

Abstract
We report the synthesis and characterization of covalent organic frameworks (COFs) incorporating thiophene-based building blocks. We show that these are amenable to reticular synthesis, and that bent ditopic monomers, such as 2,5-thiophenediboronic acid, are defect-prone building blocks that are susceptible to synthetic variations during COF synthesis. The synthesis and characterization of an unusual charge transfer complex between thieno[3,2-b]thiophene-2,5-diboronic acid and tetracyanoquinodimethane enabled by the unique COF architecture is also presented. Together, these results delineate important synthetic advances toward the implementation of COFs in electronic devices.
Keywords: electronic materials, polymers, porous materials
Covalent organic frameworks (COFs) have emerged as an important class of polymeric crystalline materials with potential applications that complement those of metal organic frameworks (MOFs) (1–7). In particular, 2D COFs exhibit unique architectures wherein the monomers that make up their 2D layers stack almost perfectly into infinite 1D columns that are ideal for charge and exciton transport (6–12). Such electronic properties are particularly difficult to engineer in microporous MOFs, where high charge mobility and conductivity have only recently been demonstrated (13–15). Despite their layered structures, the typical hexagonal or square lattices of 2D COFs exhibit large 1D channels with well-defined pores that can be subject to reticular chemistry and postsynthetic modification (16, 17), and where complementary donor or acceptor molecules may in principle be inserted. Clearly, the unprecedented combination of high surface area, crystallinity, chemical and pore size tunability, and unique molecular architecture make COFs tantalizing targets for a new generation of electronic devices, including sustainable batteries, field-effect transistors, and photovoltaics. The first steps toward such applications must involve the incorporation of electroactive monomers within the COFs. This has been beautifully exemplified by the insertion of porphyrins (18), phthalocyanines (9), pyrene (8), naphthalenetetracarboxydiimide (19), and benzothiadiazole (20) in such materials. Notably, however, one of the most popular monomers in conductive organic polymers, thiophene, has not been incorporated in COFs thus far. Herein, we report the synthesis and characterization of thiophene-based COFs, including materials made from thiophene-, bithiophene-, and thienothiophene-diboronic acids. We also advance a hypothesis regarding the more general synthetic tractability of COFs made from bent ligands. Finally, we show that p-type COFs are amenable to doping with electron acceptors and report an unusual charge-transfer complex with tetracyanoquinodimethane (TCNQ).
Results and Discussion
We prepared thiophene-based COF (T-COFs) under conditions mimicking those used for the synthesis of the prototypical COF-5. Thus, condensation of 2,3,6,7,10,11-hexahydroxytriphenylene (HHTP) with 2,5-thiophenediboronic acid (H4TDB) (21) in dioxane and mesitylene at 120 °C over 72 h afforded (HOTP)2(TDB)3 (T-COF 1; HOTP = 2,3,6,7,10,11-hexaoxytriphenylene) as an off-white powder. Boronate ester linkages were confirmed by fourier transform infrared spectroscopy (FT-IR) spectroscopy, which revealed stretching frequencies at 1,348, 1,323, and 1,225 cm−1, the vibrational signature of catechol boroester rings. T-COF 1 is crystalline and shows four main powder X-ray diffraction (PXRD) peaks at 2θ = 3.72°, 6.11°, 9.96°, and 26.38°. Exploration of several possible 2D structures for T-COF 1, including eclipsed, staggered, and slipped-stacked AA and AB sequences of 2D layers (SI Appendix, Fig. S3), confirmed that the best fit for these peaks corresponded to the [100], [110], [210], and [001] reflections of the eclipsed AA structure shown in Fig. 1, with unit cell parameters a = b = 28.51 Å and c = 3.4 Å. Importantly, pure samples of T-COF 1 could be obtained only by using a precise 3:2 H4TDB–HHTP stoichiometry and rigorous air-free conditions and by minimizing the exposure of the reaction mixture to water. The latter is particularly difficult to achieve with boronate-linked COFs because water is a byproduct of the condensation of boronic acids with catechols. Here, we were able to circumvent this challenge by using vacuum-sealed glass tubes with ample empty volume as the reaction media. The large empty volume left in the sealed tubes, ∼20 times greater than that of the solution, is essential because it allows water to vaporize as it is being produced during the boronate-catechol condensation.
Fig. 1.

Simulated (red) and experimental (black) PXRD patterns of T-COF 1. Inset shows its building blocks and the partial structure viewed normal and parallel to the ab plane. Yellow, red, gray, and pink spheres represent S, O, C, and B atoms, respectively. In Figs. 1 and 4, the asterisks indicate reflection peaks from H3BO3, which is a decomposition product of all T-COFs in the presence of moisture, as verified by 11B MAS NMR δiso = 19.6 ppm (SI Appendix, Fig. S17).
As shown in Table 1, deviations from these rigorous conditions resulted either in phases with poor crystallinity, or in mixed products where a second COF cocrystallizes with T-COF 1. This second product was identified as (HOTP)(TDB)3 (T-COF 2), a material that displays both HOTP and boroxine rings as the three connected nodes. The structure of T-COF 2 and its unit cell of a = b = 20.94 Å and c = 3.4 Å were assigned by matching the relatively broad PXRD pattern, shown in Fig. 2, with the expected AA eclipsed stacking, as in the case of T-COF 1. In T-COF 2, boroxine rings are formed by self-condensation of the boronic acid groups in H4TDB, similar to those formed by the self-condensation of benzene-diboronic acid found in COF-1 (1, 22). As shown in Table 1, T-COF 2 can be accessed in pure form by adding a small amount of water [1% (vol/vol)] to the reaction mixture, by using a Dean–Stark trap instead of a sealed glass tube, or by increasing the H4TDB–HHTP ratio in the reaction mixture. Although none of the pure samples of T-COF 2 exhibited the same crystallinity observed for T-COF 1, their identity and purity was confirmed by FT-IR spectroscopy, which revealed the 712 cm−1 peak characteristic of boroxine rings, and by satisfactory elemental analysis (see the experimental details and SI Appendix, Fig. S5).
Table 1.
Methodology for T-COF synthesis
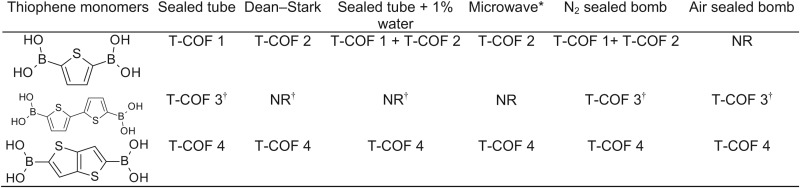 |
NR, no reaction.
Microwave power = 150 W, 30 min.
120 °C, 8 d.
Fig. 2.

Simulated (red) and experimental (black) PXRD patterns of T-COF 2. Inset shows its building blocks and the partial structure viewed normal and parallel to the ab plane. Yellow, red, gray, and pink spheres represent S, O, C, and B atoms, respectively. Extensive attempts to improve the crystallinity of this compound were unsuccessful; we assign it to the bent nature of H4TDB (vide infra and Fig. 5).
Having identified appropriate conditions for accessing thiophene-based boronate-linked COFs, we turned our attention to extended thiophene boronic acids with lower oxidation potentials that may offer better p-type hosts for electronic applications. Thus, condensation of bithiophene-2,5′-diboronic acid (H4BTDB) with HHTP under conditions similar to those used for T-COF 1 provided (HOTP)2(BTDB)3 (T-COF 3), which crystallizes in an eclipsed hexagonal 2D lattice with unit cell parameters a = b = 34.55 Å and c = 3.4 Å (Fig. 3). Notably, the formation of boroxine rings is not observed in this case, and no secondary products are obtained when using acid digestion bombs as the reaction media, although water exposure must once again be minimized (Table 1).
Fig. 3.

Simulated (red) and experimental (black) PXRD patterns of T-COF 3. Inset shows its building blocks and the partial structure viewed normal and parallel to the ab plane. Yellow, red, gray, and pink spheres represent S, O, C, and B atoms, respectively.
Our last thiophene-based COF target was one containing thieno[3,2-b]thiophene, one of the most promising monomers for organic conducting polymers (23, 24). Reaction of thieno[3,2-b]thiophene-2,5-diboronic acid (H4TTDB) (25) with HHTP produced (HOTP)2(TTDB)3 (T-COF 4) as an off-white crystalline powder in good yield. T-COF 4 also crystallizes in an eclipsed AA hexagonal 2D lattice with unit cell parameters a = b = 31.58 Å and c = 3.41 Å, and is shown in Fig. 4. We found that the synthesis of T-COF 4 is much more tolerant to various reaction conditions, and under all circumstances described in Table 1, T-COF 4 was accessible as a pure material; a sample with the highest crystallinity was obtained by using a Dean–Stark trap.
Fig. 4.

Simulated (red) and experimental (black) PXRD patterns of T-COF 4. Inset shows its building blocks and the partial structure viewed normal and parallel to the ab plane. Yellow, red, gray, and pink spheres represent S, O, C, and B atoms, respectively.
The relative insensitivity of T-COF 4 formation to reaction conditions may be attributed to the rigidity of the H4TTDB monomer, which, owing to its fewer degrees of rotational freedom, is less likely to cause defects during crystallization. Additionally, it is empirically evident that the more linear diboronic acids, H4BTDB and H4TTDB, where the two boronic acid groups—more precisely, the two B–C bonds—are parallel, are less sensitive to the reaction conditions than H4TDB. In the latter, the two boronic acid groups are disposed at an angle of 141.5° with respect to each other, which likely causes defects during the crystallization of T-COF 1 and T-COF 2 and lowers the symmetry of these materials compared with other reported 2D COFs. Indeed, of the three conformers stemming from the condensation of one HHTP with three H4TDB molecules (Fig. 5), which may serve as nucleation units for both T-COF 1 and T-COF 2, only isomer A has threefold symmetry and is productive toward the formation of crystalline phases in either case. The bent geometry of H4TDB is therefore possibly responsible for the sensitive synthesis of these two materials, which respectively crystallize in the very unusual space groups P-62m and P31m. Importantly, these observations suggest that bent linkers may generally be more difficult to incorporate into crystalline extended polymers formed by strong, less reversible bonds such as those in COFs, which allow faster defect propagation during crystallization.
Fig. 5.

Three conformers of a possible nucleation unit in T-COF 1 and 2, which stems from a three-connected HHTP node. Isomers B and C are not threefold symmetric and are therefore defect units that are not productive toward COF crystallization.
To investigate the porosity of the thiophene boronate materials, thermogravimetric analyses (TGAs) were conducted on pristine samples of T-COF 1–4. As shown in SI Appendix, Figs. S2, S6, S9, and S13, all of the samples lose guest solvent molecules upon heating to below 180–200 °C. Accordingly, all of the T-COFs were desolvated by heating under vacuum at 200 °C, and their integrity was confirmed by PXRD analysis. N2 adsorption isotherms performed on the desolvated materials revealed that all four are porous and adsorb 466, 172, 295, and 431 cm3 of N2/g at ∼760 torr and 77 K for T-COF 1 through T-COF 4, respectively (Fig. 6). Brunauer–Emmett–Teller (BET) analysis fits to the microporous region of the N2 adsorption isotherms (i.e., P/P0 < ∼0.2) revealed apparent surface areas of 927 (8), 562 (2), 544 (8), and 904 (2) m2/g for T-COF 1, 2, 3, and 4, respectively, values that are in line with those observed with other 2D COFs (26). With the smallest expected pore size of ∼13.8 Å, T-COF 2 was the only material that exhibited a true Type I isotherm (27), while the other three materials gave indication of mesoporosity characteristic of large-pore COFs (19, 28). Indeed, analysis of the N2 adsorption isotherms using the density functional theory (DFT) package gave pore size distributions centered at 2.06, 1.38, 3.24, and 2.57 nm for T-COF 1 through 4, respectively. These are in excellent agreement with the proposed structures, which give expected pore sizes of 2.04, 1.36, 3.15, and 2.75–2.94 nm for T-COF 1 through 4, respectively (see SI Appendix, Figs. S4, S7, S11, and S14 for geometrical analyses of the T-COFs).
Fig. 6.

N2 adsorption isotherms for T-COF 1–4 at 77 K.
In their native form, all of the T-COFs are off-white powders, which is indicative of wide bandgap materials and insulators. To take advantage of the electron donating ability of the thiophene units, we pursued doping experiments hoping either to form charge-transfer complexes, an important step toward promoting electron transfer for functional devices, or to provide a sufficient concentration of charge carriers to induce electric conductivity. Thus, immersing samples of T-COF 1 through T-COF 4 in solutions of strong oxidizers such as 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) and chloranyl produced dark materials, possibly indicative of charge transfer (CT) or even electron transport, but also induced amorphization of all of the T-COFs. We surmised that DDQ and chloranyl may oxidize the HOTP core itself or attack the oxidatively-prone B–C bond (29), both of which would explain the observed decomposition. Accordingly, we turned our attention to less potent oxidizers such as I2 and TCNQ, the latter being a widely used electron acceptor in CT salts and complexes. Whereas I2 also invariably caused amorphization of the T-COFs, likely through coordination of I− to the boron atom or through oxidation of the B–C bond, a 0.1 mM solution of TCNQ in CH2Cl2 gave no reaction with T-COF 1, 2, or 3, but immediately formed a crystalline black precipitate in contact with solid T-COF 4. The black material, denoted as TCNQ⊂T-COF 4, displayed virtually identical a and b unit cell parameters as T-COF 4, but a slightly elongated c axis by ∼0.2 Å, deduced from a decreased 2θ value of the [001] reflection (Fig. 7). After repeated washing with CH2Cl2 to eliminate weakly associated TCNQ molecules, elemental microanalysis of TCNQ⊂T-COF 4 revealed a formula of C54H18B6O12S6⋅(C12H4N4)0.98, which corresponds to exactly one TCNQ guest molecule per formula unit.
Fig. 7.

Simulated (green) and experimental PXRD patterns for T-COF 4 (red), and TCNQ⊂T-COF 4 (black). Insets show, in clockwise order from the Upper Left, FTIR spectra, diffuse reflectance UV/Vis-NIR spectra, and a zoom-in of the [001] reflection in the PXRD patterns. Arrows indicate peak shifts.
Ultraviolet-visible-near infrared (UV-Vis-NIR) diffuse reflectance spectroscopy suggested that the new material owes its dark color to a wide CT absorption band centered at 850 nm, shown in an inset of Fig. 7, rather than to delocalized electrons. Although planned charge mobility measurements are necessary to completely rule out electron transfer within TCNQ⊂T-COF 4, we note that control experiments aimed at preparing donor–acceptor dyads or CT complexes by mixing TCNQ with free HHTP, H4TTDB, or with thieno[3,2-b]thiophene in the same ratios and concentrations as those used for the formation of TCNQ⊂T-COF 4 did not result in the formation of dark materials and no stable CT complexes could be isolated. This suggested that TCNQ⊂T-COF 4 contains weakly associated donor–acceptor pairs that are only stabilized in the rigid COF matrix and are not isolable from solution. Indeed, attempts to evacuate the solvent molecules from TCNQ⊂T-COF 4 in preparation for gas sorption measurements to assess its porosity resulted in sublimation of TCNQ from the black material and recovery of clean T-COF 4. Further evidence for the weak association between TCNQ and T-COF 4 came from a competitive experiment in which TCNQ⊂T-COF 4 was immersed in mesitylene, which caused rapid bleaching of the black material, recovery of T-COF 4, and isolation of a red solution of the known mesitylene–TCNQ CT adduct (30, 31), which is therefore thermodynamically favored over the TCNQ⊂T-COF 4 complex. Finally, an EPR experiment of TCNQ⊂T-COF 4 did not reveal a radical species, suggesting once again that the interaction of TCNQ with T-COF 4, does not involve the complete transfer of an electron from the valence band of T-COF 4 to TCNQ and that TCNQ most likely remains neutral.
To probe the nature of the interaction of TCNQ with T-COF 4, we analyzed the black material by FT-IR spectroscopy. Shifts in the C ≡ N (νCN) and C = C (νCC) stretches, and the out-of-plane C–H bend (δCH), which occur respectively at 2,226, 1,545, and 860 cm−1 in neat TCNQ, are typically used to discern the degree of CT in TCNQ CT complexes (32). Albeit small, shifts of νCC and δCH to lower energy (1,537 and 798 cm−1, respectively) are in line with partial negative charge being transferred from T-COF 4 to TCNQ. However, accumulation of negative charge should also partially populate the π* orbital of the nitrile groups and shift νCN to lower frequency. Instead, we observe an unusual shift of νCN to higher energy, from 2,226 to 2,233 cm−1 (Fig. 7, Inset). Although hypsochromic shifts of νCN are not unprecedented for TCNQ and other nitriles, they are usually associated with sigma-only N-coordination of TCNQ to heavier metals (33, 34). However, 11B magic angle spinning (MAS) NMR spectroscopy of TCNQ⊂T-COF 4 attested that the boron atoms, the only likely candidates for TCNQ binding, remain trigonal (δiso =12–40 ppm) and are not pyramidalized (δiso = –5–10 ppm), as expected if they acted as Lewis acids toward TCNQ (SI Appendix, Fig. S18). Additionally, coordination of one of the nitrile groups typically splits the B1u and B2u normal modes of the C ≡ N stretch, which is not observed in our case, and confirms the coordinatively innocent nature of the TCNQ molecule. As such, we tentatively assign the unusual shift of νCN to a symmetric intercalation of TCNQ guests between the 2D layers of T-COF 4, which may cause a rigidification of TCNQ to the extent that it affects the CN bonds by stiffening them. This qualitative hypothesis agrees with the observed elongation of the c axis in TCNQ⊂T-COF 4 relative to T-COF 4 itself, and clearly merits further investigation, as planned in our laboratory.
In conclusion, we reported the synthesis of COFs containing thiophene-based monomers. We have shown that these are amenable to reticular synthesis principles, but that bent diboronic acids are defect-prone building blocks that are more susceptible to synthetic variations during COF synthesis. These, combined with the characterization of the first CT complex isolated postsynthetically inside a COF, are important steps toward the implementation of such materials in electronic devices. The search for adequate redox partners that will not disrupt the crystalline structure and will promote full electron transfer, thereby providing a sufficient concentration of charge carriers, will be an important challenge for eventually achieving electric conductivity in these extraordinary materials.
Materials and Methods
Starting materials were purchased from Sigma-Aldrich or TCI and used without further purification. Mesitylene and 1,4-dioxane were purchased from TCI. N,N-dimethylformamide (DMF), hexane, ethyl acetate, and silica-gel were purchased from VWR. THF was taken from an alumina column solvent purification system. NMR spectra were recorded on a Varian 300 Mercury NMR spectrometer and a Bruker Avance-400 NMR spectrometer. 1H NMR data are reported as follows: chemical shift [multiplicity (s, singlet; d, doublet; b, broad), coupling constant, number of hydrogen atoms (n H) by integration]. 1H and 13C chemical shifts are reported in ppm from TMS with the residual solvent resonances as internal standards. Peaks in infrared spectra are assigned as weak (w), medium (m), or strong (s). Elemental analyses were performed by Midwest Microlab.
PXRD patterns were recorded with a Bruker D8 Advance diffractometer equipped with a θ/2θ Bragg–Brentano geometry and nickel-filtered Cu Kα radiation (Kα1 = 1.5406 Å, Kα2 = 1.5444 Å, Kα1/Kα2 = 0.5). The tube voltage and current were 40 kV and 40 mA, respectively. Samples for PXRD were prepared by placing a thin layer of the designated materials on a zero-background silicon (510) crystal plate.
A Micromeritics ASAP 2020 Surface Area and Porosity Analyzer was used to measure the nitrogen adsorption isotherms. Oven-dried sample tubes equipped with TranSeals (Micromeritics) were evacuated and tared. The samples were transferred to the sample tubes, which were then capped by TranSeals. The samples were heated to 200 °C under a vacuum of 2 mtorr for 12 h, at which point the outgas rate was less than 2 mtorr/min. The evacuated sample tubes were weighed again, and the samples’ mass were determined by subtracting the mass of the previously tared tubes. N2 isotherms were measured using liquid nitrogen baths (77 K). Ultra high purity grade (99.999% purity) N2 and He, oil-free valves, and gas regulators were used for the free space correction and measurement. Relative pressure (P/P0) range for BET analysis was taken from 5×10−5 to ∼0.2. Pore sizes were determined using the DFT N2 model for the cylindrical pores geometry, with relative pressure (P/P0) range from 0.003 to 0.1.
Infrared spectra were obtained on a PerkinElmer Spectrum 400 Spectrometer equipped with a Pike Technologies GladiATR accessory with a germanium crystal. Diffuse reflectance UV-Vis spectra were collected on a Varian Cary 5000 UV-Vis-NIR spectrometer equipped with a Praying Mantis diffuse reflectance accessory and an environmental chamber (Harrick Scientific Products) and were referenced to BaSO4.
2,2′-Bithiophene-5,5′-Diboronic Acid (H4BTDB).
In a 250 mL round bottom flask, 3.00 g (7.18 mmol) of 5,5′-Bis(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-2,2'-bithiophene were dissolved in 20 mL of THF and 5 mL of water. The mixture was stirred for 2 h at room temperature, followed by the addition of 5 mL of a 6 M aqueous solution of HCl. The resulting mixture was stirred for 12 h. Water (150 mL) was then added to precipitate the desired product, which was recovered by filtration as a fine white powder. After air drying and drying under high vacuum (10−3 torr), we obtained 1.91 g (7.11 mmol, 98.0%) of pure H4BTDB. 1H NMR (400 MHz, DMSO-d6), δ 8.29 (s, b, 4 H), 7.60 (d, J = 3.6 Hz, 2 H), and 7.35 (d, J = 3.6 Hz, 2 H). 13C NMR (101 MHz, DMSO-d6), δ 134.1 (2 C, quaternary), 132.0 (2 C, tertiary), 126.5 (2 C, tertiary), and 122.4 (2 C, quaternary). Elemental analysis calculated for C8H8B2O4S2, C (37.84%), H (3.18%), and S (25.26%). Found, C (35.21%), H (3.14%), and S (25.25%). Low-carbon analysis is typical for boron-containing compounds due to the formation of refractory boron carbide (BC) during the analysis.
2,5-Dibromothieno[3,2-b]Thiophene.
In a 250 mL round bottom flask wrapped with aluminum foil, 5.00 g [1.00 equivalent (eq), 35.7 mmol] of thieno[3,2-b]thiophene and 19.06 g of N-bromosuccinimide (3.00 eq, 107 mmol) were dissolved in 100 mL of DMF. The colorless reaction mixture was stirred in the dark for 12 h. The resulting deep orange solution was poured in 500 mL of water. A pale pink solid precipitated and was isolated by filtration, dissolved in CH2Cl2, and mixed with 20 g of silica gel in a 500 mL flask. The solvent was evaporated under reduced pressure and dry-loaded on a chromatography column (diameter 5.5 cm, 350 g SiO2, elution:hexane). The first fractions were concentrated under reduced pressure to afford 9.1 g (30.5 mmol, 85%) of 2,5-dibromo-thieno[3,2-b]thiophene as a white powder. 1H NMR (400 MHz, DMSO-d6), δ 7.61 (s, 2 H); 13C NMR (101 MHz, DMSO-d6), δ 134.1 (2 C, quaternary), 132.0 (2 C, tertiary), and 126.5 (2 C, quaternary). The analytical data matches that reported previously (32).
2,5-Bis(4,4,5,5-Tetramethyl-1,3,2-Dioxaborolan-2-yl)Thieno[3,2-b]Thiophene.
In a 250 mL round bottom flask wrapped with aluminum foil, 2.5 g (1.0 eq, 8.6 mmol) of 2,5-dibromo-thieno[3,2-b]thiophene was loaded and then purged three times with dry nitrogen. Dry THF was then added and the mixture was cooled to –78 °C in a dry ice–acetone bath. After cooling, 7.6 mL of 2.5 M solution of n-butyllithium (n-BuLi) in hexane (2.2 eq, 18.9 mmol) was added dropwise (over 20 min). The reaction mixture was stirred for 30 min at –78 °C, and then 4.8 g (3.0 eq, 25.8 mmol) of freshly distilled 2-isopropoxy-4,4,5,5-tetramethyl-1,3,2-dioxaborolane was added to the mixture. The reaction was allowed to reach room temperature, and stirred overnight. A portion of 3 mL of anhydrous ethanol was used to quench the mixture, and then 15 g of silica was added to the flask. The solvent was removed under reduced pressure and dry-loaded on a chromatography column (diameter 5.5 cm, 350 g SiO2) wrapped in aluminum foil (elution:hexane/ethyl acetate = 20/1). The second elution gave 0.8 g (2.0 mmol, 23%) of pure 2,5-Bis(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-thieno[3,2-b-]thiophene. The desired product is sensitive to UV light, and must be stored in a dry, dark place. 1H NMR (400 MHz, DMSO-d6):, δ 7.79 (s, 2 H), 1.71 (s, 24 H). The analytical data matches that reported previously (33).
2,5-Thieno[3,2-b]Thiophene-Diboronic Acid (H4TTDB).
In a 100 mL round bottom flask, 0.8 g (2.0 mmol) of 2,5-Bis(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-thieno[3,2-b-]thiophene were dissolved in 10 mL of THF and 2.5 mL of water. The mixture was stirred for 2 h at room temperature, followed by the addition of 2.5 mL of a 6 M aqueous solution of HCl. The resulting mixture was stirred for 12 h. A portion of 50 mL of water was then added to precipitate the desired product, which was recovered by filtration as a fine white powder. After drying under air and high vacuum (10−3 torr), we isolated 354 mg (1.55 mmol, 77%) of pure H4TTDB. 1H NMR (400 MHz, DMSO-d6), δ 8.38 (s, b, 4 H), 7.85 (s, 2 H). The analytical data matches that reported previously (34).
T-COF 1.
In a 30-cm-long Pyrex glass tube (i.d. × o.d. = 0.9 × 1.1 cm), 129 mg (3.00 eq, 0.75 mmol) of H4TDB and 165 mg (2.00 eq, 0.5 mmol) of HHTP were dissolved in 5 mL of dry dioxane and 5 mL of dry mesitylene. The tube was connected to a vacuum line and the reaction mixture was subjected to four cycles of freezing (with liquid N2), vacuum pumping, and thawing, and was then flame-sealed under vacuum. The tube was heated at 120 °C for 3 d in a preheated oven, and then cooled to room temperature over 12 h. The off-white flocculent solid was washed with dry dioxane (5 mL) and dry CH2Cl2 (15 mL) and recovered by centrifugation or filtration. The remaining powder was dried under vacuum (10−3 torr) for 2 h. The material was desolvated at 200 °C under high vacuum (10−6 torr) overnight to obtain 132 mg of T-COF 1 (0.14 mmol, 56%). IR [Ge attenuated total reflectance (ATR)], 612 (w), 655 (m), 687 (m), 727 (m), 833 (m), 852 (w), 930 (w) 1,013 (m), 1,057 (m), 1,162 (m), 1,196 (m), 1,238 (s), 1,314 (m), 1,339 (s), 1,365 (s), 1,445 (s), 1,492 (s), 1,522 (s), 1,608 (w), and 2,919 (w) cm−1. Elemental analysis calculated for C48H18B6O12S3, C (60.83%), H (1.91%), and S (10.15%). Found, C (54.56%), H (1.92%), B [not determined (nd)], O (nd), and S (10.12%). Low-carbon analysis is typical for boron-containing compounds due to the formation of refractory BC during the analysis. TGA was performed before any evacuation/activation.
T–COF 2.
In a 100 mL round bottom flask, 129 mg (3.00 eq, 0.75 mmol) of H4TDB and 82.5 mg (1.00 eq, 0.25 mmol) of HHTP were dissolved in 15 mL of dry dioxane and 15 mL of dry mesitylene. A Dean–Stark apparatus was placed on top of the flask and filled with mesitylene. The setup was purged three times with N2 and heated at 120 °C for 2 d. The white flocculent solid was washed with filtration or centrifuge with dry dioxane and dry CH2Cl2. The remaining powder was dried under high vacuum (10−3 torr) for 2 h. Total desolvation can be performed at 200 °C under high vacuum (10−6 torr) overnight to obtain 139 mg of T-COF 2 (0.21 mmol, 82%). IR (Ge ATR), 662 (m), 687 (w), 712 (s), 760 (w), 831 (m), 855 (w), 995 (w), 1,013 (w), 1,038 (w), 1,117 (w), 1,161 (m), 1,187 (m), 1,264 (s), 1,316 (s), 1,492 (w), and 1,509 (m) cm−1. Elemental analysis calculated for C30H12B6O9S3, C (53.19%), H (1.79%), and S (14.20%). Found, C (50.66%), H (1.85%), and S (14.17%). Low-carbon analysis is typical for boron-containing compounds due to the formation of refractory BC during the analysis. TGA was performed before any evacuation/activation.
T-COF 3.
In a 20-cm-long Pyrex glass tube (i.d. × o.d. = 0.9 × 1.1 cm), 38 mg (3.0 eq, 0.15 mmol) of H4BTDB and 33 mg (2.0 eq, 0.10 mmol) of HHTP were disolved in 1 mL of dry dioxane and 1 mL of dry mesitylene. The tube was connected to a vacuum line, and the reaction mixture was subjected to four cycles of freezing (with liquid N2), vacuum pumping, and thawing, and was then sealed under vacuum. The tube was heated at 120 °C for 8 d in a preheated oven, and then cooled to room temperature over 12 h. The yellow flocculent solid was washed with dry dioxane (5 mL) and dry CH2Cl2 (15 mL) and recovered by centrifugation or filtration. The remaining powder was dried under vacuum (10−3 torr) for 2 h. The material was desolvated at 200 °C under high vacuum (10−6 torr) overnight to obtain 40.0 mg of T-COF 3 (0.034 mmol, 67.0%). IR (Ge ATR), 652 (s), 727 (w), 808 (w), 829 (m), 863 (s), 900 (w), 983( m), 1,071 (m), 1,160 (s), 1,244 (s), 1,350 (s), 1,431 (s), 1,495 (m), and 1,520 (m). Elemental analysis calculated for C60H24B6O12S6, C (60.35%), H (2.03%), and S (16.11%). Found, C (55.86%), H (2.40%), and S (15.98%). Low-carbon analysis is typical for boron-containing compounds due to the formation of refractory BC during the analysis. TGA analysis was performed before any evacuation/activation.
T-COF 4.
Both Dean–Stark and sealed tube setups gave high yield and crystallinity. In a 100 mL round bottom flask, 171 mg (3.00 eq, 0.75 mmol) of H4TTDB and 165 mg (2 eq, 0.5 mmol) of HHTP were dissolved in 15 mL of dry dioxane and 15 mL of dry mesitylene. A Dean–Stark apparatus was placed on top of the flask and filled with mesitylene. The setup was purged three times with N2 and heated at 120 °C for 2 d under N2. The gray flocculent solid was washed with filtration or centrifuge with dry dioxane (20 mL) and dry CH2Cl2. The remaining powder was dried under vacuum (10−3 torr) for 2 h. Total desolvation can be performed at 200 °C under high vacuum (10−6 torr) overnight to obtain 254 mg of T-COF 4 (0.23 mmol, 91%). IR (Ge ATR), 650 (w), 798 (s), 1,017 (s), 1,091 (s), 1,162 (m), 1,239 (m), 1,260 (s), 1,357 (s), 1,442 (s), 1,486 (s), and 2,962 (s). Elemental analysis calculated for C54H18B6O12S6, C (58.12%), H (1.63%), and S (17.24%). Found, C (54.55%), H (1.74%), and S (17.14%). Low-carbon analysis is typical for boron-containing compounds due to the formation of refractory BC during the analysis. TGA analysis was performed before any evacuation/activation.
TCNQ⊂T-COF 4.
TCNQ (10 mg, 0.05 mmol) and T-COF 4 (20 mg, 0.012 mmol) were mixed in a 2 mL vial. The vial was then filled with CH2Cl2 to the brim and was capped. After 2 d at room temperature in the dark, the brown–black solid was filtered and washed five times with 10 mL of dichloromethane. The product was dried for 2 h under high vacuum (10−6 torr) to give 21 mg of a black powder. IR (Ge ATR), 617 (w), 642 (w), 797 (w), 832 (w), 1,000 (m), 1,169 (s), 1,246 (s), 1,284 (s), 1,340 (s), 1,378 (s), 1,445 (s), 1,475 (m), 1,539 (s), 2,233 (s), and 2,981 (w). Elemental analysis calculated for C54H18B6O12S6·(C12H4N4)0.97 (one TCNQ per formula unit of T-COF 4), Calculated C (60.09%), H (1.73%), N (4.28%), and S (14.97%). Found, C (55.62%), H (1.85%), N (4.29%), and S (15.10%). Low-carbon analysis is typical for boron-containing compounds due to the formation of refractory BC during the analysis. TGA was performed before any evacuation/activation.
A control experiment was performed by mixing TCNQ (20.4 mg, 0.1 mmol) with H4TTDB (23.4 mg, 0.1 mmol) or thieno[3,2-b]thiophene (14.4 mg, 0.1 mmol) in CH2Cl2 (10 mL) in the dark for 48 h. In each case, the powders isolated after filtration did not show a nitrile stretch in the IR spectra, which matched the IR spectra of the neat thiophene monomers.
Supplementary Material
Acknowledgments
This work was supported by the US Department of Energy, Office of Science, Office of Basic Energy Sciences under Award DE-SC0006937 (all work except NMR studies). G.H.V.B. was supported by a Fondation Monahan postdoctoral fellowship. NMR studies were supported by the National Institute of Health through Grants EB001960 and EB002026 (to R.G.G.). V.K.M. was supported by a Natural Sciences and Engineering Research Council of Canada postdoctoral fellowship.
Footnotes
The authors declare no conflict of interest.
†This Direct Submission article had a prearranged editor.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1221824110/-/DCSupplemental.
References
- 1.Côté AP, et al. Porous, crystalline, covalent organic frameworks. Science. 2005;310(5751):1166–1170. doi: 10.1126/science.1120411. [DOI] [PubMed] [Google Scholar]
- 2.Wan S, Guo J, Kim J, Ihee H, Jiang D. A belt-shaped, blue luminescent, and semiconducting covalent organic framework. Angew Chem Int Ed Engl. 2008;47(46):8826–8830. doi: 10.1002/anie.200803826. [DOI] [PubMed] [Google Scholar]
- 3.Han SS, Furukawa H, Yaghi OM, Goddard WA., 3rd Covalent organic frameworks as exceptional hydrogen storage materials. J Am Chem Soc. 2008;130(35):11580–11581. doi: 10.1021/ja803247y. [DOI] [PubMed] [Google Scholar]
- 4.Uribe-Romo FJ, et al. A crystalline imine-linked 3-D porous covalent organic framework. J Am Chem Soc. 2009;131(13):4570–4571. doi: 10.1021/ja8096256. [DOI] [PubMed] [Google Scholar]
- 5.Furukawa H, Yaghi OM. Storage of hydrogen, methane, and carbon dioxide in highly porous covalent organic frameworks for clean energy applications. J Am Chem Soc. 2009;131(25):8875–8883. doi: 10.1021/ja9015765. [DOI] [PubMed] [Google Scholar]
- 6.Spitler EL, Dichtel WR. Lewis acid-catalysed formation of two-dimensional phthalocyanine covalent organic frameworks. Nat Chem. 2010;2(8):672–677. doi: 10.1038/nchem.695. [DOI] [PubMed] [Google Scholar]
- 7.Feng X, Ding X, Jiang D. Covalent organic frameworks. Chem Soc Rev. 2012;41(18):6010–6022. doi: 10.1039/c2cs35157a. [DOI] [PubMed] [Google Scholar]
- 8.Wan S, Guo J, Kim J, Ihee H, Jiang D. A photoconductive covalent organic framework: Self-condensed arene cubes composed of eclipsed 2D polypyrene sheets for photocurrent generation. Angew Chem Int Ed Engl. 2009;48(30):5439–5442. doi: 10.1002/anie.200900881. [DOI] [PubMed] [Google Scholar]
- 9.Ding X, et al. An n-channel two-dimensional covalent organic framework. J Am Chem Soc. 2011;133(37):14510–14513. doi: 10.1021/ja2052396. [DOI] [PubMed] [Google Scholar]
- 10.Wan S, et al. Covalent organic frameworks with high charge carrier mobility. Chem Mater. 2011;23(18):4094–4097. [Google Scholar]
- 11.Ding X, et al. Conducting metallophthalocyanine 2D covalent organic frameworks: The role of central metals in controlling π-electronic functions. Chem Commun (Camb) 2012;48(71):8952–8954. doi: 10.1039/c2cc33929c. [DOI] [PubMed] [Google Scholar]
- 12.Koo BT, Dichtel WR, Clancy P. A classification scheme for the stacking of two-dimensional boronate ester-linked covalent organic frameworks. J Mater Chem. 2012;22(34):17460. [Google Scholar]
- 13.Gándara F, et al. Porous, conductive metal-triazolates and their structural elucidation by the charge-flipping method. Chemistry. 2012;18(34):10595–10601. doi: 10.1002/chem.201103433. [DOI] [PubMed] [Google Scholar]
- 14.Narayan TC, Miyakai T, Seki S, Dincă M. High charge mobility in a tetrathiafulvalene-based microporous metal-organic framework. J Am Chem Soc. 2012;134(31):12932–12935. doi: 10.1021/ja3059827. [DOI] [PubMed] [Google Scholar]
- 15.Kobayashi Y, Jacobs B, Allendorf MD, Long JR. Conductivity, doping, and redox chemistry of a microporous dithiolene-based metal−organic framework. Chem Mater. 2010;22(14):4120–4122. [Google Scholar]
- 16.Nagai A, et al. Pore surface engineering in covalent organic frameworks. Nat Commun. 2011;2:536. doi: 10.1038/ncomms1542. [DOI] [PubMed] [Google Scholar]
- 17.Bunck DN, Dichtel WR. Internal functionalization of three-dimensional covalent organic frameworks. Angew Chem Int Ed Engl. 2012;51(8):1885–1889. doi: 10.1002/anie.201108462. [DOI] [PubMed] [Google Scholar]
- 18.Feng X, Chen L, Dong Y, Jiang D. Porphyrin-based two-dimensional covalent organic frameworks: Synchronized synthetic control of macroscopic structures and pore parameters. Chem Commun (Camb) 2011;47(7):1979–1981. doi: 10.1039/c0cc04386a. [DOI] [PubMed] [Google Scholar]
- 19.Spitler EL, et al. Lattice expansion of highly oriented 2D phthalocyanine covalent organic framework films. Angew Chem Int Ed Engl. 2012;51(11):2623–2627. doi: 10.1002/anie.201107070. [DOI] [PubMed] [Google Scholar]
- 20.Feng X, et al. An ambipolar conducting covalent organic framework with self-sorted and periodic electron donor-acceptor ordering. Adv Mater. 2012;24(22):3026–3031. doi: 10.1002/adma.201201185. [DOI] [PubMed] [Google Scholar]
- 21.Coutts GC, Goldschmid HR, Musgrave OC. Organoboron compounds, Part VIII, Aliphatic and aromatic diboronic acids. J Chem Soc. 1970;(3):488–493. [Google Scholar]
- 22.Dienstmaier JF, et al. Synthesis of well-ordered COF monolayers: Surface growth of nanocrystalline precursors versus direct on-surface polycondensation. ACS Nano. 2011;5(12):9737–9745. doi: 10.1021/nn2032616. [DOI] [PubMed] [Google Scholar]
- 23.McCulloch I, et al. Liquid-crystalline semiconducting polymers with high charge-carrier mobility. Nat Mater. 2006;5(4):328–333. doi: 10.1038/nmat1612. [DOI] [PubMed] [Google Scholar]
- 24.DeLongchamp DM, et al. High carrier mobility polythiophene thin films: Structure determination by experiment and theory. Adv Mater. 2007;19(19):833–837. [Google Scholar]
- 25.Tang W, Singh SP, Ong KH, Chen Z-K. Synthesis of thieno[3,2-b]thiophene derived conjugated oligomers for field-effect transistors applications. J Mater Chem. 2010;20(8):1497–1505. [Google Scholar]
- 26.Côté AP, El-Kaderi HM, Furukawa H, Hunt JR, Yaghi OM. Reticular synthesis of microporous and mesoporous 2D covalent organic frameworks. J Am Chem Soc. 2007;129(43):12914–12915. doi: 10.1021/ja0751781. [DOI] [PubMed] [Google Scholar]
- 27.Rouquerol J, et al. Recommendations for the characterization of porous solids. Pure Appl Chem. 1994;66(8):1739–1758. [Google Scholar]
- 28.Dogru M, Sonnauer A, Gavryushin A, Knochel P, Bein T. A Covalent Organic Framework with 4 nm open pores. Chem Commun (Camb) 2011;47(6):1707–1709. doi: 10.1039/c0cc03792c. [DOI] [PubMed] [Google Scholar]
- 29.Soderquist JA, Najafi RM. Selective oxidation of organoboranes with anhydrous trimethylamine N-oxide. J Org Chem. 1986;51(8):1330–1336. [Google Scholar]
- 30.Hanna MW, Ashbaugh AL. Nuclear magnetic resonance study of molecular complexes of 7,7,8,8-tetracyanoquinodimethane and aromatic donors. J Phys Chem. 1963;68(4):811–816. [Google Scholar]
- 31.Holm RD, Carper WR, Blancher JA. Polarographic and spectral studies of charge-transfer complexes. J Phys Chem. 1967;71(12):3960–3965. [Google Scholar]
- 32.Chappell JS, et al. TCNQ. J Am Chem Soc. 1981;103(7):2442–2443. [Google Scholar]
- 33.Kaim W, Moscherosch M. The coordination chemistry of TCNE, TCNQ and related polynitrile N acceptors. Coord Chem Rev. 1994;129(1–2):157–193. [Google Scholar]
- 34.Hartmann H, et al. Proof of innocence for the quintessential noninnocent ligand TCNQ in its tetranuclear complex with four [fac-Re(CO)3(bpy)]+ groups: Unusually different reactivity of the TCNX ligands (TCNX = TCNE, TCNQ, TCNB) Inorg Chem. 2003;42(22):7018–7025. doi: 10.1021/ic034232l. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.