

Significance
This paper reports a large-scale collaborative study of an approach for predicting the function of chain elongation prenyltransferases from genetic data. A diverse set of genes for enzymes in the isoprenoid synthase superfamily was identified for cloning, expression, X-ray structural analysis, and prediction of function by docking to homology models. Blind predictions, later verified biochemically, were accurate to within one isoprene unit for all but a few of the 74 enzymes studied, an extraordinarily high level of prediction given that the enzymes often give products whose chain lengths vary by one isoprene unit.
Keywords: chain-elongation, prenyltransferase
Abstract
The number of available protein sequences has increased exponentially with the advent of high-throughput genomic sequencing, creating a significant challenge for functional annotation. Here, we describe a large-scale study on assigning function to unknown members of the trans-polyprenyl transferase (E-PTS) subgroup in the isoprenoid synthase superfamily, which provides substrates for the biosynthesis of the more than 55,000 isoprenoid metabolites. Although the mechanism for determining the product chain length for these enzymes is known, there is no simple relationship between function and primary sequence, so that assigning function is challenging. We addressed this challenge through large-scale bioinformatics analysis of >5,000 putative polyprenyl transferases; experimental characterization of the chain-length specificity of 79 diverse members of this group; determination of 27 structures of 19 of these enzymes, including seven cocrystallized with substrate analogs or products; and the development and successful application of a computational approach to predict function that leverages available structural data through homology modeling and docking of possible products into the active site. The crystallographic structures and computational structural models of the enzyme–ligand complexes elucidate the structural basis of specificity. As a result of this study, the percentage of E-PTS sequences similar to functionally annotated ones (BLAST e-value ≤ 1e−70) increased from 40.6 to 68.8%, and the percentage of sequences similar to available crystal structures increased from 28.9 to 47.4%. The high accuracy of our blind prediction of newly characterized enzymes indicates the potential to predict function to the complete polyprenyl transferase subgroup of the isoprenoid synthase superfamily computationally.
The five-carbon molecules isopentenyl diphosphate (IPP) and dimethylallyl diphosphate (DMAPP) are the fundamental building blocks for isoprenoid compounds. Beginning with DMAPP, a series of polyprenyl diphosphates with C10 (geranyl diphosphate, GPP), C15 (farnesyl diphosphate, FPP), C20 (geranylgeranyl diphosphate, GGPP), C25 (farnesylgeranyl diphosphate, FGPP), and higher molecular weight isoprenoid chains are synthesized by polyprenyl transferases (PTSs). With only a few exceptions, PTSs provide substrates for all but a few branch point enzymes for the biosynthesis of the more than 55,000 known isoprenoid metabolites, including monoterpenes, sesquiterpenes, diterpenes, sterols, carotenoids, ubiquinones, and prenylated proteins and peptides fulfilling essential roles in cells (Fig. S1) (1, 2).
There are two distinct classes of PTSs, E-PTS forming trans bonds and Z-PTS forming cis bonds throughout chain elongation. The carbon skeletons of the great majority of isoprenoid metabolites are derived from products of E-PTSs, which share a common protein fold and two functionally important Asp-rich (DDXXD) motifs (1, 3, 4). E-PTSs synthesize linear allylic diphosphates ranging from C10 to C50, where functional assignment of these enzymes is linked to the chain length of their respective predominant products under nonforced conditions. As revealed by a variety of high-quality crystal structures, the enzymes typically are homodimers (5–7). In each monomer, the allylic binding region (S1) in the active site contains three Mg2+ ions ligated by two pairs of aspartates from both Asp-rich regions, which in turn holds the diphosphate of the allylic substrate in place, as illustrated with DMAPP in Fig. 1. The hydrocarbon tail of DMAPP extends into a pocket that accommodates the growing polyisoprenoid chain. The IPP binding region (S2) positions C4 of IPP near C1 of the allylic substrate. The isoprenoid moieties of the substrates are joined by a dissociative electrophilic alkylation initiated by cleavage of the carbon–oxygen bond of the allylic diphosphate to form a carbocation, which then attacks the C3–C4 double bond of allylic IPP to form a tertiary carbocationic intermediate that loses a proton from C2 to give the allylic product (8). The sequential addition of IPP to the growing chain proceeds through release of diphosphate from S1, rearrangement of the product from S2 to S1, and binding of another IPP in S2, followed by the same reaction as before (Fig. 1A). The polyisoprenoid chain grows into the elongation cavity flanked by helices D, F, G, and H of the protein near the dimer interface (Fig. 1 B and C) (7, 9). Following this elongation mechanism, these enzymes are named either according to their final product (e.g., farnesyl diphosphate synthase) or according to the longest ligand being transferred to the final product (in this case, geranyl transferase).
Fig. 1.

Crystal structure of GGPP synthase (PDB ID 1RQI). (A) Active site S1 with DMAPP, Mg2+ ions, and Asp-rich motifs and active site S2 with IPP are highlighted. The electrophilic attack of the C1 atom of DMAPP against the double bond of IPP after cleavage of diphosphate is indicated by the black arrow. (B and C) Side view (B) and top view (C) of the bioactive dimer with the active site and elongation cavity displayed. Helices D–H are identified by capital letters.
Considerable effort has been made to investigate how chain elongation is terminated, establishing that steric hindrance of the growing chain in the elongation cavity is the main factor (1, 3, 9, 10). However, because the number of predicted sequences in the superfamily is so large, structural and enzymatic studies can be performed on only a small fraction of the sequences with likely E-PTS activity. We identified 5,839 such sequences at the initiation of this study in May 2011 (see below). At that time, only 46 individual sequences from 35 UniProt entries were functionally annotated based on published biochemical essays and thus available in the Gene Ontology Annotation (UniProt-GOA) database (11), and 61 had been structurally characterized with crystal structures available in the Protein Data Bank (PDB) database (12). Moreover, the rate of genome sequencing continues to increase exponentially, and even with on-going advances in high-throughput structural biology and in vitro screening methods, the gap between the number of known sequences and the number characterized experimentally will continue to grow. For this reason, reliable methods of inferring function of uncharacterized sequences are urgently needed. The highest-throughput and most widely applied approaches transfer functional annotations from characterized proteins to closely related proteins, because closely related proteins frequently are iso-functional. However, there is no simple sequence-based criterion that can be used universally to define when sequences are similar enough for the inference of iso-functionality to be made confidently (13). In the case of E-PTS enzymes, it has been demonstrated experimentally that chain-length specificity can be changed dramatically by a small number of mutations of key residues lining the elongation cavity (9, 14). Thus, in principle, even very closely related sequences could have different chain-length specificities. Furthermore, the previously characterized E-PTS enzymes were insufficient to allow inferences about the function of many members of this protein family, using any reasonable sequence-similarity cutoff, in part because many of the characterized enzymes were closely related to each other, leaving large regions of “sequence space” completely uncharacterized.
We now describe a large-scale study that integrates available genomics data, in vitro experiments, X-ray crystallography, and computational approaches on a large set of representative members of the E-PTS subgroup to assign and, more importantly, to predict function for uncharacterized sequences. We show that computational tools can play a critical role in functional assignment. We use bioinformatics analysis and sequence clustering for target selection and place a particular emphasis on the use of structural information to make functional inferences. To this end, we determined 27 crystal structures and used these structures (in addition to available structures in the PDB) to create comparative models for 61 PTS enzymes for which structural information is lacking. Structures and models then were used to make predictions of chain-length specificity, using a ligand-docking method that evaluates the steric complementarity between various polyprenyl products and the elongation cavity. This structure-based approach to predicting function is validated through blind predictions on 74 PTS enzymes as well as on a subsequently obtained crystal structure in complex with a long-chain polyprenyl product.
Results
Sequence Analysis and Clustering.
In May 2011 we identified a group of 5,839 sequences from the National Center for Biotechnology Information (NCBI) protein database that, based on sequence and structural information, may have E-PTS activity. These sequences, spanning all domains of life, were stored in the Structure-Function Linkage Database (SFLD) (15) as the polyprenyl transferase-like subgroup. To visualize relationships between members of the subgroup, we generated sequence similarity networks using Pythoscape (16), where nodes represent sequences, and edges represent pairwise local alignments with BLAST e-values more significant than a specified cutoff, allowing a dynamic view of clustering patterns and sequence annotations. Sequence-similarity networks can handle thousands of sequences, are quick to compute and robust to missing data (17, 18), and have been shown to correlate well with phylogenetic trees (19, 20). Fig. 2 shows a sequence-similarity network for the E-PTS subgroup at an e-value cutoff of 1e−50 (with a 41% median sequence identity of 41% and median alignment length of287 residues), including a zoom into the densest region with an e-value cutoff of 1e−70 (median sequence identity of 46% and median alignment length of 309 residues). The map shows all 46 previously functionally characterized enzymes from GOA (small colored nodes) as well as the 79 enzymes characterized in this study (large colored nodes). Furthermore, it indicates crystal structures applied in this study by their PDB identifier. Fig. S2 shows additional networks at 1e−70 that highlight the difference in the information available before and after this study. It can be seen that previously characterized enzymes are concentrated in a small number of clusters, but the majority of the sequence clusters contained no, or only a few characterized entries.
Fig. 2.

Sequence similarity map of the E-PTS subgroup with (A) BLAST e-value cutoff = 1e−50 and (B) zoom at cutoff = 1e−70. Template sequences are tagged by PDB identifiers, and colored sequence nodes indicate experimentally assigned product chain length determined either in this study (large nodes) or previously, based on GOA (small nodes).
To characterize the sequence and structural determinants of chain-length specificity more systematically, we chose 248 sequences for large-scale protein expression and structural characterization. Targets were chosen to maximize distance from those that had solved X-ray structures and/or were functionally annotated. Additionally, targets were chosen preferentially from species with available genomes. Of the 248 sequences, we now have purified and functionally characterized 79 distinct enzymes and have determined 27 crystal structures for 19 of these enzymes.
Determination of in Vitro Biochemical Function of E-PTSs.
In vitro screening for the functions of E-PTSs is simplified by the relatively small number of potential substrates. The polyprenyl transferases require IPP and an allylic isoprenoid diphosphate: DMAPP, GPP, FPP or GGPP (Fig. S1). The preferred allylic substrates and the chain lengths of the products can be determined from incubations of each of the allylic substrates with [14C]IPP, followed by removal of the diphosphate moiety with acid phosphatase and a radio-TLC assay of the resulting isoprenoid alcohols with detection by phosphor imaging (21).
Initial screens were performed using a 4:1 or 10:1 ratio of IPP:DMAPP concentrations. In most cases, these screens gave us clear product(s) on the TLC plates (Dataset S1). Because DMAPP can be a poor substrate for long-chain (≥C40) E-PTSs (6, 22–24), we repeated the screens using GPP or FPP as allylic substrates when production of longer-chain materials was poor or not observed. Typically, similar results were obtained using DMAPP or FPP, as seen in TLCs 8 and 39. Some enzymes required screening with IPP and GPP or FPP, specifically GenBank identification nos. (GIs) 15805956, 291005007, 126460364, 126458776, 29348670, and 149238027 (for example TLCs 31–38). It is known that the ratio of the allylic to homoallylic substrates can affect the length of the product chain. For instance, FPP synthases sometimes can over-elongate to C20/C25 in the presence of an IPP excess. Thus, we reassayed those targets producing C15/C20 (short-chain) products using a 1:1 ratio of IPP:DMAPP. The flexibility of the elongation cavity is even more obvious for the long-chain E-PTSs. In our study, all the long-chain E-PTSs gave at least two or more products. In an extreme case, GI 29840764 produced a broad distribution of isoprenoid diphosphates with chain lengths from C25 to C65 when IPP:FPP = 10:1 (TLC 37). Typically, E- and Z-PTSs elongate selectively to the desired chain length by binding the intermediate products more tightly than the initial allylic primer or the final product (25, 26), as is clearly observed in our assays for long-chain E-PTSs using the IPP:DMAPP (or FPP) ratio of 4:1. The functional annotations of all E-PTS under investigation are given in Tables S8–S10.
Many of the screened enzymes were produced from constructs with N-terminal or C-terminal His tags. In several instances, the presence of a His tag substantially altered the activity of the enzymes and the chain length of the products. Typically, the C terminus in wild-type PTSs is rich in basic amino acids and participates in binding IPP in the enzyme–substrate complex. We found that product distributions of many of the C-His–tagged proteins differed from those of their N-terminal counterparts. The N-His–tagged enzymes gave a less selective distribution of longer chain products, for example GIs 19551716, 16131077, 29376566, 15640461, 39934115, 67866738, 52842862, and 60682991. The C-His–tagged enzymes typically were less active, although there were exceptions. For example, GI 16126352, an N-His–tagged enzyme, was inactive. N- and C-His–tagged short-chain PTSs typically gave the same products. Nevertheless, GI 52842540 bearing a C-His tag elongated to FGPP, whereas the N-His–tagged enzyme gave GGPP. For GI 23308904, both N- and C-His–tagged enzymes elongated to GGPP. However, the GPP-to-FPP step for the C-His–tagged enzyme was slower than the two following steps with a concomitant significant accumulation of GPP (TLC 29). A few targets also were produced as untagged enzymes. For GIs 56551751 and 83945403, both N-His–tagged and untagged enzymes gave FPP as the major product. Similarly for GI 40062988, both C-His–tagged and untagged enzymes gave GGPP as the major product. However, the activities of the untagged enzymes for GIs 83764459, 68489506, and 29840764 were low, perhaps as a result of the repurification/concentration steps after removing the His tag.
GI 153799383 is annotated as polyprenyl synthase (NapT7) from Streptomyces aculeolatus that synthesizes an intermediate in the biosynthesis of the antibiotic napyradiomycin. According to the proposed biosynthetic pathway, it is a putative GPP synthase (27). This identification was confirmed in vitro (TLC 29). However, the enzyme over-elongates up to GGPP in the presence of excess IPP (TLC 3).
GI 118468511 bearing a C-His tag gave a distinctive noncontinuous distribution of products; GGPP was the major product along with a small amount of decaprenyl diphosphate. Substantial amounts of GGPP were formed even when the ratio of IPP:FPP was 10:1 (TLC 38). However, for another short polyprenyl transferase, GI 15640906, the C-His–tagged enzyme produced GGPP, whereas the N-His–tagged protein gave FPP, GGPP, and decaprenyl diphosphate under similar conditions (TLC 26).
Finally, two enzymes in the polyprenyl transferase subgroup, GI 15645545 and GI 116333612, gave unexpected products. In addition to chain elongation, both catalyzed synthesis of presqualene diphosphate by combining two FPP molecules (TLCs 3, 9, and 34). This unexpected activity for these two enzymes currently is under further investigation.
Computational Prediction of Product Specificity.
Simultaneously, we developed and then applied a computational method for predicting chain-length specificity based on structural modeling. We reasoned that it would be possible to determine chain-length specificity by modeling progressively longer polyprenyl chains until they no longer fit.
For initial development and testing, we selected 10 crystal structures of E-PTSs that contained an ordered active site, including ligand and Mg2+ ions in the S1 active site: 1UBW, 1RQI, 2E8W, 3AQ0, 3PDE, 3P41, 3KRF, 3OYR, 3Q1O, and 3QQV. As a simple metric, we calculated the n elongation-cavity volume of the chain for each of the crystal structures with SiteMap (28). However, these volumes do not correlate strongly with the known product chain length (Fig. S3). A possible reason is that flexible side chains in the elongation cavity are not in the correct position for binding a ligand with the longest chain length for that enzyme.
As a consequence, we developed a docking strategy to model polyprenyl chains explicitly into the cavity, allowing side chains in the cavity to adjust their conformations flexibly in response to the ligand. Rather than using standard docking methods, which perform full translational and rotational conformational searches for ligands, we used a method based on a covalent docking algorithm available in the Prime software package. In this approach, we start with the diphosphate head group of the ligands positioned as observed in the crystal structures, coordinated by the magnesium ions. Then, for each chain length, we built the prenyl units in an arbitrary conformation and systematically sampled all the rotatable bonds in the ligand (three per prenyl unit), keeping the phosphate groups fixed. The computational algorithm is described in greater detail in SI Methods. To evaluate whether a given chain length fits within the cavity, we computed the Lennard–Jones energy (ELJ) of the complex; the prediction was based on the lowest (most favorable) ELJ, reflecting a lack of steric clashes, as well as favorable packing with side chains lining the cavity. More complex scoring functions incorporating electrostatics and implicit solvation performed slightly worse on average for this application (Table S8). Because the computational effort of ligand sampling increases exponentially with the number of C5 units, and because enzymes with long-chain polyprenyl products invariably produced multiple products, we stopped chain-length prediction at a ligand length of C25, resulting in product chain-length predictions of C10, C15, C20, C25, and greater than or equal C30 (C≥30).
The results of the computationally predicted and the experimentally determined product chain lengths are shown in Table 1. Predicting the chain length while keeping the protein rigidly fixed in the crystal conformation performed very poorly, with the chain-length specificity underpredicted in almost every case. However, when we allowed the side chains in the elongation cavity to adjust flexibly to the ligands, we correctly predicted the correct product chain lengths for 8 of the 10 structures. The incorrect predictions were generated for PDB 3KRF, which we predicted to be a C15-ase but experiments have shown to be primarily a C10-ase, and PDB 2E8W, for which we predicted a C25 product rather than the experimentally determined C20. 3OYR proves to be a special case. The elongation cavity of its crystal structure is too narrow to accommodate long-chain ligands. A short molecular dynamics run of 5 ns with a short-chain C15 ligand in the S1 active site let the elongation cavity relax, and the subsequent product chain length was predicted correctly to be C≥30. As a consequence, we used the “relaxed” 3OYR instead of the crystal structure when constructing homology models for the prospective predictions (see below).
Table 1.
Training set: Experimentally determined vs. predicted product chain length
| GI | PDB ID | Experimentally determined | Source | Prediction |
|
| ELJ, rigid | ELJ, flex | ||||
| 158979013 | 3KRF | C10 | (29) | C10 | C15 |
| 24111799 | 1RQI | C15 | (8) | C10 | C15 |
| 116333612 | 3PDE | C15 | EFI | C10 | C15 |
| 15645545 | 3Q1O | C15/C20 | EFI | C10 | C15 |
| 70732810 | 3P41 | C15/C20 | EFI | C10 | C20 |
| 218766585 | 2E8W | C20 | (30) | C15 | C25 |
| 23308904 | 3QQV | C20 | EFI | C10 | C20 |
| 319443461 | 3AQ0 | C35 | (6) | C20 | C≥30 |
| 3915686 | 1UBW | C35/C40 | (9) | C20 | C≥30 |
| 16126352 | 3OYR | C50-C60 | EFI | C10* | C20*/C≥30† |
*Crystal structure of 3OYR.
†Relaxed crystal structure of 3OYR.
Structures are available for only a very small fraction of isoprenoid diphosphate synthases, and in most cases these have been functionally characterized, so the practical application of this approach to predicting function requires the use of homology models. As a more realistic test, we compiled a larger set of 34 sequences (set targetsknown) that were screened and functionally assigned before the computational part of this study was completed. For 10 of these sequences, we also obtained apo crystal structures. To use these structures for the docking procedure, we manually placed diphosphate and Mg2+ ions, based on their positions in holo structures, and adjusted the conformations of the aspartates ligating the ions. For the 24 sequences with no structures, we created homology models based on templates with sequence identity as low as 34%. A plot of the error in the product-length prediction, in terms of C5 units, for our predictions compared with experiments vs. the sequence identity of the homology models used is given in Fig. 3A. The predictions were precisely correct for only 53% (18/34) of the test cases. However, nearly all the remaining predictions (38%, 13/34) are within one C5 unit of the experimentally assigned product specificity. It also should be noted that the experimental assignment of product-length specificity frequently is somewhat ambiguous, in that multiple products sometimes are observed, and the distribution of products varies according to the experimental conditions, as discussed above. The predictions were grossly incorrect for only 3/34 (9%) of the sequences (Table S9). As a point of reference, we compare these structure-based predictions with the annotations provided in UniProt Knowledgebase/Translated EMBL Nucleotide Sequence Data Library (UniProtKB/TrEMBL) (31), which are obtained using automated methods, in contrast to the manually reviewed annotations available from UniProt-GOA, which have been shown to be of especially high quality (32). The TrEMBL annotations are correct for 53% (18/34) of the cases, similar to the modeling-based predictions, but a much larger fraction, 32% (11/34), was assigned incorrectly (more than one C5 unit in error) or was annotated vaguely (i.e., “polyprenyl synthase”) so that the product chain length could not be inferred.
Fig. 3.

Error in C5 units of computationally predicted compared vs. experimentally determined product chain length for (A) training set and targetsknown, and (B) targetsblind. Circles represent predictions using homology models constructed based on holo crystal structure templates; triangles represent apo structures or homology models based on apo structures. The larger symbols indicate multiple predictions that have the same sequence identity and prediction error.
Having benchmarked the computational method using data generated in this project as well as previously published data, we next performed a blind prediction of product chain length for a set of 40 sequences (set targetsblind) for which we did not know the functional assignment during the computational prediction. At the time of the predictions, crystal structures were available for only three sequences; for the remaining sequences we constructed homology models based on sequence identity to existing structures as low as 29%. The modeling approach resulted in correct chain-length prediction (based on experimental results provided in Table S10) for 65% (26/40) of the sequences, and again, most of the other predictions (12/40, 30%) were within one C5 unit. The predictions were grossly incorrect for only 5% (2/40) of the sequences. For comparison, available functional annotations in TrEMBL are correct for only 45% (18/40), correct within one C5 unit for 15% (6/40), and incorrect or unclear for 40% (16/40) of the 40 target sequences. Detailed results are given in Table S10 and Fig. 3B.
After the creation of the homology model and chain-length prediction of GI 126460364, its crystal structure (PDB 4FP4) was solved, which includes a ligand bound in the elongation cavity, which was interpreted as a partial C20 product. The superposition of the crystal structure and the homology model with the predicted C20, given in Fig. 4, highlights the accuracy of the homology model and the ligand modeling, both with an rmsd of 2.0 Å calculated on heavy atoms present in both structures.
Fig. 4.

Superposition of the 4FP4 crystal structure and the homology model of the same protein based on PDB 3AQ0 with 29% sequence identity, created before the structure was available. The computationally predicted ligand conformation is shown in red, and side chains of the elongation cavity are in orange. The partial ligand observed in the crystal structure is shown in green, and the elongation-cavity side chains are in blue.
Structural Determinants of Specificity.
In total, we built ligand-receptor models for 79 E-PTS sequences. The predicted ligands in the 52 cases for which we predicted the precisely correct chain length are superimposed and displayed together with the general E-PTS fold in Fig. 5. The results show that the polyprenyl chains generally follow a similar cavity between helices D, F, G, and H, but there are some differences, particularly for the termini of the longer-chain products. These FGPPs, shown in green in Fig. 5, extend either between helices F and G of their own chain or into the dimer interface and eventually between helices F and G of the opposite chain.
Fig. 5.
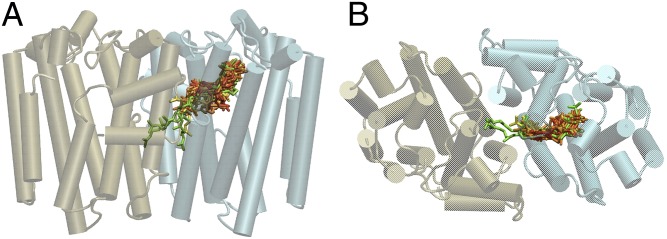
(A) Side and (B) top view of E-PTS fold with superposition of all correctly predicted ligands colored according to their chain length. Red, GPP; orange, FPP; yellow, GGPP; green, FGPP.
Although we did not attempt to assign chain-length specificity greater than C30 in the prospective predictions, we also created a model of a C50 ligand in the structure 3OYR (Fig. 6). To do so, we performed the prediction in stages, starting from the prospective prediction of the C25 ligand in the elongation cavity and then subsequently elongating this prediction to C50, keeping the first 20 carbon atoms fixed in this second round of prediction. Each chain of 3OYR has two bulky residues (Phe127 and Phe131) localized in the elongation cavity, but the growing ligand in the computational experiment is able to displace these side chains, providing a favorable hydrophobic environment for the polyprenyl tail of the ligand. Our results indicate that the ligand exit point on the protein surface is not between helices D, F, G, and H but instead is between the dimer interface, as suggested previously (9). The model also predicts that products longer than C50 would extend outside the protein.
Fig. 6.

(A) Computational model of a C50 ligand (red) in the elongation channel of polyprenyl transferase 3OYR. The cavity volume is colored according to partial charges of surrounding residues, with neutral (hydrophobic) shown in green, negative in red, and positive in blue. (B) Conformational changes of residues in the elongation channel through displacement by the ligand (chains A and B of crystal structure 3OYR are shown in blue and maroon, respectively; chains A and B of the long-chain model of 3OYR are shown in orange and yellow, respectively; C50 is shown in red). (C) Superposition of the predicting binding modes of C25 in 3OYR (orange) and C15 in a structural model of protein GI 126458776 (green).
Mapping of Function to Sequence Space.
Most E-PTSs form iso-functional clusters when clustered by their mutual sequence similarity using a stringent BLAST e-value cutoff of 1e-70 (Fig. S2). Exceptions include cluster 3 and cluster 7, highlighted by red dashed circles in Fig. S2. Within both clusters, the characterized enzymes all have long-chain products, ranging from C30 to C55. GI 126458776 is assigned as a medium-chain C20-ase that is dissimilar to other short-or medium-chain polyprenyl transferases. Although TrEMBL does not assign a specific product chain length to this enzyme, we correctly predicted it to be a C20-ase, although its closest template is the long-chain E-PTS 3OYR with a sequence identity of 32%. The superposition of covalently docked C25 in 3OYR and C15 in the structural model of GI 126458776 (Fig. 6C) demonstrates the chain-length termination mechanism of the medium-chain compared with the long-chain enzyme. As described above, 3OYR has two phenylalanines located in the chain elongation cavity, whose side chains can be displaced by the longer-chain ligands. The small side chains of neighboring residues (Ala90, Ala147 and Ser150) are important for providing space for the displaced phenylalanine side chains. In contrast, GI 126458776 does not allow displacement of the ligand-hindering side chains (Arg115) because of large side chains of its neighboring residues (Tyr78 and Glu119). Consequently, its elongation cavity is capped at the range of a C15 ligand, as highlighted in green in Fig. 6C.
Discussion
Isoprenoid carbon skeletons are constructed from the fundamental building blocks IPP and DMAPP by enzymes that catalyze chain elongation, cyclopropanation, rearrangement, and cyclization reactions. Chain elongation forms the trunk of the biosynthetic tree with cyclopropanation, rearrangement, cyclization, and prenyl transfer to nonisoprenoid substrates located at branch points to all the different classes of isoprenoid metabolites. Thus, the chain-elongation enzymes provide the C10 (monoterpenes), C15 (sesquiterpenes, sterols, heme A, farnesylated proteins), C20 (diterpenes, carotenoids, geranylgeranylated proteins, chlorophyll), and C30–C50 (ubiquinones, menaquinones) as well as other chain lengths for less common metabolites. Of the 5,839 putative DNA sequences for polyprenyl transferase enzymes identified here, 5,240 are contained in UniProtKB, and of these only 3,330 (63.5%) are functionally annotated with a specific chain length. However, a substantial proportion of those annotations are incorrect because they are only inferred from sequence homology. Especially problematic for improving annotation across the polyprenyl transferase subgroup, only 46 sequences (0.8%) have been characterized biochemically. A reliable method for determining chain-length specificity is important for predicting the types of isoprenoid compounds in the metabolome of an organism, determining their subcellular location, and studying regulation of their biosynthesis.
We predicatively have assigned function to the large class of E-PTS in the isoprenoid superfamily by a combined approach of bioinformatics and computational modeling with accuracies of 62% for correctly predicted function and 94% for correctly predicted function within one C5 unit. The accuracy of these predictions should be interpreted in the context of known problems for experimental determination of functions in this subgroup of proteins. For example, many short-chain and most medium- and long-chain E-PTSs do not synthesize a single product exclusively but instead produce a series of linear isoprenoid diphosphates (16, 31–33) and thus are especially challenging even for in vitro determination of function. Moreover, the distribution of chain lengths is sensitive to protein tags and can be driven to longer chain length by higher ratios of IPP/allylic substrate. Thus, our approach to predicting function lies within a range of precision similar to that obtained by in vitro experiments.
We have shown that chain-length specificity in E-PTSs is caused mainly by steric hindrance of the ligand within the elongation cavity but also reflects the ability of side chains to move out of the ligand’s way (Fig. 6 B and C). Consequently, we expect second-shell residues of the elongation cavity to affect chain-length specificity. Our work can be seen as a successful case study toward the more complicated functional assignment of terpene cyclases (33). Finally, even for the relatively simple case of chain-length specificity in the E-PTSs, sequence similarity alone is not a sufficient criterion for precise functional prediction, as shown by the relatively low accuracy of automated function predictions provided by TrEMBL. Likewise, many of the clusters shown in Fig. 2 clearly are not iso-functional, suggesting that functional boundaries do not correlate well with sequence similarity. The plot also supports the view that evolution of isoprenoid synthases was rapid and predominately divergent but was compounded by instances of convergent evolution (34, 35), as exemplified by the different FPP synthases spread over the sequence space. A well-known example that further illustrates the complex nature of sequence-function mapping in this group of proteins is the study of Tarshis et al. (9) on avian FPP synthase, where the mutation of only two residues in the elongation cavity changed the product profile from C15 to C35.
Another important aspect of this study has been the careful selection of targets for function assignment, which has resulted in 79 additional assignments, compared with the 46 previously available through GOA, as well as 27 additional crystal structures of 19 E-PTS. As a result of this study, the percentage of E-PTS sequences similar to functionally annotated ones (BLAST e-value ≤ 1e−70) increased from 40.6 to 68.8%, and the percentage of sequences similar to available crystal structures increased from 28.9 to 47.4%. Currently, efforts are underway to solve more holo crystal structures and to assign function experimentally in other subgroups of the isoprenyl synthase superfamily and E-PTSs in particular.
Methods
Experimental Methods.
Detailed information on methods of crystallography, protein expression and purification, determination of in vitro chain length, enzyme activity assays, and products analysis is given in the SI Methods and Tables S1–S7.
Computational Methods.
The selection of the set of proteins that defines the E-PTS subgroup is outlined in SI Methods. All sequences and their associated metadata were added to the SFLD (http://sfld.rbvi.ucsf.edu) and can be accessed using the GI and PDB identifiers reported in this article. Look-up in the SFLD will provide the link to the Enzyme Function Initiative (EFI) (36) and its experimental database (EFI-DB: http://kiemlicz.med.virginia.edu/efi/), where specific experimental information can be viewed also for each target. Visualization of the sequences was performed in Cytoscape (37) using thresholded networks in which edges correspond to worst reciprocal BLAST e-values.
The template structures used for homology modeling were prepared with Schrodinger Protein Preparation Wizard before being used for ligand docking with Prime (38). Here, the diphosphate group of the ligand is kept frozen in the S1 active site while the tail is docked flexibly into the elongation cavity. Side chains of residues within the vicinity (5 Å) of the elongation cavity are treated as conformationally flexible. The modeled ligands are DMAPP, GPP, FPP, GGPP, and FGPP, ranging from 5–25 carbon atoms. The ELJ of the complex and the molecular mechanics/generalized-Born surface area (MMGBSA) binding energy Ebind (SI Methods) are computed for each of the top three models of each docking run and are applied as the two distinct scoring functions for the chain-length prediction. For each PTS, the docked ligand with the lowest relative energy score is considered the reactant of the last step of prenylation; thus the actual product of the reaction specific for this enzyme is predicted to be one C5 unit longer. Homology models are built with Prime from alignments derived from PROMALS3D (39) using the closest (highest sequence identity) template available.
Supplementary Material
Acknowledgments
Crystal structure 3MZV was determined with the help of Christine E. Quartararo at Albert Einstein College of Medicine, New York. This research was part of the Enzyme Function Initiative supported by the National Institutes of Health Grant U54 GM093342.
Footnotes
Conflict of interest statement: M.J.P. is a consultant for Schrodinger LLC, a company whose software was used for a part of the study.
Data deposition: Crystallography, atomic coordinates, and structure factors reported in this paper have been deposited in the Protein Data Bank, www.pdb.org (PDB ID codes 3LOM, 3LVS, 3MZV, 3NF2, 3OYR, 3PDE; 3PKO; 3P8L, 3P8R, 3P41, 3QQV, 3Q1O, 3Q2Q, 3RMG, 3TS7, 3UCA, 4FP4, 4F62, 4DHD).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1300632110/-/DCSupplemental.
References
- 1.Kellogg BA, Poulter CD. Chain elongation in the isoprenoid biosynthetic pathway. Curr Opin Chem Biol. 1997;1(4):570–578. doi: 10.1016/s1367-5931(97)80054-3. [DOI] [PubMed] [Google Scholar]
- 2.Sacchettini JC, Poulter CD. 1997. Creating isoprenoid diversity. Science 277(5333):1788–1789.
- 3.Liang PH. Reaction kinetics, catalytic mechanisms, conformational changes, and inhibitor design for prenyltransferases. Biochemistry. 2009;48(28):6562–6570. doi: 10.1021/bi900371p. [DOI] [PubMed] [Google Scholar]
- 4.Liang PH, Ko TP, Wang AH. Structure, mechanism and function of prenyltransferases. Eur J Biochem. 2002;269(14):3339–3354. doi: 10.1046/j.1432-1033.2002.03014.x. [DOI] [PubMed] [Google Scholar]
- 5.Kainou T, et al. Dimer formation of octaprenyl-diphosphate synthase (IspB) is essential for chain length determination of ubiquinone. J Biol Chem. 2001;276(11):7876–7883. doi: 10.1074/jbc.M007472200. [DOI] [PubMed] [Google Scholar]
- 6.Hsieh FL, Chang TH, Ko TP, Wang AH. Structure and mechanism of an Arabidopsis medium/long-chain-length prenyl pyrophosphate synthase. Plant Physiol. 2011;155(3):1079–1090. doi: 10.1104/pp.110.168799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.No JH, et al. Lipophilic analogs of zoledronate and risedronate inhibit Plasmodium geranylgeranyl diphosphate synthase (GGPPS) and exhibit potent antimalarial activity. Proc Natl Acad Sci USA. 2012;109(11):4058–4063. doi: 10.1073/pnas.1118215109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hosfield DJ, et al. Structural basis for bisphosphonate-mediated inhibition of isoprenoid biosynthesis. J Biol Chem. 2004;279(10):8526–8529. doi: 10.1074/jbc.C300511200. [DOI] [PubMed] [Google Scholar]
- 9.Tarshis LC, Proteau PJ, Kellogg BA, Sacchettini JC, Poulter CD. Regulation of product chain length by isoprenyl diphosphate synthases. Proc Natl Acad Sci USA. 1996;93(26):15018–15023. doi: 10.1073/pnas.93.26.15018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chang TH, Guo RT, Ko TP, Wang AH, Liang PH. Crystal structure of type-III geranylgeranyl pyrophosphate synthase from Saccharomyces cerevisiae and the mechanism of product chain length determination. J Biol Chem. 2006;281(21):14991–15000. doi: 10.1074/jbc.M512886200. [DOI] [PubMed] [Google Scholar]
- 11.Camon E, Barrell D, Lee V, Dimmer E, Apweiler R. The Gene Ontology Annotation (GOA) Database–an integrated resource of GO annotations to the UniProt Knowledgebase. In Silico Biol (Gedrukt) 2004;4(1):5–6. [PubMed] [Google Scholar]
- 12.Berman H, Henrick K, Nakamura H. Announcing the worldwide Protein Data Bank. Nat Struct Biol. 2003;10(12):980. doi: 10.1038/nsb1203-980. [DOI] [PubMed] [Google Scholar]
- 13.Schnoes AM, Brown SD, Dodevski I, Babbitt PC. Annotation error in public databases: misannotation of molecular function in enzyme superfamilies. PLOS Comput Biol. 2009;5(12):e1000605. doi: 10.1371/journal.pcbi.1000605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ohnuma S, et al. Conversion of product specificity of archaebacterial geranylgeranyl-diphosphate synthase. Identification of essential amino acid residues for chain length determination of prenyltransferase reaction. J Biol Chem. 1996;271(31):18831–18837. doi: 10.1074/jbc.271.31.18831. [DOI] [PubMed] [Google Scholar]
- 15.Pegg SC, et al. Leveraging enzyme structure-function relationships for functional inference and experimental design: the structure-function linkage database. Biochemistry. 2006;45(8):2545–2555. doi: 10.1021/bi052101l. [DOI] [PubMed] [Google Scholar]
- 16.Barber AE, 2nd, Babbitt PC. Pythoscape: a framework for generation of large protein similarity networks. Bioinformatics. 2012;28(21):2845–2846. doi: 10.1093/bioinformatics/bts532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Atkinson HJ, Morris JH, Ferrin TE, Babbitt PC. Using sequence similarity networks for visualization of relationships across diverse protein superfamilies. PLoS ONE. 2009;4(2):e4345. doi: 10.1371/journal.pone.0004345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Barabási AL, Oltvai ZN. Network biology: understanding the cell’s functional organization. Nat Rev Genet. 2004;5(2):101–113. doi: 10.1038/nrg1272. [DOI] [PubMed] [Google Scholar]
- 19.Kalyanaraman C, et al. Discovery of a dipeptide epimerase enzymatic function guided by homology modeling and virtual screening. Structure. 2008;16(11):1668–1677. doi: 10.1016/j.str.2008.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lukk T, et al. Homology models guide discovery of diverse enzyme specificities among dipeptide epimerases in the enolase superfamily. Proc Natl Acad Sci USA. 2012;109(11):4122–4127. doi: 10.1073/pnas.1112081109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Koyama T, Fujii H, Ogura K. Enzymatic hydrolysis of polyprenyl pyrophosphates. Methods Enzymol. 1985;110:153–155. doi: 10.1016/s0076-6879(85)10070-4. [DOI] [PubMed] [Google Scholar]
- 22.Zahiri HS, Noghabi KA, Shin YC. Biochemical characterization of the decaprenyl diphosphate synthase of Rhodobacter sphaeroides for coenzyme Q10 production. Appl Microbiol Biotechnol. 2006;73(4):796–806. doi: 10.1007/s00253-006-0524-1. [DOI] [PubMed] [Google Scholar]
- 23.Hirooka K, et al. Functional analysis of two solanesyl diphosphate synthases from Arabidopsis thaliana. Biosci Biotechnol Biochem. 2005;69(3):592–601. doi: 10.1271/bbb.69.592. [DOI] [PubMed] [Google Scholar]
- 24.Ferella M, et al. A solanesyl-diphosphate synthase localizes in glycosomes of Trypanosoma cruzi. J Biol Chem. 2006;281(51):39339–39348. doi: 10.1074/jbc.M607451200. [DOI] [PubMed] [Google Scholar]
- 25.Pan JJ, Chiou ST, Liang PH. Product distribution and pre-steady-state kinetic analysis of Escherichia coli undecaprenyl pyrophosphate synthase reaction. Biochemistry. 2000;39(35):10936–10942. doi: 10.1021/bi000992l. [DOI] [PubMed] [Google Scholar]
- 26.Pan JJ, Kuo TH, Chen YK, Yang LW, Liang PH. Insight into the activation mechanism of Escherichia coli octaprenyl pyrophosphate synthase derived from pre-steady-state kinetic analysis. Biochim Biophys Acta. 2002;1594(1):64–73. doi: 10.1016/s0167-4838(01)00283-7. [DOI] [PubMed] [Google Scholar]
- 27.Winter JM, et al. Molecular basis for chloronium-mediated meroterpene cyclization: cloning, sequencing, and heterologous expression of the napyradiomycin biosynthetic gene cluster. J Biol Chem. 2007;282(22):16362–16368. doi: 10.1074/jbc.M611046200. [DOI] [PubMed] [Google Scholar]
- 28. (2012) SiteMap, version 2.6 (Schrödinger, LLC, New York)
- 29.Chang TH, et al. Structure of a heterotetrameric geranyl pyrophosphate synthase from mint (Mentha piperita) reveals intersubunit regulation. Plant Cell. 2010;22(2):454–467. doi: 10.1105/tpc.109.071738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Guo RT, et al. Bisphosphonates target multiple sites in both cis- and trans-prenyltransferases. Proc Natl Acad Sci USA. 2007;104(24):10022–10027. doi: 10.1073/pnas.0702254104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bairoch A, Apweiler R. The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res. 2000;28(1):45–48. doi: 10.1093/nar/28.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.UniProt Consortium. Reorganizing the protein space at the Universal Protein Resource (UniProt) Nucleic Acids Res. 2012 doi: 10.1093/nar/gkr981. 40(Database issue): D71–D75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Christianson DW. Structural biology and chemistry of the terpenoid cyclases. Chem Rev. 2006;106(8):3412–3442. doi: 10.1021/cr050286w. [DOI] [PubMed] [Google Scholar]
- 34.Trapp SC, Croteau RB. Genomic organization of plant terpene synthases and molecular evolutionary implications. Genetics. 2001;158(2):811–832. doi: 10.1093/genetics/158.2.811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sharkey TD, et al. Evolution of the isoprene biosynthetic pathway in kudzu. Plant Physiol. 2005;137(2):700–712. doi: 10.1104/pp.104.054445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gerlt JA, et al. The enzyme function initiative. Biochemistry. 2011;50:9950–9962. doi: 10.1021/bi201312u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cline MS, et al. Integration of biological networks and gene expression data using Cytoscape. Nat Protoc. 2007;2(10):2366–2382. doi: 10.1038/nprot.2007.324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. (2011) Prime, version 3.0 (Schrödinger, LLC, New York)
- 39.Pei J, Kim B-H, Grishin NV. PROMALS3D: a tool for multiple protein sequence and structure alignments. Nucleic Acids Res. 2008;36(7):2295–2300. doi: 10.1093/nar/gkn072. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.