

Abstract
Genome structure variation, including copy number variation and presence/absence variation, comprises a large extent of maize genetic diversity; however, its effect on phenotypes remains largely unexplored. Here, we describe how copy number variation underlies a rare allele that contributes to maize aluminum (Al) tolerance. Al toxicity is the primary limitation for crop production on acid soils, which make up 50% of the world’s potentially arable lands. In a recombinant inbred line mapping population, copy number variation of the Al tolerance gene multidrug and toxic compound extrusion 1 (MATE1) is the basis for the quantitative trait locus of largest effect on phenotypic variation. This expansion in MATE1 copy number is associated with higher MATE1 expression, which in turn results in superior Al tolerance. The three MATE1 copies are identical and are part of a tandem triplication. Only three maize inbred lines carrying the three-copy allele were identified from maize and teosinte diversity panels, indicating that copy number variation for MATE1 is a rare, and quite likely recent, event. These maize lines with higher MATE1 copy number are also Al-tolerant, have high MATE1 expression, and originate from regions of highly acidic soils. Our findings show a role for copy number variation in the adaptation of maize to acidic soils in the tropics and suggest that genome structural changes may be a rapid evolutionary response to new environments.
Keywords: abiotic stress, selection
Aluminum toxicity severely limits plant growth on highly acidic soils (pH <5). Under these conditions, the rhizotoxic Al species Al3+ is solubilized, inhibiting root growth and function (1), and leaving plants more vulnerable to drought and mineral nutrient deficiencies. Approximately 30% of the earth’s total land area consists of highly acid soils, and as much as 50% of the world’s potentially arable lands are acidic (2). As large areas of acid soils in the tropics and subtropics are critical food-producing regions, Al toxicity constitutes a food security threat exceeded only by drought with regard to abiotic limitations on crop production (2).
A major physiological mechanism of plant aluminum tolerance consists of the Al-activated release of organic acids from the root apex, the site of Al toxicity (1). Organic acid anions such as malate, citrate, and oxalate chelate Al3+ in the rhizosphere forming stable, nontoxic complexes (3). This “exclusion” mechanism limits Al3+ uptake by the roots, minimizing harmful interactions with Al-sensitive sites in the root apical apoplast and/or inside root cells, and has been correlated with differential Al tolerance in a large number of monocot and dicot species (1). Transporters from the multidrug and toxic compound extrusion (MATE) family have been shown to mediate the release of citrate, and contribute to Al tolerance in a number of plant species (4–9).
Maize (Zea mays) is a major food crop throughout the tropics and subtropics, where acid soils are prevalent. Therefore, maize breeding programs in these regions have focused intensely on Al tolerance. The development of Al-tolerant varieties of maize and other crops has been crucial for the agricultural development of previously unproductive regions such as the Brazilian Cerrado, an area of 205 million hectares (10). Al tolerance in maize is a quantitative trait (11–13), and quantitative trait loci (QTLs) associated with the trait have been identified. Five QTLs explaining 60% of the variance in Al tolerance were detected in a mapping population generated from a cross between a highly Al-tolerant tropical inbred line commonly used as a tolerance donor in breeding programs, Cateto Al237, and the Al-sensitive inbred line L53 (14).
We recently reported on the characterization of the maize Al tolerance gene MATE1 (15), originally identified in a microarray study (16) as the most up-regulated gene in root tips of an Al-tolerant maize line under Al stress. MATE1 was mapped to the telomeric region of chromosome 6, colocalizing with the QTL of largest effect on Al tolerance identified by Ninamango-Cárdenas et al. (14), explaining 16.2% of the phenotypic variance. MATE1 shares significant identity to its homologs in sorghum and Arabidopsis, SbMATE (52%) and AtMATE (64%). MATE1 mediates citrate efflux, as observed in [14C]citrate efflux studies in Xenopus laevis oocytes. Expressing MATE1 in transgenic Arabidopsis conferred a significant increase in Al tolerance and root citrate exudation in response to Al. In maize, MATE1 expression is concentrated in the root apex and is strongly up-regulated by Al. MATE1 expression is significantly higher in the Al-tolerant parent Al237 (and in Al-tolerant C100-6, used in the original microarray study) than in Al-sensitive L53, both in the absence or presence of Al (15).
Our recent findings suggested that functional, structural, or regulatory variation in MATE1 may underlie the large-effect Al tolerance QTL previously detected on chromosome 6. In the present study, we show that copy number variation (CNV) in the MATE1 locus is associated with both gene expression and phenotypic differences within our recombinant inbred line (RIL) population. We also demonstrate that structural variation in this locus is a rare and probably recent occurrence, and discuss its potential implications for maize adaptation to acid soils.
Results
Cloning and sequencing the MATE1 ORFs from the parents of the mapping population (Al237, Al-tolerant, and L53, Al-sensitive) previously revealed six nucleotide differences between their coding regions, of which only two resulted in amino acid substitutions (R171C and G527V) (15). We performed further functional investigations to establish whether these two structural modifications resulted in differences in the transporter’s basic functional properties (Fig. S1A). For this purpose, the proteins encoded by each of the two MATE1 alleles were functionally characterized in Xenopus laevis oocytes (Fig. S1 B–D). Oocytes expressing either allele showed similar time- and pH-dependent electrogenic activities (i.e., negative currents), which were significantly larger than those of control cells (Fig. S1 B and C). We also examined substrate recognition by testing the effect of exogenous citrate or malate added to the bath media (Fig. S1D). Addition of citrate or malate increased MATE1-mediated inward currents, with the changes in current magnitude being dependent on the extracellular concentration of organic acid supplied. Again, no differences between oocytes injected with either allele were observed under these conditions.
We did not identify any significant structural or functional differences of the MATE1 proteins encoded by the parental alleles in our analysis that could explain the difference in Al tolerance. However, we previously showed that MATE1 expression is higher in Al237 than in L53, both in the absence and presence of Al (15). These results suggest that the causal polymorphism underlying the QTL must somehow affect this gene’s expression levels. Therefore, we looked for sequence polymorphisms in potential cis regulatory regions, by cloning an 8-kbp fragment upstream of the MATE1 ORFs from both parental lines (Fig. S2). The sequences revealed extremely low levels of polymorphism (99.9% identity) between the two lines, suggesting that sequence polymorphism in cis regulatory regions are also not likely to explain the Al tolerance QTL.
CNV in MATE1.
To assess whether a structural polymorphism is associated with the expression differences observed between the parental lines, we quantified gene copy number using quantitative real-time PCR (qPCR). The method was validated using sequences with known copy number in the reference maize line B73 and indicated that, although B73 and L53 have a single copy of MATE1, three copies are found in the Al237 genome (Fig. S3). The location of the MATE1 gene copies on maize chromosomes of inbred lines Al237 and L53 was investigated using fluorescent in situ hybridization (FISH). We localized MATE1 on the short arm of chromosome 6 in both lines, distal to the nucleolar organizing region (Fig. 1), in agreement with our mapping results (15). Minor signals were also consistently detected on 1L, 7S, and 9L. Weak or infrequent signals were observed on 2L, 3L, 7L, 8L (Al237 only), and 10L. The sequence locations of partial gene transpositions of MATE1 located on chromosomes 2, 7, and 10 (Fig. S3) are consistent with the minor FISH signals observed on these chromosomes; however, their precise locations cannot be determined at this FISH resolution level. Nevertheless, these results indicate that the MATE1 copies in Al237 must be located near each other on the short arm of chromosome 6, and that no gene copies undetected by qPCR exist at other genomic locations. In addition, although only one signal per chromatid was observed in all L53 images (n = 17), in over 70% of Al237 images (15 of 21), partially overlapping MATE1 signals were observed on a single chromatid (Fig. 1C). These results are consistent with multiple copies of the gene being present at that location.
Fig. 1.

Localization of MATE1 in maize lines L53 and Al237 using FISH. Homologous metaphase chromosomes from root meristem cells are aligned as pairs. (A) L53, Al-sensitive, one copy of MATE1. (B) Al237, Al-tolerant, three copies of MATE1. A large arrowhead indicates the location of MATE1 on 6S; small arrowheads indicate minor signals. (C) Multiple MATE1 FISH signals are observed on Al237 chromosome 6S. Multicolor image and grayscale version of the respective red channel are shown. The arrowheads indicate regions with two MATE1 signals per chromatid. The MATE1 probe is labeled with Texas Red. Green fluorescein isothiocyanate-labeled probes include centromere C repeat sequences (found on all chromosomes but copy number highly variable), a TAG microsatellite repeat (chromosome arms 1L, 2S, 2L, and 4S), and the ribosomal gene cluster (6S at the nucleolar organizing region). Blue DAPI counterstain was used to visualize heterochromatic 180-bp knob regions (line-specific locations). The second karyotype shown in each panel is the grayscale version of the MATE1 probe (red) channel in the respective multicolor karyotype. Adjustment of individual color channels was performed on original images. (Scale bars, 3 μm.)
MATE1 CNV and Al Tolerance.
Taking into account that the parents of the population used to map Al tolerance QTL vary in both MATE1 copy number and MATE1 expression levels, we set out to investigate the relationship between these genotypic differences and the Al tolerance phenotype. First, we determined the distribution of MATE1 copy number in the Al237×L53 RILs using qPCR (Fig. 2A and Dataset S1A). The proportion of progeny with one versus three gene copies conformed to a 1:1 segregation ratio: 57 RILs have a single MATE1 copy, and 53 RILs have three copies (P value of χ2 test = 0.7029). These results are in agreement with what was observed using FISH and suggest that the paralog copies are in tight genetic linkage. Then, we examined the relationship between CNV in the MATE1 locus and phenotypic variation for Al tolerance in the Al237×L53 RILs. MATE1 copy number is positively correlated with Al tolerance in the RIL population (P = 5.46 × 10−7, R2 = 0.20), such that individuals with three copies have a significantly higher mean Al tolerance than those inheriting a single copy (Fig. 2B).
Fig. 2.

MATE1 CNV in Al237×L53 RILs correlates with aluminum tolerance. (A) MATE1 copy number in the RIL population derived from a cross between Al237 (Al-tolerant, three copies of MATE1) and L53 (Al-sensitive, one copy of MATE1), determined by qPCR. RILs are arranged based on copy number in ascending order (from one copy to three copies). The red bars denote single-copy RILs (n = 57); the blue bars denote RILs with three copies of MATE1 (n = 53). Error bars indicate 1 SD. (B) One-way ANOVA of RRG (standard phenotypic index of Al tolerance) by MATE1 copy number in the Al237×L53 RIL population. Mean diamonds represent 95% confidence intervals, and midbars in diamonds represent the group mean. The gray line represents the overall mean.
The results above strongly suggest that greater gene copy number is the basis for higher MATE1 expression levels in the RILs, which in turn results in increased Al tolerance. Thus, we also examined how MATE1 expression relates directly to gene copy number in the Al237×L53 population. Before designing gene expression assays, we performed extensive Sanger sequencing of the entire coding region of MATE1 from Al237 genomic DNA to identify SNPs or indels between the three gene copies. Strikingly, no polymorphisms were identified anywhere in the sequence: the exons, introns, 5′- and 3′-UTRs of the three MATE1 copies in Al237 are 100% identical. Therefore, any measurement of MATE1 expression will capture the combined expression levels of the three copies. With this in mind, we used qPCR to determine root tip MATE1 expression in the Al237×L53 population, in plants grown under control and Al stress conditions (Fig. 3A and Dataset S1A). MATE1 expression in the RILs varies continuously under both growth conditions. In control conditions, the parents represent the extremes of the distribution (L53, low expression; Al237, high expression). Under Al stress, there is a greater number of RILs in which we observed MATE1 expression lower than L53 or higher than Al237, indicating that some degree of transgressive segregation exists for MATE1 expression under Al. Because MATE1 expression is up-regulated under Al stress, variation in trans-acting elements involved in the regulation of this response is likely to account for this effect. MATE1 copy number is also strongly correlated with MATE1 expression (Fig. 3B), such that RILs carrying three copies of the gene have significantly higher expression levels than RILs carrying one copy, both in the absence (P = 1.48 × 10−33, R2 = 0.74) and in the presence (P = 1.23 × 10−42, R2 = 0.82) of Al. Thus, differences in MATE1 expression between RILs can be largely explained by differences in gene copy number, both under control and Al stress conditions. This observation is also supported by the fact that, within the RIL population, MATE1 expression levels under Al stress are highly correlated with those under control conditions (R2 = 0.64; Fig. S4), so that RILs with higher expression in the absence of Al tend to also display higher expression when exposed to the stress.
Fig. 3.

MATE1 expression in the Al237×L53 RIL population correlates with gene copy number. (A) MATE1 relative expression in root tips quantified via qPCR from maize seedlings grown under control conditions (Left), or treated with 39 µM Al3+ activity (Right) for 24 h. In each graph, maize lines are arranged based on MATE1 relative expression in ascending order. The bars are color-coded based on MATE1 copy number: the red bars denote one-copy RILs, and the blue bars denote three-copy RILs. The gray bars denote the parents of the population, Al237 (Al-tolerant, three copies of MATE1) and L53 (Al-sensitive, one copy of MATE1). Samples were calibrated against L53 (relative expression = 1) within each dataset. Error bars indicate 1 SD. (B) One-way ANOVA of MATE1 relative expression by MATE1 copy number in the Al237×L53 RIL population, in control and Al stress conditions. Mean diamonds represent 95% confidence intervals, and the midbars in diamonds represent the group mean. The gray line represents the overall mean.
Genetic Factors Controlling MATE1 Expression.
We investigated the genetic factors controlling MATE1 expression via expression QTL (eQTL) mapping in the Al237×L53 population. For this purpose, a high density of markers was generated via genotyping-by-sequencing (GBS), a restriction enzyme-based method for constructing reduced representation genomic libraries for genotyping (17). Using this new marker set, we repeated the mapping of Al tolerance QTL and also mapped eQTL controlling MATE1 expression (Fig. 4). eQTL mapping was performed independently for MATE1 expression both in the presence and in the absence of Al. The mapping of loci controlling Al tolerance in the Al237×L53 population confirmed our previous results showing that a genomic region on the distal end of chromosome 6, colocalizing with the MATE1 locus, represents a major Al tolerance QTL. Mapping of MATE1 expression identified a single genomic region controlling the trait, coincident with the location of the gene, under both control and Al stress conditions. These results indicate that MATE1 expression is controlled mainly in cis. Trans factors involved in the up-regulation of MATE1 expression under Al stress may exist, but their smaller effect is overshadowed by the large effects in cis; thus, they were not detected by eQTL mapping. Data from near-isogenic lines (NILs) also corroborate these results. NILs generated by introgressing the three-copy MATE1 allele from Al237 into the Al-sensitive L53 background are nearly twice as tolerant as L53 (averaging 50% root growth inhibition, versus 80% in L53; Dataset S1B). The level of MATE1 expression in root tips of these NILs is comparable to those of Al237, indicating that no other loci acting in trans are necessary to retain the high levels of expression observed in the Al-tolerant parent, Al237.
Fig. 4.

A major aluminum tolerance QTL and eQTL for MATE1 expression colocalize with MATE1. A GLM analysis identified GBS-generated SNP markers that are associated with Al tolerance (expressed as RRG; red circles, y axis on Left) and with MATE1 expression (open triangles: expression under control conditions; solid triangles: expression under Al treatment; y axis on Right). The level of statistical association for each polymorphism is expressed as –Log P. x axis: physical position on maize chromosome 6. Inset is a magnification of a 40-Mb window on the top of chromosome 6. A gray bar indicates the location of the MATE1 locus.
Genomic Organization of the MATE1 Locus.
To examine the genomic structure of the MATE1 locus and determine how the three gene copies are organized, a genomic BAC library from the Al-tolerant parent Al237 was constructed. A BAC library pooling and screening strategy was developed that did not require individual BACs to be arrayed (SI Materials and Methods). A positive BAC clone identified from the library screening was shown to carry three copies of MATE1 and was fully sequenced via single-molecule real-time sequencing technology, using a combination of small- and large-insert sequencing libraries (600 bp and 8 kb; SI Materials and Methods). Although the long, single-molecule reads facilitate the assembly of repetitive DNA regions, the typical error rates generated by this technology are high. Therefore, the sequences from the small-insert library were used to error-correct the long reads generated by sequencing of the large-insert library, using a hybrid approach (18). The error rate of the long reads was reduced to less than 1%, as estimated based on the known vector sequence. After error correction, the BAC sequence was assembled manually from the corrected long reads to circumvent the limitation of existing assemblers in generating contigs from long repetitive sequences, with supporting data from long-range PCR and from depth-of-coverage analysis of the error-corrected long reads aligned along the BAC sequence (SI Materials and Methods) (Fig. S5).
Sequencing the genomic BAC clone revealed that the three MATE1 copies are part of a tandem triplication, with each repeat unit totaling nearly 30 kbp in size (Fig. 5). Except for MATE1-1, -2, and -3, no other maize genes are predicted within the BAC sequence. Instead, each copy of MATE1 is flanked by various transposon insertions, mainly upstream of each ORF. The three 30-kbp repeats share high sequence identity with each other. The second repeat region differs from the first and third repeats, in that a class I long terminal repeat (LTR) Gypsy retrotransposon is inserted just upstream of MATE1-2. This insertion is not present in the upstream sequence cloned from L53 (Fig. S2). Compared with its syntenic region, the BAC sequence shares no substantial sequence identity to the B73 reference sequence except within the MATE1 coding region. This is not surprising, given the degree of heterogeneity in genome organization observed in maize. In fact, extensive noncolinearity in intergenic regions has been observed between different inbred lines for other loci (19–22).
Fig. 5.
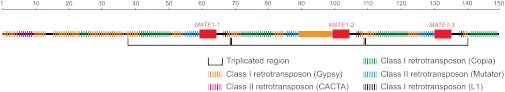
Genomic organization of the MATE1 locus shows three tandemly arrayed gene copies. Diagram showing Al237 genomic BAC ZMMCBa0006o22. The triplicated regions are indicated by brackets. The three copies of MATE1 are indicated by solid red boxes. The dashed boxes of different colors indicate different families of retrotransposons (annotated using http://maizetedb.org/~maize). The unique Gypsy retrotransposon insertion upstream of MATE1-2 is indicated by a solid orange box. Except for MATE1-1, -2, and -3, no other genes are predicted within the BAC sequence according to MAKER (45). The scale bar is in kilobase pairs.
Pervasiveness of MATE1 CNV.
Taking into account the effect of MATE1 CNV on Al tolerance in the Al237×L53 population, we sought to probe the prevalence of this variation in a broad maize genetic pool. For this purpose, we surveyed a panel of 166 diverse maize inbred lines and 16 teosinte lines (23) for MATE1 CNV (Dataset S1C). From a maize association panel (24) phenotyped for Al tolerance (Fig. 6A), we determined MATE1 copy number via qPCR in lines with intermediate-to-high levels of Al tolerance [root growth inhibition below 50%, i.e., relative root growth (RRG) of >0.5]. We also included maize inbreds known as “founder” lines, which capture most of maize genetic diversity (25), regardless of their level of Al tolerance. The results of our survey show that MATE1 CNV is rare: only two maize inbred lines with more than one copy of MATE1 were identified (C100-6 and Il677a; three MATE1 copies). Similar to the Al-tolerant parent of our population, Al237, these lines are Al-tolerant, ranking 6th and 29th (respectively) out of the 279 maize lines that were phenotyped for Al tolerance (Dataset S1C and Fig. 6A). Remarkably, maize lines with three MATE1 copies can be traced to the same regional origin, the acid soils of the South American tropics. Al237 (formerly L1327) and C100-6 are Cateto inbreds originally from Brazil. The other maize inbred line found to carry three MATE1 copies, Il677a, is a sweet corn inbred with a South American parent. This line (pedigree [(Bolivia 1035*Il44b)*Il422a]) is derived from a cross between Illinois sweet corn and a Bolivian line, donor of the sugary enhancer allele se1 (26, 27). Last, we measured MATE1 expression in root tips of these maize inbred lines carrying three MATE1 copies in comparison with lines with only one copy of MATE1 (Fig. 6B). Both under control and Al stress conditions, lines carrying three copies of MATE1 (Al237, C100-6, and Il677a) have high MATE1 expression compared with one-copy lines (L53 and B73). MATE1 expression levels in C100-6 and Il677a are comparable with those of Al237 under both conditions. These results are in agreement with the hypothesis that greater MATE1 copy number leads to higher gene expression levels.
Fig. 6.

Maize inbred lines carrying three MATE1 copies also show high MATE1 expression. (A) Distribution of Al tolerance (i.e., RRG) in maize diversity panel. The Al tolerance of lines in which MATE1 expression was quantified are indicated (L = L53; B = B73; IL = Il677a; Al = Al237; C = C100-6). The number in parenthesis indicates MATE1 copy number. (B) MATE1 relative expression quantified via qPCR in root tips of maize inbred lines from a survey for MATE1 CNV. Seedlings were grown under control conditions (Upper), or treated with 39 µM Al3+ activity (Lower) for 24 h. The red bars denote lines with one MATE1 copy (L53 and B73, Al-sensitive), and the blue bars denote three-copy lines (Al237, C100-6, and IL677a, Al-tolerant). Error bars indicate 1 SD.
Discussion
In humans, copy number variants encompass more nucleotides and arise more frequently than SNPs, and to a larger extent have been shown to drive human evolution and diversity between individuals (28, 29). Genome structural variation was discovered to also be pervasive in plants (21, 29–31), challenging the notion that a plant genome can be understood through the reference sequence of a single genotype (32). One recent maize study identified thousands of structural variants between the reference genome of B73 and another North American inbred line, Mo17 (31). As the authors point out, the high level of structural variation and differences in genome content observed in maize are unprecedented among higher eukaryotes. In the recently released maize HapMap2 study, structural variation was probed through a global analysis of read-depth variants in over 100 maize lines. Results showed that more than 90% of the maize genome shows some degree of CNV between lines (33). These structural variations are enriched among genes related to stress and stimulus responses, suggesting a role in maize phenotypic diversity and adaptation. Similar associations were observed in soybean, where structural variants are enriched in genomic regions harboring genes involved in plant biotic defense (34).
Despite these results from large-scale studies, examples of the effect of structural variants on plant phenotypes are scarce. Rather than CNVs, instances described to date involve transposable element insertions that modify the gene’s level and/or pattern of expression. This is the case for the maize domestication gene tb1 (35), where a Hopscotch insertion in a regulatory region acts as an enhancer of gene expression. Another example involves another Al tolerance gene, HcAACT1 from barley (36). In this case, a small CACTA-like transposon inserted 5 kb upstream of the ORF both enhances and alters the tissue localization of HcAACT1 expression. The results presented here strongly suggest that CNV for MATE1 is the genetic basis for a major Al tolerance QTL mapped in the Al237×L53 population, therefore providing a direct link between CNV and phenotype in maize. In plants, this is a landmark example of a QTL explained by CNV of the gene responsible for the molecular mechanism that is the QTL’s mode of action.
The majority of CNVs described in maize are present in multiple inbred lines (33) and are thought to predate domestication, as suggested by their presence in the ancestor of maize, teosinte (29). CNV for MATE1, however, is rare among maize lines and possibly occurred after domestication, as it was not observed in teosinte. This suggests that de novo copy number variants are likely to still be arising in the maize genome, and may have significant roles in phenotypic variation and adaptation. Interestingly, only two alleles were found for the MATE1 locus, and the three-copy allele appears at a very low frequency among maize lines. In addition, sequencing of the BAC clone containing the three copies of MATE1 from maize line Al237 showed that the 30-kbp repeat regions share high sequence identity with each other. Taken together, these results suggest the triplication at the MATE1 locus is a very recent event.
It is interesting to note that all three maize lines that carry the three-copy allele share the same geographical origin, in regions of acid soils of the South American tropics. Al237 and C100-6 are Cateto inbreds, the most widespread racial group in South America and the only native Brazilian race used in local maize hybrid breeding programs (37). Cateto constitutes a group of landraces, originally cultivated by the native Indians living in coastal areas from Argentina to the Guianas. Maize lines and hybrids derived from Cateto races have been identified as important sources of Al tolerance since the early 1980s (38, 39). Thus, Al tolerance can be considered an important adaptive trait carried by Cateto, which may have contributed to its overall acceptance, particularly in Brazil. In summary, maize lines carrying the three-copy allele of MATE1 are all originally from a region where Al tolerance has a strong adaptive value, and where breeding for acid soil tolerance has been intense. The fact that the three-copy allele was fixed in these lines suggests that it may provide a specific advantage for growth on acid soils. Taken together, our data indicate that structural variation in the maize genome has contributed to local adaptation of this crop to the highly acidic soils of the tropics.
Materials and Methods
Plant Growth and Treatment, DNA and RNA Isolations.
Maize seeds were germinated and seedlings were grown in full nutrient solution at pH 4.0 in a growth chamber at 26 °C/24 °C (light/dark, 16/8 h), as previously described (40). Free Al3+ activities were calculated using the chemical speciation software GEOCHEM-EZ (41). Phenotyping for Al tolerance based on RRG was performed using RootReader2D software (www.plantmineralnutrition.net/rootreader.htm), as described (42). For RNA extraction, root tips (1 cm) were collected and flash-frozen in liquid N2, and then stored at −80 °C. Total RNA was isolated using the RNeasy Plant Mini Kit (Qiagen) according to the manufacturer’s instructions and quantified spectrophotometrically in a Nanodrop 1000 (Thermo Scientific). Genomic DNA was isolated from young shoot tissue using the DNeasy Plant 96 and Mini kits (Qiagen), and quantified using PicoGreen dsDNA quantitation assay (Life Technologies); fluorescence was read in a DTX880 microplate reader (Beckman Coulter).
Gene Copy Number Determination via qPCR.
Primers for qPCR were designed using Primer Express software (Applied Biosystems) with default parameters, or obtained from previously published material. Primer sequences and sources are listed in Table S1. Real-time PCR was performed with Power SYBR Green PCR mastermix (Applied Biosystems) and conducted in a 7900HT Sequence Detection System (Applied Biosystems) using a relative standard curve method. In each run, samples were assayed for both the gene-of-interest (target) and for actin, a gene with known copy number (endogenous control). A set of standards was built from a serial dilution of genomic DNA; standard curves were run for both assays (target and endogenous control), and samples were quantified against the curves using 6 ng of genomic DNA as template. Relative quantities (RQs) were calculated by dividing the target sample quantity by the endogenous control sample quantity, and then normalizing against a calibrator sample (indicated in the text). Therefore, RQ values represent gene copy number relative to the calibrator genotype. Three technical replicates of each sample were averaged per assay (i.e., target and endogenous control). Because RQs are the ratio of two means, SDs were calculated from the coefficients of variation (cv, defined as the ratio between the SD s and the mean 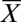 ). The cv for RQ values is as follows:
). The cv for RQ values is as follows: 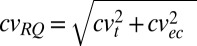 , where t is target and ec is endogenous control. Once cvRQ is known, then SD is calculated by resolving
, where t is target and ec is endogenous control. Once cvRQ is known, then SD is calculated by resolving 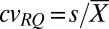 for s.
for s.
Gene Expression Analysis via qPCR.
First-strand cDNA was synthesized from total RNA using the High Capacity RNA-to-cDNA Kit (Applied Biosystems) according to the manufacturer’s instructions. MATE1 expression was determined using a custom-designed TaqMan assay (forward primer, 5′-CACCCGCTTAGCGTATTCCT-3′; reverse primer, 5′-GCACCGCGATCCTCATGAT-3′; and probe, 5′-TCTGAATGCGAGCCTCG-3′). A predesigned TaqMan assay for Eukaryotic 18S (Applied Biosystems) was used as the endogenous control. Real-time PCR was conducted in a 7900HT Sequence Detection System (Applied Biosystems). Relative expression was calculated using a relative standard curve method (see above), using standard curves built from a bulked cDNA pool. When determining MATE1 expression in the RILs, each dataset (i.e., control and Al-treated) was generated independently. The Al-sensitive parent L53 was set as calibrator sample; therefore, RQ values represent expression relative to L53 = 1 within each dataset. Three technical replicates of each sample were averaged per assay; SDs were calculated as described above.
FISH.
Procedures are as previously described (43). Details are described in SI Materials and Methods.
GBS of the Al237×L53 Population.
GBS was performed as previously described (17). Details are described in SI Materials and Methods and Fig. S6.
QTL and eQTL Mapping.
The phenotypic index used for mapping Al tolerance QTL was RRG as previously described (15). For mapping MATE1 expression (eQTL), two phenotypes were used: relative expression under control conditions and relative expression under Al stress. Mean RQ values were obtained as described above. Before single marker analysis, lines and SNP loci with more than 10% of missing data were excluded. Furthermore, markers with the least amount of missing data were selected within 2-Mbp windows, among 1,894 SNPs distributed along 165 Mbp of chromosome 6. Association between SNP markers generated by GBS and phenotypic data were evaluated using a general linear model (GLM) in TASSEL (44), and P values for the F tests were generated.
Supplementary Material
Acknowledgments
We thank Doreen Ware and Joshua Stein for their help and suggestions. We also thank Julia Vrebalov and Carlos Fasane for technical assistance; Jayson Talag for BAC library cloning; Wolfgang Golser and Raju Jetty for expertise and suggestions on BAC library strategies; and Jon Shaff, Randy Clark, and Eric Craft for providing phenotypic data for the maize association panel. This work was supported by Generation Challenge Programme Grants G3008.02 and G7010.03.02 (to L.V.K.).
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1220766110/-/DCSupplemental.
References
- 1.Kochian LV, Hoekenga OA, Pineros MA. How do crop plants tolerate acid soils? Mechanisms of aluminum tolerance and phosphorous efficiency. Annu Rev Plant Biol. 2004;55:459–493. doi: 10.1146/annurev.arplant.55.031903.141655. [DOI] [PubMed] [Google Scholar]
- 2. von Uexküll HR, Mutert E (1995) Global extent, development and economic-impact of acid soils. Plant Soil 171(1):1–15.journal.
- 3.Ma JF, Ryan PR, Delhaize E. Aluminium tolerance in plants and the complexing role of organic acids. Trends Plant Sci. 2001;6(6):273–278. doi: 10.1016/s1360-1385(01)01961-6. [DOI] [PubMed] [Google Scholar]
- 4.Magalhaes JV, et al. A gene in the multidrug and toxic compound extrusion (MATE) family confers aluminum tolerance in sorghum. Nat Genet. 2007;39(9):1156–1161. doi: 10.1038/ng2074. [DOI] [PubMed] [Google Scholar]
- 5.Furukawa J, et al. An aluminum-activated citrate transporter in barley. Plant Cell Physiol. 2007;48(8):1081–1091. doi: 10.1093/pcp/pcm091. [DOI] [PubMed] [Google Scholar]
- 6.Liu JP, Magalhaes JV, Shaff J, Kochian LV. Aluminum-activated citrate and malate transporters from the MATE and ALMT families function independently to confer Arabidopsis aluminum tolerance. Plant J. 2009;57(3):389–399. doi: 10.1111/j.1365-313X.2008.03696.x. [DOI] [PubMed] [Google Scholar]
- 7.Yokosho K, Yamaji N, Ma JF. An Al-inducible MATE gene is involved in external detoxification of Al in rice. Plant J. 2011;68(6):1061–1069. doi: 10.1111/j.1365-313X.2011.04757.x. [DOI] [PubMed] [Google Scholar]
- 8.Ryan PR, Raman H, Gupta S, Horst WJ, Delhaize E. A second mechanism for aluminum resistance in wheat relies on the constitutive efflux of citrate from roots. Plant Physiol. 2009;149(1):340–351. doi: 10.1104/pp.108.129155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yang XY, et al. A de novo synthesis citrate transporter, Vigna umbellata multidrug and toxic compound extrusion, implicates in Al-activated citrate efflux in rice bean (Vigna umbellata) root apex. Plant Cell Environ. 2011;34(12):2138–2148. doi: 10.1111/j.1365-3040.2011.02410.x. [DOI] [PubMed] [Google Scholar]
- 10. Bahia Filho AFC, Magnavaca R, Schaffert RE, Alves VMC (1997) Plant-Soil Interactions at Low pH: Sustainable Agriculture and Forestry Production, ed Moniz AC (Brazilian Soil Science Society, Campinas/Viçosa, Brazil), pp 59–70.
- 11.Magnavaca R, Gardner C, Clark R. In: Genetic Aspects of Plant Mineral Nutrition. Gabelman HW, Loughman BC, editors. Dordrecht, The Netherlands: Martinus Nijhoff; 1987. pp. 255–265. [Google Scholar]
- 12.Pandey S, et al. Genetics of tolerance to soil acidity in tropical maize. Crop Sci. 1994;34(6):1511–1514. [Google Scholar]
- 13.Borrero JC, Pandey S, Ceballos H, Magnavaca R, Bahia AFC. Genetic variances for tolerance to soil acidity in a tropical maize population. Maydica. 1995;40(3):283–288. [Google Scholar]
- 14.Ninamango-Cárdenas FE, et al. Mapping QTLs for aluminum tolerance in maize. Euphytica. 2003;130(2):223–232. [Google Scholar]
- 15.Maron LG, et al. Two functionally distinct members of the MATE (multi-drug and toxic compound extrusion) family of transporters potentially underlie two major aluminum tolerance QTLs in maize. Plant J. 2010;61(5):728–740. doi: 10.1111/j.1365-313X.2009.04103.x. [DOI] [PubMed] [Google Scholar]
- 16.Maron LG, et al. Transcriptional profiling of aluminum toxicity and tolerance responses in maize roots. New Phytol. 2008;179(1):116–128. doi: 10.1111/j.1469-8137.2008.02440.x. [DOI] [PubMed] [Google Scholar]
- 17.Elshire RJ, et al. A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS One. 2011;6(5):e19379. doi: 10.1371/journal.pone.0019379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Koren S, et al. Hybrid error correction and de novo assembly of single-molecule sequencing reads. Nat Biotechnol. 2012;30(7):693–700. doi: 10.1038/nbt.2280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Song R, Messing J. Gene expression of a gene family in maize based on noncollinear haplotypes. Proc Natl Acad Sci USA. 2003;100(15):9055–9060. doi: 10.1073/pnas.1032999100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brunner S, Fengler K, Morgante M, Tingey S, Rafalski A. Evolution of DNA sequence nonhomologies among maize inbreds. Plant Cell. 2005;17(2):343–360. doi: 10.1105/tpc.104.025627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morgante M, et al. Gene duplication and exon shuffling by helitron-like transposons generate intraspecies diversity in maize. Nat Genet. 2005;37(9):997–1002. doi: 10.1038/ng1615. [DOI] [PubMed] [Google Scholar]
- 22.Wang QH, Dooner HK. Remarkable variation in maize genome structure inferred from haplotype diversity at the bz locus. Proc Natl Acad Sci USA. 2006;103(47):17644–17649. doi: 10.1073/pnas.0603080103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wright SI, et al. The effects of artificial selection on the maize genome. Science. 2005;308(5726):1310–1314. doi: 10.1126/science.1107891. [DOI] [PubMed] [Google Scholar]
- 24.Flint-Garcia SA, et al. Maize association population: A high-resolution platform for quantitative trait locus dissection. Plant J. 2005;44(6):1054–1064. doi: 10.1111/j.1365-313X.2005.02591.x. [DOI] [PubMed] [Google Scholar]
- 25.Yu J, Holland JB, McMullen MD, Buckler ES. Genetic design and statistical power of nested association mapping in maize. Genetics. 2008;178(1):539–551. doi: 10.1534/genetics.107.074245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gonzales JW, Rhodes AM, Dickinson DB. A new inbred with high sugar content in sweet corn. HortScience. 1974;9:79–80. [Google Scholar]
- 27.Liu K, et al. Genetic structure and diversity among maize inbred lines as inferred from DNA microsatellites. Genetics. 2003;165(4):2117–2128. doi: 10.1093/genetics/165.4.2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stankiewicz P, Lupski JR. Structural variation in the human genome and its role in disease. Annu Rev Med. 2010;61:437–455. doi: 10.1146/annurev-med-100708-204735. [DOI] [PubMed] [Google Scholar]
- 29.Swanson-Wagner RA, et al. Pervasive gene content variation and copy number variation in maize and its undomesticated progenitor. Genome Res. 2010;20(12):1689–1699. doi: 10.1101/gr.109165.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fu H, Dooner HK. Intraspecific violation of genetic colinearity and its implications in maize. Proc Natl Acad Sci USA. 2002;99(14):9573–9578. doi: 10.1073/pnas.132259199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Springer NM, et al. Maize inbreds exhibit high levels of copy number variation (CNV) and presence/absence variation (PAV) in genome content. PLoS Genet. 2009;5(11):e1000734. doi: 10.1371/journal.pgen.1000734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Morgante M, De Paoli E, Radovic S. Transposable elements and the plant pan-genomes. Curr Opin Plant Biol. 2007;10(2):149–155. doi: 10.1016/j.pbi.2007.02.001. [DOI] [PubMed] [Google Scholar]
- 33.Chia JM, et al. Maize HapMap2 identifies extant variation from a genome in flux. Nat Genet. 2012;44(7):803–807. doi: 10.1038/ng.2313. [DOI] [PubMed] [Google Scholar]
- 34.McHale LK, et al. Structural variants in the soybean genome localize to clusters of biotic stress-response genes. Plant Physiol. 2012;159(4):1295–1308. doi: 10.1104/pp.112.194605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Studer A, Zhao Q, Ross-Ibarra J, Doebley J. Identification of a functional transposon insertion in the maize domestication gene tb1. Nat Genet. 2011;43(11):1160–1163. doi: 10.1038/ng.942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fujii M, et al. Acquisition of aluminium tolerance by modification of a single gene in barley. Nat Commun. 2012;3:713. doi: 10.1038/ncomms1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Paterniani E, Goodman MM. Races of Maize in Brazil and Adjacent Areas. Mexico City: CIMMYT; 1977. p. 95. [Google Scholar]
- 38.Miranda LT, Furlani PR, Miranda LEC, Sawazaki E. Genetics of environmental resistance and super-genes: Latente aluminum tolerance. Maize Genet Coop News Lett. 1984;58:46–48. [Google Scholar]
- 39.Sawazaki E, Furlani PR. Genetics of aluminum tolerance in maize cateto. Bragantia. 1987;46(2):269–278. [Google Scholar]
- 40.Piñeros MA, Magalhaes JV, Carvalho Alves VM, Kochian LV. The physiology and biophysics of an aluminum tolerance mechanism based on root citrate exudation in maize. Plant Physiol. 2002;129(3):1194–1206. doi: 10.1104/pp.002295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shaff JE, Schultz B, Craft E, Clark R, Kochian LV. GEOCHEM-EZ: A chemical speciation program with greater power and flexibility. Plant Soil. 2010;330(1):207–214. [Google Scholar]
- 42.Famoso AN, et al. Development of a novel aluminum tolerance phenotyping platform used for comparisons of cereal aluminum tolerance and investigations into rice aluminum tolerance mechanisms. Plant Physiol. 2010;153(4):1678–1691. doi: 10.1104/pp.110.156794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kato A, et al. Chromosome painting for plant biotechnology. Methods Mol Biol. 2011;701:67–96. doi: 10.1007/978-1-61737-957-4_4. [DOI] [PubMed] [Google Scholar]
- 44.Bradbury PJ, et al. TASSEL: Software for association mapping of complex traits in diverse samples. Bioinformatics. 2007;23(19):2633–2635. doi: 10.1093/bioinformatics/btm308. [DOI] [PubMed] [Google Scholar]
- 45.Cantarel BL, et al. MAKER: An easy-to-use annotation pipeline designed for emerging model organism genomes. Genome Res. 2008;18(1):188–196. doi: 10.1101/gr.6743907. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.