

Abstract
The role of pro-inflammatory cytokine production in the pathogenesis of malaria is well established, but the identification of the parasite products that initiate inflammation is not complete. Hemozoin is a crystalline metabolite of hemoglobin digestion that is released during malaria infection. In the present study, we characterized the immunostimulatory activity of pure synthetic hemozoin (sHz) in vitro and in vivo. Stimulation of naïve murine macrophages with sHz results in the MyD88-independent activation of NF-κB and ERK, and the release of the chemokine MCP-1; these responses are augmented by IFN-γ. In macrophages pre-stimulated with IFN-γ, sHz also results in a MyD88-dependent release of TNF-α. Endothelial cells, which encounter hemozoin after schizont rupture, respond to sHz by releasing IL-6 and the chemokines MCP-1 and IL-8. In vivo, the introduction of sHz into the peritoneal cavity produces an inflammatory response characterized by neutrophil recruitment and the production of MCP-1, KC, IL-6, IL-1α and IL-1β. MCP-1 and KC are produced independently of MyD88, TLR 2/4 and 9, and components of the inflammasome, however neutrophil recruitment, the localized production of IL-1β, and the increase in circulating IL-6 require MyD88 signaling, the IL-1R pathway and the inflammasome components ICE, ASC and NALP3. Of note, inflammasome activation by sHz is reduced by allopurinol, which is an inhibitor of uric acid synthesis. This data suggests that uric acid is released during malaria infection and may serve to augment the initial host response to hemozoin via activation of the NALP3 inflammasome.
Keywords: Chemokine, Cytokine, Inflammasome, Plasmodium, TLR9, NALP3
Introduction
Malaria has profound worldwide impact, infecting millions of people annually and resulting in at least 2 million deaths per year (1, 2). The highest mortality rates occur in the children of Sub-Saharan Africa infected with Plasmodium falciparum, where severe anemia and cerebral malaria are common sequelae of infection (3). Unifying the disparate sequelae of P. falciparum infection is the understanding that the host immune response is important not only for parasite control, but also much of the pathology of malaria (4, 5). Indeed, the excessive production of pro-inflammatory cytokines such as TNF-α (6), IFN-γ (7), lymphotoxin (8) and MIF (9) have been implicated in many manifestations of the disease including the neurologic dysfunction of cerebral malaria and the dyserythropoiesis of severe malarial anemia.
Despite progress in understanding disease pathogenesis using mouse models of malarial infection, the host and parasite factors that mediate early cytokine production are still poorly understood (10). As much of the parasite life cycle occurs within infected erythrocytes and symptoms of pro-inflammatory excess such as fever and rigors accompany schizont rupture, investigators have focused on parasite products released during schizogeny (4). One such product is hemozoin, a polymer of heme that is produced as a result of the parasite’s digestion of host hemoglobin within the erythrocyte (11, 12). Hemozoin is released by the rupture of parasitized red cells, reaches high concentrations in the circulation, and is actively phagocytized by circulating monocytes and neutrophils as well as by tissue resident macrophages of the liver and spleen (13, 14).
The role of hemozoin in the activation of cytokine responses by the innate immune system is controversial. Several investigators have found that both parasite-derived hemozoin and synthetically produced hemozoin (sHz) activate the release of various pro-inflammatory mediators including TNF-α, IL-1β, and the chemokines MIP-1α and MIP-1β by monocytes/macrophages (15, 16). Other investigators however, have suggested that hemozoin plays an inhibitory role on the maturation and function of immature dendritic cells and may be responsible for certain features of the immune suppression that is characteristic of malarial infection (17, 18). The injection of hemozoin into mice nevertheless results in an acute pro-inflammatory response with the strong recruitment of leukocytes and the release of pro-inflammatory chemokines and cytokines (19).
Innate immune receptors mediate many aspects of the pro-inflammatory response to parasite invasion (20), and the Toll-like receptor (TLR) system is considered to be a likely candidate for mediating the host response to hemozoin. Indeed, the TLR pathway has been shown to be essential for the inflammatory response to parasite lysates (21, 22). A specific role for TLRs in the host response to hemozoin was first defined by Coban et al., who demonstrated that recognition of both parasite purified and synthetic hemozoin required TLR9 (23), which has also previously been shown to recognize the unmethylated CpG motifs in microbial DNA (24).
A recent study by Parroche et al. has questioned the immunomodulatory activity of hemozoin via TLR9 (25). The authors utilized a highly pure preparation of synthetic hemozoin and showed that it had negligible pro-inflammatory activity on FLT-3 ligand derived dendritic cells (FL-DCs). It was proposed instead that hemozoin binds and loads malarial DNA into the endosome for recognition via TLR9. A role for hemozoin in modulating the responses of other innate immune cells nevertheless remains to be clarified, and the importance of this issue was highlighted by Dewalick et al. who found that deletion of plasmacytoid dendritic cells had no effect on the development of cerebral malaria, but that conventional dendritic cells were important mediators of this disease (26). Furthermore, both murine and human studies suggest that macrophages are essential mediators of cerebral malaria and key cells in the production of the inflammatory cytokines such as TNF-α and lymphotoxin that are important in cerebral malaria pathogenesis (27).
In this study, we produced sHz from FPLC purified hemin to eliminate possible contamination with pro-inflammatory molecules. We noted that while stimulation with sHz did not result in the production of IL-6 and TNF-α by naïve dendritic cells or macrophages, chemokines, most notably MCP-1 are produced in vitro by macrophages stimulated with sHz. More importantly, in vivo, the peritoneal injection of sHz results in a strong inflammatory response characterized by recruitment of neutrophils and the production of inflammatory cytokines and chemokines including KC, MCP-1, IL-1β and IL-6. This inflammatory response appears dependent on intact IL-1R signaling through MyD88 and is augmented by the inflammasome components ASC, ICE and NALP3 via the release of uric acid.
Materials and Methods
Reagents and Mice
The TLR agonist CpG DNA ODN 1826 and Poly (I:C) was purchased from Invivogen. Recombinant IFN-γ and FLT-3 ligand, GM-CSF were from Peprotech, uric acid, uricase and allopurinol were from Sigma Aldrich. Monosodium urate crystals were prepared as described previously (28). ELISA kits for the mouse cytokines TNF-α, MCP-1, IL-1β and IL-6 were purchased from eBioscience, and the ELISA kit for mouse KC was purchased from R&D. C57BL/6 mice were obtained from Jackson Labs. MyD88−/−, TRIF−/−, TLR2/4−/− and TLR9−/− mice were generated previously (29–31) and backcrossed with C57BL/6 mice for 9, 7, 9 and 9 generations respectively. The ICE−/− and the IPAF−/− mice were provided by Richard Flavell (Yale University), and the ASC−/− and the NALP3−/− mice were from Lexicon Genetics Inc. These mice were housed in pathogen-free conditions according to institutional protocols of Yale University.
Synthesis of Pure Hemozoin
Synthetic hemozoin (sHz) was prepared as previously described (15) except that FPLC purified hemin-chloride (Fluka) was used for synthesis instead of the less pure porcine hematin from Sigma. Briefly, 40 mg hemin was solubilized in 8 ml of 0.1M NaOH and then treated with 49 mmol of acetic acid. This suspension was heated overnight at 70°C and then washed with 0.1M NaHCO3, ddH20, and 70% EtOH. The hemozoin was resuspended at the indicated concentrations in appropriate medium and sonicated in a bath sonicator for 1 min prior to use. The final preparation of hemozoin contained less that 0.005EU/mL of LPS as measured by the Limulus amoebocyte assay.
Purification of Malarial DNA
Plasmodium falciparum strain FCR3 was cultivated in leukocyte-free erythrocytes of blood group A+ (American Red Cross) at 5% hematocrit in RPMI 1640 medium (Invitrogen) at 37°C in a humidified gas mixture of 90%N2, 5% CO2, and 5%O2. Culture medium was supplemented with 25 mM HEPES, sodium bicarbonate (2 g/L), gentamicin (1μg/ml), hypoxanthine (92 μM) and human serum (10%), type AB. Mixed late stage cultures (trophozoites and schizonts at 2 to 10% parasitemia) were incubated with 0.05% saponin for 5 min at 25°C followed by centrifugation at 2500 × g for 10 min. The resulting parasite pellet was washed twice by centrifugation in 1xPBS and stored at −70°C until DNA purification by Easy-DNA kit (Invitrogen) according to the manufacturer’s instructions.
Cells
FL-DCs were generated by culturing bone marrow in 100 ng/ml of FLT-3 ligand for 8 days in DMEM/10% FCS and penicillin/streptomycin (23). cDCs were generated by culturing bone marrow in 10ng/ml GM-CSF for 8 days in DMEM/10%FCS. Bone marrow macrophages were generated by culturing bone marrow in 30% L929 supernatant and 20% FCS for five days. Peritoneal macrophages were isolated after washing the peritoneal cavity with 10 ml of cold PBS and cells were plated in 24 well plates in DMEM 10% FCS with penicillin/streptomycin. Human umbilical vein endothelial cells (HUVECs) were cultured on gelatin-coated plates in M199 media supplemented with 20% FCS, L-glutamine, penicillin/streptomycin and endothelial cell growth factor (ECGF). THP-1 cells were stimulated with 0.5 μM PMA in RPMI 10% FCS overnight prior to use (32).
In vivo Cell Recruitment and Cytokine Production
Synthetic hemozoin (2.5 mg) in 200 μl PBS or PBS alone was injected i.p. into mice. After 12 hrs, the peritoneal cavity was washed with 10 ml of cold PBS and cells were counted. 100,000 cells were cytospun and stained with the DiffQuick staining reagent according to the manufacturer’s instructions (Protocol). The percentage of neutrophils was determined by enumeration of cells with the characteristic, multi-lobed nuclei of neutrophils using a Nikon Microphot-FXA microscope at (40X). For cytokine analysis, 2.5 mg of synthetic hemozoin was injected and the peritoneal cavity was washed with 1 ml of cold PBS at the indicated time. The concentration of multiple cytokines was analyzed using the Beadlyte cytokine multiplex technology (Upstate) analyzed on the luminex platform (Biorad). In subsequent experiments, the concentration of MCP-1, IL-1β, IL-6 and KC in the lavage fluid were quantified by ELISA (eBioscience for MCP-1, IL-6 and IL-1β and R&D for KC).
Flow Cytometry Analysis
At determined time points after hemozoin injection, peritoneal lavage was performed with 1ml PBS. The total recovered cells were counted and 1×106 cells were stained with FITC-conjugated Gr-1 neutrophil surface marker (eBioscience). Flow cytometry with FACSCalibur was used to determine the percentage of Gr-1 positive cells.
Western Blotting
Mice were injected i.p. with 2 ml of 4% sterile thioglycolate. Five days after injection, peritoneal lavage cells were plated at 2.5×106 cells/well in 6 well plates in DMEM media supplemented with 10% FCS. After 4 hours, the adherent macrophages were washed 3 times with PBS and the media was replaced with DMEM/low glucose and 0.1% FCS for 16 hrs. Cells then were stimulated with 100 μg/ml sHz and at the indicated times and harvested in RIPA lysis buffer. Equal amounts of protein were electrophoresed on SDS-PAGE gels and Western blotted with anti-phospho-ERK antibody (Santa Cruz Biotechnology) or anti-phospho p38, anti-phospho JNK, or anti-Iκ-B antibodies (Cell Signaling). The specific bands were visualized using the ECL chemiluminescence reagent kit (Amersham Biosciences). Membranes were stripped using Blot Restore (Chemicon International) according to the manufacture’s instructions and reprobed for total ERK (Santa-Cruz), p38, JNK (Cell Signaling) and β-actin (Sigma) using the above protocol.
Luciferase Assays
RAW264.7 murine monocytes were stably transfected with a plasmid containing the luciferase gene linked to 3 tandem NF-κB promoters. Cells were stimulated for 12 hrs and then harvested in Passive Lysis Buffer (Promega) according to the manufacturer’s instructions. Lysates were mixed with the Luciferase substrate LAR (Promega) and analyzed with a Luminometer according to the manufacturer’s instructions.
Results
Synthetic hemozoin (sHz) does not activate naïve dendritic cells or macrophages, but enhances the response to malarial DNA
In order to study the role of hemozoin free of possible contaminants, we employed for these studies only sHz produced from FPLC-purified hemin chloride. To ensure that our preparation of sHz was comparable in activity to that used by Parroche et al. (25) we first stimulated FLT3 ligand derived mouse plasmacytoid DCs (FL-DCs) with either sHz or unmethylated CpG DNA following their published procedure. In agreement with their report, sHz did not induce the production of either IL-6 or TNF-α from FL-DCs when studied under the same experimental conditions in which stimulation with CpG DNA induced a strong cytokine response (Fig. 1A). Because this observation may have been specific to FLT-3 ligand DCs, we also stimulated bone marrow-derived, GM-CSF matured conventional DCs (Fig 1B), bone marrow-derived macrophages (Fig 1C), and thioglycolate-elicited peritoneal macrophages (Fig 1D) with hemozoin and measured the IL-6 and TNF-α responses by these cells. As can be seen in the corresponding figures, pure hemozoin does not result in the production of either IL-6 or TNF-α by any of these cells types, while control stimulation with various TLR ligands did result in production of these cytokines by all cell types tested. Parroche et al. suggested that the stimulatory activity of non-synthetic preparations of hemozoin (i.e. purified from Plasmodium) was due to the presence of malarial DNA. In support of this hypothesis, we found that the addition of malarial DNA (mDNA) together with sHz to FL-DCs results in a TNF-α response that is augmented over that seen with either mDNA or hemozoin alone (Fig. 2A). Of note, we also stimulated peritoneal-derived macrophages by adding sHz together with the synthetic TLR ligands, CpG DNA and Poly (I:C). We tested these model ligands instead of malaria DNA to ensure a lack of contamination by parasite products, which may occur with malarial DNA preparations (25). In accordance with the results observed with malarial DNA (Fig. 2A), sHz significantly augments the release of TNF-α by macrophages when added together with the nucleic acid TLR ligands CpG DNA (Fig. 2B) and Poly (I:C) (Fig. 2C).
Figure 1. Naïve DCs or macrophages are not stimulated by pure sHz.
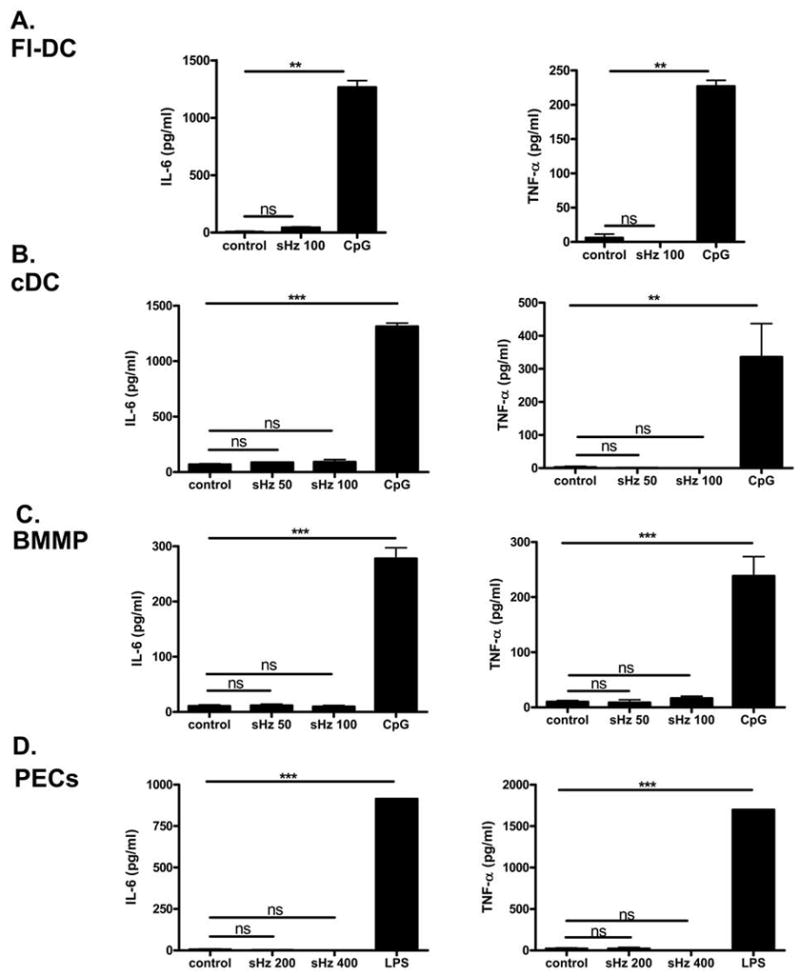
(A) FL-DCs (B) bone marrow derived conventional DCs (C) bone marrow derived macrophages or (D) peritoneal derived macrophages were stimulated with either control media, the indicated amount of synthetic hemozoin (sHz) ranging from 50 to 400 μg/ml or 2.5 μM CpG or 10ng/ml LPS as labeled for 24 hrs and the production of IL-6 and TNF-α was assessed by ELISA. In all cell types tested sHz did not significantly increase the production of IL-6 or TNF-α above unstimulated controls. *P<0.05, **P<0.01, ***P<0.001. Each figure is representative of three independent experiments.
Figure 2. sHz augments the inflammatory response to malarial DNA or TLR ligands.
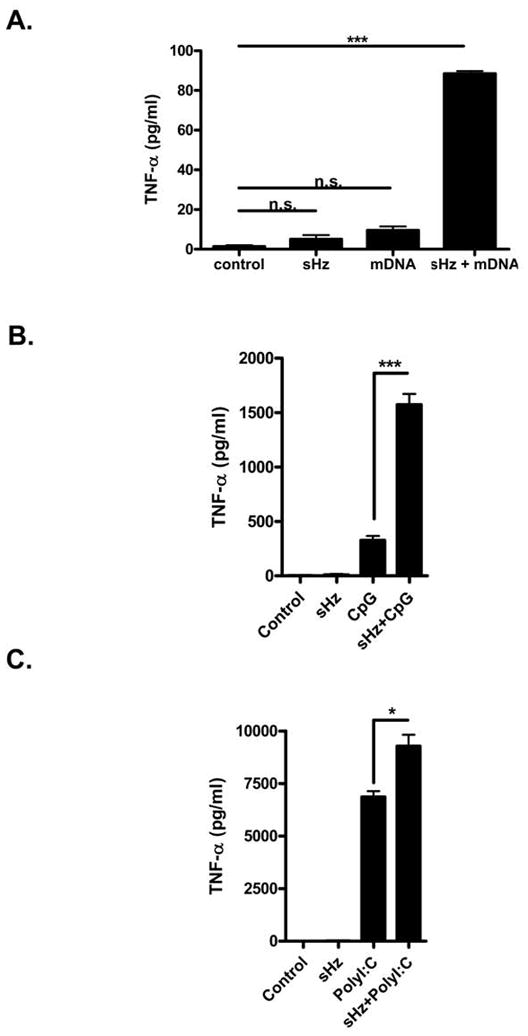
(A) PECs were stimulated with either control media, 100 μg/ml sHz, 5 μg/ml of malarial DNA (mDNA) alone, or both sHz and mDNA for 24 hrs and the production of TNF-α was assessed by ELISA. (B) Peritoneal macrophages from WT mice were stimulated with either 100 μg/ml sHz, 2.5μM CpG DNA or both together for 24 hrs and the production of TNF-α was measured by ELISA. (C) Peritoneal macrophages from WT mice were stimulated with either 100 μg/ml sHz, 25 μg/ml Poly (I:C) or both together for 24 hrs and the production of TNF-α was measured by ELISA. *P<0.05, **P<0.01, ***P<0.001. Each figure is representative of three independent experiments.
Macrophages produce the chemokine MCP-1 when stimulated with hemozoin in vitro
Although stimulation of various dendritic cells and macrophages did not result in production of IL-6 and TNF-α, we did observe the release of the chemokine MCP-1 when macrophages were stimulated with sHz. RAW 264.7 macrophages stimulated with sHz released a large quantity of MCP-1 (Fig. 3A). Because both IL-6 and TNF-α are typically released by activation of the TLR system, while the chemokine MCP-1 may be released under a large variety of stimuli besides TLR activation (33), we hypothesized that sHz may activate MCP-1 production by non-TLR pathways. To test this hypothesis, we isolated peritoneal macrophages from wild-type (WT) and MyD88−/− mice, stimulated them with sHz, and observed a significant, MyD88-independent production of the chemokine MCP-1 from these cells (Fig. 3B). This increase in MCP-1 production was augmented when macrophages were activated with IFN-γ and still occurred in WT and MyD88−/− macrophages. Of note, while naïve macrophages stimulated with sHz did not release TNF-α, we did note that sHz did result in a small but significant TNF-α release that occurred only when macrophages were activated with IFN-γ (Fig. 3C). This TNF-α release occurred in a TLR9 independent but MyD88-dependent manner.
Figure 3. Peritoneal macrophages release the chemokine MCP-1 upon sHz stimulation and TNF-α upon stimulation with both IFN-γ and sHz.
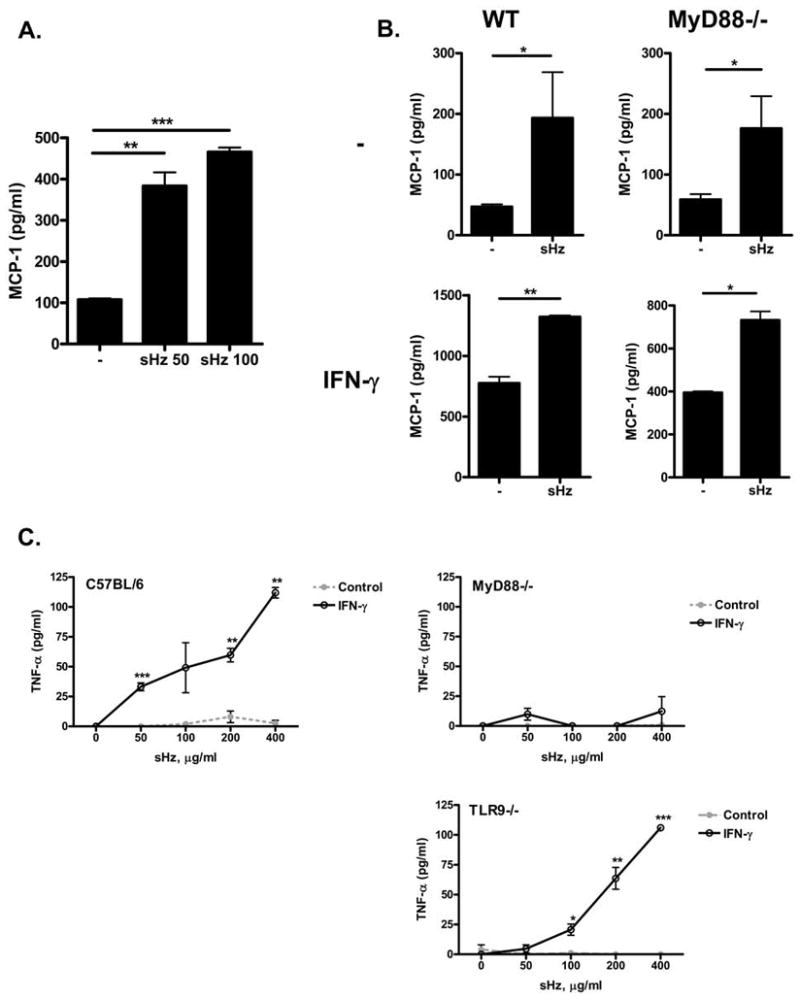
(A) RAW 264.7 macrophages were stimulated with increasing concentrations of sHz for 24 hours and the levels of MCP-1 released in the supernatant were measured by ELISA. (B) Naïve or IFN-γ stimulated peritoneal macrophages from WT or MyD88−/− C57BL/6 mice were stimulated with either 50 μg/ml of sHz or control media (control) and the levels of MCP-1 in the supernatant was analyzed by ELISA. (C) Peritoneal macrophages from WT, MyD88−/−, or TLR9−/− C57BL/6 mice were stimulated with increasing concentrations of sHz in the presence (black line) or absence (gray dotted line) of IFN-γ and the levels of TNF-α in the supernatant after 12 hrs were assessed by ELISA. All values are compared with control media: * P<0.05, **P<0.01, ***P<0.001. Each figure is representative of at least 3 independent experiments.
Hemozoin activates pro-inflammatory responses by endothelial cells
To further delineate the innate cells that may contribute to the inflammatory response to hemozoin, we assessed the in vitro responses of endothelial cells to sHz because these cells are among the first to be exposed to hemozoin after schizont rupture (34). We found that in vitro cultures of human umbilical vein endothelial cells (HUVECs) significantly up-regulated expression of the pro-inflammatory cytokines IL-6, IL-8 and MCP-1 in response to sHz (Fig. 4A–C). This data further supports the concept that while FL-DCs may not participate in hemozoin recognition, innate immune cells such as macrophages and endothelial cells nevertheless contribute to the host inflammatory response induced by this malarial product.
Figure 4. Human endothelial cells produce cytokines in response to sHz stimulation.
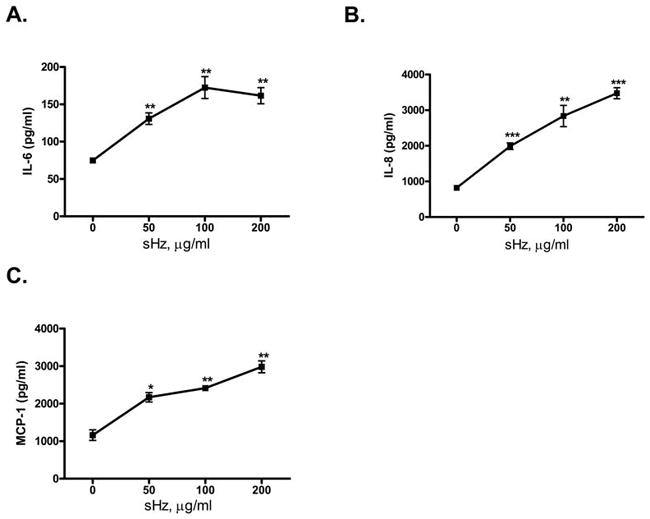
HUVECs were stimulated for 24 hrs with increasing concentrations of sHz and the levels of IL-6 (A), IL-8 (B), MCP-1 (C) in the supernatant were measured by ELISA. *P<0.05, **P<0.01, ***P<0.001 for control versus sHz stimulation. Each figure is representative of at least 4 separate experiments.
Macrophages activate ERK and NF-κB signaling independently of MyD88
To determine if MyD88 is important in the initial recognition of hemozoin, or is more important in modulating downstream stimuli such as IL-1β and IFN-γ signaling, we studied the early response of the mitogen activated protein kinase (MAPK) and NF-κB pathways in sHz treated macrophages. We stimulated peritoneal elicited macrophages from WT mice with sHz and assessed activation of the MAPK (ERK, JNK, p38) and NF-κB signaling pathways. As assessed by Western blotting for specific downstream MAPK phosphorylated products, the ERK1/2 MAPK was activated within 15 minutes of sHz addition with the response peaking at 30 minutes and then decaying after 60 minutes (Fig. 5A). We did not observe an activation effect on the p38 MAPK in WT macrophages and did note that the phosphorylation of JNK was decreased but still observable after sHz stimulation (Fig. 5A). Supporting the hypothesis that hemozoin recognition occurs independently of MyD88 and TLR9, we noted that ERK1/2 phosphorylation is upregulated in MyD88−/− and TLR9−/− macrophages in response to sHz with exactly the same kinetics as in WT macrophages (Fig. 5B).
Figure 5. sHz activates ERK and NF-κB signaling pathways independent of MyD88.

(A) Thioglycolate elicited peritoneal macrophages from WT C57BL/6 mice were stimulated with 100 μg of sHz and the activation of the ERK1/2, JNK and p38 MAPK pathways was assessed by western blotting for phosphorylation of ERK1/2, JNK and p38 (upper panels) compared to total protein controls (lower panels). (B) Thioglycolate elicited peritoneal macrophages from MyD88−/− and TLR9−/− C57BL/6 mice were stimulated with 100 μg of sHz and the activation of the ERK1/2 pathway was assessed by western blotting for phosphorylation of ERK1/2 (upper panels) compared to total ERK protein (lower panels). (C) RAW 264.7 cells with a stable NF-κB luciferase promoter were stimulated with 100 ug/ml of sHz in the presence or absence of IFN-γ for 12 hrs. For control versus sHz stimulation: **P<0.01 and ***P<0.001. (D) WT or MyD88−/− peritoneal macrophages were stimulated for the indicated times and Iκ-B degradation, a marker for NF-κB activation was assessed by western blotting. Each blot was stripped and restained with β-actin as a loading control. Each figure is representative of at least three independent experiments.
As a sensitive assay for NF-κB activation, we used a RAW264.7 macrophage cell line that had been stably transfected with a NF-κB sensitive luciferase reporter (35). The addition of sHz to these cells produced an activation of the NF-κB promoter, and this response was further enhanced by IFN-γ (Fig. 5C). We also assessed the activation of the NF-κB pathway in MyD88 deficient cells. WT or MyD88−/− peritoneal macrophages were stimulated with sHz and the degradation of Iκ-B, which is an essential step for canonical NF- κB activation (36), was assessed by Western blotting. Both WT and MyD88−/− macrophages rapidly degraded Iκ-B in response to sHz, indicating that the initial steps of NF- κB signaling occur independently of MyD88 (Fig. 5D).
Synthetic hemozoin is an inflammatory stimulus in vivo
Given the diversity of cell types that participate in innate immune responses and the chemokine response observed in vitro, we reasoned that sHz will initiate inflammation in vivo. To test this hypothesis, we examined the recruitment of inflammatory cells into the peritoneal cavity of mice after the direct injection of sHz. The intraperitoneal injection into WT C57BL/6 mice of 2.5 mg of sHz, which is an amount comparable to that released in malaria-infected hosts (13), results in the recruitment of 3-fold more cells than after a control injection of PBS alone (Fig. 6A). The majority of recruited cells after visualization were neutrophils, which increased in percentage from <5% to >60% after sHz injection (Figs. 6B). This observation was confirmed by FACs analysis of peritoneal cells, which demonstrated that the percentage of neutrophils identified as being Gr-1 positive increased with sHz injection from <14% to >60% (Fig. 6C). When the resultant increase in neutrophils was quantified directly using the visualization method, we noted a large and statistically significant increase in the number of neutrophils 6 hours after the injection of sHz (Fig. 6D).
Figure 6. Intraperitoneal injection of sHz mediates inflammatory recruitment of neutrophils.

(A) sHz (2.5 mg) in PBS or PBS alone was injected i.p. into WT C57BL/6 mice. 12 hrs later, peritoneal cells were harvested in 10 ml of cold PBS and counted. For PBS vs. sHz injection: ***P<0.001. (B) 12 hrs after injection of PBS or 2.5 mg sHz in PBS into WT mice, peritoneal cells were harvested, stained and visualized at 40X magnification. (C) FAC analysis of the recruited cells demonstrates an increase in the percentage of GR-1 positive cells (a marker for neutrophils) from 13% to approximately 60% at 6 and 12 hrs after the injection of sHz into the peritoneal cavity of mice. (D) Total number of recruited neutrophils determined after sHz intraperitoneal injection into WT mice (n=5). For sHz injection into WT mice versus MyD88−/− mice: ***P<0.001. Each figure is representative of at least three independent experiments.
Intraperitoneal injection of synthetic hemozoin results in local and systemic inflammatory responses
Neutrophil recruitment upon the intraperitoneal injection of sHz suggests that hemozoin activates inflammatory signaling. To better identify the inflammatory pathways activated, we injected sHz into the peritoneal cavities of WT C57/BL6 mice and harvested both serum and peritoneal fluid for cytokine analysis. Figure 7A demonstrates the plasma cytokine response of 8 selected cytokines and chemokines 2 hours after the peritoneal injection of sHz (grey bars) compared with mock injected mice (black bars). At this early time point, we noted a statistically significant increase in the serum levels of the chemokine KC and the cytokines IL-1β and IL-6, with no change in the levels of RANTES, MCP-1, IL- 1α, TNF-α and IL-12. At the same time point of 2 hours, the local concentrations of chemokines and cytokines in the peritoneal cavity were significantly increased not only for KC, IL-1β, and IL-6, but also for MCP-1 and IL-1α (Fig 7B). Four hours after injection of sHz (gray bars) both plasma (Fig 7C) and peritoneal (Fig 7D) levels of KC, MCP-1, IL-1α, IL-1β, and IL-6 were significantly increased compared to mock PBS injection (black bars) while there was no increase in the levels of the chemokine RANTES or the cytokines IL-12 or TNF-α.
Figure 7. Injection of sHz into the peritoneal cavity results in the local and systemic production of the chemokines KC and MCP-1 and cytokines IL-1α, IL-1β and IL-6.

sHz (2.5 mg in PBS, grey bars) or PBS alone (black bars) was injected i.p. into 3–6 WT C57BL/6 mice per group. At 2 (A, B) and 4 hrs (C, D) after injection, serum (A, C) and peritoneal fluid (B,D) were harvested from the mice and analyzed for the production of multiple cytokines as indicated using the Luminex platform. *P<0.05, **P<0.01, ***P<0.001 for control versus sHz stimulation.
Neutrophil recruitment in response to hemozoin requires MyD88 but not TLR9 or TLR2/4
TLRs are innate immune pattern recognition receptors that recognize microbial products and orchestrate the early inflammatory responses to infection (37), and thus are likely candidates for mediating the inflammatory response to hemozoin. As all known TLRs use either MyD88 or TRIF as essential intracellular adaptors for downstream signaling (38), we injected mice genetically deficient in either MyD88 or TRIF with sHz and reassessed the subsequent recruitment of inflammatory cells. Neutrophil recruitment into the peritoneal cavity was markedly reduced in MyD88−/− mice when compared to WT mice, while TRIF−/− mice showed levels of recruitment that were not significantly different than WT mice (Fig. 8A). This data suggests that hemozoin may mediate inflammation via the TLR receptor system. Thus, we tested likely TLR receptors for inflammatory responses. TLR9 has previously been implicated in the inflammatory response to hemozoin, however, as can be seen in Figure 8B, the recruitment of neutrophils in response to sHz injection remains intact in TLR9−/− mice. Similarly, Figure 8C demonstrates that mice deficient in both TLR2 and TLR4 also are able to mount a statistically significant neutrophil recruitment response to intra-peritoneal sHz injection. This data suggests that likely TLR receptors do not influence sHz mediated inflammation.
Figure 8. MyD88 but not TRIF, TLR9 or TLR2/4 is required for neutrophil recruitment in response to sHz.
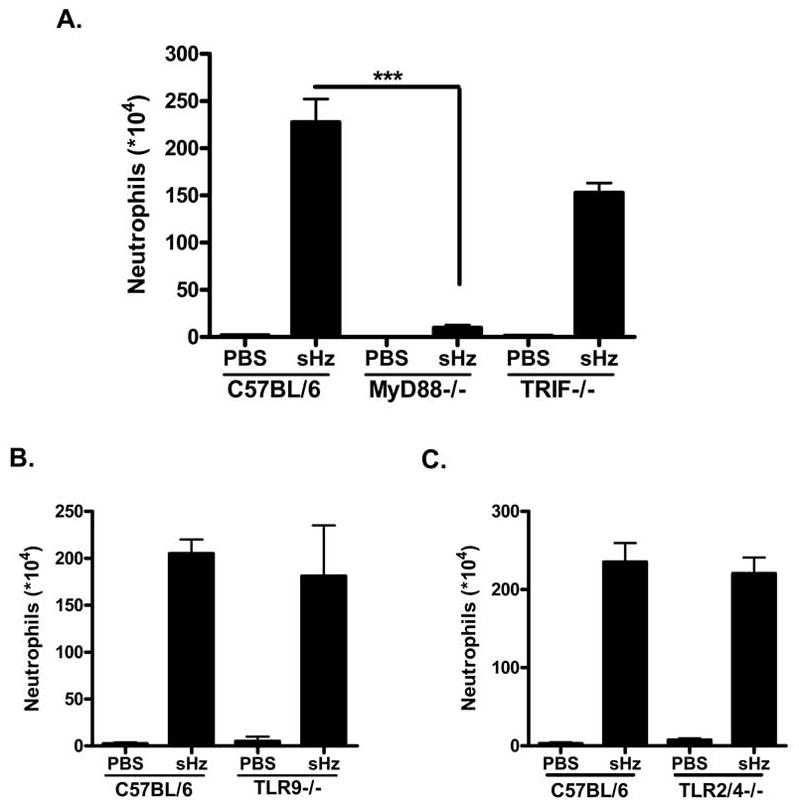
(A) Total number of recruited neutrophils determined after sHz intraperitoneal injection into WT (n=5), MyD88−/− (n=5) and TRIF−/− (n=3) mice. For sHz injection into WT mice versus MyD88−/− mice: ***P<0.001. (B) sHz (2.5 mg in PBS) or PBS alone was injected into either WT (n=3) or TLR9−/− (n=4) C57BL/6 mice and the recruitment of neutrophils was determined 12 hrs after injection. (C) sHz (2.5 mg in PBS) or PBS alone was injected i.p into either WT (n=4) or TLR2/4 DKO mice (n=5) and the recruitment of neutrophils was determined 12 hrs after injection. Each figure is representative of at least three independent experiments.
Hemozoin results in MyD88 independent chemokine release
To further examine the possibility of TLR mediated recruitment of inflammatory cells into the peritoneal cavity by sHz, we compared the local production of inflammatory mediators by WT and MyD88−/− after peritoneal sHz injection. We examined the peritoneal production of MCP-1, KC, IL-6 and IL-1β by ELISA 6 hours after injection of sHz. In accord with the results shown in Figure 7, WT mice injected with hemozoin produced elevated amounts of the chemokines MCP-1 (Fig. 9A) and KC (Fig. 9B) and the cytokines IL-6 (Fig. 9C) and IL-1β (Fig. 9D) when compared to mice that received PBS alone. In addition, when sHz was injected into mice deficient in MyD88 or TRIF, these mice produced comparable amounts of MCP-1 and KC as WT mice (Fig. 9A, 9B). The levels of IL-6 and IL-1β, however, were reduced in MyD88−/− compared to WT and TRIF−/− mice (Fig. 9C and 9D). These data indicate that sHz is recognized in vivo by a MyD88 and TRIF independent pathway resulting in chemokine production, but that the IL-1β and IL-6 response requires MyD88 dependent signaling.
Figure 9. sHz mediates MyD88 independent local production of KC and MCP-1 but MyD88 deficient mice release decreased amounts of IL-1β and IL-6 in vivo.
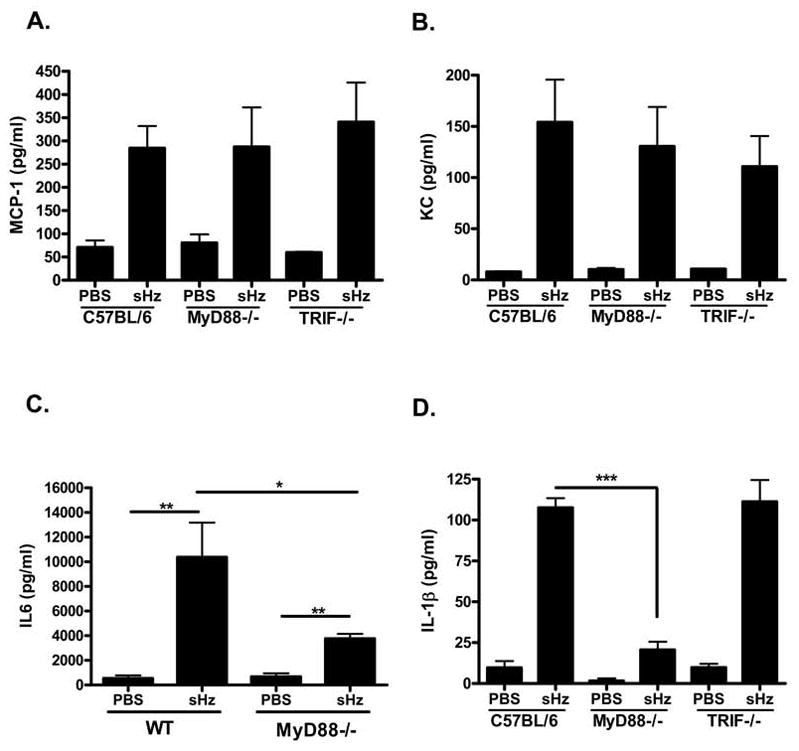
(A, B, C)
sHz (2.5 mg in PBS) or PBS alone was injected i.p. into WT (n=6), MyD88−/− (n=8) and TRIF−/− (n=4) C57BL/6 mice and the concentration of MCP-1 (A), KC (B), IL-6 (C) and IL- 1β (D) in the peritoneal fluid was determined by ELISA. *P<0.05, **P<0.01, ***P<0.001. Each figure represents one of four independent experiments.
This data, taken together, support the conclusion that with the exception of IL-1β, hemozoin activates chemokine production independently of TLR initiated pathways. Indeed, it is known that the production of IL-1β requires two signals. One signal is provided by activation of the TLR system, which initiates the transcription of pro-IL-1β mRNA, and a second signal is provided by activation of the inflammasome, which leads to the proteolytic processing and release of mature IL-1β (39). We therefore hypothesized that hemozoin activates the inflammasome-mediated stage of IL-1β release. The noted decrease of secreted IL-1β in MyD88 deficient mice compared with WT mice likely results from decreased levels of basal pro-IL-1β levels in MyD88−/− mice and not in a defect in activation of the inflammasome.
Neutrophil recruitment in response to sHz requires IL-1R and components of the inflammasome
In order to determine the role of IL-1β release in the host response to sHz, we compared neutrophil recruitment in WT and IL-1R−/− mice. As can be seen in Figure 10A, the recruitment of neutrophils in response to sHz is abrogated in IL-1R−/− mice, while there was no significant difference detected in IL-18−/− mice. This data supports the hypothesis that the role of MyD88 in the neutrophil recruitment response to sHz is not via direct TLR signaling but by increasing basal levels of pro-IL-1β and, perhaps more importantly, signaling through the IL-1R pathway.
Figure 10. The recruitment of neutrophils in response to sHz injection requires intact IL-1R signaling and the components of the NALP3 inflammasome.

(A) sHz (2.5 mg in PBS) was injected into either WT (n=4) or IL-1R−/− (n=4) or IL-18−/− (n=4) B6/129 mice and the recruitment of neutrophils was determined 12 hrs after injection. ***P<0.001 for the difference in recruitment from WT to IL-1R−/− mice. (B) sHz (2.5 mg in PBS) or PBS alone was injected into either WT (n=5), ICE−/− (n=6) or ASC−/− (n=3) C57BL/6 mice and the recruitment of neutrophils 12 hrs after injection was assessed as previously described. **P<0.01, ***P<0.001 compared with WT. (C, D) sHz (2.5 mg in PBS) or PBS alone was injected into either WT or ICE−/− C57BL/6 mice and the concentration of the chemokines MCP-1 (C) and KC (D) in the peritoneal fluid was assessed by ELISA. (E, F) sHz (2.5 mg in PBS) was injected into either WT or NALP3−/− (E) or IPAF−/− mice (F) and the number of neutrophils recruited was determined 12 hrs after injection. **P<0.01 for the difference in recruitment from WT to IL-1R−/− mice. Each figure is representative of two or three separate experiments.
The dependence of sHz-induced neutrophil recruitment on IL-1R and MyD88 signaling, as well on as the production of IL-1β, suggested to us that sHz activates the inflammasome. Crystalline materials that have been identified to activate the inflammasome include monosodium urate and calcium pyrophosphate, which cause gout and pseudogout respectively (32). Accordingly, we studied the recruitment of neutrophils in mice lacking components of the inflammasome. We used mice genetically deficient in either the IL-1-converting enzyme (ICE−/−) or the apoptosis-associated, speck-like protein containing CARD (ASC−/−) gene. Both of these genes are essential for the processing of the inactive pro-form of IL-1β into its biologically active form (40). When ICE−/− and ASC−/− mice were injected with sHz, the recruitment of neutrophils to the peritoneal cavity was significantly reduced in comparison with WT mice (Fig. 10B), but not totally abrogated as seen in MyD88 and IL-1R−/− mice. This reduced response in ICE−/− and ASC−/− mice indicates that activated IL-1β signaling via MyD88 is essential for a complete hemozoin-mediated, inflammatory cell recruitment response. Furthermore, the defect in neutrophil recruitment in ICE−/− mice occurred despite intact production of the chemokines MCP-1 (Fig. 10C) and KC (Fig. 10D), which is a finding that occurred in MyD88−/− mice as well (Figs. 9A and 9B). While ASC and ICE are key components of the inflammasome processing machinery, another group of proteins of the nucleotide-binding domain leucine-rich repeat-containing (NLR) family, are the proposed cytoplasmic sensors of pathogen associated molecular patterns(39, 41). Thus, we reasoned that sHz signaling may be mediated by one of these receptors. Two of the best studied NLR family members are IPAF and NALP3 and these have been shown to be indispensable for the production of IL-1β in response to a large number of pathogens and endogenous molecules (41). Interestingly, NALP3 has been shown to mediate the recognition of a large number of crystals including uric acid (32), silica (42), and aluminum adjuvants (43). Thus, we reasoned that NALP3 may be important in recognition of sHz crystals. As can be seen in Figure 10E, NALP3−/− mice showed a significant reduction in neutrophil recruitment in response to sHz when compared with WT mice. No defects in neutrophil recruitment were observed in the case of IPAF−/− mice (Figure 10F). These data, taken together, support the view that sHz activates IL-1β release by the NALP3 inflammasome, leading to the inflammatory recruitment of neutrophils.
Serum chemokine expression in response to sHz is independent of TLR and inflammasome signaling while systemic IL-6 release depends on inflammasome and IL-1R signaling
As demonstrated in Figure 7 the chemokines MCP-1 and KC and the cytokines IL-6 and IL-1β are released into the circulation after the peritoneal injection of sHz. This data is in accord with that of Coban et al., who demonstrated that both MCP-1 and IL-6 are expressed in the serum after the injection of sHz, and that this response was dependent on intact MyD88 and TLR9 signaling(23). However, the interpretation of this data is complicated by the possible contamination of the sHz preparation with DNA that activates TLR9 (25). We measured the production of serum chemokines and cytokines after the injection of pure sHz preparation into the peritoneal cavity of mice with genetic deletions of either MyD88, TLR9 or components of the inflammasome pathway that are important for sHz signaling. The injection of sHz into MyD88−/−, TLR9−/−, ASC−/− or NALP3−/− mice resulted in no significant changes in the serum levels of the chemokines MCP-1 and KC (Fig 11A–C), although a small increase in KC production was detected in the NALP3−/− mice. These data, taken together, are in accord with that of Figures 9–10, which demonstrated that the local production of these cytokines occurred independently of MyD88 and ICE.
Figure 11. Peritoneal injection of sHz results in systemic chemokine production independent of TLR or inflammasome signaling while systemic IL-6 production relies upon IL-1β production and signaling.
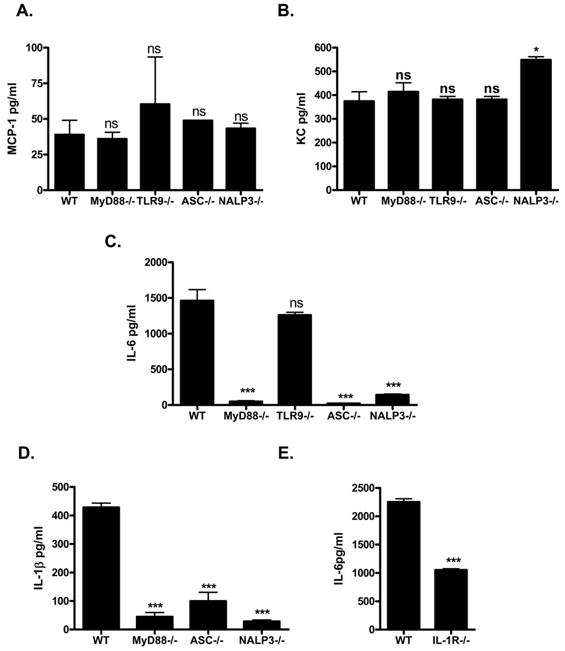
sHz (2.5mg in PBS) was injected into the peritoneal cavity of either WT (n=5) or MyD88−/− (n=5), TLR9−/− (n=4), ASC−/− (n=4) or NALP3−/− (n=3) mice and the levels of MCP-1 (A), KC (B), IL-6 (C), and IL-1β (D) were measured in the serum by multiplex cytokine analysis 4–5 hours after injection. (E) 2.5mg sHz in PBS was injection into the peritoneal cavity of either WT or IL-1R−/− mice and the serum levels of the cytokine IL-6 was measured. *P<0.1, **P<0.01, ***P<0.001 for the difference form WT and the indicated mouse strain. Each figure is representative of three separate experiments.
Of note, when we measured the serum concentration of IL-6 after sHz injection, we did observe a statistically significant reduction in IL-6 levels in MyD88, ASC and NALP3 deficient mice but not in TLR9−/− mice. This suggested to us that systemic IL-6 production relies upon systemic IL-1β signaling. We next measured systemic IL-1β production in mice with these genetic deletions and observed a significant decrease in the systemic production of IL-1β in all three strains (Fig. 11D). These data support the hypothesis that systemic IL-1β production is important for an optimal serum IL-6 response to sHz. We confirmed this possibility by studying the serum IL-6 response of IL-1R−/− mice after sHz injection. As shown in Figure 11E, mice deficient in IL-1R−/− had decreased circulating levels of IL-6 when compared to WT mice. These data indicate that the systemic production of the chemokines MCP-1 and KC occurs independently of TLR and inflammasome signaling but that IL-6 production relies upon IL-1β signaling mediated by the inflammasome components ASC and NALP3.
sHz activates the NALP3 inflammasome via release of uric acid
A commonly used cellular model for examining inflammasome activation and IL-1β release is the PMA-matured, human THP-1 monocyte (32). We added increasing amounts of sHz to these cells in vitro together with monosodium urate crystals as a positive control. Interestingly, as can be seen in Figure 12A, we were able to detect significant IL-1β release in response to an increasing dose of urate crystals (gray bars), but no release in response to sHz despite using high concentrations (black bars). This result suggested to us that sHz mediates inflammasome activation by an intermediary present in vivo but not in vitro. Of note, when we measured the concentration of uric acid in the peritoneal cavity of mice that had been injected with sHz, a significantly increased level was observed (Fig. 12B). To test the possibility that uric acid contributes to sHz signaling in vivo, we pre-treated mice with allopurinol and uricase, which deplete uric acid, prior to the injection of sHz. As can be seen in Figure 12C, treatment with allopurinol or allopurinol plus uricase decreased neutrophil recruitment to a level that was comparable to that observed with ASC, ICE or NALP3 deficient mice. Thus, sHz likely activates two pathways, one that mediates the release of chemokines MCP-1, KC and another that leads to the release of uric acid and the activation of the NALP3 inflammasome.
Figure 12. sHz stimulation does not result in IL-1β release by THP-1 cells in vitro, but may activate the inflammasome in vivo by increasing local concentrations of uric acid.
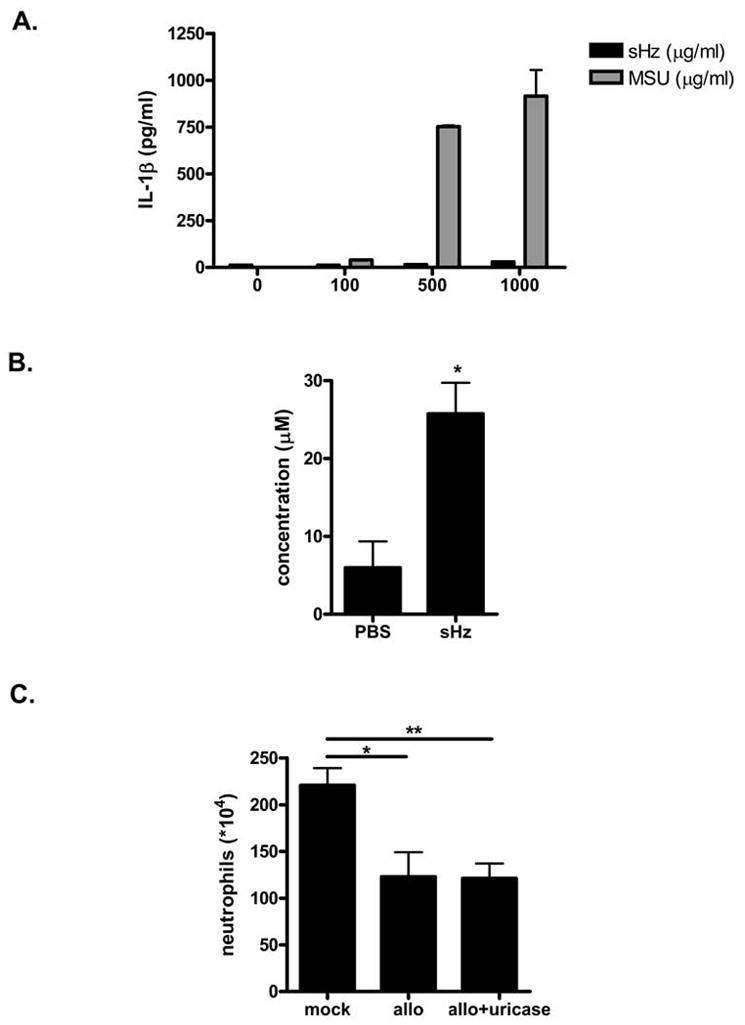
(A) THP-1 cells were stimulated overnight with PMA. Adherent cells then were incubated for 6 hrs with increasing concentrations of sHz or MSU crystals and the levels of IL-1β released into the supernatant was determined by ELISA. (B) sHz (2.5 mg in PBS) or PBS alone was injected into the peritoneal cavity of WT C57/BL6 mice. 4 hrs after injection, the peritoneal cavity was lavaged with 500 μL of PBS and the concentration of uric acid in the lavage fluid was determined. *P<0.05. (C) WT C57/BL6 mice were treated i.p. with either PBS, 500 μg of allopurinol or 500 μg of allopurinol plus 10μg of uricase on day -1 and day 0 prior to injection with sHz. 12 hrs after sHz administration, the recruitment of neutrophils to the peritoneal cavity was determined. *P<0.05, **P<0.01. Each experiment was conducted 2–3 times.
Discussion
Hemozoin is a by-product of parasite hemoglobin digestion that has been studied in malaria for many years. Although hemozoin has been considered a benign byproduct of parasite metabolism (14), its pathogenic role has been suggested by clinical data showing that the intraleukocytic hemozoin content correlates more closely with mortality than peripheral parasitemia (44, 45), and by the observation that tissue hemozoin correlates both with malarial anemia (46) and with survival from cerebral malaria (47). Subsequent in vitro studies documented an inflammatory response to hemozoin (15), suggesting that this parasite product may contribute to the excessive inflammation of severe malaria. The molecular mechanism by which hemozoin activates the inflammatory response was unknown until Coban et al. reported that hemozoin’s pro-inflammatory actions required TLR9 and the TLR adaptor molecule MyD88 (23). Subseqently, Parroche et al (25), suggested that hemozoin preparations were contaminated by DNA and that the structure of hemozoin acts to bind and load DNA complexes into plasmacytoid DCs so that they are recognized by TLR9.
Several lines of evidence nevertheless suggest that pure hemozoin may exhibit additional inflammatory properties. While the data of Parroche et al. suggest that pure hemozoin does not activate plasmacytoid DCs (25), recent studies in mouse models of malaria have found that cerebral disease occurs despite the in vivo depletion of these cells (26). This data supports the importance of other inflammatory cell types in the mediation of malarial disease. Likewise, a recent report by Akman-Anderson et al. demonstrates that pure hemozoin activates cell signaling and the expression of nitric oxide synthetase in the Anopheles mosquito (48). Thus, we attempted to broadly characterize in vitro and in vivo the inflammatory response to FPLC pure synthetic hemozoin.
In accord with Parroche et al., we found that sHz does not activate naïve FL-DCs to produce IL-6 or TNF-α in vitro (25). Furthermore, when conventional bone marrow derived DCs and bone marrow and peritoneal derived macrophages were stimulated with sHz, no increase in the production of either IL-6 or TNF-α over unstimulated cells was detected. Supporting the conclusions of Parroche et al. that hemozoin loads nucleic acid complexes for increased recognition by TLRs, we did observe a significant increase in the production of TNF-α when sHz was combined with either malarial DNA or pure CpG DNA, and we also observed an augmentation of cytokine production when sHz was added to PolyI:C, a TLR3 ligand. Recent evidence suggests that both TLR3 and TLR9 reside in the same intracellular compartment (49, 50). Thus, sHz may serve to load nucleic acid TLR ligands into specific cellular compartments for the augmentation of recognition by different intracellular TLRs.
Previous studies of the inflammatory properties of hemozoin in vitro have reported that the stimulation of macrophages with this product results in increased mRNA production for various chemokines (16). In accord with these prior results, we found that sHz induces significant MCP-1 production by peritoneal and RAW 264.7 macrophages; moreover, the production of MCP-1 was augmented by IFN-γ. Surprisingly, the release of MCP-1 in response to sHz was independent of MyD88 in both naïve and IFN-γ stimulated cells. These data suggest that sHz is recognized by an as yet unknown pathway that is independent of MyD88, and they support the study by Jaramillo et al, which has suggested that IFN-γ stimulation is essential for the generation of NO in response to sHz (51).
While naïve macrophages stimulated with sHz only produced MCP-1, IFN-γ stimulated macrophages also produced detectable levels of TNF-α. Interestingly, unlike the production of MCP-1, which was independent of MyD88, the production of TNF-α by IFN-γ stimulated macrophages required MyD88 but not TLR9. The reason for this discrepancy in MCP-1 versus TNF-α production is unclear, however recent studies indicate that MyD88 plays a role in mediating the activation effects of IFN-γ and may specifically stabilize TNF-α mRNA transcripts (52–54). In one study, the IFN-γ mediated upregulation of TNF and nitric oxide synthase was compromised in MyD88−/− mice during infection with Mycobacterium tuberculosis (53). This phenomenon occurred independently of known TLR signaling. In another study, MyD88 was found to interact with the IFN-γR and to stabilize the mRNA of various inflammatory genes including TNF via p38 signaling (54). Therefore, the observed effect of MyD88 in our study may represent the effects of MyD88 on the upregulation of TNF-α production by IFN-γ and not a direct effect of MyD88 mediated TLR signaling.
While macrophages are important cellular mediators of the response to malarial infection (55), other cell types also encounter parasite products during the intra-vascular lysis of parasitized RBCs (5). We observed increased production of IL-6, IL-8 and MCP-1 by sHz-stimulated human endothelial cells; thus multiple cell types may recognize and respond to sHz in vivo. The cellular receptors in endothelial cells that recognize sHz are unknown, although endothelial cells are known to express innate immune receptors including TLRs 2,4 and 9 (56, 57) and scavenger receptors (58).
Analysis of the downstream signaling events induced by Hz stimulation of macrophages showed an upregulation of the ERK MAP kinase cascade and NF-κB signaling, no significant effect p38 signaling, and a minor down-regulation of JNK phosphorylation. Furthermore, we found that both the ERK and NF-κB activation was independent of MyD88 and TLR9, which supports the conclusion that sHz does not initiate TLR signaling.
To better clarify the inflammatory action of sHz, we also studied the in vivo response to sHz. We observed that the injection of pure sHz into the peritoneal cavity of mice resulted in a strong inflammatory response characterized by the influx of neutrophils. Clinical studies of malaria infection have noted that neutrophils ingest large amounts of hemozoin (59), and in murine studies, neutrophil depletion was found to protect from the development of cerebral disease (60). Analysis of the mediators induced by sHz injection into the peritoneal cavity showed a significant increase in the local and systemic production of the chemokines KC and MCP-1 and the pro-inflammatory cytokines IL-1α, IL-1β and IL-6.
Given the early cytokine production and neutrophil predominant inflammation, we reasoned that innate immune receptors were likely critical for initiation of these responses. As two of the best characterized innate immune system receptors are the TLRs and inflammasomes, we injected sHz into mice with genetic deletions of components of both these pathways. We found that MyD88 was required for neutrophil influx in response to sHz while neither the adaptor molecule TRIF nor the cellular receptors TLR 2/4 or TLR9 were necessary. The local production of MCP-1 and KC was intact in MyD88−/− mice while the release of IL-1β and to a lesser extent IL-6 was reduced in MyD88−/− mice. These results support the conclusion that chemokine production in response to sHz occurs independently of MyD88 while production of other inflammatory mediators essential to neutrophil influx require MyD88−/−. Because TLR 2/4 and TLR9 were not involved in neutrophil recruitment, we suspected that the MyD88 dependence observed was related to IL-1 signaling instead of TLR signaling. Indeed, the importance of IL-1 signaling for the recruitment of neutrophils in response to sHz was demonstrated by examining IL-1R−/− mice, which showed a complete abrogation of neutrophil recruitment that was comparable to that observed in MyD88−/− mice. These data thus led to hypothesis that an essential component of sHz induced neutrophil recruitment is the release of IL-1β via activation of the inflammasome.
The processing and release of IL-1β is regulated at multiple cellular levels (39). The production of IL-1β mRNA and the translation of the inactive pro-form of the protein can be induced by TNF-α or the activation of TLR signaling. A second signal then is provided by intracellular pattern recognition receptors of the NLR family that associate with molecules such as CARD, ASC, and ICE to form a signaling complex called the inflammasome, which leads to the cleavage of the inactive pro-IL-1β into its active form (39). This complex is activated by a large number of signals including bacterial components such as muranyl dipeptide (61), environmental products such as silica and asbestos (62), and endogenous stimuli such as ATP and uric acid (63). The best studied NLR is NALP3, which has been shown to recognize a variety of substances including monosodium urate crystals, alum, silica and asbestos (32, 42, 62). Given that many of these ligands are crystalline or insoluble, we reasoned that sHz also activates the NALP3 inflammasome and we tested this hypothesis by assessing the neutrophil recruitment response in mice lacking different components of the inflammasome. Mice genetically deficient in either ASC or ICE showed impaired neutrophil recruitment upon sHz stimulation despite intact MCP-1 and KC production. The reduction in neutrophil recruitment in the ASC or ICE deficient mice was not as complete as that observed in the absence of IL-1R or MyD88; the reason for this is unclear but suggests that other inflammatory pathways may become activated to produce a low level of inflammatory cell recruitment.
To identify the particular NLR responsible for inflammasome activation, we studied both NALP3 −/− and IPAF−/− mice. Mice lacking NALP3 showed a significant reduction in the recruitment of neutrophils in response to sHz when compared to WT or IPAF−/− mice. This data suggests that NALP3 either recognizes sHz or that sHz leads to the release of a secondary mediator that then activates this NLR. In order to address this question, we stimulated THP-1 monocytes directly with either sHz or monosodium urate crystals using an experimental protocol that was developed to examine inflammasome activation (32, 43). No detectable release of IL-1β was observed in response to sHz; however, in noting the study of Orengo et al. which showed the release of uric acid during malaria infection (64) as well as the recent report that the adjuvant activity of alum is augmented by uric acid mediated activation of NALP3 (65), we reasoned that hemozoin lead to the release of uric acid and the inflammasome-dependent production of IL-1β. Indeed, the injection of sHz into the peritoneal cavity of mice resulted in a 3-fold increase in uric acid concentration, and mice pre-treated with allopurinol or allopurinol and uricase showed a significant reduction in the recruitment of neutrophils in response to sHz.
We also examined systemic cytokine production after peritoneal injection of sHz into mice deficient in inflammasome and TLR components. We found that the systemic production of MCP-1 and KC were intact despite deletion of various TLR and inflammasome signaling pathways including MyD88, TLR9, ICE, ASC and NALP3. This observation is in accord with our in vivo data and with the local peritoneal cytokine profiles noted in the peritoneum after sHz injection. On the other hand, systemic IL-6 levels were significantly decreased in mice deficient in MyD88, ASC or NALP but not TLR9. While local, peritoneal production of IL-6 was reduced in MyD88−/− mice, the effect of MyD88 deletion was much more significant on systemic IL-6 production. These data suggest that IL-6 is strongly upregulated in the circulation as a consequence of inflammasome activation and IL-1 signaling. We confirmed this observation with IL-1R−/− mice, which also showed a significant reduction in the systemic IL-6 response when compared with WT mice.
In conclusion, the present findings indicate that hemozoin is an inflammatory molecule with multiple effects on the host response (Fig. 13). Our data support the results of Parroche et al (25) that hemozoin can load malarial DNA into the appropriate cellular compartment for recognition by TLR9. Moreover, hemozoin activates macrophages and endothelial cells directly by an unknown mechanism to induce production of the chemokines MCP-1 and KC. Finally, hemozoin stimulation leads to the release of uric acid, which then activates IL-1β production via the NALP3 inflammasome. IL-1β upregulates inflammatory responses via IL-1R activation of MyD88, resulting in IL-6 production and neutrophil recruitment.
Figure 13. Proposed scheme for the inflammatory action of sHz.
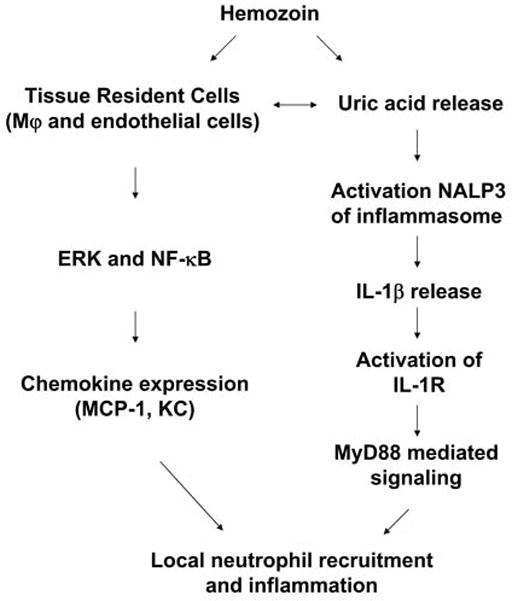
sHz induces the local production of chemokines by macrophages (Mφ) and endothelial cells via upregulation of ERK MAPkinase and NF-κB signaling pathways. sHz also results in the release of uric acid that leads to activation of the NALP3 inflammasome, the release of IL-1β, and further inflammatory effects via the IL-1R and MyD88.
The role of MyD88 in the host response to malaria infection has been suggested previously (66–68); however, the relative contributions of TLR and IL-1R signaling are still controversial. Though various studies have suggested an important role for TLR signaling in response to Plasmodium berghei (66, 67) a recent study by Cramer et al., determined that MyD88 signaling via IL-1R is important for control of Plasmodium yoelii infection while deletion of TLR2/4/9 did not influence parasite numbers (68). We have previously demonstrated that mice genetically deficient in the inflammasome component Caspase 1 do not have statistically significant protection from cerebral malaria mediated by P. berghei, although a trend towards increased survival was observed (67). However, these observations do not necessarily exclude an important role for the inflammasome or IL-1 signaling in the pathogenesis of malaria. The P. berghei model of cerebral malaria is highly lethal with a mortality approaching 80–100% over a period of <2 weeks, and such acute lethality suggests that multiple and overlapping inflammatory cascades become activated so that genetic deletion of one component may not be sufficient to effect survival. Of note, a recent study has found that mice genetically deficient in the NLR family members NOD1 and NOD2 show no difference in survival or parasitemia during P. berghei infection but manifest a reduction in cytokine production (69). Likewise, TLR9−/− mice infected with P. berghei show improved survival but not complete protection when compared to WT mice (67), which suggests an important role for other inflammatory pathways besides TLR signaling in severe complications. These observations indicate that in the P. berghei model of cerebral malaria multiple inflammatory pathways are activated that may not be detected by genetic deletion but which are still important mediators of the host response. Thus, further study of the role of the inflammasome and hemozoin in malaria pathogenesis is required to better delineate the hierarchy of the inflammatory response, especially in the context of lethal complications in murine models and in human disease.
While the relative contribution of these pathways to the pathogenesis of malarial complications in humans remains to be determined, two recent studies have implicated a significant role for uric acid in the human inflammatory response to malaria infection (64,70). Furthermore, human genetic data have suggested that polymorphisms in the IL-1β promoter correlate with susceptibility of severe malaria anemia (71). Given the high concentrations of hemozoin present in tissues during malaria (13, 45), we hypothesize that the release of uric acid by hemozoin as well as the other inflammatory effects of this molecule play a critical role in the clinical expression of severe malaria. Whether these individual pathways may be amenable to pharmacological intervention warrants additional study.
Acknowledgments
This work was supported by NIH grants AI051306, AI042310-07 (RB), MSTP TG 5T32GM07205 (JG), and a fellowship from the NSF (TS).
We would like to thank R. Medzhitov, F. Sutterwal and T. Town for providing KO mice and reagents used in this study. We are grateful to K. Moore and M. Freeman for provision of the CD36 and SRA knockout mice and for assistance with those studies.
Abbreviations
- ASC
apoptosis-associated, speck-like protein containing CARD
- ERK1/2
extracellular-signal regulated kinase1/2
- FL-DC
FLT-3 ligand derived mouse dendritic cell
- ICE
IL-1β-converting enzyme
- IPAF
IL-1β-converting enzyme protease activating factor
- IL-1R
IL-1 receptor
- JNK
c-Jun N-terminal kinase
- MAPK
MAP kinase or mitogen-activated protein kinase
- MyD88
myeloid differentiation factor 88
- NLR
nucleotide-binding domain leucine-rich repeat
- sHz
synthetic hemozoin
- TLR
Toll-like receptor
- TRIF
Toll/IL-1R domain-containing adaptor-inducing IFN-β
Footnotes
The authors have no conflicting financial interests.
References
- 1.Snow RW, Guerra CA, Noor AM, Myint HY, Hay SI. The global distribution of clinical episodes of Plasmodium falciparum malaria. Nature. 2005;434:214–217. doi: 10.1038/nature03342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Guerra CA, Snow RW, Hay SI. Mapping the global extent of malaria in 2005. Trends in parasitology. 2006;22:353–358. doi: 10.1016/j.pt.2006.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rowe AK, Rowe SY, Snow RW, Korenromp EL, Schellenberg JR, Stein C, Nahlen BL, Bryce J, Black RE, Steketee RW. The burden of malaria mortality among African children in the year 2000. International journal of epidemiology. 2006;35:691–704. doi: 10.1093/ije/dyl027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Miller LH, Baruch DI, Marsh K, Doumbo OK. The pathogenic basis of malaria. Nature. 2002;415:673–679. doi: 10.1038/415673a. [DOI] [PubMed] [Google Scholar]
- 5.Hunt NH, Golenser J, Chan-Ling T, Parekh S, Rae C, Potter S, Medana IM, Miu J, Ball HJ. Immunopathogenesis of cerebral malaria. International journal for parasitology. 2006;36:569–582. doi: 10.1016/j.ijpara.2006.02.016. [DOI] [PubMed] [Google Scholar]
- 6.Grau GE, Fajardo LF, Piguet PF, Allet B, Lambert PH, Vassalli P. Tumor necrosis factor (cachectin) as an essential mediator in murine cerebral malaria. Science (New York, NY. 1987;237:1210–1212. doi: 10.1126/science.3306918. [DOI] [PubMed] [Google Scholar]
- 7.Grau GE, Heremans H, Piguet PF, Pointaire P, Lambert PH, Billiau A, Vassalli P. Monoclonal antibody against interferon gamma can prevent experimental cerebral malaria and its associated overproduction of tumor necrosis factor. Proceedings of the National Academy of Sciences of the United States of America. 1989;86:5572–5574. doi: 10.1073/pnas.86.14.5572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Engwerda CR, Mynott TL, Sawhney S, De Souza JB, Bickle QD, Kaye PM. Locally up-regulated lymphotoxin alpha, not systemic tumor necrosis factor alpha, is the principle mediator of murine cerebral malaria. The Journal of experimental medicine. 2002;195:1371–1377. doi: 10.1084/jem.20020128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McDevitt MA, Xie J, Shanmugasundaram G, Griffith J, Liu A, McDonald C, Thuma P, Gordeuk VR, Metz CN, Mitchell R, Keefer J, David J, Leng L, Bucala R. A critical role for the host mediator macrophage migration inhibitory factor in the pathogenesis of malarial anemia. The Journal of experimental medicine. 2006;203:1185–1196. doi: 10.1084/jem.20052398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Coltel N, Combes V, Hunt NH, Grau GE. Cerebral malaria -- a neurovascular pathology with many riddles still to be solved. Current neurovascular research. 2004;1:91–110. doi: 10.2174/1567202043480116. [DOI] [PubMed] [Google Scholar]
- 11.Rosenthal PJ, Meshnick SR. Hemoglobin catabolism and iron utilization by malaria parasites. Molecular and biochemical parasitology. 1996;83:131–139. doi: 10.1016/s0166-6851(96)02763-6. [DOI] [PubMed] [Google Scholar]
- 12.Pandey AV, V, Babbarwal K, Okoyeh JN, Joshi RM, Puri SK, Singh RL, Chauhan VS. Hemozoin formation in malaria: a two-step process involving histidine-rich proteins and lipids. Biochemical and biophysical research communications. 2003;308:736–743. doi: 10.1016/s0006-291x(03)01465-7. [DOI] [PubMed] [Google Scholar]
- 13.Newton CR, Taylor TE, Whitten RO. Pathophysiology of fatal falciparum malaria in African children. The American journal of tropical medicine and hygiene. 1998;58:673–683. doi: 10.4269/ajtmh.1998.58.673. [DOI] [PubMed] [Google Scholar]
- 14.Arese P, Schwarzer E. Malarial pigment (haemozoin): a very active ‘inert’ substance. Annals of tropical medicine and parasitology. 1997;91:501–516. doi: 10.1080/00034989760879. [DOI] [PubMed] [Google Scholar]
- 15.Sherry BA, Alava G, Tracey KJ, Martiney J, Cerami A, Slater AF. Malaria-specific metabolite hemozoin mediates the release of several potent endogenous pyrogens (TNF, MIP-1 alpha, and MIP-1 beta) in vitro, and altered thermoregulation in vivo. Journal of inflammation. 1995;45:85–96. [PubMed] [Google Scholar]
- 16.Jaramillo M, Godbout M, Olivier M. Hemozoin induces macrophage chemokine expression through oxidative stress-dependent and -independent mechanisms. J Immunol. 2005;174:475–484. doi: 10.4049/jimmunol.174.1.475. [DOI] [PubMed] [Google Scholar]
- 17.Morakote N, Justus DE. Immunosuppression in malaria: effect of hemozoin produced by Plasmodium berghei and Plasmodium falciparum. International archives of allergy and applied immunology. 1988;86:28–34. doi: 10.1159/000234602. [DOI] [PubMed] [Google Scholar]
- 18.Skorokhod OA, Alessio M, Mordmuller B, Arese P, Schwarzer E. Hemozoin (malarial pigment) inhibits differentiation and maturation of human monocyte-derived dendritic cells: a peroxisome proliferator-activated receptor-gamma-mediated effect. J Immunol. 2004;173:4066–4074. doi: 10.4049/jimmunol.173.6.4066. [DOI] [PubMed] [Google Scholar]
- 19.Jaramillo M, Plante I, Ouellet N, Vandal K, Tessier PA, Olivier M. Hemozoin-inducible proinflammatory events in vivo: potential role in malaria infection. J Immunol. 2004;172:3101–3110. doi: 10.4049/jimmunol.172.5.3101. [DOI] [PubMed] [Google Scholar]
- 20.Gazzinelli RT, Denkers EY. Protozoan encounters with Toll-like receptor signalling pathways: implications for host parasitism. Nature reviews. 2006;6:895–906. doi: 10.1038/nri1978. [DOI] [PubMed] [Google Scholar]
- 21.Adachi K, Tsutsui H, Kashiwamura S, Seki E, Nakano H, Takeuchi O, Takeda K, Okumura K, Van Kaer L, Okamura H, Akira S, Nakanishi K. Plasmodium berghei infection in mice induces liver injury by an IL-12- and toll-like receptor/myeloid differentiation factor 88-dependent mechanism. J Immunol. 2001;167:5928–5934. doi: 10.4049/jimmunol.167.10.5928. [DOI] [PubMed] [Google Scholar]
- 22.Pichyangkul S, Yongvanitchit K, Kumarb U, Hemmi H, Akira S, Krieg AM, Heppner DG, Stewart VA, Hasegawa H, Looareesuwan S, Shanks GD, Miller RS. Malaria blood stage parasites activate human plasmacytoid dendritic cells and murine dendritic cells through a Toll-like receptor 9-dependent pathway. J Immunol. 2004;172:4926–4933. doi: 10.4049/jimmunol.172.8.4926. [DOI] [PubMed] [Google Scholar]
- 23.Coban C, Ishii KJ, Kawai T, Hemmi H, Sato S, Uematsu S, Yamamoto M, Takeuchi O, Itagaki S, Kumar N, Horii T, Akira S. Toll-like receptor 9 mediates innate immune activation by the malaria pigment hemozoin. The Journal of experimental medicine. 2005;201:19–25. doi: 10.1084/jem.20041836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hemmi H, Kaisho T, Takeda K, Akira S. The roles of Toll-like receptor 9, MyD88, and DNA-dependent protein kinase catalytic subunit in the effects of two distinct CpG DNAs on dendritic cell subsets. J Immunol. 2003;170:3059–3064. doi: 10.4049/jimmunol.170.6.3059. [DOI] [PubMed] [Google Scholar]
- 25.Parroche P, Lauw FN, Goutagny N, Latz E, Monks BG, Visintin A, Halmen KA, Lamphier M, Olivier M, Bartholomeu DC, Gazzinelli RT, Golenbock DT. Malaria hemozoin is immunologically inert but radically enhances innate responses by presenting malaria DNA to Toll-like receptor 9. Proc Natl Acad Sci U S A. 2007;104:1919–1924. doi: 10.1073/pnas.0608745104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.deWalick S, Amante FH, McSweeney KA, Randall LM, Stanley AC, Haque A, Kuns RD, MacDonald KP, Hill GR, Engwerda CR. Cutting edge: conventional dendritic cells are the critical APC required for the induction of experimental cerebral malaria. J Immunol. 2007;178:6033–6037. doi: 10.4049/jimmunol.178.10.6033. [DOI] [PubMed] [Google Scholar]
- 27.Clark IA, Virelizier JL, Carswell EA, Wood PR. Possible importance of macrophage-derived mediators in acute malaria. Infection and immunity. 1981;32:1058–1066. doi: 10.1128/iai.32.3.1058-1066.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen CJ, Shi Y, Hearn A, Fitzgerald K, Golenbock D, Reed G, Akira S, Rock KL. MyD88-dependent IL-1 receptor signaling is essential for gouty inflammation stimulated by monosodium urate crystals. J Clin Invest. 2006;116:2262–2271. doi: 10.1172/JCI28075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Adachi O, Kawai T, Takeda K, Matsumoto M, Tsutsui H, Sakagami M, Nakanishi K, Akira S. Targeted disruption of the MyD88 gene results in loss of IL-1- and IL-18-mediated function. Immunity. 1998;9:143–150. doi: 10.1016/s1074-7613(00)80596-8. [DOI] [PubMed] [Google Scholar]
- 30.Yamamoto M, Sato S, Hemmi H, Hoshino K, Kaisho T, Sanjo H, Takeuchi O, Sugiyama M, Okabe M, Takeda K, Akira S. Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science (New York, NY. 2003;301:640–643. doi: 10.1126/science.1087262. [DOI] [PubMed] [Google Scholar]
- 31.Hemmi H, Takeuchi O, Kawai T, Kaisho T, Sato S, Sanjo H, Matsumoto M, Hoshino K, Wagner H, Takeda K, Akira S. A Toll-like receptor recognizes bacterial DNA. Nature. 2000;408:740–745. doi: 10.1038/35047123. [DOI] [PubMed] [Google Scholar]
- 32.Martinon F, Petrilli V, Mayor A, Tardivel A, Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. 2006;440:237–241. doi: 10.1038/nature04516. [DOI] [PubMed] [Google Scholar]
- 33.Melgarejo E, Medina MA, Sanchez-Jimenez F, Urdiales JL. Monocyte chemoattractant protein-1: A key mediator in inflammatory processes. Int J Biochem Cell Biol. 2008 doi: 10.1016/j.biocel.2008.07.018. [DOI] [PubMed] [Google Scholar]
- 34.Pasloske BL, Howard RJ. Malaria, the red cell, and the endothelium. Annual review of medicine. 1994;45:283–295. doi: 10.1146/annurev.med.45.1.283. [DOI] [PubMed] [Google Scholar]
- 35.Stetson DB, Medzhitov R. Recognition of cytosolic DNA activates an IRF3-dependent innate immune response. Immunity. 2006;24:93–103. doi: 10.1016/j.immuni.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 36.Hayden MS, Ghosh S. Signaling to NF-kappaB. Genes & development. 2004;18:2195–2224. doi: 10.1101/gad.1228704. [DOI] [PubMed] [Google Scholar]
- 37.Iwasaki A, Medzhitov R. Toll-like receptor control of the adaptive immune responses. Nature immunology. 2004;5:987–995. doi: 10.1038/ni1112. [DOI] [PubMed] [Google Scholar]
- 38.Akira S, Takeda K. Toll-like receptor signalling. Nature reviews. 2004;4:499–511. doi: 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]
- 39.Mariathasan S, Monack DM. Inflammasome adaptors and sensors: intracellular regulators of infection and inflammation. Nat Rev Immunol. 2007;7:31–40. doi: 10.1038/nri1997. [DOI] [PubMed] [Google Scholar]
- 40.Fantuzzi G, Dinarello CA. Interleukin-18 and interleukin-1 beta: two cytokine substrates for ICE (caspase-1) Journal of clinical immunology. 1999;19:1–11. doi: 10.1023/a:1020506300324. [DOI] [PubMed] [Google Scholar]
- 41.Ting JP, Willingham SB, Bergstralh DT. NLRs at the intersection of cell death and immunity. Nat Rev Immunol. 2008;8:372–379. doi: 10.1038/nri2296. [DOI] [PubMed] [Google Scholar]
- 42.Cassel SL, Eisenbarth SC, Iyer SS, Sadler JJ, Colegio OR, Tephly LA, Carter AB, Rothman PB, Flavell RA, Sutterwala FS. The Nalp3 inflammasome is essential for the development of silicosis. Proc Natl Acad Sci U S A. 2008;105:9035–9040. doi: 10.1073/pnas.0803933105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Eisenbarth SC, Colegio OR, O’Connor W, Sutterwala FS, Flavell RA. Crucial role for the Nalp3 inflammasome in the immunostimulatory properties of aluminium adjuvants. Nature. 2008;453:1122–1126. doi: 10.1038/nature06939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nguyen PH, Day N, Pram TD, Ferguson DJ, White NJ. Intraleucocytic malaria pigment and prognosis in severe malaria. Transactions of the Royal Society of Tropical Medicine and Hygiene. 1995;89:200–204. doi: 10.1016/0035-9203(95)90496-4. [DOI] [PubMed] [Google Scholar]
- 45.Metzger WG, Mordmuller BG, Kremsner PG. Malaria pigment in leucocytes. Transactions of the Royal Society of Tropical Medicine and Hygiene. 1995;89:637–638. doi: 10.1016/0035-9203(95)90423-9. [DOI] [PubMed] [Google Scholar]
- 46.Casals-Pascual C, Kai O, Cheung JO, Williams S, Lowe B, Nyanoti M, Williams TN, Maitland K, Molyneux M, Newton CR, Peshu N, Watt SM, Roberts DJ. Suppression of erythropoiesis in malarial anemia is associated with hemozoin in vitro and in vivo. Blood. 2006;108:2569–2577. doi: 10.1182/blood-2006-05-018697. [DOI] [PubMed] [Google Scholar]
- 47.Taylor TE, Fu WJ, Carr RA, Whitten RO, Mueller JS, Fosiko NG, Lewallen S, Liomba NG, Molyneux ME. Differentiating the pathologies of cerebral malaria by postmortem parasite counts. Nature medicine. 2004;10:143–145. doi: 10.1038/nm986. [DOI] [PubMed] [Google Scholar]
- 48.Akman-Anderson L, Olivier M, Luckhart S. Induction of nitric oxide synthase and activation of signaling proteins in Anopheles mosquitoes by the malaria pigment, hemozoin. Infect Immun. 2007;75:4012–4019. doi: 10.1128/IAI.00645-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nishiya T, Kajita E, Miwa S, Defranco AL. TLR3 and TLR7 are targeted to the same intracellular compartments by distinct regulatory elements. J Biol Chem. 2005;280:37107–37117. doi: 10.1074/jbc.M504951200. [DOI] [PubMed] [Google Scholar]
- 50.Kajita E, Nishiya T, Miwa S. The transmembrane domain directs TLR9 to intracellular compartments that contain TLR3. Biochem Biophys Res Commun. 2006;343:578–584. doi: 10.1016/j.bbrc.2006.03.014. [DOI] [PubMed] [Google Scholar]
- 51.Jaramillo M, Gowda DC, Radzioch D, Olivier M. Hemozoin increases IFN-gamma-inducible macrophage nitric oxide generation through extracellular signal-regulated kinase- and NF-kappa B-dependent pathways. J Immunol. 2003;171:4243–4253. doi: 10.4049/jimmunol.171.8.4243. [DOI] [PubMed] [Google Scholar]
- 52.Reiling N, Ehlers S, Holscher C. MyDths and un-TOLLed truths: sensor, instructive and effector immunity to tuberculosis. Immunol Lett. 2008;116:15–23. doi: 10.1016/j.imlet.2007.11.015. [DOI] [PubMed] [Google Scholar]
- 53.Shi S, Nathan C, Schnappinger D, Drenkow J, Fuortes M, Block E, Ding A, Gingeras TR, Schoolnik G, Akira S, Takeda K, Ehrt S. MyD88 primes macrophages for full-scale activation by interferon-gamma yet mediates few responses to Mycobacterium tuberculosis. J Exp Med. 2003;198:987–997. doi: 10.1084/jem.20030603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sun D, Ding A. MyD88-mediated stabilization of interferon-gamma-induced cytokine and chemokine mRNA. Nature immunology. 2006;7:375–381. doi: 10.1038/ni1308. [DOI] [PubMed] [Google Scholar]
- 55.Pais TF, Chatterjee S. Brain macrophage activation in murine cerebral malaria precedes accumulation of leukocytes and CD8+ T cell proliferation. J Neuroimmunol. 2005;163:73–83. doi: 10.1016/j.jneuroim.2005.02.009. [DOI] [PubMed] [Google Scholar]
- 56.Faure E, Thomas L, Xu H, Medvedev A, Equils O, Arditi M. Bacterial lipopolysaccharide and IFN-gamma induce Toll-like receptor 2 and Toll-like receptor 4 expression in human endothelial cells: role of NF-kappa B activation. J Immunol. 2001;166:2018–2024. doi: 10.4049/jimmunol.166.3.2018. [DOI] [PubMed] [Google Scholar]
- 57.Li J, Ma Z, Tang ZL, Stevens T, Pitt B, Li S. CpG DNA-mediated immune response in pulmonary endothelial cells. Am J Physiol Lung Cell Mol Physiol. 2004;287:L552–558. doi: 10.1152/ajplung.00436.2003. [DOI] [PubMed] [Google Scholar]
- 58.Adachi H, Tsujimoto M. Endothelial scavenger receptors. Prog Lipid Res. 2006;45:379–404. doi: 10.1016/j.plipres.2006.03.002. [DOI] [PubMed] [Google Scholar]
- 59.Huy NT, Trang DT, Kariu T, Sasai M, Saida K, Harada S, Kamei K. Leukocyte activation by malarial pigment. Parasitol Int. 2006;55:75–81. doi: 10.1016/j.parint.2005.10.003. [DOI] [PubMed] [Google Scholar]
- 60.Chen L, Zhang Z, Sendo F. Neutrophils play a critical role in the pathogenesis of experimental cerebral malaria. Clin Exp Immunol. 2000;120:125–133. doi: 10.1046/j.1365-2249.2000.01196.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Monack DM. The inflammasome: a key player in the inflammation triggered in response to bacterial pathogens. J Pediatr Gastroenterol Nutr. 2008;46(Suppl 1):E14. doi: 10.1097/01.mpg.0000313827.16713.5e. [DOI] [PubMed] [Google Scholar]
- 62.Dostert C, Petrilli V, Van Bruggen R, Steele C, Mossman BT, Tschopp J. Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science. 2008;320:674–677. doi: 10.1126/science.1156995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Petrilli V, Dostert C, Muruve DA, Tschopp J. The inflammasome: a danger sensing complex triggering innate immunity. Curr Opin Immunol. 2007;19:615–622. doi: 10.1016/j.coi.2007.09.002. [DOI] [PubMed] [Google Scholar]
- 64.Orengo JM, Evans JE, Bettiol E, Leliwa-Sytek A, Day K, Rodriguez A. Plasmodium-induced inflammation by uric acid. PLoS Pathog. 2008;4:e1000013. doi: 10.1371/journal.ppat.1000013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kool M, Soullie T, van Nimwegen M, Willart MA, Muskens F, Jung S, Hoogsteden HC, Hammad H, Lambrecht BN. Alum adjuvant boosts adaptive immunity by inducing uric acid and activating inflammatory dendritic cells. J Exp Med. 2008;205:869–882. doi: 10.1084/jem.20071087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Coban C, Ishii KJ, Uematsu S, Arisue N, Sato S, Yamamoto M, Kawai T, Takeuchi O, Hisaeda H, Horii T, Akira S. Pathological role of Toll-like receptor signaling in cerebral malaria. Int Immunol. 2007;19:67–79. doi: 10.1093/intimm/dxl123. [DOI] [PubMed] [Google Scholar]
- 67.Griffith JW, O’Connor C, Bernard K, Town T, Goldstein DR, Bucala R. Toll-like receptor modulation of murine cerebral malaria is dependent on the genetic background of the host. J Infect Dis. 2007;196:1553–1564. doi: 10.1086/522865. [DOI] [PubMed] [Google Scholar]
- 68.Cramer JP, Lepenies B, Kamena F, Holscher C, Freudenberg MA, Burchard GD, Wagner H, Kirschning CJ, Seeberger PH, Jacobs T. MyD88/IL-18-dependent pathways rather than TLRs control early parasitaemia in non-lethal Plasmodium yoelii infection. Microbes Infect. 2008;10:1259–65. doi: 10.1016/j.micinf.2008.07.024. [DOI] [PubMed] [Google Scholar]
- 69.Finney CAM, Lu Z, LeBourhis L, Philpott DJ, Kain KC. Disruption of Nod-like receptors alters inflammatory response to infection but does not confer protection in experimental cerebral malaria. Am J Trop Med Hyg. 2009;80:718–722. [PubMed] [Google Scholar]
- 70.Orengo JM, Leliwa-Sytek A, Evans JE, Evans B, van de Hoef D, Nyako M, Day K, Rodriquez A. Uric Acid is a mediator of the Plasmodium falciparum- induced inflammatory response. PLoS ONE. 2009;4:e5194. doi: 10.1371/journal.pone.0005194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ouma C, Davenport GC, Awandare GA, Keller CC, Were T, Otieno MF, Vulule JM, Martinson J, Ong’echa JM, Ferrell RE, Perkins DJ. Polymorphic variability in the interleukin (IL)-1beta promoter conditions susceptibility to severe malarial anemia and functional changes in IL-1beta production. J Infect Dis. 2008;198:1219–26. doi: 10.1086/592055. [DOI] [PMC free article] [PubMed] [Google Scholar]