

Abstract
This study was focused on molecular profiling of prostate cancer (PCa) using scant amounts of both frozen and formalin-fixed paraffin-embedded (FFPE) PCa tissue specimens. DNA and RNA were extracted and interrogated for: (1) whole-genome gene expression profiling, (2) miRNA expression analysis, (3) SNP analysis, and (4) mutation analysis. Data was statistically analyzed and correlated with clinical and pathologic variables. Expression profiling of 47,224 genes revealed 74 genes that were significant in predicting high tumor grade in PCa (p<0.0001). These were involved in many cellular processes as analyzed by Ingenuity Pathway Analysis (IPA). Using novel high throughput technologies, we identified a specific oncogenomic and miRNA signatures showing loss of miR-34 expression. Interestingly, p53 was at the center hub of the signaling pathways, and the loss of miR-34a expression was consistent with the central role of p53 in PCa. Analysis of 731,442 SNP’s, revealed 638 SNP’s that were significant in predicting high tumor grade (p<0.0001; logistic regression analysis). We also found, for the first time, a novel hot spot mutation in MET oncogene, variant T992I, suggesting that our findings would be useful in further defining the role of specific regulatory genes and miRNAs in the pathological evolution of PCa, and could also have potential clinical utility in improving diagnostic accuracy, refining prognostic and predictive capabilities and may serve as therapeutic targets.
Keywords: Molecular, oncogenomic, miRNA, profiling, prostate cancer
Introduction
Therapeutic decision-making in clinical oncology is currently undergoing a transition towards personalized pharmaco-therapeutics. The treatment paradigm is being dictated more by genetic and/or molecular characteristics than the anatomic site of tumor origin. Oncogene mutations are targets of anticancer therapies. With the proliferation of targeted agents in clinical practice, it is important to determine susceptibility to these targets to specific agents. EGFR mutations in non-small cell lung cancers are sensitive to erlotinib [1-3]; KIT mutations in gastrointestinal stromal tumors (GISTs) predict response to imatinib or nilotinib [4]; KRAS oncogene mutations are unresponsive to treatment with anti-EGFR agents in colorectal cancers [5]; and BRAF mutations predict a strong correlation with preclinical sensitivity to targeted agents [6]; HER2 amplification predicts a response to trastuzumab [7]. Widespread clinical application of this approach in prostate cancer (PCa) is hampered by a lack of comprehensive knowledge of the oncogenes that are activated either by point mutation or over-expression due to DNA copy number increases or transcriptional deregulation.
PCa remains the most frequently diagnosed male malignancy and the 2nd leading cause of cancer mortality among American mens. This is mainly due to the lack of curative therapies, inherent complex heterogeneity of the tumors, and thus making it difficult to determine which patient would respond to anticancer therapies. Informative and predictive molecular biomarkers are being analyzed in this regard. Mutations frequently described at the molecular level in PCa include PTEN, KRAS, BRAF, CTNNB1 and HRAS and the infrequent ones include EGFR, FGFR3, NRAS, CDKN2A, RB1, APC, TP53, FBXW7, PIK3CA, IDH1, SMARCA4, AKT1, CEBPA, STK11, MLH1 and NF1 [8]. However, as of yet it is not known which of these are clinically relevant to serve as molecular signatures for early diagnosis, prognosis and as potential therapeutic targets, and no attempt has been made in predicting disease outcome.
Therefore, there is a dire need to comprehensively characterize PCa using point mutation analysis, whole genome gene expression analysis and Single Nucleotide Polymorphism (SNP) analysis. These novel high-throughput technologies will allow reliable oncogenomic profiling of small biopsies of PCa samples for multiple pivotal cancer genes simultaneously. Comprehensive molecular oncogenomic and microRNA (miRNA) profiling of tumors can also provide tumor specific oncogenomic and miRNA signatures which can potentially improve diagnostic accuracy, refine prognostic and predictive capabilities, and may serve as therapeutic targets. These will allow reliable oncogenomic profiling of small biopsies for multiple pivotal cancer genes simultaneously.
Oncogene mutations do not usually occur randomly, but are more frequent in certain regions of each oncogene. The Oncocarta assay for mutation analysis could simultaneously identify both wild type and up to 3 different mutant alleles, showed a high sensitivity exceeding that of traditional Sanger sequencing (which remains the gold standard for many genetic diagnostic approaches); and is highly concordant with Sanger sequencing, pyrosequencing, and allele-specific PCR [9-11] . The fresh frozen tissue allows us to get RNA of sufficient quality to allow gene expression profiling by micorarray. Emerging evidence is accumulating suggesting that molecular markers of tumor aggressiveness could have potential use in the clinical scenario for different malignancies [12,13] including PCa [14].
Recently microRNAs (miRNAs) are being investigated for their role in the progression and biological evolution of cancer [15]. These non-coding small RNA molecules have been proposed to regulate cancer onset, progression and metastasis. They are being evaluated for their role as biomarkers of cancer since they are down or up-regulated in different malignancies [15] as well as being investigated for their possibility as targets to reduce tumor burden in cancer therapeutics [16]. Therefore, a comprehensive analysis of these genomic and miRNA signatures of tumor aggressiveness in PCa would be helpful to delineate potential utility of these in clinical management and diagnosis of PCa.
Materials and methods
Patient and tumor tissue collection
After obtaining Institutional Review Board (IRB) approval and patients’ consent, prostate cancer (PCa) cases were identified from the computerized records from the department of pathology.
Clinical and pathologic characteristics
In each case, the clinical details of the patients were obtained from the computerized hospital database. Pathologic features based on microscopic evaluation of tumor slides were recorded including Gleason’s score, pathologic stage and final histopathologic diagnosis. Cases were then grouped into (a) High grade and (b) Low Grade: based on Gleason’s score.
Specimen selection
Fresh, frozen tissue specimens
From the Biorepository Core at the Karmanos Cancer Institute, Detroit, MI, fresh frozen PCa tissue specimens were retrieved. Each specimen was dissected by a uropathologist, snap frozen within 1 hour of surgical excision, and stored at −80°C until extraction of DNA/RNA. For confirmation of tumor, cryo sections were cut in each case and stained with hematoxylin and eosin stain and microscopically examined to confirm the presence of tumor. Only samples containing >70% tumor cells were included in the study. Seven tissue sections each of 10 microns were cut from the fresh frozen tumor tissue samples each case and placed into sterile eppendorf tubes.
Formalin fixed paraffin embedded (FFPE) tissue specimens
Histopathology slides from PCa cases were microscopically reviewed to select representative tumor blocks containing >70% tumor cells. Five tumor tissue sections, 10 microns in thickness each, were cut from the selected blocks and placed into sterile eppendorf tubes.
Nucleic acid extraction
DNA extraction
DNA was extracted by using QIAamp DNA FFPE Tissue Kit from Qiagen according to the manufacturer’s protocol. The integrity and concentration of DNA was determined spectrophotometrically.
RNA extraction
RNA was similarly extracted using standard methods by column purification (Qiagen) with an included DNA degradation step to ensure that the only nucleic acid present is RNA. RNA purity and quality was verified by separating on an Agilent 2100 Bioanalyzer.
MicroRNA (miRNA) extraction
Total RNA was extracted from the FFPE samples using the Qiagen kit as per the manufacturer’s protocol especially designed to capture smaller size RNA for miRNA analysis as previously described by our laboratory [16].
Whole-genome gene expression analysis
Using Illumina Human HT-12 v4 Bead Chip Microarrays on DNA samples, we performed the expression profiles of 47,224 genes per array. These experiments use 60-mer oligonucleotide arrays which have the most complete coverage of the whole human transcriptome. Two color hybridizations interrogates the cancer cells from the collected specimens against a common standardized reference sample (Universal Human Reference RNA, Stratagene) which permits the comparison of our results to transcriptional profiles generated by other researchers. Alexa dyes (Alexa647-red and Alexa555-green) was used for fluorescent labeling since these dyes are more resistant to oxidation and do not quench as easily as traditional Cy dyes. Labeled targets were synthesized from the purified RNA using linear amplification and indirect labeling by incorporation of aminoallyl-labeled nucleotide, which was subsequently modified by the covalent addition of Alexa dye (Epicentre Technologies). In this way, dye incorporation bias was minimized. In addition, experiments were performed in duplicate using dye-reversed replicates where possible.
Point mutation analysis
DNA was interrogated for mutation analysis using a mass spectroscopy based method, Sequenom Mass-array. OncoCarta panel v1.0 was used that profiles 238 common cancer mutations in 19 oncogenes (known predictors of response or resistance to targeted therapies) [9]. It also determines deletions, insertions as well as single-base pair changes. Briefly, an initial PCR reaction was performed to amplify a small region (between ~80 to 120 base pairs) which includes the potential point mutation site. Next, a 10 base DNA oligonucleotide primer binds immediately upstream of the mutation site and is extended by one base into the potential mutation site. The oligo-nucleotide primers are subsequently separated on a matrix-assisted laser desorption/ionization time of flight (MALDI-TOF) mass spectrometer which is able to quantitatively discern the specific nucleotide that was extended. This method can reproducibly detect mutations at a frequency of 15% within a given sample.
Single nucleotide polymorphism (SNP) analysis
SNP analysis was done using Illumina Omni microarrays which have a genome-wide coverage as per the manufacturer’s protocol.
MicroRNA (miRNA) analysis
miRNA analysis was done using expression microarrays and RT-PCR as described previously by our group [16].
Data analysis
Data obtained was statistically analyzed and correlated with clinical and pathologic variables.
For mutation analysis
Statistical analysis of the relationship between oncogene mutations in samples and their clinical properties was performed using either the Kruskal-Wallis test or Fisher’s Exact test. The Kruskal-Wallis test is used to test the relationship between a nominal variable (the mutation) and a parametric variable (a measured clinical feature, e.g. survival in days). The null hypothesis assumes that the parametric variable comes from identical populations (defined by the nominal variable). Thus, the Kruskal-Wallis test indicates if a mutation is associated with a measured clinical feature of the sample. This is a non-parametric test since this study consists of small samples sizes that may not be normally distributed. Fisher’s Exact test was used to test the relationship between two nominal variables such as a mutation and a categorical clinical feature (e.g. tumor stage). Similar to the Kruskal-Wallis test, Fisher’s Exact test indicates if a mutation is associated with a categorical clinical feature of the tumor and is suited to small samples sizes.
For gene expression analysis
Gene expression profiles were preprocessed using the R Statistical Package and Bioconductor libraries. Gene expression microarray experiments were first normalized using lowess normalization. Dye-reversed replicate array samples were combined using linear modeling techniques provided by the Bioconductor library limma. The statistical significance of these changes was determined primarily by the implementation of a Z-score as well as the use of a regularized T-test provided by limma. When multiple samples are found to harbor amplified regions at similar regions of the genome, statistical methods was used to determine whether genes within the commonly amplified region exhibit a bias towards over-expression relative to samples without the same amplified region. Moreover, this type of statistical analysis determines which genes within the amplicon give the greatest contribution towards the over-expression bias. This type of analysis provides statistical validity to changes that are observed in multiple samples as well as indicate which genes within the amplicon are more over expressed, and therefore more likely to be driving selection for that amplicon. This statistical analysis was carried out with two Bioconductor libraries, globalTest and Gene Set Enrichment Analysis (GSEA). These methods were applied to multiple samples with similar amplicons which provides a basis for determining the fold change threshold required for an individual human tumor biopsy to indicate meaningful over-expression harboring unique genetic amplification events.
Gene analysis
Genes significant in predicting high PCa tumor grade were stratified by ordinal regression analysis and logistic regression analysis.
Pathway analysis
To identify the genes which were significant in predicting high PCa tumor grade, we used the novel software, Ingenuity Pathway Analysis (Ingenuity Systems http://www.ingenuity.com), to determine which biologic cellular pathways were the significant genes involved.
SNP analysis
To identify SNPs which were significant in predicting high PCa tumor grade, ordinal and logistic regression analysis was used.
Results
A total of 40 frozen and 120 FFPE prostate cancer cases were included in the study. All cases evaluated in the present study were histopathologically confirmed cases of prostate adenocarcinoma (PCa). The age range of the patients was 45 to 70 years (mean 59.57 years).
Whole-genome gene expression analysis for identifying alterations in 47,224 genes revealed several genes which were able to predict high tumor grade. Upon statistical analysis using ordinal regression analysis, 2,986 genes were significant in predicting high tumor grade at the 0.05 level; 660 at the 0.01 level; 349 at the 0.005 level; and 74 at the 0.0001 level. The most significant gene was ILMN_1754102; TGIF1.
Genes which were significant predictors of high tumor grade were further analyzed using the novel software, Ingenuity Pathway Analysis (Ingenuity Systems http://www.ingenuity.com), to determine the biologic pathways in which significant genes were involved. The statistically significant predictor genes (p<0.005) were involved in the pathways, ‘Gene Expression, Cell Cycle, Cancer’, ‘Inflammatory Response, Cell Death, Infection Mechanism’ and ‘Cellular Assembly and Organization, Gene Expression, Cancer’ (Figures 1 and 2). The ingenuity network analysis was used to display an interactive graphical representation of the interrelationships between molecules. There was considerable overlap of the significant predictor genes between the significant biologic pathways (Figure 3). The tumor suppressor gene, p53, was found to be at the center hub of the significantly predicting biologic pathways.
Figure 1.
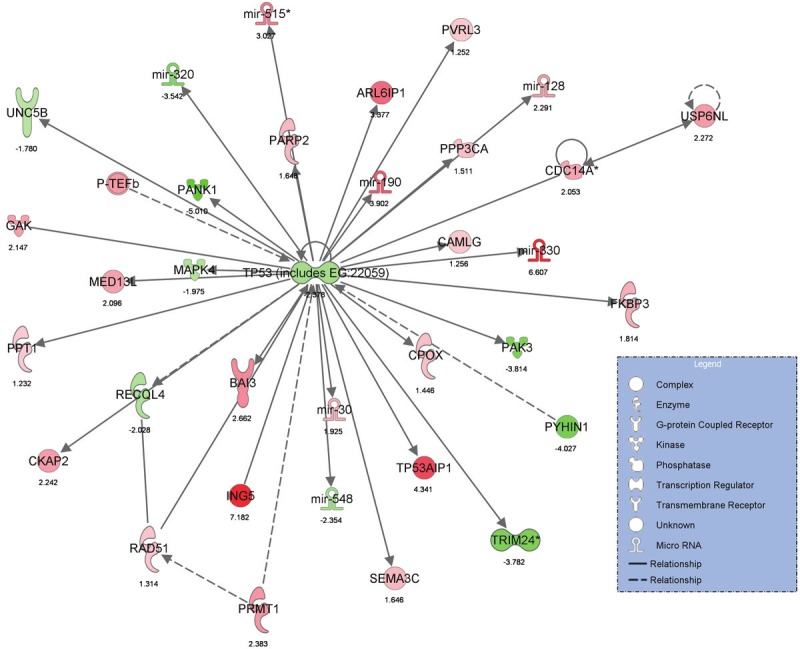
Graphical representation of Ingenuity Pathway Analysis results showing significant predictors genes in ‘Gene Expression, Cell Cycle, Cancer’ pathway.
Figure 2.
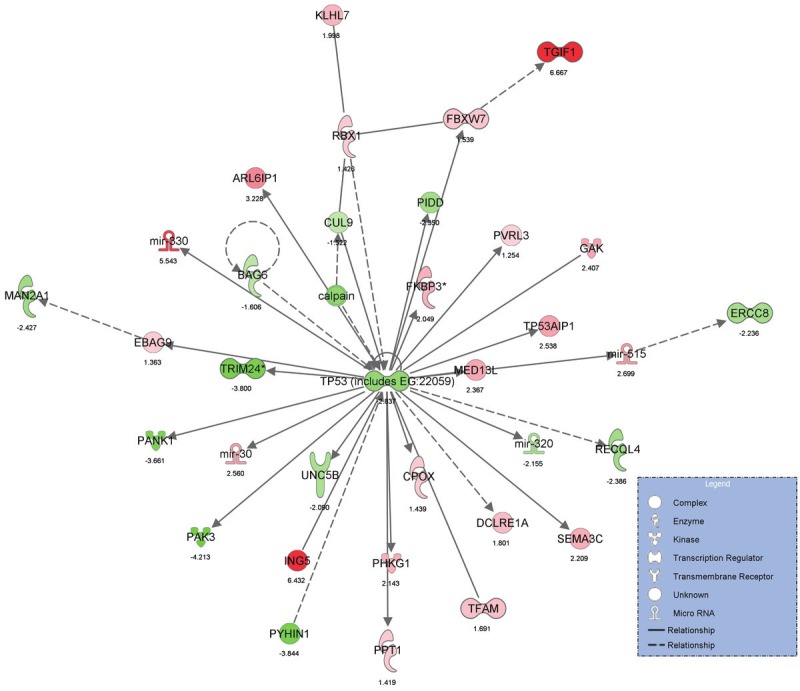
Graphical representation of Ingenuity Pathway Analysis results showing significant predictor genes in ‘InflammatoryResponse, Cell Death, Infection Mechanism’ pathway.
Figure 3.
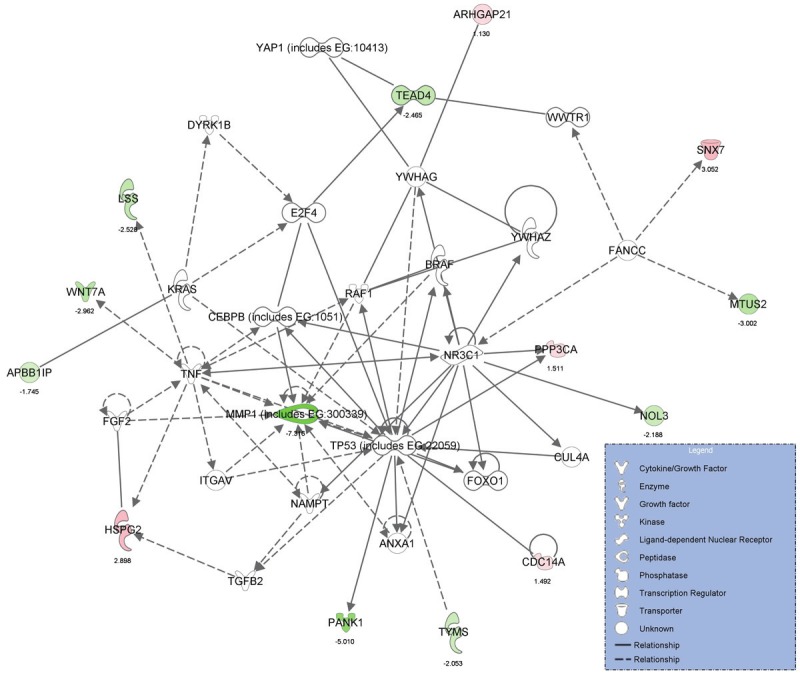
Graphical representation of Ingenuity Pathway Analysis results showing considerable overlap of the significantpredictor genes in the significant biologic pathways.
A total of 731,442 Single Nucleotide Polymorphisms (SNP) were analyzed in the prostate cancer cohort in this study. Using ordinal regression analysis, 48,091 SNPs were significant in predicting the tumor grade at the 0.05 level; 9,776 at the 0.001 level; 4,918 at the 0.005 level; and 1,022 at the 0.01 level. By logistic regression analysis, 41,456 SNPs were significant at the 0.05 level, 7,180 at the 0.001 level, 3,266 at the 0.005 level and 638 at the 0.0001 level in predicting the tumor grade.
Further, the SNP/gene pair interactions were statistically evaluated to determine if there was any interaction between them. A total of 531 SNP/gene pairs showed significant interaction between genes and SNPs (p<0.05). ‘The number of genes, SNPs and miRNA were analyzed and their ability to predict tumor grade are detailed in the figures obtained through IPA (Figures 1, 2 and 3). MicroRNA expression analysis displayed loss of miR-34a in PCa. This finding was validated by RT-PCR as published previously [17,18].
DNA from each case was interrogated for deletions, insertions and base pair changes using the OncoCarta panel comprising of 238 mutations across 19 common oncogenes (Table 1). The mass-spectroscopy based data obtained from the study samples was plotted in a graphic representation to compare the study samples with the normal controls run with each assay. The mass-spectroscopic peak intensity of oncogene MET was found to be altered in PCa case # 21 when compared with the normal control (Figure 4). This was the only gene which showed significant alteration. The alteration was observed in the MET oncogene, variant T992I. The estimated MET mutation frequency in overall samples was 3% with Wilson’s 95% confidence interval (0%, 13%). However, it is not statistically significant (Fisher’s exact test p value of 0.23). All study samples were repeated in duplicate and similar observations were seen. The patient with the mutation was a Caucasian American male aged 49 years with the PCa Gleason score 7 (4+3).
Table 1.
Genes interrogated using the OncoCarta panel
| Genes | Mutations |
|---|---|
| ABL | 13 |
| AKT1 | 7 |
| AKT2 | 2 |
| BRAF | 29 |
| CDK4 | 2 |
| EGFR | 44 |
| ERBB2 | 8 |
| FGFR1 | 2 |
| FGFR3 | 9 |
| FLT3 | 3 |
| HRAS | 12 |
| JAK2 | 1 |
| KIT | 32 |
| KRAS | 18 |
| MET | 5 |
| NRAS | 18 |
| PDGFRA | 11 |
| PIK3CA | 16 |
| RET | 6 |
Figure 4.
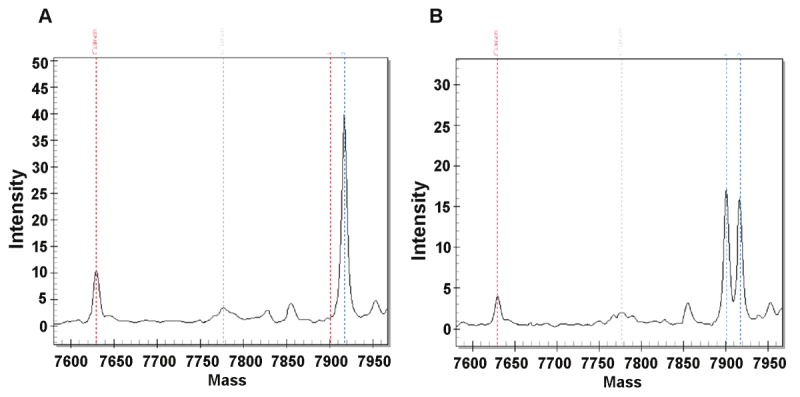
OncoCarta results showing alteration in mass-spectroscopic peak intensity of MET oncogene, variant T992I in PCa case (B) when compared to normal control (B).
Discussion
Prostate cancer (PCa) remains the most frequently diagnosed male malignancy and the 2nd leading cause of cancer mortality among American men. This is mainly due to the lack of curative therapies, inherent complex heterogeneity of the tumors, and thus making it difficult to determine which patient would respond to anticancer therapies. Despite years of research in this field, as of yet it is not known which markers are clinically relevant to serve as molecular signatures for early diagnosis, prognosis and as potential therapeutic targets, and in predicting disease outcome.
In the present study, we focused on molecular profiling of PCa cases using novel, high throughput molecular techniques on scant amounts of fresh-frozen and FFPE tumor samples. Both tissue samples were able to reveal excellent DNA and RNA both quantitatively and qualitatively. The data obtained appears very significant for potential clinical utility in the future.
Whole-genome gene expression analysis revealed several statistically significant genes as predictors of high tumor grade with the most significant gene being TGIF1.
As in previous studies, we found several genes were altered in PCa including PTEN, KRAS, BRAF, CTNNB1, HRAS, EGFR, FGFR3, NRAS, CDKN2A, RB1, APC, TP53, FBXW7, PIK3CA, IDH1, SMARCA4, AKT1, CEBPA, STK11, MLH1 and NF1 [8]; the IPA results shown in the Figures 1, 2 and 3 demonstrate these findings. In a recent study, K Yano using gene expression correlation analysis evaluated the genes associated with the risk of PCa and found the high-confidence risk genes [genes enriched in fetal prostate stem cells (PSCs) and ectoderm development genes, related to squamous metaplasia] are associated with an early stage of prostate carcinogenesis and the low-confidence genes may be involved in a later stages of carcinogenesis [19].
The Ingenuity Pathway Analysis revealed genes which were significant predictors of PCa aggressiveness were involved in biologic pathways “Gene Expression, Cell Cycle, Cancer”, “Inflammatory Response, Cell Death, Infection Mechanism” and “Cellular Assembly and Organization, Gene Expression, Cancer”. Our findings are similar to a recent study demonstrating a general activated process of signaling pathways, specifically shown in the cell cycle related biological processes in PCa namely “Gene Expression, Cellular Growth and Proliferation, Cellular Development, Cell Cycle, Cell Death, Cellular Movement, Cell-To-Cell Signaling and Interaction, Cell Signaling, Cellular Assembly and Organization, and Cellular Compromise” pathways [20].
The tumor suppressor gene p53 was found to be at the center hub of all significant pathways. The p53 plays a crucial role in maintaining genomic stability and tumor prevention. The p53 pathway is important in PCa development and progression. Our findings are in agreement with previous studies analyzing the role of common polymorphisms in the p53 pathway and found association with more aggressive PCa indicating that these findings could point to the relevance of this pathway in the development of aggressive PCa and may lead to a consideration of using p53 genetic variants as part of a multi-genic model for identifying high-risk subgroups which may benefit from intensive therapeutic strategies [21].
The miRNA expression analysis revealed loss of miR-34a. The miRNAs form a class of non-coding small RNA molecules considered to be key regulators of gene expression [15]. Their dysregulation has been shown to play important roles in cancer onset, progression and metastasis, and thus miRNAs represent a promising new class of cancer biomarkers. Down- and up-regulated miRNAs in PCa could provide potential biomarkers and/or therapeutic targets for the treatment of PCa aggressiveness. The loss of miR-34a found in this study was consistent with p53 function in PCa. Our findings are in direct agreement with the biological role of miR-34a. The miR-34a is a p53 target. Enforced expression of miR-34a in bulk or purified CD44(+) PCa cells inhibited clonogenic expansion, tumor regeneration, and metastasis [22], which supports the rationale for developing miR-34a as a novel therapeutic agent against prostate CSCs [16].
Using the Sequenom Mass-array, we identified mutation in MET oncogene in the present study. The OncoCarta assay simultaneously identifies both wild type and up to 3 different mutant alleles, has a high sensitivity exceeding that of traditional Sanger sequencing, which remains the gold standard for many genetic diagnostic approaches; and is highly concordant with Sanger sequencing, pyrosequencing, and allele-specific PCR [10,23].
The MET oncogene mutation identified in this study is a novel finding in PCa and has not been previously reported in PCa patients. The MET gene is the hepatocyte growth factor receptor MET, identified as part of the fusion oncogene, TPR-MET [24,25]. Point mutations in MET have been identified in renal papillary carcinoma [26-28]. Although 2 variants have been identified for this oncogene, T992I and R970C, it was the MET variant T992I (also designated T1010I), which was recently found in lung cancer cell lines, as well as individuals with lung, thyroid, renal, breast cancer, chronic lymphocytic leukemia (CLL), and lymphoma [29-35]. These variants have been characterized by Ba/F3 transformation and phospho-tyrosine immunoblots, among other assay systems, and were concluded to be transforming [31]. However, more recent study showed that MET mutations are not transforming in several types of malignancies including leukemia, colorectal cancer, thyroid cancer, endometrial cancer, melanoma, etc. [36]. Therefore, the malignancy-driven role of MET mutations is not universal but might be cancer-type specific. MET variant T992I was first identified by Schmidt and colleagues in 1999 [32] and thought to represent a rare polymorphism, owing to lack of disease segregation and failure to induce focus formation or phosphorylation in NIH3T3 cells. Lee and colleagues observed slightly faster tumor growth in nude mice with MET variants [30]. MET variants have a role in cytoskeletal function and have not been identified in samples from healthy individuals [31]. The mutation in MET oncogene seen in the present study may have clinical and translational relevance because small-molecule inhibitors for MET are emerging on the clinical cancer therapeutics horizon [37]. Therefore, the MET mutation identified in the present study, has the potential to be used a molecular signature for targeted therapies. Since anti-cancer therapeutic agents may have potentially toxic effects, before we can potentially use such novel targets for personalized tailored medicine, it is important to carefully validate our findings of MET allele, T992I. Future planned studies based on a larger retrospective cohort of PCa patients with survival data to derive a clinical algorithm to identify its role in prostate oncogenesis are warranted.
Our study has its limitations. This is a survey of global profiling of various genes, SNPs, mutation and miRNAs using small number of samples. More cases need to be analyzed to determine the clinical significance of our findings in prostate cancer. Furthermore, although 1200 miRNAs were analyzed only miR-34 was found to be lost significantly in prostate cancer.
Overall, we conclude that using novel, high throughput molecular techniques one would be able to comprehensively profile the genes and miRNAs from scant amounts of clinical tumor tissue samples from PCa patients. Whole genome gene expression analysis revealed several significant genes as predictors of tumor aggressiveness. The most significant predictor gene was TGIF1. The relevant target genes presumably increase the opportunities of tumor aggressiveness in PCa. Network analysis confirmed the protein alterations affecting biologic pathways upon interaction due to the deregulation of DNA. The p53 was found to be at the center hub of all significant pathways. It was also observed that many of the genes interacted with p53 gene and contributed to the modulation of cellular pathways involved in the PCa progression and aggressiveness. Loss of miR-34a was consistent with p53 function in PCa. The molecular signatures identified in the present study may have a significant clinical impact on improving diagnostic and predictive capabilities and in designing targeted therapies. Additionally it will help in understanding the biological pathways involved in the PCa tumor aggressiveness. Lastly, the MET oncogene, mutation in variant T992I, is a unique finding in PCa which may have clinical relevance hot spot mutation with promise as a potential molecular signature for targeted therapy for personalized medicine.
Acknowledgments
This work was presented at the Annual Meeting of the United States and Canadian Academy of Pathologists, Vancouver, BC, March 2012 and was awarded the Stowell-Orbison Award.
Disclosure/conflict of interest
The authors have no disclosure/conflict of interest.
References
- 1.Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, Harris PL, Haserlat SM, Supko JG, Haluska FG, Louis DN, Christiani DC, Settleman J, Haber DA. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350:2129–2139. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 2.Paez JG, Janne PA, Lee JC, Tracy S, Greulich H, Gabriel S, Herman P, Kaye FJ, Lindeman N, Boggon TJ, Naoki K, Sasaki H, Fujii Y, Eck MJ, Sellers WR, Johnson BE, Meyerson M. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304:1497–1500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- 3.Pao W, Miller V, Zakowski M, Doherty J, Politi K, Sarkaria I, Singh B, Heelan R, Rusch V, Fulton L, Mardis E, Kupfer D, Wilson R, Kris M, Varmus H. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci U S A. 2004;101:13306–13311. doi: 10.1073/pnas.0405220101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Heinrich MC, Corless CL, Demetri GD, Blanke CD, von MM, Joensuu H, McGreevey LS, Chen CJ, Van den Abbeele AD, Druker BJ, Kiese B, Eisenberg B, Roberts PJ, Singer S, Fletcher CD, Silberman S, Dimitrijevic S, Fletcher JA. Kinase mutations and imatinib response in patients with metastatic gastrointestinal stromal tumor. J. Clin. Oncol. 2003;21:4342–4349. doi: 10.1200/JCO.2003.04.190. [DOI] [PubMed] [Google Scholar]
- 5.Khambata-Ford S, Garrett CR, Meropol NJ, Basik M, Harbison CT, Wu S, Wong TW, Huang X, Takimoto CH, Godwin AK, Tan BR, Krishnamurthi SS, Burris HA, III, Poplin EA, Hidalgo M, Baselga J, Clark EA, Mauro DJ. Expression of epiregulin and amphiregulin and K-ras mutation status predict disease control in metastatic colorectal cancer patients treated with cetuximab. J. Clin. Oncol. 2007;25:3230–3237. doi: 10.1200/JCO.2006.10.5437. [DOI] [PubMed] [Google Scholar]
- 6.Solit DB, Garraway LA, Pratilas CA, Sawai A, Getz G, Basso A, Ye Q, Lobo JM, She Y, Osman I, Golub TR, Sebolt-Leopold J, Sellers WR, Rosen N. BRAF mutation predicts sensitivity to MEK inhibition. Nature. 2006;439:358–362. doi: 10.1038/nature04304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vegran F, Boidot R, Coudert B, Fumoleau P, Arnould L, Garnier J, Causeret S, Fraise J, Dembele D, Lizard-Nacol S. Gene expression profile and response to trastuzumab-docetaxel-based treatment in breast carcinoma. Br J Cancer. 2009;101:1357–1364. doi: 10.1038/sj.bjc.6605310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Forbes SA, Tang G, Bindal N, Bamford S, Dawson E, Cole C, Kok CY, Jia M, Ewing R, Menzies A, Teague JW, Stratton MR, Futreal PA. COSMIC (the Catalogue of Somatic Mutations in Cancer): a resource to investigate acquired mutations in human cancer. Nucleic Acids Res. 2010;38:D652–D657. doi: 10.1093/nar/gkp995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thomas RK, Baker AC, Debiasi RM, Winckler W, Laframboise T, Lin WM, Wang M, Feng W, Zander T, MacConaill L, Lee JC, Nicoletti R, Hatton C, Goyette M, Girard L, Majmudar K, Ziaugra L, Wong KK, Gabriel S, Beroukhim R, Peyton M, Barretina J, Dutt A, Emery C, Greulich H, Shah K, Sasaki H, Gazdar A, Minna J, Armstrong SA, Mellinghoff IK, Hodi FS, Dranoff G, Mischel PS, Cloughesy TF, Nelson SF, Liau LM, Mertz K, Rubin MA, Moch H, Loda M, Catalona W, Fletcher J, Signoretti S, Kaye F, Anderson KC, Demetri GD, Dummer R, Wagner S, Herlyn M, Sellers WR, Meyerson M, Garraway LA. High-throughput oncogene mutation profiling in human cancer. Nat Genet. 2007;39:347–351. doi: 10.1038/ng1975. [DOI] [PubMed] [Google Scholar]
- 10.Vivante A, Amariglio N, Koren-Michowitz M, Ashur-Fabian O, Nagler A, Rechavi G, Cohen Y. High-throughput, sensitive and quantitative assay for the detection of BCR-ABL kinase domain mutations. Leukemia. 2007;21:1318–1321. doi: 10.1038/sj.leu.2404635. [DOI] [PubMed] [Google Scholar]
- 11.van PM, Dierssen JW, Stanssens P, van ER, Cleton-Jansen AM, van WT, Morreau H. Mass spectrometry-based loss of heterozygosity analysis of single-nucleotide polymorphism loci in paraffin embedded tumors using the MassEXTEND assay: single-nucleotide polymorphism loss of heterozygosity analysis of the protein tyrosine phosphatase receptor type J in familial colorectal cancer. J Mol Diagn. 2005;7:623–630. doi: 10.1016/S1525-1578(10)60596-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sethi S. Molecular diagnosis of respiratory tract infection in acute exacerbations of chronic obstructive pulmonary disease. Clin Infect Dis. 2011;52(Suppl 4):S290–S295. doi: 10.1093/cid/cir044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ali-Fehmi R, Semaan A, Sethi S, Arabi H, Bandyopadhyay S, Hussein YR, Diamond MP, Saed G, Morris RT, Munkarah AR. Molecular typing of epithelial ovarian carcinomas using inflammatory markers. Cancer. 2011;117:301–309. doi: 10.1002/cncr.25588. [DOI] [PubMed] [Google Scholar]
- 14.Sethi S, Macoska J, Chen W, Sarkar FH. Molecular signature of epithelial-mesenchymal transition (EMT) in human prostate cancer bone metastasis. Am J Transl Res. 2010;3:90–99. [PMC free article] [PubMed] [Google Scholar]
- 15.Hassan O, Ahmad A, Sethi S, Sarkar FH. Recent updates on the role of microRNAs in prostate cancer. J Hematol Oncol. 2012;5:9. doi: 10.1186/1756-8722-5-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kong D, Heath E, Chen W, Cher ML, Powell I, Heilbrun L, Li Y, Ali S, Sethi S, Hassan O, Hwang C, Gupta N, Chitale D, Sakr WA, Menon M, Sarkar FH. Loss of let-7 up-regulates EZH2 in prostate cancer consistent with the acquisition of cancer stem cell signatures that are attenuated by BR-DIM. PLoS One. 2012;7:e33729. doi: 10.1371/journal.pone.0033729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kashat M, Azzouz L, Sarkar SH, Kong D, Li Y, Sarkar FH. Inactivation of AR and Notch-1 signaling by miR-34a attenuates prostate cancer aggressiveness. Am J Transl Res. 2012;4:432–442. [PMC free article] [PubMed] [Google Scholar]
- 18.Kong D, Heath E, Chen W, Cher M, Powell I, Heilbrun L, Li Y, Ali S, Sethi S, Hassan O, Hwang C, Gupta N, Chitale D, Sakr WA, Menon M, Sarkar FH. Epigenetic silencing of miR-34a in human prostate cancer cells and tumor tissue specimens can be reversed by BR-DIM treatment. Am J Transl Res. 2012;4:14–23. [PMC free article] [PubMed] [Google Scholar]
- 19.Yano K. Gene expression correlation analysis predicts involvement of high- and low-confidence risk genes in different stages of prostate carcinogenesis. Prostate. 2010;70:1746–1759. doi: 10.1002/pros.21210. [DOI] [PubMed] [Google Scholar]
- 20.Chen JH, He HC, Jiang FN, Militar J, Ran PY, Qin GQ, Cai C, Chen XB, Zhao J, Mo ZY, Chen YR, Zhu JG, Liu X, Zhong WD. Analysis of the specific pathways and networks of prostate cancer for gene expression profiles in the Chinese population. Med Oncol. 2012 Sep;29:1972–84. doi: 10.1007/s12032-011-0088-5. [DOI] [PubMed] [Google Scholar]
- 21.Sun T, Lee GS, Oh WK, Pomerantz M, Yang M, Xie W, Freedman ML, Kantoff PW. Single-nucleotide polymorphisms in p53 pathway and aggressiveness of prostate cancer in a Caucasian population. Clin Cancer Res. 2010;16:5244–5251. doi: 10.1158/1078-0432.CCR-10-1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu C, Kelnar K, Liu B, Chen X, Calhoun-Davis T, Li H, Patrawala L, Yan H, Jeter C, Honorio S, Wiggins JF, Bader AG, Fagin R, Brown D, Tang DG. The microRNA miR-34a inhibits prostate cancer stem cells and metastasis by directly repressing CD44. Nat Med. 2011;17:211–215. doi: 10.1038/nm.2284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.van PM, Dierssen JW, Stanssens P, van ER, Cleton-Jansen AM, van WT, Morreau H. Mass spectrometry-based loss of heterozygosity analysis of single-nucleotide polymorphism loci in paraffin embedded tumors using the MassEXTEND assay: single-nucleotide polymorphism loss of heterozygosity analysis of the protein tyrosine phosphatase receptor type J in familial colorectal cancer. J Mol Diagn. 2005;7:623–630. doi: 10.1016/S1525-1578(10)60596-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cooper CS, Park M, Blair DG, Tainsky MA, Huebner K, Croce CM, Vande Woude GF. Molecular cloning of a new transforming gene from a chemically transformed human cell line. Nature. 1984;311:29–33. doi: 10.1038/311029a0. [DOI] [PubMed] [Google Scholar]
- 25.Park M, Dean M, Kaul K, Braun MJ, Gonda MA, Vande WG. Sequence of MET protooncogene cDNA has features characteristic of the tyrosine kinase family of growth-factor receptors. Proc Natl Acad Sci U S A. 1987;84:6379–6383. doi: 10.1073/pnas.84.18.6379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jeffers M, Schmidt L, Nakaigawa N, Webb CP, Weirich G, Kishida T, Zbar B, Vande Woude GF. Activating mutations for the met tyrosine kinase receptor in human cancer. Proc Natl Acad Sci U S A. 1997;94:11445–11450. doi: 10.1073/pnas.94.21.11445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schmidt L, Duh FM, Chen F, Kishida T, Glenn G, Choyke P, Scherer SW, Zhuang Z, Lubensky I, Dean M, Allikmets R, Chidambaram A, Bergerheim UR, Feltis JT, Casadevall C, Zamarron A, Bernues M, Richard S, Lips CJ, Walther MM, Tsui LC, Geil L, Orcutt ML, Stackhouse T, Lipan J, Slife L, Brauch H, Decker J, Niehans G, Hughson MD, Moch H, Storkel S, Lerman MI, Linehan WM, Zbar B. Germline and somatic mutations in the tyrosine kinase domain of the MET proto-oncogene in papillary renal carcinomas. Nat Genet. 1997;16:68–73. doi: 10.1038/ng0597-68. [DOI] [PubMed] [Google Scholar]
- 28.Schmidt L, Junker K, Weirich G, Glenn G, Choyke P, Lubensky I, Zhuang Z, Jeffers M, Vande WG, Neumann H, Walther M, Linehan WM, Zbar B. Two North American families with hereditary papillary renal carcinoma and identical novel mutations in the MET proto-oncogene. Cancer Res. 1998;58:1719–1722. [PubMed] [Google Scholar]
- 29.Brown JR, Levine RL, Thompson C, Basile G, Gilliland DG, Freedman AS. Systematic genomic screen for tyrosine kinase mutations in CLL. Leukemia. 2008;22:1966–1969. doi: 10.1038/leu.2008.222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee JH, Han SU, Cho H, Jennings B, Gerrard B, Dean M, Schmidt L, Zbar B, Vande Woude GF. A novel germ line juxtamembrane Met mutation in human gastric cancer. Oncogene. 2000;19:4947–4953. doi: 10.1038/sj.onc.1203874. [DOI] [PubMed] [Google Scholar]
- 31.Ma PC, Kijima T, Maulik G, Fox EA, Sattler M, Griffin JD, Johnson BE, Salgia R. c-MET mutational analysis in small cell lung cancer: novel juxtamembrane domain mutations regulating cytoskeletal functions. Cancer Res. 2003;63:6272–6281. [PubMed] [Google Scholar]
- 32.Schmidt L, Junker K, Nakaigawa N, Kinjerski T, Weirich G, Miller M, Lubensky I, Neumann HP, Brauch H, Decker J, Vocke C, Brown JA, Jenkins R, Richard S, Bergerheim U, Gerrard B, Dean M, Linehan WM, Zbar B. Novel mutations of the MET proto-oncogene in papillary renal carcinomas. Oncogene. 1999;18:2343–2350. doi: 10.1038/sj.onc.1202547. [DOI] [PubMed] [Google Scholar]
- 33.Tengs T, Lee JC, Paez JG, Zhao X, Laframboise T, Giannoukos G, Thomas RK. A transforming MET mutation discovered in non-small cell lung cancer using microarray-based resequencing. Cancer Lett. 2006;239:227–233. doi: 10.1016/j.canlet.2005.08.007. [DOI] [PubMed] [Google Scholar]
- 34.Tjin EP, Groen RW, Vogelzang I, Derksen PW, Klok MD, Meijer HP, van ES, Pals ST, Spaargaren M. Functional analysis of HGF/MET signaling and aberrant HGF-activator expression in diffuse large B-cell lymphoma. Blood. 2006;107:760–768. doi: 10.1182/blood-2005-05-1929. [DOI] [PubMed] [Google Scholar]
- 35.Wasenius VM, Hemmer S, Karjalainen-Lindsberg ML, Nupponen NN, Franssila K, Joensuu H. MET receptor tyrosine kinase sequence alterations in differentiated thyroid carcinoma. Am J Surg Pathol. 2005;29:544–549. doi: 10.1097/01.pas.0000156103.37756.e2. [DOI] [PubMed] [Google Scholar]
- 36.Tyner JW, Fletcher LB, Wang EQ, Yang WF, Rutenberg-Schoenberg ML, Beadling C, Mori M, Heinrich MC, Deininger MW, Druker BJ, Loriaux MM. MET receptor sequence variants R970C and T992I lack transforming capacity. Cancer Res. 2010;70:6233–6237. doi: 10.1158/0008-5472.CAN-10-0429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Comoglio PM, Giordano S, Trusolino L. Drug development of MET inhibitors: targeting oncogene addiction and expedience. Nat Rev Drug Discov. 2008;7:504–516. doi: 10.1038/nrd2530. [DOI] [PubMed] [Google Scholar]