

Abstract
Wilson disease (WD) is an autosomal recessive disorder of copper transport caused by alteration of the adenosine triphosphatase 7B gene. It is rare to diagnose WD below the age of three years. Molecular genetic testing is one of the most important diagnostic methods and may confirm the diagnosis in equivocal cases. We report a case of a 9-mo old boy with WD who presented as chronic hepatitis. Genetic analysis showed compound heterozygotes of p.G1186S and c.4006delA.
Keywords: Hepatolenticular degeneration, Wilson disease, Early diagnosis, Molecular genetics, Mutation
INTRODUCTION
Wilson disease (WD) is an autosomal recessive disorder of copper metabolism that results in an accumulation of copper in the liver, brain and other tissues. The prevalence of WD is between 1 in 30 000 and 1 in 100 000 persons worldwide and in a nationwide survey, the prevalence of WD in South Korea is 1:37 000[1,2]. It has been demonstrated that children with WD usually have no symptoms below the age of 3 years. However, cases of WD presenting as a liver disease at 13 mo[3] and 2 years[4] have been reported, as well as diagnosis by asymptomatic sibling screening. Recently, molecular genetic testing has shown to be one of the most important diagnostic methods and may confirm the diagnosis in equivocal cases. We report a case of a 9-mo old infant with WD diagnosed by direct molecular genetic testing, whose hypertransaminasemia was normalized with penicillamine therapy.
CASE REPORT
A 9-mo old male infant was found to have hypertransaminasemia on routine laboratory investigation when he visited for acute diarrhea. Although clinical symptoms of enteritis improved, aminotransferase levels remained persistently high. He had no other symptoms and his development was normal for his age. All the etiological investigations of hypertransaminasemia (hepatitis A virus, hepatitis B virus, hepatitis C virus, cytomegalovirus, rubella virus, herpes virus, toxoplasmosis, autoimmune hepatitis, myopathies) were negative. However, the serum ceruloplasmin level was below normal (< 3 mg/dL) and serum copper level was 37.4 μg/dL. He had no abnormal neurological findings and a Kayser-Fleischer corneal ring was not found on a slit-lamp examination. Laboratory investigations revealed a hemoglobin level of 12.8 g/dL, with normal white blood cell and platelet counts. The serum total bilirubin was 0.3 mg/dL, albumin level was measured at 4.1 g/dL. Prothrombin time was 12.7 s and the international normalized ratio was 6.54. However, a 24-h urinary copper excretion was normal in a repeated test (Table 1). Percutaneous liver biopsy was performed. Histological evaluation revealed mild lobular activity, mild portoperiportal activity and periportal fibrosis with frequent portal to portal bridging fibrosis (Figure 1). Quantitative determination of hepatic copper level was elevated (748 μg/g dry weight of liver tissue). On electron microscopic examination, there was no evidence of abnormal mitochondria. Some fat vacuoles were found. In the portal area, a few inflammatory cells and collagen depositions were observed (Figure 2). The diagnosis of WD was confirmed by molecular analysis of the adenosine triphosphatase 7B (ATP7B) gene, which showed that the child had a compound heterozygote of p.G1186S and c.4006delA. The mutation p.G1186S has previously been described in a Japanese patient[5]. The mutation c.4006delA was a novel frameshift mutation, resulting in a stop codon (Figures 3 and 4).
Table 1.
Clinical and biochemical features of the patient
| Age (mo) | 9 | 22 | 23 | 24 | 26 | 27 | 30 |
| AST/ALT (IU/L) | 102/122 | 113/144 | 90/105 | 171/194 | 185/285 | 262/401 | 111/162 |
| Cu in urine (μg/24 h) | 14.9 | 15.2 | 62.2 | 421.4 | |||
| Ceruloplasmin (mg/dL) | < 9 | < 9 | < 8 | < 8 | |||
| Mutation | p.G1186S/c.4006delA | ||||||
| K-F ring | No | ||||||
| Cu in the liver (μg/g dry weight) | 748 | ||||||
AST: Aspartate aminotransferase; ALT: Alanine aminotransaminase.
Figure 1.
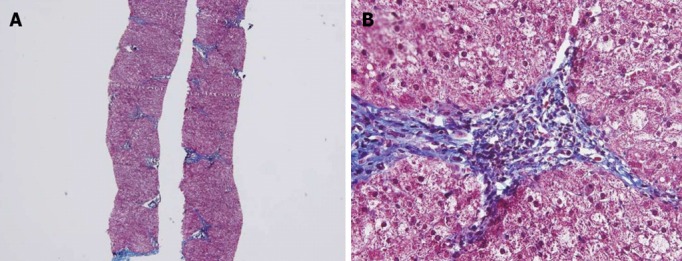
Histological findings. Biopsy specimen showing mild lobular activity, mild portoperiportal activity and periportal fibrosis with frequent portal to portal bridging fibrosis. A: Masson’s Trichrome stain, 40×; B: Masson’s Trichrome stain, 400×.
Figure 2.
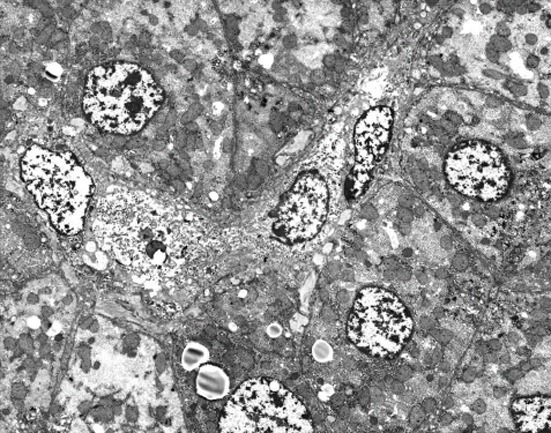
Electron microscopic examination. There was no evidence of abnormal mitochondria. Some fat vacuoles are found. In the portal area, a few inflammatory cells and collagen depositions are observed (original magnification 2000×).
Figure 3.
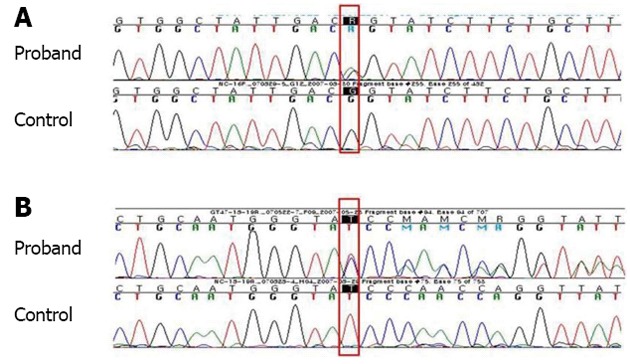
Adenosine triphosphatase 7B sequencing. A: c.3556G>A, p.G1186S, heterozygote; B: c.4006delA, p.Ile1336TyrfsX57 (reverse complementary sequence, STOP 1392), heterozygote.
Figure 4.
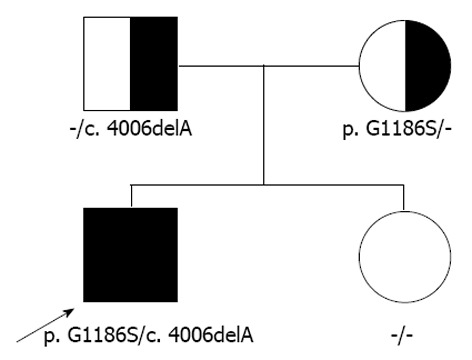
Pedigree. The patient has a compound heterozygote of p.G1186S and c.4006delA.
Therapy with zinc 24 mg twice a day was immediately started, as well as a low copper diet. However, after commencing zinc treatment, aminotransferase level increased; aspartate transminase 262 IU/L; alanine transminase 401 IU/L (Table 1). The therapeutic regimen was switched to D-penicillamine 125 mg twice a day (22 mg/kg per day), pyridoxine 12.5 mg once a day and zinc. At regular follow-ups under medical therapy, hypertransaminasemia was normalized and currently he continues to do well.
DISCUSSION
WD is an autosomal recessive disorder of copper transport that results in an accumulation of copper, primarily in the liver, brain and the cornea, and was first described as a syndrome by Wilson[6] in 1912. WD is the most common inherited liver disease, with a prevalence of 1:37 000 in the pediatric population in South Korea[2,7].
In recent years, molecular testing for ATP7B mutations has been used to aid in diagnosis. More than 500 mutations in the ATP7B gene are now recognized (www.medicalgenetics.med.ualberta.ca/wilson/index.php). The p.R778L (an allele frequency of 37%), p.A874V (13%), p.L1083F (8%) and p.N1270S (6%) are the common major mutations in South Korea[2,7]. The identification of one mutation may be adequate to confirm the diagnosis if characteristic clinical symptoms and at least some of the biochemical features are present and if the one mutation detected is clearly established as a disease-causing mutation[8]. Direct DNA sequencing did confirm WD in 98% of the Korean patients. Two mutations were detected in 70% and one mutation in 28% of the patients who showed characteristic biochemical and clinical findings of WD[2]. Iorio et al[3] reported a case of a 13-mo old child with WD who was diagnosed by biochemical family screening. However, the molecular analysis had not revealed known mutations in either the patient or her 8-year old brother. The 9-mo old infant in the present study represents the youngest patient confirmed by two ATP7B mutations, even in the absence of a family history of WD.
The diagnosis of WD is based upon the clinical setting combined with compatible biochemical, histological and physical findings. None of the laboratory parameters alone allows for a definite diagnosis of WD, particularly in infants and young children. At a consensus meeting, a scoring system was devised by Ferenci et al[9] in 2003, based on a composite of biochemical, histological and genetic features. In 2008, the American Association for Study of Liver Diseases guidelines provided diagnostic approaches and specific recommendations on the basis of published data and the researcher’s experience in caring for patients with WD[10]. The biochemical diagnosis relies upon demonstration of abnormal copper parameters, such as low serum ceruloplasmin concentration, high urinary copper and increased hepatic copper concentration. A combination of any two of these three findings is strong support for a diagnosis of WD[8]. In South Korea, in a nation-wide survey of WD, low serum ceruloplasmin (< 20 mg/dL), high 24-h urine copper excretion (> 100 μg), high hepatic copper contents (> 250 μg/g of dry liver) and Kayser-Fleischer (KF) rings were found in 96%, 86%, 88% and 73% of the patients with WD respectively[2,7].
There are numerous pitfalls in the diagnosis of WD because of the lack of any single sensitive and specific diagnostic test. Serum ceruloplasmin concentrations are elevated by acute inflammation and may be low in conditions associated with a hypoproteinemic state. Particularly in early infancy to the age of 6 mo, serum ceruloplasmin concentrations are physiologically very low. In one series of WD in children, 2 of 57 cases had a normal ceruloplasmin level and the diagnosis was confirmed by genetic analysis[11].
A 24-h urinary copper excretion is useful for the diagnosis of WD and for monitoring treatment. However the interpretation of 24-h urinary copper excretion can be difficult because similar results may be seen in chronic liver diseases, including autoimmune hepatitis. Furthermore, heterozygote carriers could also have intermediate levels. Twenty-four hours urinary copper excretion may show false negative results at presentation in patients diagnosed with WD and it may also show false negative results in asymptomatic children.
Hepatic copper content greater than 250 μg/g dry weight is usual in patients with WD but the concentration can be falsely low due to an inhomogeneous distribution of copper. In a pediatric study, sampling error was common in rendering this test unreliable in patients with cirrhosis and clinically evident WD[12] and hepatic copper concentrations can be highly increased in long-term cholestasis. KF rings also are present in of 44%-62% of patients with mainly hepatic disease at the time of diagnosis[9]. Especially in children presenting with liver disease, KF rings are usually absent. In the genetically confirmed present case, the serum ceruloplasmin level was below normal and the amount of hepatic copper was elevated, but 24-h urinary copper excretion was normal at presentation and a KF ring was absent. Specific ultrastructural changes may be visible in WD. Typical findings include variability in size and shape, increased density of material, inclusions of lipid and granular material, and increased intracristal space with dilatation of the tips of cristae[13] but ultrastructural analysis of liver specimens revealed no evidence of abnormal mitochondria in our case.
The treatment of first choice among chelators and zinc in specific clinical situations of WD is still debated. Not only the agent, but the age at which presymptomatic patients with WD should be treated is still a matter of debate. Brewer et al[14] recommended an early treatment of presymptomatic patients during pediatric years with zinc. Iorio et al[3] had treated the 13-mo old child with zinc. In this case, although therapy with zinc was continued, his transaminase levels increased but after changing the regimen to penicillamine, the liver enzyme levels normalized. Further research on management of very young children with WD is needed.
In conclusion, even in infancy, elevation of transaminase should lead to exclusion of WD if other etiologies of hypertransaminasemia are ruled out. Genetic testing is the only reliable tool to identify very young patients with WD. We report a case of a 9-mo old boy with WD presenting as chronic hepatitis with compound heterozygotes of p.G1186S and c.4006delA.
Footnotes
P- Reviewer Stefanovic B S- Editor Zhang DN L- Editor Roemmele A E- Editor Li JY
References
- 1.Ala A, Walker AP, Ashkan K, Dooley JS, Schilsky ML. Wilson’s disease. Lancet. 2007;369:397–408. doi: 10.1016/S0140-6736(07)60196-2. [DOI] [PubMed] [Google Scholar]
- 2.Seo JK. Wilson disease: an update. Korean J Hepatol. 2006;12:333–363. [PubMed] [Google Scholar]
- 3.Iorio R, D’Ambrosi M, Mazzarella G, Varrella F, Vecchione R, Vegnente A. Early occurrence of hypertransaminasemia in a 13-month-old child with Wilson disease. J Pediatr Gastroenterol Nutr. 2003;36:637–638. doi: 10.1097/00005176-200305000-00009. [DOI] [PubMed] [Google Scholar]
- 4.Beyersdorff A, Findeisen A. Morbus Wilson: Case report of a two-year-old child as first manifestation. Scand J Gastroenterol. 2006;41:496–497. doi: 10.1080/00365520500389453. [DOI] [PubMed] [Google Scholar]
- 5.Shimizu N, Nakazono H, Takeshita Y, Ikeda C, Fujii H, Watanabe A, Yamaguchi Y, Hemmi H, Shimatake H, Aoki T. Molecular analysis and diagnosis in Japanese patients with Wilson’s disease. Pediatr Int. 1999;41:409–413. doi: 10.1046/j.1442-200x.1999.01092.x. [DOI] [PubMed] [Google Scholar]
- 6.Wilson SAK. Progressive lenticular degeneration: a familial nervous disease associated with cirrhosis of the liver. Brain. 1912;34:295–509. doi: 10.1093/brain/awp193. [DOI] [PubMed] [Google Scholar]
- 7.Seo JK, Kim YS, Hahn CJ, Baik SK. A nationwide survey for prevalence and clinical characteristics of Wilson disease in Korea. Korean J Hepatol. 2004;10(Suppl 6):5–15. [Google Scholar]
- 8.Cox DW, Roberts E. Wilson Disease. In: Pagon RA, Bird TD, Dolan CR, Stephens K, Adam MP, et al., editors. GeneReviews™ (Internet) Suppl 6. Seattle (WA): University of Washington; 1993. [Google Scholar]
- 9.Ferenci P, Caca K, Loudianos G, Mieli-Vergani G, Tanner S, Sternlieb I, Schilsky M, Cox D, Berr F. Diagnosis and phenotypic classification of Wilson disease. Liver Int. 2003;23:139–142. doi: 10.1034/j.1600-0676.2003.00824.x. [DOI] [PubMed] [Google Scholar]
- 10.Roberts EA, Schilsky ML. Diagnosis and treatment of Wilson disease: an update. Hepatology. 2008;47:2089–2111. doi: 10.1002/hep.22261. [DOI] [PubMed] [Google Scholar]
- 11.Manolaki N, Nikolopoulou G, Daikos GL, Panagiotakaki E, Tzetis M, Roma E, Kanavakis E, Syriopoulou VP. Wilson disease in children: analysis of 57 cases. J Pediatr Gastroenterol Nutr. 2009;48:72–77. doi: 10.1097/MPG.0b013e31817d80b8. [DOI] [PubMed] [Google Scholar]
- 12.Martins da Costa C, Baldwin D, Portmann B, Lolin Y, Mowat AP, Mieli-Vergani G. Value of urinary copper excretion after penicillamine challenge in the diagnosis of Wilson’s disease. Hepatology. 1992;15:609–615. doi: 10.1002/hep.1840150410. [DOI] [PubMed] [Google Scholar]
- 13.Sternlieb I. Fraternal concordance of types of abnormal hepatocellular mitochondria in Wilson’s disease. Hepatology. 1992;16:728–732. doi: 10.1002/hep.1840160319. [DOI] [PubMed] [Google Scholar]
- 14.Brewer GJ, Dick RD, Johnson VD, Fink JK, Kluin KJ, Daniels S. Treatment of Wilson’s disease with zinc XVI: treatment during the pediatric years. J Lab Clin Med. 2001;137:191–198. doi: 10.1067/mlc.2001.113037. [DOI] [PubMed] [Google Scholar]