

Abstract
The construction of synthetic gene circuits relies on our ability to engineer regulatory architectures that are orthogonal to the host’s native regulatory pathways. However, as synthetic gene circuits become larger and more complicated, we are limited by the small number of parts, especially transcription factors, that work well in the context of the circuit. The current repertoire of transcription factors consists of a limited selection of activators and repressors, making the implementation of transcriptional logic a complicated and component-intensive process. To address this, we modified bacteriophage T7 RNA polymerase (T7 RNAP) to create a library of transcriptional AND gates for use in Escherichia coli by first splitting the protein and then mutating the DNA recognition domain of the C-terminal fragment to alter its promoter specificity. We first demonstrate that split T7 RNAP is active in vivo and compare it with full-length enzyme. We then create a library of mutant split T7 RNAPs that have a range of activities when used in combination with a complimentary set of altered T7-specific promoters. Finally, we assay the two-input function of both wild-type and mutant split T7 RNAPs and find that regulated expression of the N- and C-terminal fragments of the split T7 RNAPs creates AND logic in each case. This work demonstrates that mutant split T7 RNAP can be used as a transcriptional AND gate and introduces a unique library of components for use in synthetic gene circuits.
Keywords: protein fragment complementation, H-loop
Synthetic gene circuits provide valuable insights into biophysical phenomena by enabling the construction and characterization of genetic systems from the ground up (1–3). Further, synthetic gene circuits are rapidly becoming core components of biotechnologies in metabolic engineering and in medicine (4–7). The continued development of synthetic gene circuits calls for the ability to construct larger and more complex circuits, which in turn necessitates the development of additional parts and component libraries with which to build them (8, 9).
Recent efforts to address the “component problem” (10) (ie, the lack of well-characterized orthogonal parts with which to build synthetic gene circuits) have led to the development of novel transcriptional and translational regulators. The effort to develop transcriptional regulators has, for instance, involved transplanting transcriptional regulators from genetically distant microorganisms into a particular chassis organism such as Escherichia coli. This reimagining of transcriptional regulatory systems has led to novel ligand-sensitive transcription factor–promoter pairs (11) and transcriptional logic gates (12). In addition, recent work has made it possible to synthetically regulate protein translation through, for example, modulating access to the ribosome binding site (RBS) or varying the stability of the transcript (5, 13–16).
At the core of many synthetic gene circuits lie transcriptional logic gates. Transcriptional logic gates mirror digital logic gates in that the value of the output is determined by the values of two separate inputs. Engineering transcriptional logic gates is difficult, and this difficulty is further compounded by the component problem.
Transcriptional logic gates require at least two transcriptional regulators of a single promoter (17). Therefore, creating circuits containing multiple logic gates quickly exhausts the current library of transcriptional components. Further, engineering hybrid promoters that respond to two transcription factors is a difficult and inexact science. One method for solving this problem involves mining other organisms for orthogonal transcriptional components that act in tandem at a promoter (11, 12). Here, we attack the problem from another angle by engineering, at the protein level, a unique functionality into an already well-known and well-characterized synthetic component. Specifically, we first split and then modify T7 RNAP to create a library of transcriptional AND gates in E. coli that are highly specific, as outlined in Fig. 1. In addition, the differences between the on and off states of the outputs of these AND gates is large, which is a desirable characteristic of transcriptional logic gates that makes them easy to use in larger circuits.
Fig. 1.

Outline of the steps needed to create a library of transcriptional AND gates from T7 RNAP. (A) T7 RNAP acting on its cognate promoter PT7 creates a single-input expression system. (B) Splitting T7 RNAP in vivo creates a transcriptional AND gate whereby both fragments of the split protein will be needed to drive transcription from PT7. (C) Mutating the specificity loop of T7 RNAP modifies the specificity of the protein for various promoters and hence creates unique T7 expression systems. When applied to split T7 RNAP, these mutations create orthogonal transcriptional AND gates, vastly increasing the computational power of the current component library available to researchers.
T7 RNAP is a single-subunit RNA polymerase that is a strong driver of transcription. It is functionally orthogonal to most hosts, acting only on its cognate promoter, PT7. During the purification of T7 RNAP, the protein was sometimes found cleaved between amino acids 179 and 180 to create a 20-kDa N-terminal fragment and an 80-kDa C-terminal fragment (18, 19). Only when mixed together would the two fragments drive transcription from PT7 in vitro. Further, the promoter specificity of T7 RNAP is determined by a specificity loop near the C terminus that forms specific contacts with PT7 (20). Point mutations in the specificity loop have been demonstrated to significantly alter the promoter specificity of full-length T7 RNAP, targeting the mutants to versions of PT7 that have been mutated between base pairs −11 to −8 (20–24).
To construct the libraries of transcriptional AND gates, we first show that the split T7 RNAP mutant that was previously reported to be active in vitro (18, 19) also functions in vivo. Then, we create mutants of the C-terminal fragment that contain point mutations known to alter its promoter specificity. We show that these mutations, when used in conjunction with their respective promoters, function in vivo. Finally, we create a library of transcriptional AND gates by placing the split T7 RNAP mutants behind different inducible promoters and demonstrate that each drives transcription if and only if both halves are induced.
Results and Discussion
In Vivo Activity of Split T7 RNAP.
During the purification of T7 RNAP, it was found that the protein can be nicked between amino acids 179 and 180, located in the H-loop domain (25, 26). After T7 RNAP binds to its promoter, the H-loop domain is known to refold during the enzyme’s transition from an initiation complex into an elongation complex (25, 27). The C-terminal fragment of nicked T7 RNAP (amino acids 180–880) was found to bind PT7 on its own but was unable to synthesize full-length mRNA (25). However, the addition of the N-terminal domain (amino acids 1–179) was able to rescue the ability to transcribe mRNA, albeit with reduced activity. However, although the activity of nicked T7 RNAP in vitro has been described (18, 19), its in vivo activity has not.
We first sought to demonstrate that split T7 RNAP can drive transcription in vivo and compare its activity to its full-length counterpart, as shown in Fig. 2. To assess the activity of full-length T7 RNAP, we cotransformed the plasmids pTara:500 and pET28:GFP (Fig. 2A). The plasmid pTara:500 contains the T7 RNAP coding sequence downstream of the arabinose inducible promoter PBAD with the protein-coding sequence of T7 RNAP driven by a RBS with a putative strength of 500, according to the RBS calculator (28). The plasmid pET28:GFP is the pET28b expression vector with GFP downstream of the T7-specific promoter PT7/LacO, which contains binding sites for the lactose repressor, LacI. We then assayed the expression of GFP as a function of arabinose inducer (Materials and Methods and Fig. S1).
Fig. 2.
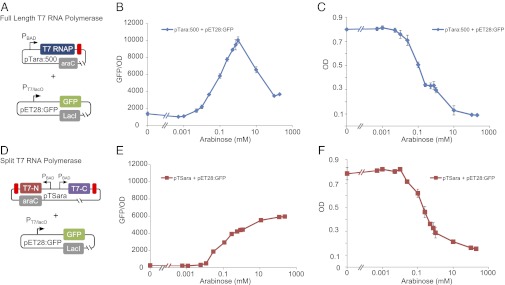
Comparing the in vivo functionality of full-length and split T7 RNAP. (A) To assess the activity of full-length T7 RNAP, we cotransformed a plasmid containing an arabinose-inducible T7 RNAP with a plasmid containing GFP driven by PT7/lacO. (B) GFP fluorescence of the full-length system described in A normalized to OD600 as a function of arabinose concentration after growing for 10 h. As expected, we saw high levels of GFP expression with increasing amounts of arabinose despite not adding IPTG to de-repress PT7/LacO. However, at high levels of arabinose, the expression of GFP drops dramatically because of a seven–base pair insertion into the T7 RNAP protein coding sequence that prematurely truncates the protein. (C) OD of the full-length system as a function of arabinose concentration. The drop in OD corresponds to the dramatic rise in GFP expression at the same concentrations of arabinose. (D) To characterize the activity of split T7 RNAP, we cotransformed a plasmid containing the two halves of the T7 RNAP (both driven by the arabinose-inducible promoter PBAD) with the plasmid containing GFP described in (A). (E) GFP fluorescence of the split system described in D normalized to OD600 as a function of arabinose concentration after growing for 10 h. Note that unlike the full-length system, no decrease in fluorescence is observed at high arabinose levels. (F) OD of the split system as a function of arabinose concentration.
As expected, when full-length T7 RNAP was used to drive gene expression, we saw exceedingly high levels of GFP fluorescence with increasing amounts of inducer, despite repression resulting from the transcriptional regulator LacI (Fig. 2B). Interestingly, at high concentrations of arabinose, the GFP expression drops dramatically. At 10 mM arabinose and higher, we observed a consistent frameshift mutation that truncates the last 165 amino acids of the polymerase. In addition, at high amounts of arabinose, the OD of the culture drops precipitously, suggesting significant amounts of cell stress (Fig. 2C).
We then tested the in vivo activity of split T7 RNAP. We modified the plasmid pTara:500 to create the plasmid pTSara (Fig. 2C). On pTSara, the coding regions of amino acids 1–179 and 180–880 of T7 RNAP are downstream of separate, antiparallel copies of PBAD for symmetric, inducible, and independent expression of the N- and C-terminal fragments. To match the translational activity of the full-length system, and hence the N-terminal fragment, we designed a RBS site with a putative strength of 500 to drive translation of the C-terminal fragment of the split protein (28). We then cotransformed the plasmids pTSara and pET28:GFP and assessed the expression of GFP as a function of arabinose as before.
Compared with the full-length system, the split system produced a dramatically different induction curve. The split system demonstrated lower and saturable GFP expression (Fig. 2E). Unlike the full-length system, high concentrations of arabinose did not induce a drop in expression of GFP in the split system, and no frameshift mutations were ever found. Further, growth of cells containing the split system was comparable to the growth in the full-length system, indicating that cells were able to tolerate the split T7 RNAP fragments (Fig. 2F). The stability at high inducer concentrations and saturable gene expression may be desirable features in synthetic gene networks. Dynamically, both full-length and split-protein systems exhibit similar characteristics on the introduction and removal of inducer from the media, as shown in Fig. S2.
To determine whether both fragments of split T7 RNAP are necessary to drive gene transcription in our assays, we created plasmids containing just the N-terminal fragment, just the C-terminal fragment, or both. A PBAD promoter was used to drive expression in each of these fragments. The RBSs were also unchanged. We then cotransformed these plasmids with a reporter plasmid and assayed GFP expression with or without 10 mM arabinose (Fig. 3A). GFP expression above background was found only in the case in which both halves of split T7 RNAP was present, as shown in Fig. 3B. This demonstrates that both halves of the split T7 RNAP are required.
Fig. 3.

Both fragments of split T7 RNAP are required for activity. (A) Plasmids containing just the N-terminal fragment, just the C-terminal fragment, or both were cotransformed with a reporter plasmid. (B) GFP fluorescence normalized to OD after 10 h incubation in either 0 or 10 mM arabinose for each version described in (A). Cells containing either half of split T7 RNAP show no GFP expression. When both halves of split T7 RNAP are present, however, high levels of GFP were found.
In our comparison of full vs. split T7 RNAP, we did not use isopropyl β-d-1-thiogalactopyranoside (IPTG) to de-repress PT7/lacO on pET28:GFP. Although de-repressing PT7/LacO should deliver higher levels of GFP from our expression system, we found that de-repression made the expression system unstable. Specifically, when the full-length system was induced with IPTG, we consistently observed a frameshift mutation that truncated the last 165 amino acids of T7 RNAP, even in the absence of arabinose. This was not the case with the split system. As a result, for this comparison we did not use IPTG. Further, in subsequent assays of the activity of split T7 RNAP, we use an unregulated PT7.
Characterizing the Promoter Specificity of Split T7 RNAP Mutants.
After characterizing the in vivo activity of split T7 RNAP, we sought to modify the promoter specificity of the split protein. Earlier studies of the determinants of promoter specificity of T7 RNAP highlight the role of a specificity loop between amino acids 742 and 773 that makes specific nucleotide contacts in the T7 promoter (20). Single–amino acid mutations to this specificity loop have been demonstrated to alter the specificity of T7 RNAP to versions of PT7 that have been mutated between base pairs −11 to −8 (22, 23, 25). The bulk of these studies were done in vitro, and quantification of T7 RNAP activity was done by northern blotting, which, although providing a direct measure of transcriptional activity of a T7 expression system, does not give an accurate depiction of in vivo activity. A more recent study found that mutating the specificity loop according to multisequence alignment with distant T7 RNAP homologs significantly alters the specificity of full-length T7 in vivo (29). Here, we propose that mutations of a single amino acid are sufficient for modifying the specificity of T7 RNAP to create orthogonal expression systems using split T7 RNAP.
We implemented point mutations Q758C, R756K, R756S, and N748D in the coding sequence of the C-terminal fragment of the split T7 RNAP (note that these mutations are annotated relative to the full-length protein). Each point mutation has been described to alter the specificity of full-length T7 RNAP to a modified T7 promoter (21–23). We also modified our reporter plasmid by first deleting the LacI binding site downstream of PT7 in PT7/LacO. In addition, we mutated base pairs −11 to −8 of PT7 to GACG, GCAT, CACT, CCCT, and ACAT. Each modified T7 promoter that we tested was previously described to be recognized preferentially over WT PT7 by a T7 RNA polymerase with a modified specificity loop.
To characterize the specificity of mutant split T7 RNAP, we cotransformed each version of mutant split T7 RNAP with each version of our altered reporter plasmid. We then assayed the induction of GFP in each of the 30 pairwise combinations of mutant split T7 RNAP and altered T7 promoters either in the absence or presence of 10 mM arabinose. This amount of inducer was chosen because we found it to saturate GFP expression with split T7 RNAP (Fig. 2E). We reasoned that by assaying expression of GFP with that much inducer, one would detect even weak functional interactions between mutant polymerase and mutant promoter.
Depending on which version of T7 promoter was used to drive expression of GFP, each mutant split T7 RNAP demonstrated a different ability to transcribe GFP, as shown in Fig. 4. Although split T7 RNAP with a WT specificity loop is able to express GFP from all of the T7 promoters tested, split T7 RNAPs with a mutant specificity loop demonstrate much narrower preferences for particular T7 promoters (Fig. 4B). It should be noted that WT pTSara working on its cognate promoter PT7:GACT on the plasmid pET:GACT:GFP is abnormally low because of increased levels of GFP expression with 0 mM arabinose (Fig. S3). Of the mutant polymerase and promoters tested, we found the mutant polymerase and mutant promoter pairs [Q758C,GACG], [R756S,ACAT], and [N748D,CACT] to demonstrate orthogonal specificities. These parts could create orthogonal split T7 expression systems whereby a particular mutant split T7 RNAP will target a particular specific mutant promoter and no other. Further, if orthogonality is not required, or perhaps not desired, additional pairs of split T7 RNAPs and promoters could be used for more complicated expression systems.
Fig. 4.

Promoter specificity of split T7 RNAP mutants. (A) To test the specificity of mutant split T7 RNAPs, the plasmid pTSara-Mut was transformed with the plasmid pET:NNNN:GFP, where Mut denotes a particular point mutation in the specificity loop and NNNN denotes the four nucleotides between −11 and −8 on PT7. (B) For each pair of mutant split T7 RNAP and mutant PT7, we assayed the expression of GFP with or without 10 mM arabinose. The fold GFP/OD, with or without 10 mM arabinose, after 10 h is plotted in the heat map. Split T7 RNAP with the wild-type specificity loop was able to express moderate to high levels of GFP from each mutant promoter that we tested. However, mutant split T7 RNAPs demonstrate various activities, depending on the promoter. The fold normalized fluorescence of WT pTSara with pET:GACT:GFP is substantially reduced in this case because of increased levels of GFP fluorescence observed with 0 mM arabinose (Fig. S3). The normalized fluorescence values used to create the heat map in B are plotted in Fig. S3.
We also sought to assess the toxicity associated with using mutant Split T7 RNAP in vivo. Similar studies of split protein fragment complementation have demonstrated that a large portion of split proteins may be insoluble and cause unwanted cellular stress (30). To determine whether mutant split T7 RNAP is toxic on its own, we tested for a significant drop in OD when T7 RNAP protein is highly expressed in the absence of a reporter plasmid. We individually transformed the plasmid Full and all of the variants of Split into cells and assayed the OD with or without 10 mM arabinose, as shown in Fig. 5. Little or no change in OD was observed despite the addition of gratuitous amounts of arabinose inducer. This suggests that the reduction in OD observed in our earlier experiments (Fig. 5B) is associated with the expression of GFP, rather than T7 RNAP protein.
Fig. 5.

To assess the relative toxicity of mutant Split T7 RNAP, we assayed the reduction in steady-state OD when T7 protein was fully induced. (A) To assess the toxicity associated with expressing mutant split T7 RNAP, we transformed plasmids containing either full-length or mutant split T7 RNAP and then assayed the OD after 10 h with or without 10 mM arabinose. (B) Of the T7 RNAP tested, gratuitous arabinose inducer does not induce a significant reduction in OD despite being able to drive large amounts of GFP expression from a PT7-regulated ORF.
Characterizing the Two-Input Function of Split T7 RNAP.
After determining split T7 RNAP to be functional in vivo and then characterizing the relative activities of specificity mutants with altered T7 promoters, we next wanted to determine whether each of the split T7 RNAP mutants could be used as transcriptional AND gates.
To assess the activity of split T7 RNAP as a transcriptional AND gate, we created the plasmid pTSlb-WT, which contains the N-terminal fragment of split T7 RNAP downstream of the IPTG-inducible PLac promoter and the C-terminal fragment of split T7 RNAP downstream of the arabinose inducible PBAD promoter. We then cotransformed this plasmid with the reporter plasmid pET:GACT:GFP and assessed the expression of GFP as a function of the inducers IPTG and arabinose, as shown in Fig. 6B. In the case of WT split T7 RNAP acting on its cognate promoter on the plasmid pET:GACT:GFP, we see strong induction of GFP if and only if both inducers are present, while observing only basal levels of GFP fluorescence if and only if one inducer is present (Fig. 6B). The GFP expression observed with just one inducer is most likely a result of leaky expression from the promoter driving the complimentary fragment, as no GFP expression is observed when either fragment is expressed alone (Fig. 3).
Fig. 6.
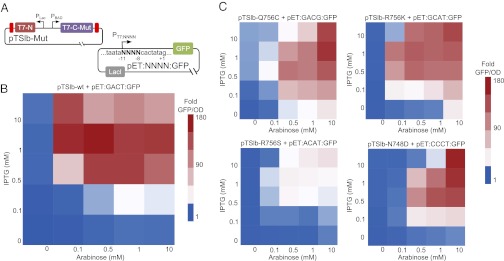
Testing transcriptional AND gates using the split T7 RNAP mutants. (A) To assess the two-input transfer function of split T7 RNAP, we cotransformed a plasmid containing the N-terminal and C-terminal fragment of split T7 RNAP downstream of PLac and PBAD, respectively, with another plasmid containing PT7 driving the expression of GFP. Here, Mut denotes a particular point mutation in the specificity loop and NNNN denotes the four nucleotides between −11 and −8 on PT7. (B) Heat map showing the relative fluorescence of the WT split T7 RNAP system described in A as a function of arabinose and IPTG. (C) Heat maps of the relative induction levels for different mutant T7 RNAP/promoter pairs (Upper Left, [Q756C,GACG]; Upper Right, [R756K,GCAT]; Lower Left, [R756S,ACAT]; and Lower Right, [N748D,CCCT]). Despite mutations to the specificity loop, each mutant split T7 RNAP preserved the two-input function observed in the wild-type case. For each heat map, values are recorded as fluorescence/OD for each value of inducer concentrations relative to the fluorescence/OD at 0 mM arabinose and 0 mM IPTG. The normalized fluorescence values used to create the extremes of the heat map in B and in C are plotted in Fig. S4.
We next tested the two-input function of mutant split T7 RNAP. To do this, we first mutated the specificity loop in the T7 RNAP coding sequence on pTSlb-wt to incorporate point mutations that we tested earlier (Fig. 4). We then assessed the two-input function of each mutant split T7 RNAP when cotransformed with a version of the reporter plasmid pET:NNNN:GFP that contains the T7 promoter that demonstrated the highest activity with that particular mutant (Fig. 6C). Similar to the WT case, the mutant split polymerases and mutant promoter pairs showed maximal GFP expression if and only if both inducers were present in high amounts. It is interesting to note, however, that different mutant split polymerase variants produced different levels of maximal GFP expression. This suggests another modality with which to modulate gene expression using mutant split T7 RNAP.
Conclusions
Two-input transcriptional logic is currently difficult to implement, as each two-input logic gate requires at least two transcriptional regulators. Given the limited amount of transcriptional regulators that are available to synthetic biologists, the implementation of multiple logic gates rapidly exhausts the existing component library. Furthermore, using two transcriptional regulators to regulate one promoter can involve the difficult task of promoter engineering, in which the design and implementation of a two-input promoter of a specific output is an inexact and resource-intensive process. The ability to support two-input combinatorial logic in a single functional protein complex will dramatically simplify the process of implementing transcriptional logic.
As a two-component transcriptional activator, split T7 RNAP has the potential to support two-input transcriptional logic through the regulated expression of the N- and C-terminal fragments. Split T7 RNAP lends itself, in particular, to AND transcriptional logic, in which output is observed if and only if both inputs as present, as the N- and C-terminal fragments are both required to form a functional transcriptional complex to drive gene expression. Although modifying the activity and specificity of full-length T7 RNAP has been recently demonstrated (29), our work reveals that T7 RNAP can be split to create an in vivo transcriptional AND gate and that its specificity can be significantly modified by changing a single amino acid.
In this study, we described the creation of a library of transcriptional AND gates by splitting and mutating T7 RNA polymerase. We confirmed the activity of the split protein and found that, although its activity is not as high as for full-length T7 RNAP, the split expression system is stable and saturable. In one sense, these features may make split T7 RNAP more valuable than full-length T7 RNAP for use in synthetic gene networks, as full-length T7 RNAP can often cause cellular stress that leads to mutations that affect the functionality of the overlying gene circuit.
We also tested the effects of specificity, altering point mutations of T7 RNAP in vivo by cotransforming an array of split T7 RNAP modified at the specificity loop with an array of modified PT7 and assaying for changes in gene expression. Of the mutant split polymerases and mutant PT7 we tested, we identified three mutant polymerase promoter pairs that demonstrate orthogonal specificities. In effect, these mutant polymerase and mutant promoters create three orthogonal expression systems. We next tested the two-input function of split T7 RNAP, hypothesizing that the split protein would lend itself in particular to AND transcriptional logic. This was confirmed in our assays, as we observed maximal gene expression if and only if both fragments of split T7 RNAP were fully induced. This was also the case when we assayed the two-input function of mutant split T7 RNAP in which each mutant polymerase/promoter pair generated a different level of maximal GFP expression.
As a two-component transcriptional activator, split T7 RNAP creates opportunities to create novel gene transcription networks. When combined with the all-or-nothing response expression systems driven by PLac or PBAD, split T7 RNAP creates a unique opportunity to generate heterogenous populations of active genetic circuits (31, 32). Split T7 RNAP might also be integrated with existing protein fragment complementation assays to create novel synthetic regulatory schemes or pathway screens (33).
Overall, this study identified mutant split T7 RNAPs that can be used as transcriptional AND gates in synthetic gene circuits. Because this library of AND gates was derived from a single component, it increases the number of available parts with which to build synthetic gene circuits. This, in turn, will enable the construction of larger and more complex gene circuits.
Materials and Methods
Plasmid Construction and Strains.
All cloning was done using standard cloning methods (34, 35). The plasmid pTara was used as a template for all constructs in this article containing either full-length or split versions of T7 RNAP (36). The RBS controlling translation of full-length T7 was modified to have a relative strength of 500, according to the RBS calculator (28). To facilitate the independent and inducible expression of amino acids 1–179 and 180–880 of T7 RNAP, the N- and C-terminal fragments of T7 RNAP downstream from two separate copies of PBAD oriented antiparallel to each other to create the plasmid pTSara. The RBSs of the N- and C-terminal of split T7 RNAP were modified to have a relative strength of 500. The reporter plasmid pET28:GFP is the expression plasmid pET-28b with GFP inserted into the multicloning site of pET-28b. To create the reporter plasmids pET:NNNN:GFP, where NNNN designates base pairs −11 to −8 of PT7, the plasmid pET28:GFP was modified. The lacI operator site in the promoter PT7/LacO on the plasmid was deleted to create the plasmid pET:GACT:GFP. Base pairs −11 to −8 of PT7 on pET:GACT:GFP were then modified to GACG, GCAT, CACT, CCCT, and ACAT to create the plasmids pET:GACG:GFP, pET:GCAT:GFP, pET:CACT:GFP, pET:CCCT:GFP, and pET:ACAT:GFP, respectively. More detailed maps of each plasmid used in this study are included in Fig. S5 and Table S1.
The strain DH10B-ALT is the E. coli strain DH10B with PlacIq:lacI and constitutive ORFs driving expression of araC and tetR integrated into the attB site on the chromosome.
Assays of Fluorescence and Growth.
For the assay of the in vivo activity of split T7 RNAP, either pTara:500 or pTSara was cotransformed with pET28:GFP into DH10B cells. These cells were then grown to stationary phase by inoculating a colony into selective LB media and incubating at 37 °C while shaking at 200 rpm for 12 h. Stationary-phase cell culture was then inoculated into 5 mL selective LB media with or without inducer at 1% (vol/vol). Each sample was then aliquoted in triplicate into a 96-well plate (BD Falcon 35117). The plate was then incubated with the cover overnight at 37 °C with 1 mm orbital shaking for 10 h as GFP fluorescence (ex:488, em:510) and OD600 were assayed (Infinite M1000, Tecan). This induction scheme was used for all assays of GFP fluorescence and growth unless otherwise noted.
To assess the transcription activity associated with each fragment of split T7 RNAP, the plasmids pTSara-N and pTSara-C were cotransformed with pET:GACT:GFP into DH10B cells. Cells were then grown to stationary phase and assayed for growth and fluorescence with or without inducer, as previously described.
For the characterization of promoter specificity of split T7 RNAP mutants, each version of the plasmid pTSara-Mut was cotransformed with each version of the plasmid pET:NNNN:GFP for 30 pairwise combinations into DH10B cells. Each cotransformation was grown to stationary phase and then assayed for induction of GFP fluorescence as a function of arabinose, as described for the assay of the in vivo activity of split T7 RNAP.
For the characterization of the two-input transfer function of mutant split T7 RNAP, the following plasmid pairs were cotransformed into the strain DH10B-ALT: [pTSlb-WT pET:GACT:GFP], [pTSlb-Q758C, pET:GACG:GFP], [pTSlb-R756K, pET:GCAT:GFP], [pTSlb-R756S, pET:ACAT:GFP], and [pTSlb-N748D, pET:CCCT:GFP]. Each cotransformation was grown to stationary phase and then assayed for induction of GFP fluorescence as a function of arabinose and IPTG inducer in the manner described for the in vivo activity of split T7 RNAP.
Supplementary Material
Acknowledgments
We thank Joff Silberg and Michael Diehl for generous access to their equipment and materials. We also thank Jeff Tabor, Faiza Hussain, and Yangyang Dong for illuminating discussions. This work was supported by National Institutes of Health Grant R01GM104974 as part of the joint National Science Foundation/National Institute of General Medical Sciences Mathematical Biology Program, and Welch Foundation Grant C-1729.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1220157110/-/DCSupplemental.
References
- 1.Basu S, Gerchman Y, Collins CH, Arnold FH, Weiss R. A synthetic multicellular system for programmed pattern formation. Nature. 2005;434(7037):1130–1134. doi: 10.1038/nature03461. [DOI] [PubMed] [Google Scholar]
- 2.Stricker J, et al. A fast, robust and tunable synthetic gene oscillator. Nature. 2008;456(7221):516–519. doi: 10.1038/nature07389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sohka T, et al. An externally tunable bacterial band-pass filter. Proc Natl Acad Sci USA. 2009;106(25):10135–10140. doi: 10.1073/pnas.0901246106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Holtz WJ, Keasling JD. Engineering static and dynamic control of synthetic pathways. Cell. 2010;140(1):19–23. doi: 10.1016/j.cell.2009.12.029. [DOI] [PubMed] [Google Scholar]
- 5.Xie Z, Wroblewska L, Prochazka L, Weiss R, Benenson Y. Multi-input RNAi-based logic circuit for identification of specific cancer cells. Science. 2011;333(6047):1307–1311. doi: 10.1126/science.1205527. [DOI] [PubMed] [Google Scholar]
- 6.Zhang F, Carothers JM, Keasling JD. Design of a dynamic sensor-regulator system for production of chemicals and fuels derived from fatty acids. Nat Biotechnol. 2012;30(4):354–359. doi: 10.1038/nbt.2149. [DOI] [PubMed] [Google Scholar]
- 7.Dietrich JA, Shis DL, Alikhani A, Keasling JD. Transcription Factor-Based Screens and Synthetic Selections for Microbial Small-Molecule Biosynthesis. ACS Synth Biol. 2013;2:47–58. doi: 10.1021/sb300091d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lu TK, Khalil AS, Collins JJ. Next-generation synthetic gene networks. Nat Biotechnol. 2009;27(12):1139–1150. doi: 10.1038/nbt.1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Keasling JD. Synthetic biology and the development of tools for metabolic engineering. Metab Eng. 2012;14(3):189–195. doi: 10.1016/j.ymben.2012.01.004. [DOI] [PubMed] [Google Scholar]
- 10.Bennett MR, Hasty J. Overpowering the component problem. Nat Biotechnol. 2009;27(5):450–451. doi: 10.1038/nbt0509-450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang B, Kitney RI, Joly N, Buck M. Engineering modular and orthogonal genetic logic gates for robust digital-like synthetic biology. Nat Commun. 2011;2:508. doi: 10.1038/ncomms1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Moon TS, Lou C, Tamsir A, Stanton BC, Voigt CA. Genetic programs constructed from layered logic gates in single cells. Nature. 2012;491(7423):249–253. doi: 10.1038/nature11516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Carothers JM, Goler JA, Juminaga D, Keasling JD. Model-driven engineering of RNA devices to quantitatively program gene expression. Science. 2011;334(6063):1716–1719. doi: 10.1126/science.1212209. [DOI] [PubMed] [Google Scholar]
- 14.Ausländer S, Ausländer D, Müller M, Wieland M, Fussenegger M. Programmable single-cell mammalian biocomputers. Nature. 2012;487(7405):123–127. doi: 10.1038/nature11149. [DOI] [PubMed] [Google Scholar]
- 15.Callura JM, Cantor CR, Collins JJ. Genetic switchboard for synthetic biology applications. Proc Natl Acad Sci USA. 2012;109(15):5850–5855. doi: 10.1073/pnas.1203808109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Qi L, Haurwitz RE, Shao W, Doudna JA, Arkin AP. RNA processing enables predictable programming of gene expression. Nat Biotechnol. 2012;30(10):1002–1006. doi: 10.1038/nbt.2355. [DOI] [PubMed] [Google Scholar]
- 17.Tamsir A, Tabor JJ, Voigt CA. Robust multicellular computing using genetically encoded NOR gates and chemical ‘wires’. Nature. 2011;469(7329):212–215. doi: 10.1038/nature09565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ikeda RA, Richardson CC. Interactions of a proteolytically nicked RNA polymerase of bacteriophage T7 with its promoter. J Biol Chem. 1987;262(8):3800–3808. [PubMed] [Google Scholar]
- 19.Ikeda RA, Richardson CC. Enzymatic properties of a proteolytically nicked RNA polymerase of bacteriophage T7. J Biol Chem. 1987;262(8):3790–3799. [PubMed] [Google Scholar]
- 20.Rong M, He B, McAllister WT, Durbin RK. Promoter specificity determinants of T7 RNA polymerase. Proc Natl Acad Sci USA. 1998;95(2):515–519. doi: 10.1073/pnas.95.2.515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Raskin CA, Diaz GA, Joho K, McAllister WT. Substitution of a single bacteriophage T3 residue in bacteriophage T7 RNA polymerase at position 748 results in a switch in promoter specificity. J Mol Biol. 1992;228(2):506–515. doi: 10.1016/0022-2836(92)90838-b. [DOI] [PubMed] [Google Scholar]
- 22.Raskin CA, Diaz GA, McAllister WT. T7 RNA polymerase mutants with altered promoter specificities. Proc Natl Acad Sci USA. 1993;90(8):3147–3151. doi: 10.1073/pnas.90.8.3147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chelliserrykattil J, Cai G, Ellington A. A combined in vitro / in vivo selection for polymerases with novel promoter specificities. BMC Biotechnol. 2001;1:1–13. doi: 10.1186/1472-6750-1-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ikeda RA, Warshamana GS, Chang LL. In vivo and in vitro activities of point mutants of the bacteriophage T7 RNA polymerase promoter. Biochemistry. 1992;31(37):9073–9080. doi: 10.1021/bi00152a051. [DOI] [PubMed] [Google Scholar]
- 25.Muller DK, Martin CT, Coleman JE. Processivity of proteolytically modified forms of T7 RNA polymerase. Biochemistry. 1988;27(15):5763–5771. doi: 10.1021/bi00415a055. [DOI] [PubMed] [Google Scholar]
- 26.Yin YW, Steitz TA. Structural basis for the transition from initiation to elongation transcription in T7 RNA polymerase. Science. 2002;298(5597):1387–1395. doi: 10.1126/science.1077464. [DOI] [PubMed] [Google Scholar]
- 27.Bandwar RP, et al. The transition to an elongation complex by T7 RNA polymerase is a multistep process. J Biol Chem. 2007;282(31):22879–22886. doi: 10.1074/jbc.M702589200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Salis HM, Mirsky EA, Voigt CA. Automated design of synthetic ribosome binding sites to control protein expression. Nat Biotechnol. 2009;27(10):946–950. doi: 10.1038/nbt.1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Temme K, Hill R, Segall-Shapiro TH, Moser F, Voigt CA. Modular control of multiple pathways using engineered orthogonal T7 polymerases. Nucleic Acids Res. 2012;40(17):8773–8781. doi: 10.1093/nar/gks597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Paschon DE, Patel ZS, Ostermeier M. Enhanced catalytic efficiency of aminoglycoside phosphotransferase (3′)-IIa achieved through protein fragmentation and reassembly. J Mol Biol. 2005;353(1):26–37. doi: 10.1016/j.jmb.2005.08.026. [DOI] [PubMed] [Google Scholar]
- 31.Novick A, Weiner M. Enzyme Induction as an All-Or-None Phenomenon. Proc Natl Acad Sci USA. 1957;43(7):553–566. doi: 10.1073/pnas.43.7.553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Siegele DA, Hu JC. Gene expression from plasmids containing the araBAD promoter at subsaturating inducer concentrations represents mixed populations. Proc Natl Acad Sci USA. 1997;94(15):8168–8172. doi: 10.1073/pnas.94.15.8168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Michnick SW, Ear PH, Manderson EN, Remy I, Stefan E. Universal strategies in research and drug discovery based on protein-fragment complementation assays. Nat Rev Drug Discov. 2007;6(7):569–582. doi: 10.1038/nrd2311. [DOI] [PubMed] [Google Scholar]
- 34.Ho SN, Hunt HD, Horton RM, Pullen JK, Pease LR. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene. 1989;77(1):51–59. doi: 10.1016/0378-1119(89)90358-2. [DOI] [PubMed] [Google Scholar]
- 35.Li MZ, Elledge SJ. Harnessing homologous recombination in vitro to generate recombinant DNA via SLIC. Nat Methods. 2007;4(3):251–256. doi: 10.1038/nmeth1010. [DOI] [PubMed] [Google Scholar]
- 36.Wycuff DR, Matthews KS. Generation of an AraC-araBAD promoter-regulated T7 expression system. Anal Biochem. 2000;277(1):67–73. doi: 10.1006/abio.1999.4385. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.