2-F. IX to skeletal muscle achieves long-term correction of hemophilia B in a large animal model. Blood. 2005;105:3458–3464. doi: 10.1182/blood-2004-07-2908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Herzog RW, Hagstrom JN, Kung SH, Tai SJ, Wilson JM, Fisher KJ, High KA. Stable gene transfer and expression of human blood coagulation factor IX after intramuscular injection of recombinant adeno-associated virus. Proc Natl Acad Sci U S A. 1997;94:5804–5809. doi: 10.1073/pnas.94.11.5804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jiang H, Lillicrap D, Patarroyo-White S, Liu T, Qian X, Scallan CD, Powell S, Keller T, McMurray M, Labelle A, Nagy D, Vargas JA, Zhou S, Couto LB, Pierce GF. Multiyear therapeutic benefit of AAV serotypes 2, 6, and 8 delivering factor VIII to hemophilia A mice and dogs. Blood. 2006;108:107–115. doi: 10.1182/blood-2005-12-5115. [DOI] [PubMed] [Google Scholar]
- 32.Mount JD, Herzog RW, Tillson DM, Goodman SA, Robinson N, McCleland ML, Bellinger D, Nichols TC, Arruda VR, Lothrop CD, Jr, High KA. Sustained phenotypic correction of hemophilia B dogs with a factor IX null mutation by liver-directed gene therapy. Blood. 2002;99:2670–2676. doi: 10.1182/blood.v99.8.2670. [DOI] [PubMed] [Google Scholar]
- 33.Mingozzi F, Liu YL, Dobrzynski E, Kaufhold A, Liu JH, Wang Y, Arruda VR, High KA, Herzog RW. Induction of immune tolerance to coagulation factor IX antigen by in vivo hepatic gene transfer. J Clin Invest. 2003;111:1347–1356. doi: 10.1172/JCI16887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang LJ. Liver stem cell-derived beta-cell surrogates for treatment of type 1 diabetes. Autoimmun Rev. 2006;5:409–413. doi: 10.1016/j.autrev.2005.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Efrat S, Tal M, Lodish HF. The pancreatic beta-cell glucose sensor. Trends Biochem Sci. 1994;19:535–538. doi: 10.1016/0968-0004(94)90056-6. [DOI] [PubMed] [Google Scholar]
- 36.Ferber S, BeltrandelRio H, Johnson JH, Noel RJ, Cassidy LE, Clark S, Becker TC, Hughes SD, Newgard CB. GLUT-2 gene transfer into insulinoma cells confers both low and high affinity glucose-stimulated insulin release. Relationship to glucokinase activity. J Biol Chem. 1994;269:11523–11529. [PubMed] [Google Scholar]
- 37.Cao O, Dobrzynski E, Wang L, Nayak S, Mingle B, Terhorst C, Herzog RW. Induction and role of regulatory CD4+CD25+ T cells in tolerance to the transgene product following hepatic in vivo gene transfer. Blood. 2007;110:1132–1140. doi: 10.1182/blood-2007-02-073304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dobrzynski E, Fitzgerald JC, Cao O, Mingozzi F, Wang L, Herzog RW. Prevention of cytotoxic T lymphocyte responses to factor IX-expressing hepatocytes by gene transfer-induced regulatory T cells. Proc Natl Acad Sci U S A. 2006;103:4592–4597. doi: 10.1073/pnas.0508685103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dobrzynski E, Mingozzi F, Liu YL, Bendo E, Cao O, Wang L, Herzog RW. Induction of antigen-specific CD4+ T-cell anergy and deletion by in vivo viral gene transfer. Blood. 2004;104:969–977. doi: 10.1182/blood-2004-03-0847. [DOI] [PubMed] [Google Scholar]
- 40.Yechoor V, Liu V, Paul A, Lee J, Buras E, Ozer K, Samson S, Chan L. Gene therapy with neurogenin 3 and betacellulin reverses major metabolic problems in insulin-deficient diabetic mice. Endocrinology. 2009;150:4863–4873. doi: 10.1210/en.2009-0527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Martino AT, Nayak S, Hoffman BE, Cooper M, Liao G, Markusic DM, Byrne BJ, Terhorst C, Herzog RW. Tolerance induction to cytoplasmic beta-galactosidase by hepatic AAV gene transfer: implications for antigen presentation and immunotoxicity. PLoS One. 2009;4:e6376. doi: 10.1371/journal.pone.0006376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shternhall-Ron K, Quintana FJ, Perl S, Meivar-Levy I, Barshack I, Cohen IR, Ferber S. Ectopic PDX-1 expression in liver ameliorates type 1 diabetes. J Autoimmun. 2007;28:134–42. doi: 10.1016/j.jaut.2007.02.010. [DOI] [PubMed] [Google Scholar]