

Abstract
Reports of emerging resistance to first-line artemisinin antimalarials make it critical to define resistance mechanisms and identify in vitro correlates of resistance. Here we combine unique in vitro experimental and analytical approaches to mimic in vivo drug exposure in an effort to provide insight into mechanisms of drug resistance. Tightly synchronized parasites exposed to short drug pulses exhibit large stage-dependent differences in their drug response that correlate with hemoglobin digestion throughout most of the asexual cycle. As a result, ring-stage parasites can exhibit >100-fold lower sensitivity to short drug pulses than trophozoites, although we identify a subpopulation of rings (2–4 h postinvasion) that exhibits hypersensitivity. We find that laboratory strains that show little differences in drug sensitivity in standard in vitro assays exhibit substantial (>95-fold) difference in sensitivity when exposed to short drug pulses. These stage- and strain-dependent differences in drug sensitivity reflect differential response lag times with rings exhibiting lag times of up to 4 h. A simple model that assumes that the parasite experiences a saturable effective drug dose describes the complex dependence of parasite viability on both drug concentration and exposure time and is used to demonstrate that small changes in the parasite’s drug response profile can dramatically alter the sensitivity to artemisinins. This work demonstrates that effective resistance can arise from the interplay between the short in vivo half-life of the drug and the stage-specific lag time and provides the framework for understanding the mechanisms of drug action and parasite resistance.
Keywords: endoperoxide, Plasmodium, qinghaosu, effective dose model
Artemisinin or qinghaosu (QHS) and its derivatives [collectively referred to as artemisinins (ARTs)] are considered a last line of defense against malaria and are the only approved and effective antimalarial drug class to which widespread resistance has not yet developed. Clinically, ARTs are fast acting due to their broad spectrum activity against blood-stage parasites, providing a prompt response that is critical for treating severe malaria (1).
ARTs are administered in combination with other more stable drugs to compensate for their short in vivo half-lives and to minimize the development of parasite resistance (2). Unfortunately, resistance of Plasmodium falciparum to ARTs has emerged along the Thailand–Cambodian border, manifested as delayed clearance of blood parasites (3), and recent reports indicate that the prevalence of the resistance phenotype is increasing (4). A phenomenon that is frustrating researchers is that parasites isolated from patients showing delayed clearance do not exhibit a consistent decrease in sensitivity, using standard in vitro assays (which determine parasite killing after exposure to drugs for 3 d in culture) (3). This complicates efforts to monitor and control the development of resistance.
We previously demonstrated that potent activity of ARTs against mature blood-stage parasites (trophozoites) is dependent on hemoglobin uptake and digestion (5). Thus, young ring-stage parasites exhibiting no hemoglobin uptake (6) might be expected to show low sensitivity to ARTs. Indeed, a recent intrahost stage-specific modeling study suggested delayed parasite clearance results from decreased drug sensitivity of ring-stage parasites (7). However, although ring-stage parasites are reported to be less sensitive than trophozoites to ARTs in some in vitro studies (8), others report little differences (9) or even increased sensitivity (10). We have therefore reexamined the stage specificity of drug action across the entire asexual blood cycle, using tightly synchronized parasite cultures. By exposing these cultures to short drug pulses that more closely mimic clinical exposure and using new analytical approaches, we provide important insights into mechanisms of drug action and resistance.
Results
Stage-Dependent Activity of ARTs.
We first examined the sensitivity of a tightly synchronized P. falciparum culture (3D7 strain) to 4-h drug pulses covering the entire asexual blood cycle (Fig. 1A). For most of the ring stage [6–20 h postinvasion (p.i.)] parasites were insensitive to QHS, with 50% lethal dose [LD50(4 h)] values for killing >100-fold those observed for the more mature trophozoites and schizonts (>20 h p.i.). Similar trends were observed with the clinically relevant dihydroartemisinin (DHA) although the decrease in ring-stage sensitivity was more modest (∼10-fold). In addition, parasites exhibited progressive increases in sensitivity to DHA from mid- to late rings that correlated with the appearance of dark puncta in Giemsa-stained parasites (Fig. 1B). These are likely predigestive vacuoles indicating the initial stages of hemoglobin digestion and the formation of hemozoin pigment (6). These results are consistent with the importance of hemoglobin digestion in explaining the relative potency of ARTs for most of the parasite’s life cycle (5).
Fig. 1.

P. falciparum exhibits large fluctuations in sensitivity to short pulses of ARTs. (A) A tightly synchronized 3D7 culture (>90% of parasites within a 1-h time window) was subjected to 4-h drug pulses every 4 h. Horizontal bars correspond to the range of ages (of ≥50% of the parasite population) during each assay. The gray vertical lines indicate where a significant fraction (indicated above the lines) progressed to the next cycle. (B) Hemozoin formation during the ring to trophozoite transition. (Left) Color images of Giemsa-stained parasites from A. (Right) Blue channel of the Giemsa images showing dark puncta that correspond to hemozoin (arrows). The percentages of represented morphologies are indicated (n ≥ 19).
Subpopulation of Early Rings Exhibits Drug Hypersensitivity.
Surprisingly the youngest rings assayed in Fig. 1A (<6 h p.i.) exhibited hypersensitivity to ARTs. We considered the possibility that this might be due to stress arising from the synchronization protocol rather than an intrinsic difference in sensitivity. We tested this by correlating drug sensitivity with the age distribution of rings during assays of a culture not subjected to the very tight synchronization procedure. For this, we used a culture of 3D7 parasites consisting of late trophozoites/early schizonts and performed 4-h drug pulses every 3 h beginning at late schizogony and continuing into the next parasite cycle. The LD50(4 h) measured in a particular assay reflects the LD50(4 h) of the predominant parasite population in the culture. We therefore monitored the appearance of the next generation of rings in the control culture by flow cytometry (SI Appendix, Fig. S1) and used this information to calculate the range of ring ages of ≥50% of the parasite population during the course of each assay (Materials and Methods and SI Appendix). This approach permits the analysis of the drug sensitivity of both late schizonts (i.e., rings with negative ages) and early rings in a culture going through the schizont–ring transition. Thus, the low LD50(4 h) values in the assays represented in black in Fig. 2A show that schizonts within a few hours of forming rings remain sensitive to the drugs whereas the low LD50(4 h) values measured in the assay represented in red can be attributed to rings <3 h p.i. and/or to late schizonts. The assay represented in blue demonstrates that early rings (somewhere between 2 and 6 h p.i.) are indeed highly sensitive to both artemisinin and DHA. We denote this population as hypersensitive rings (hRings).
Fig. 2.

A small subpopulation of rings is hypersensitive to ARTs. (A) Correlation of drug sensitivity with ring age. A 3D7 culture consisting of late trophozoites/early schizonts was subjected to 4-h drug pulses every 3 h, continuing into the next parasite cycle. The LD50(4 h) values are shown as a function of the range of ages for the middle 50% of the parasites during the course of each assay, as described in Materials and Methods (also SI Appendix). Rings with negative ages, a, correspond to trophozoites/schizonts that will form new rings in a hours. (B) Identification of the ring subpopulation that is hypersensitive to ARTs. A tightly synchronized 3D7 culture (>70% parasites within a 30-min window) was subjected to 75-min drug pulses every hour. Horizontal bars correspond to the range of ages (of ≥50% of the parasite population) during each assay. The shaded region corresponds to the hypersensitive window (∼2–4 h p.i.).
We next examined the time window of ring hypersensitivity with improved time resolution by subjecting a tightly synchronized culture to a series of short (75 min) drug pulses. The youngest and oldest rings examined (∼1 h and >4 h p.i., respectively) were insensitive to both QHS and DHA (Fig. 2B). Drug hypersensitivity was observed in rings only over a narrow age window so that hRings correspond to rings 2–4 h p.i. (Fig. 2B, shaded region). This window of ring-stage hypersensitivity appears to be specific to the ARTs as 2- to 4-h rings did not exhibit hypersensitivity to the quinoline drugs, chloroquine and quinine (SI Appendix, Fig. S6).
Concentration and Time Dependence of Drug Action.
During ART chemotherapy in vivo, parasites are exposed to fluctuating drug concentrations and varying lengths of drug exposure (Cmax = 0.5–11 µM and elimination half-life of 24–95 min for DHA) (1, 11, 12). We therefore examined the influence of both drug concentration and exposure time (te) on parasite viability (defined as the fraction of the parasite population that survives drug treatment). Parasite viability was markedly dependent on exposure time with rings effectively resistant to short drug pulses (Fig. 3A). The resultant sigmoidal dependence of parasite viability on te is evident at all parasite stages (Fig. 3B) with ring-stage parasites (red squares) exhibiting an extended lag time in their drug response of up to 4 h. Interestingly, the marked difference in drug sensitivity between rings and trophozoites [as measured by the LD50(4 h) values in Fig. 1A] is reduced with extended treatment (Fig. 3C, red squares). Measuring only LD50(6 h) values (Fig. 3C, compare blue circles and red squares) would not reveal the marked differences between rings and trophozoites. The higher sensitivity of rings in a 6-h assay cannot be explained by the progression of the parasites into the more sensitive trophozoite stage during the course of the assay, as the rings in Fig. 3 (7.5 h p.i. at start of treatment) remain refractory to 4-h QHS pulses up to 20 h p.i. (Fig. 1A). Taken together these data show that the markedly lower sensitivity of rings to short drug pulses is a reflection of a delayed drug response (increased lag time).
Fig. 3.

Dependence of drug action on drug concentration and exposure time. A tightly synchronized culture of 3D7 parasites (>70% of parasites within a 1-h time window) was subjected to pulses of QHS (Left) or DHA (Right) of varying duration at different stages of the parasite life cycle. (A) Dose–response of rings (7.5 h p.i.) exposed to drugs for 1–6 h. See SI Appendix, Fig. S2 for other stages. (B) Influence of drug pulse duration and parasite stage on parasite viability at low and high drug concentrations. (C) Influence of drug pulse duration and parasite stage on LD50(te) values. The average age of the parasite population at the beginning of each drug pulse is given in Table 1. Error bars in A and B correspond to the range of duplicates. Curves correspond to the best fits using the cumulative ED model (SI Appendix) with parameters given in Table 1.
Different Parasite Strains Exhibit Different Response to Short Drug Pulses.
We compared the drug responses of two additional laboratory strains, 7G8 and D10. These strains express different alleles of the pfmdr1 gene encoding P-glycoprotein homolog-1 (Pgh1) (13, 14) and exhibit small but inconsistent differences in ART sensitivities. For example, the D10 strain has been reported to be ∼2-fold less sensitive to QHS but ∼2-fold more sensitive to DHA than the 7G8 strain (14, 15). We found the D10 strain was slightly less sensitive than 7G8 to both QHS and DHA [3.3 ± 0.5-fold and 1.8 ± 0.2-fold higher LD50(3 d) values, respectively (n = 3, ±SD)].
In contrast, these strains exhibited large stage-dependent differences in drug sensitivity when subjected to 4-h drug pulses at different stages of the asexual blood cycle (Fig. 4A). The largest differences were observed in late rings (∼20 h p.i.) with D10 exhibiting >90-fold and 8- to 10-fold decreased sensitivity to QHS and DHA, respectively. These differences could not be attributed to different rates of progression through the intraerythrocytic stages as D10 underwent the ring to trophozoite transition ∼2 h earlier than 7G8 (Fig. 4A, Top). Interestingly, the stage-dependent LD50(4 h) values for D10 were similar to those observed for 3D7 (Fig. 1A), consistent with the same Pgh1 (wild-type) allele in these strains (16). This analysis shows that standard 3-d drug assays may not always reveal important differences in the sensitivity of parasite strains to the short-lived ARTs.
Fig. 4.

P. falciparum strains differ in their susceptibility to short exposure to ARTs. (A) Synchronized cultures of 7G8 (red) and D10 (blue) parasites (>80% within a 1-h time window) were exposed to QHS or DHA for 4 h at different stages of the intraerythrocytic cycle. Horizontal bars correspond to the range of ages (of ≥50% of the parasite population) during each assay. (Top) Images show typical parasite morphology from Giemsa smears during the time period encompassing the ring–trophozoite transition (16–28 h p.i.). The percentages of represented morphologies are indicated (n ≥ 20). (B) The effect of exposure time on sensitivity of mid-rings (14 h p.i.) and mid-trophozoites (28 h p.i.). 7G8 (red) and D10 (blue) parasites were exposed to DHA for 6 h (open symbols, dashed lines) or 2 h (Upper, solid symbols and solid lines) or 1 h (Lower, solid symbols and solid lines). Error bars indicate the range of duplicates. Additional data can be found in SI Appendix, Fig. S5A.
We further examined the effect of time of exposure to DHA on the viability of 7G8 and D10 strains. Mid-trophozoite-stage D10 parasites were effectively resistant to a 1-h exposure, whereas the LD50(1 h) value for 7G8 parasites was 28 nM (sensitivity ratio >>35; Fig. 4B, Lower). By contrast, the difference in LD50(6 h) values was only 12-fold. Similarly mid-ring-stage D10 parasites were effectively resistant to a 1-µM DHA exposure for 2 h, whereas the LD50(2 h) value for 7G8 parasites was 180 nM (sensitivity ratio >>5.5; Fig. 4B, Upper). The difference in susceptibility was less for a 6-h exposure (3.5-fold). These results further demonstrate that long exposure times can conceal what may turn out to be clinically relevant differences in drug sensitivities.
Cumulative Effective Dose Model of Drug Action.
We developed a semiempirical model to describe the complex dependence of the in vitro susceptibility on drug concentration and exposure time (see SI Appendix for full derivation). In this model, we distinguish between the drug concentration (C) administered to the parasite culture and the effective dose (ED) experienced by the parasite. This effective dose arises from the interaction of the drug with the parasite to produce a product or affect a biological process that is ultimately responsible for parasite death. We model ED as saturable in nature, so that ED = EDmaxC/(Km + C), where EDmax represents the maximum ED achieved at saturating drug concentrations and Km the drug concentration resulting in half the maximum effective dose. To account for the presence of extended lag times, parasite viability (V) is assumed to be a sigmoidal function of the cumulative ED (EDcum = EDte; SI Appendix),
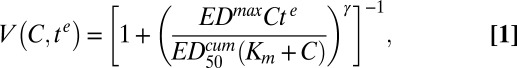 |
where γ is the slope of the sigmoidal function and 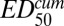 is the cumulative ED required to achieve 50% killing. In practice,
is the cumulative ED required to achieve 50% killing. In practice,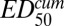 and EDmax can be determined only as a ratio,
and EDmax can be determined only as a ratio,
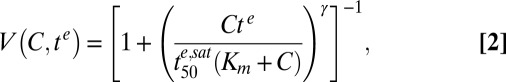 |
where 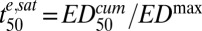 is the minimum time required to kill 50% of the parasites (at saturating drug concentrations).
is the minimum time required to kill 50% of the parasites (at saturating drug concentrations).
Analysis of parasite viability data using Eq. 2 demonstrates the V-EDcum profiles for 3D7 parasites are independent of te, confirming that viability is primarily dependent on EDcum (Fig. 5A and SI Appendix, Fig. S2) and that the cumulative ED model describes the complex relationship between V and C and te (see Fig. 5A for ring data and SI Appendix, Fig. S2 for other stages). This shows that the different response of rings and trophozoites to short pulses is a consequence of the saturable nature of the effective dose. This analysis also demonstrates that the different 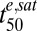 values at different stages (Table 1) are predominantly responsible for the differential lag times. A similar analysis of the 7G8 and D10 strains (SI Appendix, Fig. S5B) shows the increased
values at different stages (Table 1) are predominantly responsible for the differential lag times. A similar analysis of the 7G8 and D10 strains (SI Appendix, Fig. S5B) shows the increased 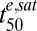 values of the D10 strain are predominantly responsible for its decreased drug sensitivity at the mid-ring and mid-trophozoite stages.
values of the D10 strain are predominantly responsible for its decreased drug sensitivity at the mid-ring and mid-trophozoite stages.
Fig. 5.

Analysis of in vitro data for 3D7 ring-stage parasites according to the cumulative ED model and simulation of in vivo-like responses to DHA treatment. (A) Dependence of parasite viability on the cumulative ED. The ring-stage viability data (from Fig. 3A) in the presence of QHS and DHA are plotted as a function of Cte/(Km + C), which is equivalent to EDcum/EDmax and hence proportional to EDcum (Eq. 1). The Km values are taken from Table 1. Symbols are as in Fig. 3A. See SI Appendix, Fig. S2 for analyses of other stages. (B) Simulation of the viability of 3D7 ring-stage parasites (blue) subjected to a DHA treatment that mimics the pharmacokinetic profile experienced in vivo. The DHA pharmacokinetic profile (Cmax = 3 µM) is shown normalized in red. (C) Simulation of the influence of γ (red squares) and 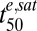 (blue circles) on Vres values for parasites subjected to an in vivo-like DHA pulse. The vertical dashed lines correspond to the γ (red) and
(blue circles) on Vres values for parasites subjected to an in vivo-like DHA pulse. The vertical dashed lines correspond to the γ (red) and 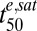 (blue) values of ring-stage parasites (Table 1). The simulations in B and C were carried out as described in SI Appendix.
(blue) values of ring-stage parasites (Table 1). The simulations in B and C were carried out as described in SI Appendix.
Table 1.
Parameter estimates from analysis of viability data vs. drug concentration and length of exposure according to the cumulative ED model
| Parameter |
||||
| Drug | Stage | Km, nM |
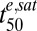 , h , h |
γ |
| QHS | hRing(2h) | 65 (6) | 0.97 (0.05) | 2.2 (0.1) |
| Ring(7.5h) | 59 (5) | 4.84 (0.05) | 6.2 (0.3) | |
| Troph(24h) | 230 (20) | 1.72 (0.05) | 5.4 (0.6) | |
| Troph(34h) | 390 (20) | 1.53 (0.04) | 5.3 (0.4) | |
| DHA | hRing(2h) | 33 (4) | 0.55 (0.05) | 2.2 (0.2) |
| Ring(7.5h) | 53 (3) | 2.08 (0.05) | 4.1 (0.2) | |
| Troph(24h) | 80 (10) | 0.53 (0.04) | 3.2 (0.3) | |
| Troph(34h) | 81 (7) | 0.68 (0.04) | 3.0 (0.2) | |
The stages and postinvasion ages of the parasites (3D7 strain) at the start of each assay are indicated. Numbers in parentheses denote SEs. Correlation coefficients between the parameter estimates are presented in SI Appendix, Table S1. Km, drug concentration resulting in half the maximum effective dose; 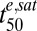 , Exposure time required for 50% parasite killing at saturating drug concentrations; γ, slope of the sigmoidal function relating parasite viability and cumulative ED.
, Exposure time required for 50% parasite killing at saturating drug concentrations; γ, slope of the sigmoidal function relating parasite viability and cumulative ED.
Simulation of in Vitro Assays.
We used the cumulative ED model to simulate the effect of Km, γ, and 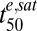 on parasite survival in in vitro drug assays. We examined their effect on the LD50(te) values and on the fraction of parasites that survives short-term exposure to high drug concentrations (the minimum viability, Vmin). This analysis (SI Appendix, Fig. S3) demonstrates that in vitro assays, in which ring-stage parasites are exposed to drug for 3 and 6 h, would enhance efforts to detect strain-specific differences in responses to the clinically relevant drug, DHA. This analysis also indicates that measurement of a single parameter such as the LD50(te) value, which reflects the average sensitivity of the population, may not always be sufficient to classify a strain as resistant or sensitive.
on parasite survival in in vitro drug assays. We examined their effect on the LD50(te) values and on the fraction of parasites that survives short-term exposure to high drug concentrations (the minimum viability, Vmin). This analysis (SI Appendix, Fig. S3) demonstrates that in vitro assays, in which ring-stage parasites are exposed to drug for 3 and 6 h, would enhance efforts to detect strain-specific differences in responses to the clinically relevant drug, DHA. This analysis also indicates that measurement of a single parameter such as the LD50(te) value, which reflects the average sensitivity of the population, may not always be sufficient to classify a strain as resistant or sensitive.
Simulation of a Single-Dose Treatment.
We next considered the clinical implications of our findings, focusing on rings, because this is considered the stage most likely to survive a single-dose treatment (7). Parasite viability was simulated for the course of a single DHA pulse, using a typical pharmacokinetic profile for the drug (1) and the in vitro model parameters derived for 3D7 parasites (Fig. 5B and SI Appendix, Fig. S4). We found that 1% of 3D7 parasites would survive such a treatment in vitro. We found that the fraction of surviving parasites (the residual viability, Vres) decreases by 50% for every 0.3 h increase in drug half-life (SI Appendix, Fig. S4A), showing that small improvements in drug stability may substantially reduce the number of parasites surviving treatment. This indicates that, other things being equal, new synthetic endoperoxides with longer exposure in the bloodstream (17) will be much more effective in clearing parasites.
We also examined the influence of various model parameters on Vres and the time it takes to kill 50% of the parasites (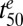 ). The latter parameter is similar to
). The latter parameter is similar to 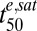 if the parasites are subjected to drug concentrations (C) >> Km for a sufficient time, which represents the situation in vivo (SI Appendix, Fig. S4). We found that relatively small changes in parasite responses to drug treatment, particularly as exemplified by the model parameters γ and
if the parasites are subjected to drug concentrations (C) >> Km for a sufficient time, which represents the situation in vivo (SI Appendix, Fig. S4). We found that relatively small changes in parasite responses to drug treatment, particularly as exemplified by the model parameters γ and 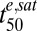 , can lead to substantial increases in the number of parasites that survive a drug pulse with a clinically relevant profile (Fig. 5C and SI Appendix, Fig. S4). Importantly, this analysis demonstrates that a parameter such as
, can lead to substantial increases in the number of parasites that survive a drug pulse with a clinically relevant profile (Fig. 5C and SI Appendix, Fig. S4). Importantly, this analysis demonstrates that a parameter such as 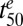 , which measures an average property of the parasite population, may not be a reliable predictor of the fraction of the parasite population that survives drug treatment; hence it may be only poorly correlated with delayed parasite clearance.
, which measures an average property of the parasite population, may not be a reliable predictor of the fraction of the parasite population that survives drug treatment; hence it may be only poorly correlated with delayed parasite clearance.
Simulation of a Multidose Treatment.
We also performed a simulation of a multidose treatment in vivo, using the model parameters determined for 7G8 and D10 parasites (SI Appendix, Fig. S5B) to determine parasite load following a 3-d drug treatment. This idealized simulation assumes a tightly synchronized infection of mid-ring-stage parasites (14 h p.i.) at the beginning of treatment and assumes that the parasites remain tightly synchronized during treatment. The analysis predicts that treatment of an infection with the D10 strain would reduce the parasite load to 0.004% of the starting parasite load after 48 h of treatment (SI Appendix, Fig. S5C), which approaches the range (0.01%) reported from field measurements for strains that show standard clearance times (3). After 72 h, the predicted parasite load for the D10 infection is 50-fold greater than for the 7G8 infection (SI Appendix, Fig. S5C). This suggests that assays using short pulses in vitro, combined with our simple analytical model, could potentially provide a useful correlate with parasite clearance times in the field.
Discussion
Stage- and strain-specific drug sensitivities have been documented for a number of different antimalarial drug classes (8, 10, 18). However, the relatively long in vivo half-lives of many of these drugs [e.g., 1–2 mo for chloroquine (19)] mean that stage-dependent effects do not influence the in vivo efficacy of these drugs. Our work confirms the broad spectrum activity of the ARTs (8, 20) but reveals this is dependent on the drugs being present for a sufficient period. Using tightly synchronized parasite cultures, we demonstrate that parasite drug susceptibility for most (>95%) of the asexual life cycle depends on the differential susceptibility of relatively insensitive rings and sensitive trophozoites/schizonts. This difference manifests itself as an extended lag time in the drug response of rings, which reflects their higher 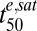 values. The lower
values. The lower  values of trophozoites (Table 1) are consistent with a proposed mechanism of action of ARTs that posits a requirement for drug activation by iron-containing species (21). In particular, an increase in EDmax (∝ 1/
values of trophozoites (Table 1) are consistent with a proposed mechanism of action of ARTs that posits a requirement for drug activation by iron-containing species (21). In particular, an increase in EDmax (∝ 1/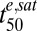 ) in trophozoites is expected due to increased flux and steady-state levels of an iron-containing activator arising from hemoglobin degradation (5).
) in trophozoites is expected due to increased flux and steady-state levels of an iron-containing activator arising from hemoglobin degradation (5).
We also identified a subpopulation of rings (2–4 h p.i.) that defies the above trends by exhibiting hypersensitivity to short drug pulses. The low 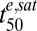 values of hRings cannot be explained by increased hemoglobin degradation, indicating this hypersensitivity involves a different as yet unidentified mechanism of action of ARTs that is specific to this class of antimalarials. Clearly, the presence of hRings in a culture containing ring-stage parasites will complicate investigations of drug sensitivity and may be responsible for the variability in reports of the drug sensitivity of ring-stage parasites (8, 10). Similarly some studies of stage-specific effects have used parasites at ∼20 h p.i. and would be expected to show a high degree of variability depending on the degree of synchronicity and the precise age distribution of parasites during the course of an assay (Fig. 1 A and B).
values of hRings cannot be explained by increased hemoglobin degradation, indicating this hypersensitivity involves a different as yet unidentified mechanism of action of ARTs that is specific to this class of antimalarials. Clearly, the presence of hRings in a culture containing ring-stage parasites will complicate investigations of drug sensitivity and may be responsible for the variability in reports of the drug sensitivity of ring-stage parasites (8, 10). Similarly some studies of stage-specific effects have used parasites at ∼20 h p.i. and would be expected to show a high degree of variability depending on the degree of synchronicity and the precise age distribution of parasites during the course of an assay (Fig. 1 A and B).
Current within-host pharmacokinetic–pharmacodynamic models of antimalarial activity do not incorporate lag times of drug action (7). A recent study of DHA response in Plasmodium berghei allowed for a delay in the onset of parasite killing but did not delineate stage-dependent effects (22). Our semiempirical cumulative ED model accounts for the complex in vitro dependence of parasite viability on drug concentration and exposure time and permits the interpretation of stage- and strain-dependent differences in drug action in terms of easy to understand underlying parameters (e.g., EDmax and Km). We also demonstrated the utility of this model as the basis for performing simulations of in vitro and in vivo drug response profiles. In the case of the ARTs, ED is defined in terms of a saturable process. This definition, as well as the sigmoidal nature of the V-EDcum relationship, is likely applicable to other systems. We believe that the general concepts used here can also be extended to other systems exhibiting nonsaturable EDs and alternative V-EDcum relationships.
Our simulations show that the extended lag time associated with the action of ARTs against ring-stage parasites will result in a small but significant fraction of rings surviving single-dose treatment. Clinically, ARTs are coadministered with long-acting partner drugs (2), and any surviving parasites would usually be killed by the partner drug and/or by the use of multiple-dose regimens. We demonstrate that relatively small changes in the response of parasites to ARTs could result in a level of residual viability that could lead to treatment failure. Importantly, our simulations of in vitro assays show that clinically relevant differences in the responses of strains may not be picked up in standard assays. Even when short pulse assays are used it may not be sufficient to measure a single parameter such as LD50(te) that represents an average property of the parasite population. We suggest measuring multiple parameters such as LD50(te) and Vmin(te) when screening for resistant isolates in the field.
It is important to note that the approaches we have used do not address the question of whether subpopulations of dormant or tolerant parasites (23–25) are involved in resistance to ARTs. The ability of a subpopulation of parasites to enter a quiescent phase could contribute to artemisinin resistance; however, there is no need to invoke the presence of such subpopulations to explain the sensitivity differences that we observe; the behavior is that of the population as a whole and represents the stochastic drug response of the parasites.
We show that common laboratory strains exhibit substantial differences in their sensitivities to QHS and DHA, when administered as single-pulse treatments. 7G8 has a mutant pfmdr1 gene that confers resistance to chloroquine but moderately enhanced sensitivity to hydrophobic drugs such as mefloquine and QHS in standard assays (14). By contrast, chloroquine-sensitive 3D7 and D10 possess a wild-type pfmdr1 gene that is associated with moderately lower sensitivity to mefloquine and QHS (13–15). The very small differences in ART sensitivity observed using standard assays are markedly amplified using our short pulse assays.
Mutations in the pfmdr1 gene and changes in pfmdr1 copy number clearly affect the in vitro response to ARTs; however, pfmdr1 has proved of limited use as a molecular marker of resistance to ARTs in the field (26, 27). This indicates that field resistance is much more complex than mutations in a single gene product. Indeed, recent studies suggest that resistance in P. falciparum is associated with an altered temporal pattern of transcription (28) and may be linked to a region on chromosome 13 (29). In vitro selection of resistance to artemisinins also points to the involvement of a range of different genes (23, 30). We predict that different molecular changes will affect the responses of different parasite stages, but that in many cases the alterations will affect the length of the lag time of the response, for example by decreasing the reactive iron level or the level of a downstream target molecule.
In summary, the short pulse assays we have developed, coupled with our unique analytical methods, facilitate the identification of parasites with different levels of sensitivity to ARTs. We anticipate that successful application of these assays to the analysis of field strains will aid efforts to monitor and combat drug resistance. Identifying in vitro correlates of the phenotypic alteration that lead to prolonged clearance times in patients is critical to global efforts to battle malaria.
Materials and Methods
Culturing and Tight Synchronization of Parasites.
To generate tightly synchronized 3D7, 7G8, and D10 parasites (31), cultures were presynchronized to an 8- to 10-h window with two sorbitol treatments (5). The culture was monitored and the schizonts harvested using a 70% Percoll cushion (32) when the ring:schizont ratio was between 2:1 and 6:1 or when the flux of ring formation was judged to be sufficient for tight synchronization. The harvested schizonts were added to precultured red blood cells (RBCs; 5% hematocrit) and incubated for 0.5–1.5 h on a shaker to maximize the generation of singly infected rings. Excess schizonts were removed by loading the culture on a VarioMACS system (Miltenyi Biotec) and collecting the flow-through. The tightly synchronized ring culture was washed and adjusted to 1–2% parasitemia, 1–4% hematocrit and incubated on a shaker. The quality of the synchronization was judged by measuring the percentage of rings immediately after the addition of schizonts to RBCs and following the collection of rings from the magnetic column. We define the fractional difference of these two parameters as the quality of the synchronization.
Drug Pulse Assays.
Drugs were serially diluted in complete medium in v-bottomed microplates. The culture was added to the drugs (0.2% final hematocrit; 1–2% final parasitemia) and incubated for 1–6 h before washing three to five times with 200 µL of complete medium. The cultures were incubated normally until assessment of parasitemia. Unwashed samples containing drugs at >10× the LD50(3 d) concentration acted as background controls for 100% parasite killing. All assays were performed in duplicate.
Parasite viability was determined by measuring parasitemia in the cycle (cycle 2) following drug treatment by flow cytometry, using SYTO 61 (31). Culture medium was removed, and the cell pellets were resuspended in 20 µL SYTO 61 (2 µM in PBS), incubated 15 min at room temperature, diluted to 200 µL, and incubated for 0.5–2 h before being measured. In cases where a significant fraction of rings from the parasite cycle following synchronization were present during the drug pulse, the cultures were split fivefold with RBCs and the parasitemia measured in cycle 3. Parasite viability (V) was calculated using the measured parasitemia (Pt):
Standard Drug Assays.
Asynchronous cultures containing rings were subjected to a single sorbitol treatment and assayed as above without removal of the drugs and with replenishment of the medium and drug after 24 h. Flow cytometry was performed ∼3 d (64–80 h) after the start of the assay.
Analytical Methods.
Nonlinear regression models were fitted to the data, using the nls function in the R Project statistical software package (33) or in Microsoft Excel using the Solver add-in. LD50 values correspond to the drug concentration (i.e., the lethal dose) producing 50% parasite viability in a particular assay and were determined by fitting a simple sigmoidal function to the data.
Age Range of Parasites During the Course of a Drug Pulse.
In this work, LD50 values from drug pulse assays are presented as a function of the range of ages of ≥50% of the parasite population during the course of the drug pulse. For a culture synchronized as described above this range corresponds to the average p.i. age of parasites at the beginning and end of the assay. For a culture assayed during the schizont–ring transition, the range of ring ages was calculated by first measuring the appearance of the next generation of rings by flow cytometry and fitting an empirical sigmoidal model to the data (e.g., SI Appendix, Fig. S1). During the course of an assay running from start time ts to end time te (with time 0 defined as the time when 50% of the total rings are formed), the time-averaged probability density of rings of age a is given by
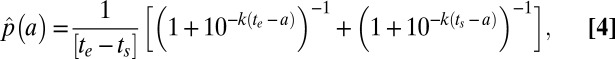 |
where k is the skewness of the sigmoidal function (see SI Appendix for full derivation). Thus, for each assay, the interquartile range of the time-averaged probability density of the age of the parasites corresponds to the range of ages of ≥50% of the parasite population.
Note Added in Proof.
While this paper was under review, Witkowski et al. (34) reported that ring stage parasites isolated from the Pailin region in Cambodia show increased viability following a 6 h exposure to 700 nM DHA, consistent with our predictions.
Supplementary Material
Acknowledgments
We thank Sam Deed, Heather Lewis, and Shannon Kenny, University of Melbourne, for technical support and Angela McLean, Oxford University, for discussions. We acknowledge support from the Australian Research Council and the Australian National Health and Medical Research Council.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
See Commentary on page 4866.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1217452110/-/DCSupplemental.
References
- 1.White NJ. Qinghaosu (artemisinin): The price of success. Science. 2008;320(5874):330–334. doi: 10.1126/science.1155165. [DOI] [PubMed] [Google Scholar]
- 2.Enserink M. Malaria’s drug miracle in danger. Science. 2010;328(5980):844–846. doi: 10.1126/science.328.5980.844. [DOI] [PubMed] [Google Scholar]
- 3.Dondorp AM, et al. Artemisinin resistance in Plasmodium falciparum malaria. N Engl J Med. 2009;361(5):455–467. doi: 10.1056/NEJMoa0808859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Phyo AP, et al. Emergence of artemisinin-resistant malaria on the western border of Thailand: A longitudinal study. Lancet. 2012;379(9830):1960–1966. doi: 10.1016/S0140-6736(12)60484-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Klonis N, et al. Artemisinin activity against Plasmodium falciparum requires hemoglobin uptake and digestion. Proc Natl Acad Sci USA. 2011;108(28):11405–11410. doi: 10.1073/pnas.1104063108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Abu Bakar NA, Klonis N, Hanssen E, Chan C, Tilley L. Digestive-vacuole genesis and endocytic processes in the early intraerythrocytic stages of Plasmodium falciparum. J Cell Sci. 2010;123(Pt 3):441–450. doi: 10.1242/jcs.061499. [DOI] [PubMed] [Google Scholar]
- 7.Saralamba S, et al. Intrahost modeling of artemisinin resistance in Plasmodium falciparum. Proc Natl Acad Sci USA. 2011;108(1):397–402. doi: 10.1073/pnas.1006113108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.ter Kuile F, White NJ, Holloway P, Pasvol G, Krishna S. Plasmodium falciparum: In vitro studies of the pharmacodynamic properties of drugs used for the treatment of severe malaria. Exp Parasitol. 1993;76(1):85–95. doi: 10.1006/expr.1993.1010. [DOI] [PubMed] [Google Scholar]
- 9.Benoit-Vical F, et al. Trioxaquines are new antimalarial agents active on all erythrocytic forms, including gametocytes. Antimicrob Agents Chemother. 2007;51(4):1463–1472. doi: 10.1128/AAC.00967-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Skinner TS, Manning LS, Johnston WA, Davis TM. In vitro stage-specific sensitivity of Plasmodium falciparum to quinine and artemisinin drugs. Int J Parasitol. 1996;26(5):519–525. doi: 10.1016/0020-7519(96)89380-5. [DOI] [PubMed] [Google Scholar]
- 11.German PI, Aweeka FT. Clinical pharmacology of artemisinin-based combination therapies. Clin Pharmacokinet. 2008;47(2):91–102. doi: 10.2165/00003088-200847020-00002. [DOI] [PubMed] [Google Scholar]
- 12.Eastman RT, Fidock DA. Artemisinin-based combination therapies: A vital tool in efforts to eliminate malaria. Nat Rev Microbiol. 2009;7(12):864–874. doi: 10.1038/nrmicro2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Duraisingh MT, Cowman AF. Contribution of the pfmdr1 gene to antimalarial drug-resistance. Acta Trop. 2005;94(3):181–190. doi: 10.1016/j.actatropica.2005.04.008. [DOI] [PubMed] [Google Scholar]
- 14.Reed MB, Saliba KJ, Caruana SR, Kirk K, Cowman AF. Pgh1 modulates sensitivity and resistance to multiple antimalarials in Plasmodium falciparum. Nature. 2000;403(6772):906–909. doi: 10.1038/35002615. [DOI] [PubMed] [Google Scholar]
- 15.Cui L, et al. Mechanisms of in vitro resistance to dihydroartemisinin in Plasmodium falciparum. Mol Microbiol. 2012;86(1):111–128. doi: 10.1111/j.1365-2958.2012.08180.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Duraisingh MT, Roper C, Walliker D, Warhurst DC. Increased sensitivity to the antimalarials mefloquine and artemisinin is conferred by mutations in the pfmdr1 gene of Plasmodium falciparum. Mol Microbiol. 2000;36(4):955–961. doi: 10.1046/j.1365-2958.2000.01914.x. [DOI] [PubMed] [Google Scholar]
- 17.Charman SA, et al. Synthetic ozonide drug candidate OZ439 offers new hope for a single-dose cure of uncomplicated malaria. Proc Natl Acad Sci USA. 2011;108(11):4400–4405. doi: 10.1073/pnas.1015762108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Paguio MF, Bogle KL, Roepe PD. Plasmodium falciparum resistance to cytocidal versus cytostatic effects of chloroquine. Mol Biochem Parasitol. 2011;178(1–2):1–6. doi: 10.1016/j.molbiopara.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Krishna S, White NJ. Pharmacokinetics of quinine, chloroquine and amodiaquine. Clinical implications. Clin Pharmacokinet. 1996;30(4):263–299. doi: 10.2165/00003088-199630040-00002. [DOI] [PubMed] [Google Scholar]
- 20.Geary TG, Divo AA, Jensen JB. Stage specific actions of antimalarial drugs on Plasmodium falciparum in culture. Am J Trop Med Hyg. 1989;40(3):240–244. doi: 10.4269/ajtmh.1989.40.240. [DOI] [PubMed] [Google Scholar]
- 21.Meshnick SR, Taylor TE, Kamchonwongpaisan S. Artemisinin and the antimalarial endoperoxides: From herbal remedy to targeted chemotherapy. Microbiol Rev. 1996;60(2):301–315. doi: 10.1128/mr.60.2.301-315.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Patel K, et al. Mechanism-based model of parasite growth and dihydroartemisinin pharmacodynamics in murine malaria. Antimicrob Agents Chemother. 2013;57(1):508–516. doi: 10.1128/AAC.01463-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tucker MS, Mutka T, Sparks K, Patel J, Kyle DE. Phenotypic and genotypic analysis of in vitro-selected artemisinin-resistant progeny of Plasmodium falciparum. Antimicrob Agents Chemother. 2012;56(1):302–314. doi: 10.1128/AAC.05540-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Teuscher F, et al. Artemisinin-induced dormancy in plasmodium falciparum: Duration, recovery rates, and implications in treatment failure. J Infect Dis. 2010;202(9):1362–1368. doi: 10.1086/656476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Witkowski B, et al. Increased tolerance to artemisinin in Plasmodium falciparum is mediated by a quiescence mechanism. Antimicrob Agents Chemother. 2010;54(5):1872–1877. doi: 10.1128/AAC.01636-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bustamante C, et al. In vitro-reduced susceptibility to artemether in P. falciparum and its association with polymorphisms on transporter genes. J Infect Dis. 2012;206(3):324–332. doi: 10.1093/infdis/jis359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.O’Brien C, Henrich PP, Passi N, Fidock DA. Recent clinical and molecular insights into emerging artemisinin resistance in Plasmodium falciparum. Curr Opin Infect Dis. 2011;24(6):570–577. doi: 10.1097/QCO.0b013e32834cd3ed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mok S, et al. Artemisinin resistance in Plasmodium falciparum is associated with an altered temporal pattern of transcription. BMC Genomics. 2011;12:391. doi: 10.1186/1471-2164-12-391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cheeseman IH, et al. A major genome region underlying artemisinin resistance in malaria. Science. 2012;336(6077):79–82. doi: 10.1126/science.1215966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chavchich M, et al. Role of pfmdr1 amplification and expression in induction of resistance to artemisinin derivatives in Plasmodium falciparum. Antimicrob Agents Chemother. 2010;54(6):2455–2464. doi: 10.1128/AAC.00947-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fu Y, Tilley L, Kenny S, Klonis N. Dual labeling with a far red probe permits analysis of growth and oxidative stress in P. falciparum-infected erythrocytes. Cytometry A. 2010;77(3):253–263. doi: 10.1002/cyto.a.20856. [DOI] [PubMed] [Google Scholar]
- 32.Ranford-Cartwright LC, Sinha A, Humphreys GS, Mwangi JM. New synchronization method for Plasmodium falciparum. Malar J. 2010;9:170. doi: 10.1186/1475-2875-9-170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. R Development_Core_Team (2011) R: A Language and Environment for Statistical Computing (R Foundation, Vienna)
- 34.Witkowski B, et al. Reduced artemisinin susceptibility of Plasmodium falciparum ring stages in western Cambodia. Antimicrob Agents Chemother. 2013;57(2):914–923. doi: 10.1128/AAC.01868-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.