

Abstract
Patients with type 2 diabetes (T2D) have disease-associated changes in B-cell function, but the role these changes play in disease pathogenesis is not well established. Data herein show B cells from obese mice produce a proinflammatory cytokine profile compared with B cells from lean mice. Complementary in vivo studies show that obese B cell–null mice have decreased systemic inflammation, inflammatory B- and T-cell cytokines, adipose tissue inflammation, and insulin resistance (IR) compared with obese WT mice. Reduced inflammation in obese/insulin resistant B cell–null mice associates with an increased percentage of anti-inflammatory regulatory T cells (Tregs). This increase contrasts with the sharply decreased percentage of Tregs in obese compared with lean WT mice and suggests that B cells may be critical regulators of T-cell functions previously shown to play important roles in IR. We demonstrate that B cells from T2D (but not non-T2D) subjects support proinflammatory T-cell function in obesity/T2D through contact-dependent mechanisms. In contrast, human monocytes increase proinflammatory T-cell cytokines in both T2D and non-T2D analyses. These data support the conclusion that B cells are critical regulators of inflammation in T2D due to their direct ability to promote proinflammatory T-cell function and secrete a proinflammatory cytokine profile. Thus, B cells are potential therapeutic targets for T2D.
Keywords: immunometabolism, lymphocytes
Multiple studies support the concept that inflammation strongly associates with insulin resistance (IR), which, in addition to loss of islet function, defines type 2 diabetes (T2D) (1). Work implicating B cells in IR/T2D is limited. We showed B cells from T2D subjects secrete a proinflammatory cytokine profile, including an extraordinary inability to secrete the potent anti-inflammatory cytokine IL-10 and an elevated production of proinflammatory IL-8 compared with B cells from non-T2D subjects (2). Given the importance of B-cell IL-10 in preventing numerous inflammatory diseases (3, 4) and the links between IL-8 and T2D (5, 6), these data suggest that altered B-cell cytokine production plays an important role in initiating or promoting IR/T2D. Published analyses further support a role for B cells in IR and include studies of B cell–null New Zealand Obese (NZO) mice, which, in contrast to B cell–sufficient NZOs, fail to develop IR in response to obesity (7). These findings have been recently reproduced in studies showing obese B cell–null or B cell–depleted mice have less inflammation and IR than obese WT mice (8). Interestingly, T-cell cytokine production is decreased in obese B cell–null mouse adipose tissue (AT) (8), which raises the possibility that, in addition to production of a proinflammatory cytokine profile, B cells may function in IR by regulating the T cell–mediated inflammation known to drive disease pathogenesis (9, 10). We identified a proinflammatory T-cell ratio [defined by increased Th17 cells plus decreased regulatory T cells (Tregs)] in T2D patients that mirrors findings in obese mice (10–13). However, the likelihood that obesity-associated B-cell changes dictate T-cell function in human T2D is untested.
To test the possibility that B cells promote metabolic disease by supporting T cell–mediated inflammation and thus IR, we compared lymphocyte function, inflammation, and metabolic outcomes in obese WT and B cell–null mice. Data herein show B cells from obese mice, like those from T2D humans, secrete a proinflammatory cytokine profile, including a defect in the ability to produce anti-inflammatory anti-IR IL-10 (14). Furthermore, B cell–null mice are protected from pathogenic outcomes of obesity and inflammation, consistent with our demonstration that obese B cell–null mice have an increased percentage of anti-inflammatory Tregs. We tested the significance of this finding with human lymphocyte analyses in vitro, which demonstrated that human B cells, but not monocyte/macrophages, promote T2D-associated T-cell inflammation through a contact-dependent mechanism. Taken together, our data identify functional similarities between obese/IR mouse and human immune cells and demonstrate that B cells increase inflammation in obesity through two pathways: regulation of an inflammatory T-cell ratio and production of a proinflammatory cytokine profile.
Results
B Cells from Obese Mice Secrete a Proinflammatory Cytokine Profile.
B cells have been proposed to promote obesity-associated metabolic disease through their ability to infiltrate the expanding AT and to produce autoimmune prodiabetogenic IgG (8, 15). Our studies support the originally published analysis of early B-cell infiltration into murine epididymal AT in response to a high-fat diet (HFD; Fig. S1A) (8, 15). However, autoimmune IgGs were extremely low/absent in sera from obese mice, as measured by antinuclear antibody assay, a clinical indicator of autoimmune antibodies (Fig. S1B). These data question the importance of a common class of autoantibodies in IR and raise the alternative possibility that the proinflammatory changes in B-cell cytokines reported in human T2D (2) play roles in individuals that lack autoantibodies yet have IR-associated inflammation.
To test the possibility that B cells from obese mice produce a proinflammatory cytokine profile that recapitulates the elevated IL-8 and dramatically decreased IL-10 from T2D patients’ B cells (2), we stimulated total splenocytes from obese or lean mice and then assayed cytokine secretion. Splenocytes from obese compared with lean mice secreted higher amounts of generally inflammatory IL-6 and IFN-γ and lower amounts of anti-inflammatory IL-10 in response to multiple stimuli (Fig. 1A). Splenocytes from obese mice also secreted less IL-5 (Fig. S2A), consistent with the demonstration of a lower percentage of IL-5–producing Th2 cells and the role of IL-5 in improving glucose tolerance (9, 16).
Fig. 1.

B cells secrete a proinflammatory cytokine profile in response to obesity. (A) Cytokine production by total splenocytes from lean (low-fat diet–fed) or obese (high-fat diet–fed) mice with cells stimulated as indicated. LPS, purified Escherichia coli lipopolysaccharide (TLR4 ligand); Pam3, Pam3CSK4 (TLR2 ligand); BCR, anti-IgM; CD40, α-CD40 antibody. (B) Cytokine production by purified splenic B cells from lean and obese mice with cells stimulated as indicated. n = 6–8 per group. Bars show mean and SEM. Significantly different groups are indicated by *P < 0.05 in comparison of obese and lean group for the same treatment; #P < 0.05, ##P < 0.01, stimulated compared with respective unstimulated (media) control with the same diet group, calculated by two-way ANOVA.
To determine whether obesity-associated changes in splenocyte cytokine profiles reflect changes in B cells, we measured cytokine production in splenic B cells (Fig. S2B). Purified B cells, and specifically follicular B cells (rather than marginal zone), secreted higher amounts of IL-6 and lower amounts of IL-10 in response to multiple stimuli (Fig. 1B; Figs. S3 and S4). Additionally, LPS-stimulated B cells from obese vs. lean mice secreted higher amounts of macrophage inflammatory 2 alpha (MIP-2) (Fig. S2C), a murine ortholog of human IL-8. We conclude the proinflammatory B-cell cytokine profile in obese mice mirrors that in T2D subjects (2). Thus, B cells contribute to chronic inflammation in IR/T2D by constitutively and inducibly producing a proinflammatory cytokine profile.
B Cells Promote Adipocyte Hypertrophy, Hyperglycemia, and Insulin Resistance in Obesity.
To test the possibility that proinflammatory responses of B cells to obesity promote disease, we measured metabolic parameters in obese WT and B cell–null (μMT) mice. μMT mice lack all pre- and mature B cells and plasma cells, as previously described (17). WT and μMT mice gain similar amounts of weight on a HFD (Fig. 2A). These data indicate that HFD-associated differences between WT and μMT mice in the present study are independent of weight. However, both epididymal and s.c. (subcutaneous inguinal) AT adipocytes were significantly smaller in obese μMT compared with obese WT mice (means were 15% and 40% larger in WT, respectively; Fig. 2B; Fig. S5A). Coincidentally, adipocyte numbers were significantly greater in both epididymal and s.c. AT from μMT mice (Fig. S5B). Whole AT explants from obese μMT mice secreted lower amounts of the proinflammatory adipokine leptin, consistent with the lower serum leptin concentrations in μMT mice (Fig. 2 C and D). Furthermore, attenuated inflammation in epididymal AT of μMT mice was shown by the relative absence of staining for the macrophage marker F4/80, decreased expression of proinflammatory macrophage genes (F4/80, CD11c, and CCR2), and increased expression of noninflammatory tissue remodeling macrophage genes (Arg1 and Ym1; Fig. S5 C and D). Despite similar whole body adiposity and lean mass, epididymal AT and the pericardial fat pad weighed less in obese μMT than in WT mice (Fig. S5 E and F). However, serum-free fatty acids, serum triglycerides, hepatosteatosis, and respiratory exchange ratio (a measure of whole body fatty acid oxidation) were similar in obese μMT and WT mice (Fig. S5 G and H), suggesting no differences in overall lipid metabolism between the two strains. Taken together, our data indicate that B cells do not alter total AT accretion in obesity but do promote development of unhealthy fat characterized by fewer and larger adipocytes, coincident with increased leptin and the presence of proinflammatory macrophages in epididymal AT.
Fig. 2.

Absence of B cells associates with protection from adipose tissue (AT) hypertrophy, insulin resistance, and impaired fasting glucose. (A) Weight gain in lean WT mice (black triangle); lean B cell–null μMT mice (gray triangle); obese WT mice (black circle); obese μMT mice (gray circle). Error bars are obscured by symbols at most points. All lean mice were fed a low-fat diet (LFD), and all obese mice were fed a high-fat diet (HFD). (B) Cell size distribution (by analysis of H&E-stained slides) of epididymal AT adipocytes as indicated. On average, adipocytes were 15% smaller in µMT compared with WT tissue. (C) Leptin secretion from ex vivo epididymal AT organ cultures or (D) in 6-h fasting serum. B–D are results from 16-wk HFD/obese mouse samples. (E) Six-hour fasting blood glucose in obese (circles) or lean (triangles) WT (black) or μMT (gray) mice at 15 wk of diet. (F) Insulin tolerance tests (ITTs) of obese WT (black) and μMT (gray) mice after 10 wk on HFD (circles) or LFD (triangles). Analysis after 15-wk HFD feeding gave similar results. Shown are absolute blood glucose levels. n = 6–8 for each panel, and when appropriate, mean and SEM are shown. *WT and μMT data are significantly different (P < 0.05) by Student t test (C–E) or two-way repeated measures ANOVA (F). In F, area under the curve analysis confirmed differences (P < 0.05).
To further examine the metabolic repercussions of proinflammatory B-cell functions in obesity, we measured fasting serum glucose and performed i.p. insulin and glucose tolerance tests (ITT and GTT, respectively). As expected, 15 wk of HFD increased fasting glucose in WT mice. In contrast, serum glucose was unchanged in obese μMT mice (Fig. 2E), coincident with lower fasting serum insulin (5.2 ng/mL in μMT vs. 7.6 ng/mL in WT; P < 0.05). These results suggest enhanced whole body insulin action in obese μMT vs. WT mice, a possibility confirmed by ITT (Fig. 2F). Glucose tolerance was improved in µMT mice independent of diet (Fig. S5I). Circulating total adiponectin was unchanged by obesity in µMT mice in contrast to decreased total adiponectin in obese vs. lean WT mice. However, circulating high-molecular-weight adiponectin was comparable in the two strains (Fig. S5J). Overall, these data support the conclusion that B cells promote hypertrophic obesity, dysregulated glucose–insulin homeostasis, and AT inflammation in obesity, but may restrain adipocyte hyperplasia.
B Cells Promote Systemic and T Cell–Mediated Inflammation in Obese/IR Mice.
To test the possibility that B cells associate with systemic inflammation in obesity, we quantified serum cytokines in obese/IR WT and μMT mice. Serum proinflammatory cytokines were uniformly higher in obese vs. lean mice (Fig. 3A), and multiple proinflammatory cytokines including IL-6, TNF-α, and MCP-1 were higher in obese WT compared with obese μMT sera (Fig. 3A). Thus, B cells promote systemic inflammation in obesity.
Fig. 3.
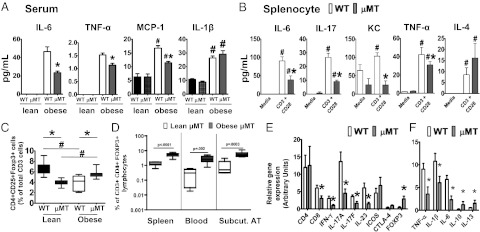
Absence of B cells associates with lower systemic inflammation and an increased percentage of regulatory T cells (Tregs) in response to obesity. (A) Serum cytokines in lean and obese WT and μMT mice as indicated. (B) Cytokines in supernatants from total splenocytes prepared from obese WT and μMT mice and cultured for 40 h either unstimulated (media) or stimulated with α-CD3/α-CD28. For A and B, *μMT are significantly different from WT (P < 0.05); #significant difference between stimulated and unstimulated (P < 0.05). Differences were determined by two-way ANOVA. In μMT mice, increases in T-cell percentages compensate for lack of B cells, indicating that T-cell cytokine production in μMT mice is much lower than shown when calculated on a “per cell” basis. (C) Flow cytometric analysis of Tregs from spleen of the indicated mice. Tregs were identified as CD3+CD4+CD25+Foxp3+ with gating strategy shown in Fig. S6. *Difference (P < 0.05) between WT and μMT mice under same diet conditions; #difference between lean and obese mice of same genotype. (D) Treg analysis from lean (white) or obese (gray) μMT mice from indicated tissues. P values for differences calculated by a two-tailed Student t test are as shown. (E) Relative mRNA expression of the indicated T cell–associated or (F) pro-/anti-inflammatory genes in epididymal AT from obese WT (white) or μMT (gray) mice. *Difference (P < 0.05) between WT and μMT as determined by a two-tailed Student t test. n = 6–8 for all panels.
Demonstrations that B cells directly regulate T cells (18) raise the possibility that B cells control systemic inflammation in obesity through their ability to direct T-cell function, either dependent or independent of their contribution to a proinflammatory cytokine profile. To test this possibility, we stimulated splenocytes from obese WT and μMT mice with T cell–specific stimuli and measured cytokine production. T-cell activation with α-CD3/α-CD28 elicited higher amounts of inflammatory cytokines in WT than in μMT splenocytes (Fig. 3B; IL-6, IL-17, KC, and TNF-α). In contrast, the Th2-associated cytokine IL-4, which generally counters proinflammatory T cells and supports anti-IR M2 macrophages (19, 20), was not increased in WT compared with μMT splenocytes (Fig. 3B). These data indicate B cells support proinflammatory T cells and thus IR in obesity.
B cells also regulate IR-protective Tregs (21, 22); therefore, we tested the possibility that obesity-associated changes in B-cell function are related to the decrease in Tregs that characterizes obese WT mice (10). Surprisingly, analysis of multiple tissues (spleen, blood, s.c. AT) showed obese μMT mice had universally higher percentages of Tregs (identified per Fig. S6) compared with lean μMT mice (Fig. 3 C and D). This result sharply contrasted with the Treg decrease in obese compared with lean WT mice (Fig. 3C). Importantly, the lower percentages of Tregs in lean μMT mice compared with lean WT mice do not predispose lean mice to increased obesity or systemic inflammation (Figs. 2A and 3A). These data concur with gene expression analysis of epididymal AT from obese μMT mice, which showed higher expression of the Treg signature gene Foxp3, lower expression of proinflammatory T-cell genes including IFNγ, IL-17A, and IL-17F, and lower expression of the Th17 survival cytokine IL-23 (Fig. 3E). CD8 expression was also lower in epididymal AT of obese μMT mice (Fig. 3E), consistent with immunohistochemical analysis showing less CD8 protein in epididymal AT of obese μMT mice (Fig. S7). As previously demonstrated, CD8 (but not CD4) staining associated with macrophage rich crown-like structures in all AT samples (23). Importantly, inducible T cell costimulator (ICOS, CD278), a marker of Treg function (24), was similarly expressed on Tregs from lean and obese WT spleen (Fig. S8), consistent with similar ICOS gene expression in epididymal AT from obese μMT and WT mice (Fig. 3E). Thus, decreased Treg proportions, rather than function, characterize obesity. Analysis of T-cell signature genes and cytokines mirrored the general decrease in proinflammatory cytokine expression (TNF-α, IL-1β, and IL-6) and increase in anti-inflammatory cytokine expression (IL-10 and IL-13) by epididymal AT from obese μMT compared with obese WT mice (Fig. 3F). Taken together, our findings indicate that B cells promote a proinflammatory T-cell ratio in obesity, characterized by the same increased Th17/Th1 function and decreased Treg percentages we identified in T2D patients (11).
Human B Cells, but Not Monocytes, Support Proinflammatory T Cells in T2D.
To confirm that B cells promote a proinflammatory T-cell ratio in obesity and test the relevance of B cells to human disease, we stimulated purified T cells from T2D and body mass index (BMI)-matched nondiabetic subjects (Table S1) in the presence of purified B cells and/or monocytes. Fig. S9 shows evidence of cell purity by flow cytometric analysis of lineage-specific markers. T-cell stimulation in the presence of monocytes and B cells (MBT) qualitatively recapitulated the T2D-associated T-cell inflammation, in this case IL-17 production, measured in peripheral blood mononuclear cells (PBMCs; Fig. 4A). Importantly, Fig. 4A confirms our previous demonstration that highly purified T cells (T) from T2D subjects fail to produce disease-associated amounts of IL-17 (11). Importantly, only CD3+CD4+ T cells (i.e., bona fide Th17s) produce IL-17 under these conditions (11). We conclude that interaction between T cells and B cells and/or monocytes drives T2D-associated proinflammatory Th17 function in humans.
Fig. 4.
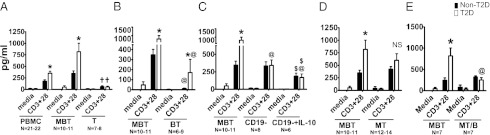
Human B cells, but not monocytes, support T2D-associated Th17 function and thus inflammation. (A) Supernatants from PBMCs (Left); cocultured monocytes, B cells, and T cells (M, B, and T, respectively; Center); or purified CD4+ T cells (T; Right) stimulated as indicated and assayed for IL-17 by ELISA. (B) IL-17 in supernatants from MBT (Left; regraphed from A) or BT cocultures of cells purified from non-T2D or T2D subjects and then stimulated as indicated. (C) IL-17 in supernatants from MBT cocultures (Left; regraphed from A), B cell–depleted PBMCs (Center; CD19−), or B cell–depleted PBMCs with recombinant IL-10 supplementation (Right; CD19−+IL-10). (D) IL-17 in supernatants from MBT (Left; regraphed from A) or MT cocultures of cells from non-T2D or T2D subjects. (E) IL-17 production by MT cultures in the presence of B cells (MBT) or in the presence of B cells physically separated from MT cells by a cytokine-permeable, cell-impermable transwell membrane (MT/B). MBT and MT/B cultures in E used a subset of the samples analyzed in A–C. For all panels, *difference (P < 0.05) between T2D (white bars) and non-T2D (black bars) under the same treatment condition; †difference between purified T cells and both MBT and PBMC results (A); @difference between MBT and BT (B) or CD19−+/− IL-10 PBMCs (C) or MT/B (E) within non-T2D or T2D cohort; $CD19−+/− IL-10 PBMCs results differ within non-T2D or T2D cohort (C). All differences were calculated by three-way ANOVA. IL-17 in all stimulated cultures was elevated compared with relevant unstimulated (media) controls. N for each culture type is indicated below x axis. Differences in MT cocultures from non-T2D and T2D subjects were insignificant (NS; D).
To identify more definitively the cell(s) required for T2D-associated Th17 function, we stimulated T cells in the presence of purified B cells. B-cell–T-cell cocultures recapitulate T2D-specific Th17 function as measured by IL-17 (Fig. 4B, BT), although IL-17 concentrations in BT cocultures were quantitatively lower than in MBT cultures. Furthermore, T-cell stimulation in the context of B cell–depleted (CD19−) PBMCs showed that removal of B cells resulted in a decrease of T2D-associated Th17 function (Fig. 4C, CD19−), which approximated IL-17 production by MBT cultures from non-T2D samples. Parallel analysis of mouse splenocytes showed B-cell depletion decreased inflammatory cytokine production (IL-6 and IFN-γ) in response to T-cell stimulation (Fig. S10), although it remains possible that removal of cytokine-secreting B cells partially explains these changes. In contrast, IL-10 was reduced only on B-cell depletion of splenocytes from lean mice (Fig. S10). This result is consistent with B cells as significant sources of IL-10 in lean, but not obese/IR, mice and humans (Fig. 1B) (2).
Lower B-cell IL-10 in obesity/IR may be confounded by the lack of inflammatory T-cell responsiveness to IL-10. To test the possibility that T2D-associated changes in T-cell IL-10 response reinforce proinflammatory B-cell function, we stimulated T cells in the context of B cell (CD19)–depleted PBMCs and in the presence or absence of IL-10. IL-10 significantly decreased IL-17 amounts in all samples (Fig. 4C). We conclude that T2D-associated T-cell inflammation may arise, at least in part, because of decreased B-cell IL-10 in the absence of defective IL-10 response. Taken together, data from both human and murine studies show that B cells support proinflammatory T-cell function in IR/T2D and that obesity-associated B cell–intrinsic changes, including decreased IL-10 production, likely play dominant roles in disease-associated T-cell function.
B-cell support of proinflammatory Th17 function in T2D (Fig. 4 A and B) does not address the possibility that monocytes/macrophages, the first immune cell type implicated in IR (25, 26), also control T-cell inflammation in obesity. We therefore measured Th17 function (i.e., IL-17 concentrations) in cocultures of purified monocytes and T cells (MT). MT cultures produce similar amounts of IL-17 regardless of whether cells are purified from non-T2D or T2D subjects (Fig. 4D). These data are consistent with disease-independent IL-17 production by B cell–depleted PBMCs, which allow MT interaction (Fig. 4C, CD19−). Taken together with higher IL-17 production in response to T-cell stimulation of WT compared with μMT murine splenocytes (Fig. 3B) and results of the human MBT and BT cocultures, the data strongly support the conclusion that B cells, rather than monocytes, directly promote T2D-associated elevated proinflammatory Th17 function. These data thus complement the demonstration that B cells regulate Treg expansion in obesity in vivo (Fig. 3D) to support the overall conclusion that B cells are master regulators of T2D-associated T-cell inflammation.
To further detail mechanisms underlying B cell–mediated T-cell inflammation in T2D, we tested the possibility that contact, or at a minimum close approximation, of B and T cells is required for B cells to support T cell–mediated inflammation. We compared Th17 function in MTB cultures to cultures that separated B cells from MT cocultures by a cytokine-permeable transwell membrane (1-μm pore size). Loss of B-cell contact prevented T2D-associated elevation of Th17 function (Fig. 4E, MT/B), suggesting that B-cell cytokines alone are insufficient to support proinflammatory T-cell function. Experiments with blocking antibody demonstrated that CD80/86 and CD28 were not required for B cell–mediated Th17 function in T2D. Overall, our data indicate that multiple mechanisms, including cellular contact and decreased IL-10 production, explain the ability of B cells to promote T cell–mediated inflammation in T2D.
Discussion
Multiple lines of evidence herein that focus on functional similarities between lymphocytes from obese/IR mice and T2D patients support the conclusion that B cells promote inflammation in obesity through multiple mechanisms. First, B cells produce a proinflammatory cytokine profile. Identification of decreased B-cell IL-10 in obesity justifies future studies that will test the possibility that IL-10+ B cells play critical roles in metabolic disease etiology, as shown for other Th1/Th17-mediated diseases (3, 4, 27–29). Second, B cells support a disease-associated proinflammatory T-cell ratio as shown by both in vivo and in vitro mouse studies, complemented by ex vivo human immune cell analyses. B cells can also promote hypertrophic obesity, which, in contrast to hyperplastic obesity (due to increased adipocyte number), promotes AT and systemic inflammation and deleterious changes in glucose–insulin homeostasis (30). More work is needed to attribute these whole body effects specifically to proinflammatory B-cell cytokines, T cell–mediated outcomes of altered B-cell function, or both. Recent work shows lack of B cells alters innate immune responses and lipid absorption in the intestine (31), which may also underlie metabolic differences between obese μMT and WT mice. Taken together, cross-species and tissue-independent similarities in B cells indicate that our focus on changes in B-cell function overcomes the practical limitations of low lymphocyte numbers (32) and depot heterogeneity of mouse and human AT. Thus, our findings address different translational questions than studies that emphasize, for example, AT depot–associated differences in lymphocytes (10).
Theoretical concerns over idiosyncrasies of the B cell–null μMT model are alleviated by data demonstrating that outcomes from μMT systemic and spleen analyses are largely consistent with published mouse data (8). Differences between our mouse work and the previously published work are likely because of analysis after different weeks on HFD and the general increase in IR on HFD feeding. The strength of our model is also indicated by similar outcomes from analyses of spleen, blood, and AT from obese µMT mice and by demonstrations that murine analyses both reflect and predict outcomes from T2D patient blood and human AT (1). For example, the greater than twofold higher amounts of the Th17 signature cytokine IL-17 in splenocytes from obese WT mice compared with obese μMT mice is quantitatively similar to elevated IL-17 secretion by Th17 cells from T2D patient blood (11). Importantly, the demonstration that lower baseline percentage of Tregs in lean μMT compared with WT mice does not associate with elevated spontaneous inflammation lessens concern that clinical B-cell depletion may inadvertently trigger proinflammatory T cells in some patients. Finally, our focus on function highlights robust changes that should shift emphasis toward clinical management of B cells as a fundamentally new approach to regulate T cell–mediated and more general inflammation in IR/T2D.
Our data show that T cells from T2D subjects respond to IL-10 by decreasing IL-17 production, but a more comprehensive assessment will be required to demonstrate definitively that T2D-associated proinflammatory T cells are relatively “normal” in isolation and absolutely require cell-extrinsic influences (such as those provided by B cells) to hypersecrete pathogenic proinflammatory cytokines. Importantly, many of our studies show that B cells regulate T cells in the absence of exogenous B-cell stimulation (i.e., in cultures stimulated only with T-cell activators). These results suggest the presence of a positive feed-forward loop between T cells and B cells. Preliminary analyses indicate stimuli that directly activate B-cell cytokine production only modestly elevate Th17 function in the absence of T-cell stimulation in samples from T2D patients, indicating that cross-talk between B and T cells is vital to these functional outcomes. Whether or not B-cell cytokines also regulate anti-inflammatory Treg numbers/function cannot be tested with in vitro cultures, given that the Treg marker Foxp3 is indiscriminately increased by T-cell stimulation. Regardless, our work supports a role for obesity-associated B-cell function in regulation of all helper (CD4+) T-cell subsets (Th1, Th2, Th17, and Tregs) currently linked to IR (9–11, 33) and is distinct from previous analysis that suggested pathogenic B-cell autoantibodies cause IR (8). Importantly, balance among T-cell subsets, as indicated by percentages of CD4+ T cells in our work, is at least as critical an indicator of net inflammatory state as cell numbers, which are difficult to quantify with great accuracy in flow analyses, especially for relatively rare AT lymphocytes.
Our work convincingly demonstrates that B cells critically regulate proinflammatory T-cell function in a disease-associated mechanism. Our findings are therefore consistent with demonstrations that B-cell depletion in lupus or rheumatoid arthritis patients dramatically decreases T-cell inflammation, including Th17 function (34, 35). Our data also indicate myeloid cells play supportive, albeit disease-independent, roles in overall levels of T-cell inflammation. Monocytes from both non-T2D and T2D subjects increase IL-17 secretion to about fivefold over baseline levels produced by purified T cells. These human data are consistent with work showing obesity-associated AT macrophages support inflammatory T-cell function in mice (36), although this study did not directly test the ability of macrophages from lean mice to support inflammatory T cells as did our work. Importantly, our human monocyte data significantly extend published data showing that dendritic cells promote Th17 function in obese mice (37). Although the monocyte–T-cell coculture data highlight disease-independent effects of monocytes on T cells, it remains possible that elevated monocyte cytokines in T2D (11) influence disease-associated T-cell function. Our data therefore suggest it may be possible to moderate T-cell inflammation in T2D by alternatively attacking the “on/off switch” provided by B cells or the “rheostat” function of monocytes to counter inflammation-associated IR. These findings emphasize the overall promise offered by an increased understanding of immune cell cross-talk in IR/T2D, which will likely identify mechanisms that may be exploited to interrupt the feed-forward cycle of inflammation and alleviate T2D.
Materials and Methods
Whole Animal and Intact Tissue Studies.
All procedures were approved by the Boston University Medical Center Institutional Animal Care and Use Committee. Studies used male C57BL/6J background mice, and standard protocols are described in SI Materials and Methods.
Human Subjects.
The Boston University Medical Center Institutional Review Board approved all procedures in accordance with the Declaration of Helsinki, and samples were obtained following informed consent. T2D and BMI-matched non-T2D patients were recruited from the Center for Endocrinology, Diabetes and Nutrition at Boston University Medical Center/Boston Medical Center. Selection criteria are listed in SI Materials and Methods. Characteristics of all subjects are shown in Table S1.
Immune Cell Preparation/Culture.
Peripheral blood (50–100 mL) was collected into heparinized tubes by venous puncture, and cells were purified, cultured, stimulated, and harvested as previously described (2, 11). CD19− PBMCs were prepared by removing CD19+ B cells with positively selecting magnetic beads (Miltenyi) and were confirmed to be B cell depleted by flow analysis for CD19+ cells. Immune cells from mouse s.c. (inguinal) AT were released by manually teasing tissue between two bent needles, which did not activate immune cells (indicated by CD69 surface staining). Whole mouse splenocytes were prepared by manual teasing and red blood cell lysis, and then CD19+ B cells were purified by negatively selecting magnetic beads (Miltenyi) and cultured as described for human cells. All lymphocyte and monocyte preparations were ≥95% and ≥92% pure, respectively, and incubated at 1 million cells/mL. Purified B cells were harvested after 24 h of stimulation. All T cell–containing cultures were harvested after 40 h. Human IL-10 was added to some cultures at high physiological concentrations (2 ng/mL).
Biochemistry.
Epididymal AT mRNA was quantified as previously described (38), using primers specific for CD19 (forward: 5′CCCTGTATCTCTGGCTCTGC; reverse: 5′GGGCACATACAGGCTTTGTT) with cyclophilin B as a normalization control. ELISA was used to quantify leptin, MIP-2, adiponectin, and human IL-17. All additional cytokines were quantified by multiplex protein assays (Invitrogen).
Flow Cytometry.
Mouse blood for flow analysis was collected by heart puncture and added to an equal volume of PBS/50 mM EDTA buffer; spleen and s.c. AT were manually dissociated. Red blood cells were lysed before antibody-mediated staining. Staining strategies are listed in SI Materials and Methods. Cells were washed twice with FACS buffer (PBS with 0.5% BSA and 2 mM EDTA) after staining and then analyzed on a LSR-II Flow Cytometer (BD Biosciences). FlowJo software (version 8.7; Tree Star) was used for final data analyses. Titration of each reagent and “fluorescence minus one” controls (39) were performed to optimize panels.
Supplementary Material
Acknowledgments
We thank Drs. Jongsoon Lee and Susan Leeman for manuscript critiques; Joel Nikolajczyk for expert technical assistance; and the Evans Center for Interdisciplinary Biomedical Research and its Affinity Research Collaborative members for valuable discussion and resources. This work was supported by National Institutes of Health Grants R21DK089270, 5R21DE021154, R56 DK090455, and R56 DK096525, Immunology Training Program AI007309, Hematology Training Program HL007501, the Boston Area Diabetes Endocrinology Research Center Pilot Program, and Boston Nutrition Obesity Research Center Grant DK046200.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission. A.C. is a guest editor invited by the Editorial Board.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1215840110/-/DCSupplemental.
References
- 1.Nikolajczyk BS, Jagannathan-Bogdan M, Denis GV. The outliers become a stampede as immunometabolism reaches a tipping point. Immunol Rev. 2012;249(1):253–275. doi: 10.1111/j.1600-065X.2012.01142.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jagannathan M, et al. Toll-like receptors regulate B cell cytokine production in patients with diabetes. Diabetologia. 2010;53(7):1461–1471. doi: 10.1007/s00125-010-1730-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Carter NA, et al. Mice lacking endogenous IL-10-producing regulatory B cells develop exacerbated disease and present with an increased frequency of Th1/Th17 but a decrease in regulatory T cells. J Immunol. 2011;186(10):5569–5579. doi: 10.4049/jimmunol.1100284. [DOI] [PubMed] [Google Scholar]
- 4.Matsushita T, Yanaba K, Bouaziz JD, Fujimoto M, Tedder TF. Regulatory B cells inhibit EAE initiation in mice while other B cells promote disease progression. J Clin Invest. 2008;118(10):3420–3430. doi: 10.1172/JCI36030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fain JN. Release of interleukins and other inflammatory cytokines by human adipose tissue is enhanced in obesity and primarily due to the nonfat cells. Vitam Horm. 2006;74:443–477. doi: 10.1016/S0083-6729(06)74018-3. [DOI] [PubMed] [Google Scholar]
- 6.Jialal I, Huet BA, Kaur H, Chien A, Devaraj S. Increased toll-like receptor activity in patients with metabolic syndrome. Diabetes Care. 2012;35(4):900–904. doi: 10.2337/dc11-2375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Haskell BD, Flurkey K, Duffy TM, Sargent EE, Leiter EH. The diabetes-prone NZO/HlLt strain. I. Immunophenotypic comparison to the related NZB/BlNJ and NZW/LacJ strains. Lab Invest. 2002;82(7):833–842. doi: 10.1097/01.lab.0000018915.53257.00. [DOI] [PubMed] [Google Scholar]
- 8.Winer DA, et al. B cells promote insulin resistance through modulation of T cells and production of pathogenic IgG antibodies. Nat Med. 2011;17(5):610–617. doi: 10.1038/nm.2353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Winer S, et al. Normalization of obesity-associated insulin resistance through immunotherapy. Nat Med. 2009;15(8):921–929. doi: 10.1038/nm.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Feuerer M, et al. Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters. Nat Med. 2009;15(8):930–939. doi: 10.1038/nm.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jagannathan-Bogdan M, et al. Elevated proinflammatory cytokine production by a skewed T cell compartment requires monocytes and promotes inflammation in type 2 diabetes. J Immunol. 2011;186(2):1162–1172. doi: 10.4049/jimmunol.1002615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Goossens GH, et al. Expression of NLRP3 inflammasome and T cell population markers in adipose tissue are associated with insulin resistance and impaired glucose metabolism in humans. Mol Immunol. 2012;50(3):142–149. doi: 10.1016/j.molimm.2012.01.005. [DOI] [PubMed] [Google Scholar]
- 13.Yang H, et al. Obesity increases the production of proinflammatory mediators from adipose tissue T cells and compromises TCR repertoire diversity: Implications for systemic inflammation and insulin resistance. J Immunol. 2010;185(3):1836–1845. doi: 10.4049/jimmunol.1000021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gotoh K, et al. A novel anti-inflammatory role for spleen-derived interleukin-10 in obesity-induced inflammation in white adipose tissue and liver. Diabetes. 2012;61(8):1994–2003. doi: 10.2337/db11-1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Duffaut C, Galitzky J, Lafontan M, Bouloumié A. Unexpected trafficking of immune cells within the adipose tissue during the onset of obesity. Biochem Biophys Res Commun. 2009;384(4):482–485. doi: 10.1016/j.bbrc.2009.05.002. [DOI] [PubMed] [Google Scholar]
- 16.Wu D, et al. Eosinophils sustain adipose alternatively activated macrophages associated with glucose homeostasis. Science. 2011;332(6026):243–247. doi: 10.1126/science.1201475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kitamura D, Roes J, Kühn R, Rajewsky K. A B cell-deficient mouse by targeted disruption of the membrane exon of the immunoglobulin mu chain gene. Nature. 1991;350(6317):423–426. doi: 10.1038/350423a0. [DOI] [PubMed] [Google Scholar]
- 18.Bouaziz JD, et al. Therapeutic B cell depletion impairs adaptive and autoreactive CD4+ T cell activation in mice. Proc Natl Acad Sci USA. 2007;104(52):20878–20883. doi: 10.1073/pnas.0709205105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fernandez-Botran R, Sanders VM, Mosmann TR, Vitetta ES. Lymphokine-mediated regulation of the proliferative response of clones of T helper 1 and T helper 2 cells. J Exp Med. 1988;168(2):543–558. doi: 10.1084/jem.168.2.543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stein M, Keshav S, Harris N, Gordon S. Interleukin 4 potently enhances murine macrophage mannose receptor activity: A marker of alternative immunologic macrophage activation. J Exp Med. 1992;176(1):287–292. doi: 10.1084/jem.176.1.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ray A, Basu S, Williams CB, Salzman NH, Dittel BN. A novel IL-10-independent regulatory role for B cells in suppressing autoimmunity by maintenance of regulatory T cells via GITR ligand. J Immunol. 2012;188(7):3188–3198. doi: 10.4049/jimmunol.1103354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shah S, Qiao L. Resting B cells expand a CD4+CD25+Foxp3+ Treg population via TGF-beta3. Eur J Immunol. 2008;38(9):2488–2498. doi: 10.1002/eji.200838201. [DOI] [PubMed] [Google Scholar]
- 23.Nishimura S, et al. CD8+ effector T cells contribute to macrophage recruitment and adipose tissue inflammation in obesity. Nat Med. 2009;15(8):914–920. doi: 10.1038/nm.1964. [DOI] [PubMed] [Google Scholar]
- 24.Chen Y, Shen S, Gorentla BK, Gao J, Zhong XP. Murine regulatory T cells contain hyperproliferative and death-prone subsets with differential ICOS expression. J Immunol. 2012;188(4):1698–1707. doi: 10.4049/jimmunol.1102448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Weisberg SP, et al. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest. 2003;112(12):1796–1808. doi: 10.1172/JCI19246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xu H, et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest. 2003;112(12):1821–1830. doi: 10.1172/JCI19451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yanaba K, et al. A regulatory B cell subset with a unique CD1dhiCD5+ phenotype controls T cell-dependent inflammatory responses. Immunity. 2008;28(5):639–650. doi: 10.1016/j.immuni.2008.03.017. [DOI] [PubMed] [Google Scholar]
- 28.Mauri C, Gray D, Mushtaq N, Londei M. Prevention of arthritis by interleukin 10-producing B cells. J Exp Med. 2003;197(4):489–501. doi: 10.1084/jem.20021293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Blair PA, et al. CD19(+)CD24(hi)CD38(hi) B cells exhibit regulatory capacity in healthy individuals but are functionally impaired in systemic Lupus Erythematosus patients. Immunity. 2010;32(1):129–140. doi: 10.1016/j.immuni.2009.11.009. [DOI] [PubMed] [Google Scholar]
- 30.Kim JY, et al. Obesity-associated improvements in metabolic profile through expansion of adipose tissue. J Clin Invest. 2007;117(9):2621–2637. doi: 10.1172/JCI31021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shulzhenko N, et al. Crosstalk between B lymphocytes, microbiota and the intestinal epithelium governs immunity versus metabolism in the gut. Nat Med. 2011;17(12):1585–1593. doi: 10.1038/nm.2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McDonnell ME, et al. B lymphocytes in human subcutaneous adipose crown-like structures. Obesity (Silver Spring) 2012;20(7):1372–1378. doi: 10.1038/oby.2012.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zúñiga LA, et al. IL-17 regulates adipogenesis, glucose homeostasis, and obesity. J Immunol. 2010;185(11):6947–6959. doi: 10.4049/jimmunol.1001269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Iwata S, et al. Phenotypic changes of lymphocytes in patients with systemic lupus erythematosus who are in longterm remission after B cell depletion therapy with rituximab. J Rheumatol. 2011;38(4):633–641. doi: 10.3899/jrheum.100729. [DOI] [PubMed] [Google Scholar]
- 35.van de Veerdonk FL, et al. The anti-CD20 antibody rituximab reduces the Th17 cell response. Arthritis Rheum. 2011;63(6):1507–1516. doi: 10.1002/art.30314. [DOI] [PubMed] [Google Scholar]
- 36.Vandanmagsar B, et al. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat Med. 2011;17(2):179–188. doi: 10.1038/nm.2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bertola A, et al. Identification of adipose tissue dendritic cells correlated with obesity-associated insulin resistance and inducing Th17 responses in mice and patients. Diabetes. 2012;61(9):2238–2247. doi: 10.2337/db11-1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Strissel KJ, et al. Adipocyte death, adipose tissue remodeling, and obesity complications. Diabetes. 2007;56(12):2910–2918. doi: 10.2337/db07-0767. [DOI] [PubMed] [Google Scholar]
- 39.Roederer M. Spectral compensation for flow cytometry: Visualization artifacts, limitations, and caveats. Cytometry. 2001;45(3):194–205. doi: 10.1002/1097-0320(20011101)45:3<194::aid-cyto1163>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.