

Abstract
Amongst the different types of adverse drug reactions, drug-induced liver injury is the most prominent cause of patient morbidity and mortality. However, the current available hepatic model systems developed for evaluating safety have limited utility and relevance as they do not fully recapitulate a fully functional hepatocyte, and do not sufficiently represent the genetic polymorphisms present in the population. The rapidly advancing research in stem cells raises the possibility of using human pluripotent stem cells in bridging this gap. The generation of human induced pluripotent stem cells via reprogramming of mature human somatic cells may also allow for disease modelling in vitro for the purposes of assessing drug safety and toxicology. This would also allow for better understanding of disease processes and thus facilitate in the potential identification of novel therapeutic targets. This review will focus on the current state of effort to derive hepatocytes from human pluripotent stem cells for potential use in hepatotoxicity evaluation and aims to provide an insight as to where the future of the field may lie.
Keywords: drug toxicity, drug-induced liver injury, embryonic stem cells, induced pluripotent stem cells, stem cells
Introduction
Adverse drug reactions (ADRs) continue to feature as a major problem to the clinician, the pharmaceutical industry and the regulatory authorities [1]. In the UK, 15% of hospital in-patients have been reported to suffer from a form of ADR during admission, with 20% of these patients readmitted again within 12 months of discharge [2, 3]. These admissions resulting from ADRs were also estimated to cost the NHS £466 million annually in a prospective observational study performed in 2001–2002 [4]. It is also the leading cause of drug attrition and confers a deep financial burden on the pharmaceutical industry [5].
Amongst the different types of ADRs, drug-induced liver injury (DILI) is the most prominent cause of patient morbidity and mortality [6–8]. This is attributed to the liver's role in drug metabolism particularly in circumstances when xenobiotics cannot be sufficiently cleared, for example in overdoses [9, 10]. Various hepatocyte models have thus been developed for use in safety pharmacology and toxicology research to understand the mechanisms of DILI and to screen new chemical entities (NCEs) for their potential to cause adverse reactions [11, 12]. Freshly isolated hepatocytes, cryopreserved hepatocytes, immortalized cancer cell lines, liver tissue preparations (slices, microsomes and S9 fractions) and animal models broadly categorize the numerous hepatocyte models available for studies into the pathophysiology of DILI. However, the utility and relevance of these models are also limited. The gold standard in vitro model for the study of DILI in humans is primary culture of freshly isolated human hepatocytes. However, the use of human primary hepatocytes (hPHs) is impeded by their limited availability, inter-donor differences, variable viability following isolation and rapid dedifferentiation of the hepatocyte phenotype in culture, particularly in the loss of cytochrome P450 (CYP) enzyme expression [11, 13, 14]. The limited life span and phenotypic instability also limits the utility of the hPH model to short term studies only and compromises their use in mechanistic studies of DILI which often occurs following prolonged exposure to drugs [11, 14, 15]. Immortalized cancer cell lines have been used to overcome these problems as they have an infinite life span and are readily available. However, they suffer from a deficit in metabolic activity [11]. Transfection methods to enable overexpression of CYP enzymes in these cells have been adopted, but this approach is still limited to the expression of one CYP isoform per cell line and therefore does not fully recapitulate the metabolic capacity of a fully functional hepatocyte [11, 16–20]. Furthermore, all the currently available hepatocyte models do not sufficiently represent the genetic polymorphisms present in the population that are now acknowledged to play an important role in ADRs [21–23]. Although the use of animal models is a more amenable approach for in vivo studies, experimentation on animals raises ethical concerns, while interspecies differences limit the translation of data into the clinic [24–26]. Therefore, there is still a clear need to improve current hepatocyte models, and to adopt new advances in experimental techniques to develop new models that will enable better prediction and understanding of the mechanisms causing DILI.
With the rapidly advancing stem cell technology, it is hoped that progress will be made in bridging this gap in toxicology research through the use of human embryonic stem cells (hESCs). The pluripotent nature and the ability of the embryonic stem cells to proliferate indefinitely are the two main attractions in using ESCs not only for safety pharmacology and toxicology research, but also in regenerative medicine, tissue engineering and cell therapy [27–31]. Directed differentiation of hESCs to somatic cells with mature phenotypes in the laboratory could potentially provide a readily available source of metabolically competent cells such as mature hepatocytes with comparable functional status to freshly isolated hepatocytes for use in safety pharmacology and toxicology applications. By doing so, the problems of using the gold standard freshly-isolated hPHs such as their limited availability, inter-donor differences and variable viability following isolation, can theoretically be solved by the use of a standard protocol-driven derivation of HLCs with batch-to-batch consistency and purity.
More recently, pluripotent stem cells (PSCs) have also been produced by reprogramming of mature somatic cells and are termed as induced pluripotent stem cells (iPSCs) [32]. This approach negates the controversies surrounding the use of embryonic tissue and potentially allows for in vitro modelling of normal and variant phenotypes for safety pharmacology and toxicology evaluations. iPSCs were first generated by cellular reprogramming of murine fibroblasts using a retroviral vector that expressed transcription factors noted to be abundant in embryonic stem cells [32]. Since then, other groups have reported a variety of techniques using various human somatic cells to induce pluripotency, albeit with different efficiencies. These methods include viral-free approaches to deliver the pluripotency gene set expressing the essential transcription factors into target somatic cells using either episomal vectors, piggyBac transposons or minicircle vectors [33–37]. Reprogramming somatic cells via delivery of the reprogramming factors in their protein or messenger ribonucleic acid (RNA) form have also been reported [38–40]. Small molecules have also been used with all or some of the classical reprogramming factors in a bid to improve the efficiency of induction [41–44]. More recently, microRNAs (miRNAs) that are shown to be abundant in ESCs and known to play important roles during cellular reprogramming were used instead of the classical pluripotency factors to produce hiPSCs [45, 46].
In view of the potential of human pluripotent stem cells (hPSCs) in providing an alternative model for safety pharmacology and toxicology applications, many pharmaceutical and biotechnology companies in recent years have invested or developed joint collaborations with academia, to develop in vitro systems based on hPSCs [47, 48]. This review will focus on the current state of efforts to derive hPSCs for potential use in hepatotoxicity evaluation.
Derivation of hepatocyte-like cells (HLCs) from human pluripotent stem cells
hESC-derived HLCs
In general, studies reporting on ‘hepatocytes’ derived from hESCs have focussed on generating a closer representation of a mature hPH phenotype. However, as no reports to date have confirmed complete recapitulation of a freshly isolated hPH, the term HLCs has been used to describe them.
Many groups have attempted to improve the differentiation of hESCs to HLCs in vitro by mimicking the developmental pathway of the liver during embryogenesis. The aim is to derive mature hepatocytes from pluripotent hESCs using differentiation protocols encompassing the three main stages of hepatic development: definitive endoderm differentiation, hepatocyte progenitor specification and hepatocyte maturation [49]. Methods employed to induce differentiation of hESCs towards HLCs include the formation of embryoid bodies by aggregation of ESCs to mimic the gastrulation stage during embryogenesis before subsequent induction of hepatocyte development and addition of exogenous differentiation factors at appropriate stages of hepatic development as characterized by their gene expression profile (Table 1) [50–67]. However, refinement of the differentiation protocol to generate HLCs with a phenotype matching hPHs continues. For example, a recent report has suggested that a greater differentiation efficiency could be gained from earlier use of the hepatocyte growth factor at the stage of definitive endoderm differentiation, rather than during the hepatocyte maturation stage as currently employed [67]. To date however, the perfect differentiation protocol has remained elusive. This is also compounded by the fact that there is currently no standardization of the methods used to characterize these HLCs and in assessing their differentiation potential, though helpful recommendations for minimal criteria to allow comparison of protocols have been proposed [68].
Table 1.
Summary of recent studies with reports of HLC-derivation from human pluripotent stem cells
| Reference | Stem cell (cell line) | Differentiation method | Differentiation factors | Differentiation efficiency | ||
|---|---|---|---|---|---|---|
| % ALB +ve HLCs | % AAT +ve HLCs | Method of assessment | ||||
| Cai et al., 2007 [50] | hESC (H1, H9) | Monolayer, EB formation | AF V, AA, ITS, BMP2, FGF4, HGF, OSM, DEX | 70 | ND | ICC |
| Ek et al., 2007 [51] | hESC (SA002, SA002.5, SA167) | Monolayer | Proprietary differentiation medium, FGF2 | ND | ND | – |
| Söderdahl et al., 2007 [52] | hESC (SA001, SA002, SA002.5, AS034, SA121, and SA167) | Monolayer | Proprietary differentiation medium, bFGF | ND | ND | – |
| Hay et al., 2008 [53] | hESC (H1, H9) | Monolayer | AA, Wnt3a | 90 | ND | ICC |
| Shiraki et al., 2008 [54] | hESC (Khes-1) | Co-culture with M15 cell line | AA, BMP4, bFGF, HGF, DMSO, DEX, Ly294002 | 9 | ND | ICC |
| Agarwal et al.,2008 [55] | hESC (WA01, WA09) | Monolayer | AA, FGF4, HGF, BSA, OSM, DEX | 67.4 | 84.7 | ICC |
| Moore et al., 2009 [56] | hESC (H1) | Monolayer, EB formation | AA, Wnt3a, HGF, OSM, DEX | 72.8 | ND | ICC |
| Basma et al., 2009 [57] | hESC (H1) | Monolayer, EB formation | AA, FGF2, HGF, DMSO, DEX | 55.5 | ND | ICC |
| Song et al., 2009 [58] | hESC (H1), hiPSC | Monolayer | AF V, AA, ITS, BMP2, FGF4, OSM, DEX, KGF, B27 | 60 | ND | ICC |
| Duan et al., 2010 [59] | hESC (H9) | Monolayer | AA, sodium butyrate, BMP2, BMP4, FGF4, HGF DMSO, B27 | 75–90 | 64–75 | ICC, FACS, qRT-PCR, GFP reporter gene |
| Synnergren et al., 2010 [60] | hESC (SA002, SA167, SA461) | Monolayer | AA, ITS, FGF1, FGF2, BMP2, BMP4, HGF, OSM, DEX | ND | ND | – |
| Touboul et al., 2010 [61] | hESC (H9) | Monolayer | AA, BMP4,FGF2, FGF4, FGF10, HGF, EGF, retinoic acid, SB431542, Ly294002 | ND | ND | – |
| Brolén et al., 2010 [62] | hESC (SA001, SA002, SA002.5, SA167) | Monolayer | AA, BMP2, BMP4, FGF1, FGF2, HGF, OSM, DEX, Wnt3A | ND | ND | – |
| Ghodsizadeh et al., 2010 [82] | hiPSC | EB formation | AA, FGF2, HGF, DMSO, DEX | 50 | ND | FACS |
| Liu et al., 2010 [63] | hESC (WA01, WA09), hiPSC (*hPH-derived) | Monolayer | AA, FGF4, HGF, OSM, DEX | ND | ND | – |
| Si-Tayeb et al., 2010 [65] | hESC (H9), hiPSC | Monolayer | AA, BMP4, FGF2, OSM, B27 | 80 | ND | FACS |
| Sullivan et al., 2010 [81] | hiPSC | Monolayer | AA, HGF, Wnt3A, DMSO, OSM, hydrocortisone, tryptose phosphate broth, B27 | 70–90 | ND | ICC |
| Rashid et al., 2010 [83] | hiPSC | Monolayer | AA, BMP4, FGF2, HGF, OSM, Ly294002, CHIR99021 (GSK-3 inhibitor) | 83 | ND | FACS |
| Zhang et al., 2011 [66] | hESC (H9), hiPSC | Monolayer, EB formation | AA, BMP2, FGF4, HGF, KGF, OSM, DEX | 60–80 | 20 | ICC, FACS |
| Bone et al., 2011 [64] | hESC (Shef1, Shef3) | Monolayer | FGF4, HGF, OSM, DEX, 1 m (GSK-3 inhibitor) | ND | ND | – |
| Chen et al., 2012 [67] | hESC (H9), hiPSC | Monolayer | AA, ITS, HGF, Wnt3A, OSM, DMSO, DEX | ND | ND | – |
AA, activin A; AF V, albumin fraction V; bFGF, human recombinant basic FGF; BMP, bone morphogenic protein; BSA, bovine serum albumin; DEX, dexamethasone; DMSO, dimethyl sulfoxide; EGF, epidermal growth factor; FACS, fluorescence-activated cell sorting; FGF, fibroblast growth factor; GFP, green fluorescent protein; GSK, glycogen synthase kinase; HGF, hepatocyte growth factor; ICC, immunocytochemistry; ITS, insulin-transferrin-selenium; KGF, keratinocyte growth factor; ND, not determined; OSM, oncostatin M; qRT-PCR, quantitative real time polymerase chain reaction; Wnt3a, wingless-type MMTV integration site family, member 3a.
Currently, the measure of success (differentiation efficiency) in the derivation of ‘mature HLCs’ broadly consists of the purity of the derived HLC population with typical epithelial morphology and gene expression profiling for liver-associated markers such as albumin (ALB), and α1-antitrypsin (AAT) (Table 1). Although gene expression profiling is useful, it is the dynamic functional capabilities of these differentiated HLCs when compared with hPHs that will determine their suitability for use in safety pharmacology and toxicology. These functional assessments include hepatic enzyme activity, albumin secretion, glycogen storage, uptake of indocyanine green (ICG) and uptake of low density lipoprotein (Table 2).
Table 2.
Summary of recent studies reporting on the functional capabilities of human pluripotent stem cell-derived HLCs
| Reference | Phase I and II enzyme activity | Other comparators used (assay method) | Albumin secretion in media | Other functional activity shown | ||
|---|---|---|---|---|---|---|
| Enzyme | % of hPH comparator (assay method) | % of hPH comparator | Other comparators used | |||
| Cai et al., 2007 [50] | CYP2B6 | ND | hESC (fluorescence) | ND | Huh-7 hepatoma cell line | Glycogen storage, ICG uptake, LDL uptake |
| Ek et al., 2007 [51] | CYP1A1 | 0 (fluorescence) | – | ND | ND | – |
| CYP3A4 | 0 (fluorescence) | – | ||||
| Söderdahl et al., 2007 [52] | GST | 80 (fluorescence) | HepG2 hepatoma cell line (fluorescence) | ND | ND | Glycogen storage |
| Hay et al., 2008 [53] | CYP1A2 | 4 (LC-MS-MS) | hESC (LC-MS-MS) | ND | HLCs assayed with no comparator | Glycogen storage |
| Shiraki et al., 2008 [54] | ND | – | – | ND | – | Glycogen storage |
| Agarwal et al., 2008 [55] | ND | – | – | ND | hESC | Glycogen storage, ICG uptake |
| Moore et al., 2009 [56] | CYP1A2 | ND | hESC-derived HLCs cultured in media with different components (fluorescence) | ND | hESC-derived HLCs cultured in media with different components | ICG uptake |
| Basma et al., 2009 [57] | CYP1A | 30 (fluorescence) | – | 75 | – | – |
| CYP3A | 90 (LC-MS-MS) | |||||
| Song et al., 2009 [58] | CYP2B6 | ND | hiPSC-derived HLCs compared with hESC-derived HLCs (fluorescence) | ND | hiPSC-derived HLCs compared with hESC-derived HLCs | Glycogen storage |
| Duan et al., 2010 [59] | CYP1A2 | 100 (LC-MS-MS) | – | ND | HLCs assayed with no comparator | ICG uptake |
| CYP2C9 | 60 (LC-MS-MS) | – | ||||
| CYP2D6 | 95 (LC-MS-MS) | – | ||||
| CYP3A4 | 90 (LC-MS-MS) | – | ||||
| Touboul et al., 2010 [61] | CYP3A | ND | HLCs assayed with no comparator (bioluminescence) | ND | Thawed foetal hepatocytes | Glycogen storage, ICG uptake, LDL uptake |
| Brolén et al., 2010 [62] | CYP1A, | ND | Spontaneously differentiated hESC-derived HLCs, HepG2 (LC-MS-MS) | ND | – | Glycogen storage, ICG uptake |
| CYP2C, | ||||||
| CYP3A | ||||||
| Ghodsizadeh et al., 2010 [82] | CYP2B6 | ND | hiPSC (fluorescence) | ND | HLCs assayed with no comparator | Glycogen storage, ICG uptake, LDL uptake |
| Liu et al., 2010 [63] | CYP1A2, CYP3A4 | ND | HLCs assayed with no comparator (bioluminescence) | ND | ND | Glycogen storage |
| Si-Tayeb et al., 2010 [65] | ND | – | – | ND | hiPSC-derived HLCs compared with hESC-derived HLCs | Glycogen storage, ICG uptake, LDL uptake |
| Sullivan et al., 2010 [81] | CYP1A2 | – | HLCs assayed with no comparator (bioluminescence) | ND | ND | – |
| CYP3A4 | ||||||
| Rashid et al., 2010 [83] | CYP3A4 | ND | hiPSC (bioluminescence) | ND | hiPSC | Glycogen storage, LDL uptake |
| Zhang et al., 2011 [66] | CYP3A4 | 0.32 (bioluminescence) | hESC-derived HLCs (bioluminescence) | 60 | – | Glycogen storage |
| Bone et al., 2011 [64] | ND | – | – | 22.6 | – | – |
| Yildirimman et al., 2011 [69] | CYP1A2 | 50 (LC-MS-MS) | – | ND | – | – |
| CYP3A4 | 50 (LC-MS-MS) | |||||
| CYP2B6 | 10 (LC-MS-MS) | |||||
| CYP2C9 | 50 (LC-MS-MS) | |||||
| CYP2C19 | 50 (LC-MS-MS) | |||||
| Chen et al., 2012 [67] | CYP3A4 | 100 (bioluminescence) | hiPSC (bioluminescence) | ND | – | Glycogen storage, LDL uptake |
ELISA, enzyme-linked immunosorbent assay; GST, glutathione transferase; ICG, indocyanine green; LC-MS-MS, liquid chromatography with tandem mass spectrometry; LDL, low density lipoprotein; ND, not determined.
In the liver, drug metabolism is largely governed by phase I and II hepatic enzymes, with DILI widely accepted to be associated with the formation of reactive metabolites following metabolism by the CYP family, the most common phase I enzyme group. Therefore, one of the key tests for the functional relevance of HLCs for drug screening purposes would be the demonstration of inducible CYP activity at levels similar to freshly isolated hPHs. To the best of our knowledge, no studies using hESCs have successfully derived HLCs with adequate CYP activity in response to known reference compounds (Table 2). Although many studies of hESC-derived HLCs report on the expression of CYP either by detection of their messenger RNA or their protein, their functional activity has only been assessed in a handful of studies [50, 51, 53, 56, 57, 59, 61–63, 66, 69]. More importantly, only half of these studies have inducible CYP activity compared with the gold standard comparator of hPHs [51, 53, 57, 59, 69]. Interesting but less informative comparisons have also been made with undifferentiated hESCs or hepatocellular cancer cell lines which are known to have limited CYP activity [50, 62].
However, even if the gold standard comparators of hPHs are used, major differences in the experimental factors make comparison of results between studies difficult. For example, in the studies reporting on the activity of the CYP3A isoform, the levels have been shown to vary considerably from 0–90% of the hPHs used as comparators [51, 57, 59, 66, 69]. This vast range of reported CYP activity is likely to be reflective of the differences in experimental factors such as the multiple hESC lines used as starting cell source, the differences in the multi-stage differentiation protocols employed, the variety of methodology used to measure CYP activity and most pertinently, the variable quality of the reference hPHs being used in different studies [70, 71]. It is also important to note that full characterization of the hPH comparators used in all of these studies was not reported, making it difficult to judge the quality of the hPHs and their metabolic capacity.
As mentioned earlier, inducible CYP activity in hESC-derived HLCs has been assessed using a variety of techniques. For example, fluorescence and luminescence-based assays have been used to measure CYP3A4 activity following induction with reference inducer compounds such as rifampicin and midazolam [51, 69]. Activities of other CYP isoforms with corresponding gene expressions have also been tested using defined chemical substrates, with results similarly suggestive of detectable but variable CYP activity [50, 51, 56]. High performance liquid chromatography with tandem mass spectrometry (LC-MS-MS) has also been applied to measure the activity of four well-established human CYP isoforms (CYP1A2, CYP2C9, CYP3A4 and CYP2D6), and to conduct metabolite profiling based on the known metabolic pathways of bufuralol, a non-selective β-adrenoceptor blocking agent [59]. Using this approach, the results suggested comparable CYP activity of hESC-derived HLCs with reference hPHs. Furthermore, four new metabolic pathways of bufuralol were identified in addition to the three previously reported. These new revelations also indirectly suggest the effectiveness of the differentiation protocol employed in this study in obtaining a phenotype comparable with hPH.
In contrast to efforts to assess the activity of phase I enzymes in hESC-derived HLCs, only one study has reported the presence and activity of phase II enzymes [52]. In this study, glutathione-S-transferases (GSTs) were found to have overall comparable activity with that of the reference hPHs, though further examination of subunits has shown a more differential expression, with GSTM-1 showing the least. The quality of the reference hPHs used was again not reported and therefore their metabolic capacity uncertain.
The large variability, reported between different laboratories of the activity of these key enzymes associated with drug metabolism in hESC-derived HLCs, implies that the application of these cells for safety pharmacology and toxicology assessment is still premature. Perhaps more importantly, problems such as the lack of agreed endpoints of hepatic differentiation and maturation as well as the lack of standardized comparators for differentiated hESC-derived HLCs, need to be addressed urgently [70]. Although hPHs are the gold standard comparators, their metabolic capability can differ markedly between different preparations [72]. Hence, standardized and validated criteria to define the quality of the reference hPHs are also needed. Comparisons between the various differentiation protocols could then be addressed, with the aim of developing one that is efficient, reproducible and sufficiently robust for drug safety and toxicology screening.
hiPSC-derived HLCs
Although there are advantages in using hiPSCs compared with using hESCs as a starting cell source for differentiation into HLCs (Table 3), there are still limitations in efficient generation of hiPSCs. Established methods of inducing pluripotency have so far failed to negate the problem of low reprogramming efficiency and concerns persist regarding potential genomic insertions of exogenous sequences with the current standard technique of using viral vectors [45]. Identifying solutions to these limitations is important, especially with regards to the application of these HLCs for high throughput screening of compounds, which requires high yields of genetically uncompromised cells with high purity to generate reliable and sustainable results.
Table 3.
Comparison of advantages and disadvantages between HLCs derived from hESCs and hiPSCs
| hESC-derived hepatocyte-like cells (HLCs) | hiPSC-derived hepatocyte-like cells (HLCs) |
|---|---|
| Advantages | Advantages |
| • More experience in the literature reporting on the functional characteristics of hESC-derived HLCs and development of multi-stage differentiation protocols | • Potential to model DILI in vitro for mechanistic studies |
| • Potential to encapsulate the phenotypic variation of phase I and II enzymes present in the population by establishing a library of HLCs derived from different individuals representing the global and ethnic genotypic variation | |
| • Potential application in robust high throughput screening for DILI of new chemical entities | |
| • Human embryos not required | |
| Disadvantages | Disadvantages |
| • Use of hESCs subject to ethical debate | • Low reprogramming efficiency to hiPSCs from parental somatic cells |
| • Limited genotypic variation with all the available hESC lines | • Concerns with regards to the impact of genomic insertions from viral vectors used in the majority of methods for reprogramming parental cells |
A new technique with the hope of improving on the reprogramming efficiency, works by the expression of defined microRNAs (miRNAs) to induce pluripotency in mature human somatic cells [46]. miRNAs belong to a recently discovered class of small non-coding RNAs that play important post transcriptional regulatory roles in cellular and developmental events [73]. They act as master regulators by binding to a specific sequence motif of a target messenger RNA to induce their degradation or translational repression [74]. The use of the miRNA302/367 cluster, which plays an important role in global DNA demethylation and chromosomal modification, has been shown to induce pluripotency in human fibroblasts with a two order of magnitude increase in efficiency when compared with ‘classical’ transcription factor-based cellular reprogramming [32, 46, 75]. Whilst these results demonstrated huge improvements in reprogramming efficiency and may allow for high throughput generation of hiPSCs, this technique still uses a viral vector for delivery of the miRNA cluster. A similar approach harnessing the potential of miRNAs, uses direct transfection of mature double-stranded miRNAs without utilizing any viral vector [45]. hiPSCs were produced from human adipose stromal cells and dermal fibroblasts by repeated transfection of miRNAs that are found to be expressed more than two-fold in murine ESCs and iPSCs compared with murine adipose stromal cells. However, the authors also reported low efficiency levels similar to transcription factor-based cellular reprogramming originally described by Takahashi & Yamanaka [32, 45]. This technique may have the potential to deliver clinical and therapeutic benefits as it uses a relatively simple protocol to induce pluripotency without compromising genomic integrity, but has yet to be replicated by other groups. It also remains to be seen if the efficiency can be improved further with a view to producing hiPSC-derived HLCs for use in high throughput screening of NCEs.
The precise mechanisms by which pluripotency is induced in mature somatic cells by miRNAs remains incompletely understood. However, increasing evidence has suggested that specific miRNAs inhibit mRNAs associated with epigenetic regulation to cause global DNA demethylation and chromosomal modification, resulting in DNA methylation patterns in iPSCs which are very similar but non-identical to ESCs [75–79]. Interestingly, they also display distinct methylation patterns similar to their parental cells from which they were derived, suggesting a retained epigenetic memory in iPSCs, possibly due to incomplete reprogramming occurring during transformation from a differentiated somatic state to the undifferentiated iPSC status [78]. Paradoxically, this raises the possibility of harnessing this epigenetic memory to improve the efficiency and differentiation potential using these iPSCs, to rederive cells with a phenotype comparable with their parental cells [78]. This possibility was first highlighted by a study which used hiPSCs reprogrammed from pancreatic β cells to rederive insulin-producing cells [80]. Using these hiPSCs as the starting cell source, greater differentiation efficiency was shown compared with using hESCs or hiPSCs derived from other somatic cell types. This approach was also similarly applied to create HLCs from hiPSCs derived from human hepatocytes though in this study, the authors did not detect any greater differentiation efficiency compared with using hESC lines [63].
Similar to their hESC-derived counterparts, HLCs derived from hiPSCs not surprisingly have also been reported to have a variable gene expression profile, functional characteristics and differentiation efficiencies (Tables 1 and 2) [58, 63, 65–67, 81–84]. When compared with hESC-derived HLCs, transcriptomic analyses between hESC- and hiPSC-derived HLCs, have shown differences in the gene expression of CYP isoforms [85]. hESC-derived HLCs demonstrated increased expression of CYP19A1, CYP1A1 and CYP11A1, whilst hiPSC-derived HLCs had enriched CYP46A1 and CYP26A1. Although comparative studies such as this are useful particularly when deciding if hiPSCs can be considered suitable alternatives to hESCs to derive HLCs for use in safety pharmacology, findings generated from a particular laboratory may not be generalizable due to different protocols employed, varied laboratory environments, and inconsistencies in the definitions of endpoints, as discussed in the previous section [71, 86]. As with hESC-derived HLCs, it is important that for any given differentiation protocol using hiPSCs, a thorough gene expression and functional characterization of the ‘mature’ HLCs should be carried out, and compared against the gold standard of hPH, which should itself also be fully characterized (Figure 1).
Figure 1.
Full characterization of hPSC-derived HLCs against the gold standard hPHs is essential, particularly when these cells are to be used for modelling DILI in vitro
A major limitation associated with using hESC-derived HLCs for potential application in safety pharmacology and toxicology is their inability to encapsulate fully the phenotypic variation of key metabolic enzymes present in the population. This could in theory be overcome through the use of hiPSCs to derive HLCs. hiPSC-derived HLCs from different individuals could in theory produce a library of HLCs representing the phenotypic and genotypic variation of the global population. For example, hiPSC-derived HLCs from individuals with varied CYP polymorphisms would be invaluable for drug screening. hiPSC-derived HLCs from individuals who suffered idiosyncratic DILI could also be used as an in vitro model for mechanistic studies and to detect as yet unknown defective or variant metabolic pathways (Figure 2). The potential of hiPSC-derived HLCs to model metabolic disorders in vitro also allows for a better understanding of disease processes and thus facilitates the potential identification of novel therapeutic targets. It has been shown that it is possible to produce hiPSCs from dermal fibroblasts of patients with a metabolic disorder and subsequently derive HLCs with a retained disease phenotype. This has been shown in AAT deficiency, familial hypercholesterolaemia, glycogen storage disease, tyrosinaemia, progressive familial hereditary cholestasis and Crigler-Najjar syndrome [82, 83]. Recent studies have also shown the therapeutic potential of using hiPSC-HLCs with corrected point mutations for restoration of function, as demonstrated in AAT deficiency and Wilson's disease [66, 84].
Figure 2.
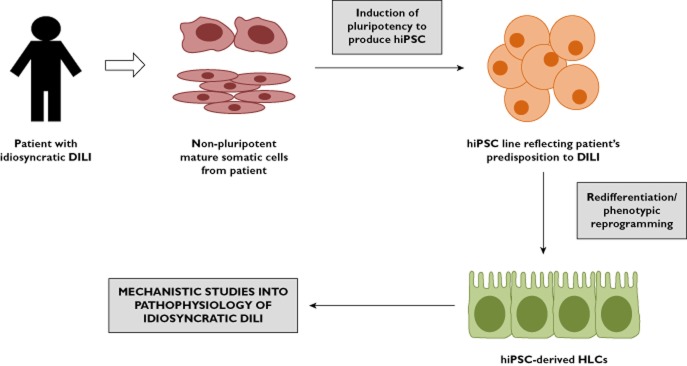
Application of hiPSC-derived HLCs for mechanistic understanding of idiosyncratic DILI in vitro
Conclusions
Recent progress in the understanding and generation of hPSCs has enabled significant progress to be made in attempts to develop a novel model system based on differentiated HLCs to study and screen for DILI. The use of hiPSCs to derive HLCs additionally offers the unique capacity to model various diseased phenotypes in vitro for mechanistic studies and identification of novel therapeutics targets, as well as the potential to study the effect of genetic polymorphism, a key factor in predicting an individual's susceptibility to ADRs and DILI. Currently, the use of the gold standard hPHs for such applications has been limited due to unpredictable availability, variable quality, phenotypic instability in culture and ethical issues surrounding procurement. However, with an optimal differentiation protocol, it is hoped that hPSCs will be able to provide an unlimited supply of HLCs for drug safety and toxicology applications. However, progress in deriving a mature HLC phenotype comparable with freshly isolated hepatocytes has been slow and HLCs obtained from these experiments have only demonstrated a phenotype which at best resembles more of a foetal liver [70]. Comparison of results between research groups has also been hampered by the lack of agreed and validated endpoints of HLC maturation and the lack of standardized characterization of hPHs as reference comparators.
It must also be borne in mind that even with the development of HLCs capable of recapitulating the function of a primary hepatocyte, various practical limitations still need to be addressed. Firstly, dedifferentiation of the HLCs in culture as observed in primary hepatocytes is likely to be an issue. With regards to using either hESC- and hiPSC-derived HLCs for modelling DILI, few studies have attempted long term culture of HLCs. This is an area of difficulty for both hPHs and HLCs and requires attention as many cases of DILI result from chronic exposure to drugs. Of the relatively few studies which have attempted to culture HLCs for a longer time period, the results are inconclusive. There is some evidence that HLCs in vitro gradually undergo maturation and display increased AAT expression, though the levels remain less than that of hPHs [87]. Similar results were also found with ALB and CYP3A4 gene expression in HLCs cultured for 50 days [54]. However, functional assessments of HLCs cultured for longer periods in vitro has never been reported to our knowledge, and therefore concerns of dedifferentiation of the HLC phenotype similar to the pattern observed with hPHs remains [70]. The predicted limited life span and phenotypic instability of the HLCs will limit their use to short term mechanistic studies of DILI, which paradoxically often occurs following prolonged exposure to drugs [11, 70]. Therefore, further understanding is still required of the differentiation patterns of HLCs in vitro. Lastly, despite the significant technological progress in miniaturization, scale-up of HLC production is mandatory if they are to be successfully deployed for high throughput screening of drug candidates by the pharmaceutical industry.
The development of the optimal protocol using either hESCs or hiPSCs to derive HLCs continues and ongoing effort should be greatly encouraged, considering the benefits this resource can potentially offer for modelling DILI in vitro.
Acknowledgments
All authors would like to thank the MRC for funding the Centre for Drug Safety Science. RK is funded by a clinical research fellowship from the Medical Research Council of United Kingdom. RLCS is funded by Stem Cells for Safer Medicine. JH is jointly funded by the Biotechnology and Biological Sciences Research Council of United Kingdom and AstraZeneca UK.
Competing Interests
JSM is a shareholder of AstraZeneca UK.
References
- 1.Olsen AK, Whalen MD. Public perceptions of the pharmaceutical industry and drug safety: implications for the pharmacovigilance professional and the culture of safety. Drug Saf. 2009;32:805–810. doi: 10.2165/11316620-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 2.Davies EC, Green CF, Mottram DR, Rowe PH, Pirmohamed M. Emergency re-admissions to hospital due to adverse drug reactions within 1 year of the index admission. Br J Clin Pharmacol. 2010;70:749–755. doi: 10.1111/j.1365-2125.2010.03751.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Davies EC, Green CF, Taylor S, Williamson PR, Mottram DR, Pirmohamed M. Adverse drug reactions in hospital in-patients: a prospective analysis of 3695 patient-episodes. PLoS ONE. 2009;4:e4439. doi: 10.1371/journal.pone.0004439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pirmohamed M, James S, Meakin S, Green C, Scott AK, Walley TJ, Farrar K, Park BK, Breckenridge AM. Adverse drug reactions as cause of admission to hospital: prospective analysis of 18 820 patients. BMJ. 2004;329:15–19. doi: 10.1136/bmj.329.7456.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Antoine DJ, Williams DP, Park BK. Understanding the role of reactive metabolites in drug-induced hepatotoxicity: state of the science. Expert Opin Drug Metab Toxicol. 2008;4:1415–1427. doi: 10.1517/17425255.4.11.1415. [DOI] [PubMed] [Google Scholar]
- 6.Davern TJ, James LP, Hinson JA, Polson J, Larson AM, Fontana RJ, Lalani E, Munoz S, Shakil AO, Lee WM. Measurement of serum acetaminophen-protein adducts in patients with acute liver failure. Gastroenterology. 2006;130:687–694. doi: 10.1053/j.gastro.2006.01.033. [DOI] [PubMed] [Google Scholar]
- 7.Kaplowitz N. Idiosyncratic drug hepatotoxicity. Nat Rev Drug Discov. 2005;4:489–499. doi: 10.1038/nrd1750. [DOI] [PubMed] [Google Scholar]
- 8.Halegoua-De Marzio D, Navarro VJ. Drug-induced hepatotoxicity in humans. Curr Opin Drug Discov Devel. 2008;11:53–59. [PubMed] [Google Scholar]
- 9.Park BK, Kitteringham NR, Maggs JL, Pirmohamed M, Williams DP. The role of metabolic activation in drug-induced hepatotoxicity. Annu Rev Pharmacol Toxicol. 2005;45:177–202. doi: 10.1146/annurev.pharmtox.45.120403.100058. [DOI] [PubMed] [Google Scholar]
- 10.Park BK, Pirmohamed M, Kitteringham NR. The role of cytochrome P450 enzymes in hepatic and extrahepatic human drug toxicity. Pharmacol Ther. 1995;68:385–424. doi: 10.1016/0163-7258(95)02013-6. [DOI] [PubMed] [Google Scholar]
- 11.Guguen-Guillouzo C, Guillouzo A. General review on in vitro hepatocyte models and their applications. Methods Mol Biol. 2010;640:1–40. doi: 10.1007/978-1-60761-688-7_1. [DOI] [PubMed] [Google Scholar]
- 12.Sahi J, Grepper S, Smith C. Hepatocytes as a tool in drug metabolism, transport and safety evaluations in drug discovery. Curr Drug Discov Technol. 2010;7:188–198. doi: 10.2174/157016310793180576. [DOI] [PubMed] [Google Scholar]
- 13.Gomez-Lechon MJ, Donato MT, Castell JV, Jover R. Human hepatocytes in primary culture: the choice to investigate drug metabolism in man. Curr Drug Metab. 2004;5:443–462. doi: 10.2174/1389200043335414. [DOI] [PubMed] [Google Scholar]
- 14.LeCluyse EL. Human hepatocyte culture systems for the in vitro evaluation of cytochrome P450 expression and regulation. Eur J Pharm Sci. 2001;13:343–368. doi: 10.1016/s0928-0987(01)00135-x. [DOI] [PubMed] [Google Scholar]
- 15.Kalgutkar AS, Soglia JR. Minimising the potential for metabolic activation in drug discovery. Expert Opin Drug Metab Toxicol. 2005;1:91–142. doi: 10.1517/17425255.1.1.91. [DOI] [PubMed] [Google Scholar]
- 16.Chen Q, Cederbaum AI. Cytotoxicity and apoptosis produced by cytochrome P450 2E1 in Hep G2 cells. Mol Pharmacol. 1998;53:638–648. doi: 10.1124/mol.53.4.638. [DOI] [PubMed] [Google Scholar]
- 17.Dai Y, Rashba-Step J, Cederbaum AI. Stable expression of human cytochrome P4502E1 in HepG2 cells: characterization of catalytic activities and production of reactive oxygen intermediates. Biochemistry. 1993;32:6928–6937. doi: 10.1021/bi00078a017. [DOI] [PubMed] [Google Scholar]
- 18.Goldring CE, Kitteringham NR, Jenkins R, Lovatt CA, Randle LE, Abdullah A, Owen A, Liu X, Butler PJ, Williams DP, Metcalfe P, Berens C, Hillen W, Foster B, Simpson A, McLellan L, Park BK. Development of a transactivator in hepatoma cells that allows expression of phase I, phase II, and chemical defense genes. Am J Physiol Cell Physiol. 2006;290:C104–115. doi: 10.1152/ajpcell.00133.2005. [DOI] [PubMed] [Google Scholar]
- 19.Pfeifer AM, Cole KE, Smoot DT, Weston A, Groopman JD, Shields PG, Vignaud JM, Juillerat M, Lipsky MM, Trump BF, Lechner JF, Harris CC. Simian virus 40 large tumor antigen-immortalized normal human liver epithelial cells express hepatocyte characteristics and metabolize chemical carcinogens. Proc Natl Acad Sci U S A. 1993;90:5123–5127. doi: 10.1073/pnas.90.11.5123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mace K, Aguilar F, Wang JS, Vautravers P, Gomez-Lechon M, Gonzalez FJ, Groopman J, Harris CC, Pfeifer AM. Aflatoxin B1-induced DNA adduct formation and p53 mutations in CYP450-expressing human liver cell lines. Carcinogenesis. 1997;18:1291–1297. doi: 10.1093/carcin/18.7.1291. [DOI] [PubMed] [Google Scholar]
- 21.Singh S, Kumar V, Singh P, Banerjee BD, Rautela RS, Grover SS, Rawat DS, Pasha ST, Jain SK, Rai A. Influence of CYP2C9, GSTM1, GSTT1 and NAT2 genetic polymorphisms on DNA damage in workers occupationally exposed to organophosphate pesticides. Mutat Res. 2011;740:101–108. doi: 10.1016/j.mrgentox.2011.11.001. [DOI] [PubMed] [Google Scholar]
- 22.Tekin D, Kayaalti Z, Soylemezoglu T. The effects of metallothionein 2A polymorphism on lead metabolism: are pregnant women with a heterozygote genotype for metallothionein 2A polymorphism and their newborns at risk of having higher blood lead levels? Int Arch Occup Environ Health. 2011 doi: 10.1007/s00420-011-0711-y. DOI: 10.1007/s00420-011-0711-y. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 23.Wilffert B, Swen J, Mulder H, Touw D. Maitland-Van der Zee AH, Deneer V. From evidence based medicine to mechanism based medicine. Reviewing the role of pharmacogenetics. Int J Clin Pharmacol. 2011;33:3–9. doi: 10.1007/s11096-011-9485-2. [DOI] [PubMed] [Google Scholar]
- 24.Celander MC, Goldstone JV, Denslow ND, Iguchi T, Kille P, Meyerhoff RD, Smith BA, Hutchinson TH, Wheeler JR. Species extrapolation for the 21st century. Environ Toxicol Chem. 2011;30:52–63. doi: 10.1002/etc.382. [DOI] [PubMed] [Google Scholar]
- 25.Lee EW, Lai Y, Zhang H, Unadkat JD. Identification of the mitochondrial targeting signal of the human equilibrative nucleoside transporter 1 (hENT1): implications for interspecies differences in mitochondrial toxicity of fialuridine. J Biol Chem. 2006;281:16700–16706. doi: 10.1074/jbc.M513825200. [DOI] [PubMed] [Google Scholar]
- 26.Saldana-Ruiz S, Soler-Martin C, Llorens J. Role of CYP2E1-mediated metabolism in the acute and vestibular toxicities of nineteen nitriles in the mouse. Toxicol Lett. 2012;208:125–132. doi: 10.1016/j.toxlet.2011.10.016. [DOI] [PubMed] [Google Scholar]
- 27.Wobus AM, Guan K, Yang HT, Boheler KR. Embryonic stem cells as a model to study cardiac, skeletal muscle, and vascular smooth muscle cell differentiation. Methods Mol Biol. 2002;185:127–156. doi: 10.1385/1-59259-241-4:127. [DOI] [PubMed] [Google Scholar]
- 28.Hamazaki T, Iiboshi Y, Oka M, Papst PJ, Meacham AM, Zon LI, Terada N. Hepatic maturation in differentiating embryonic stem cells in vitro. FEBS Lett. 2001;497:15–19. doi: 10.1016/s0014-5793(01)02423-1. [DOI] [PubMed] [Google Scholar]
- 29.Vittet D, Prandini MH, Berthier R, Schweitzer A, Martin-Sisteron H, Uzan G, Dejana E. Embryonic stem cells differentiate in vitro to endothelial cells through successive maturation steps. Blood. 1996;88:3424–3431. [PubMed] [Google Scholar]
- 30.Lumelsky N, Blondel O, Laeng P, Velasco I, Ravin R, McKay R. Differentiation of embryonic stem cells to insulin-secreting structures similar to pancreatic islets. Science. 2001;292:1389–1394. doi: 10.1126/science.1058866. [DOI] [PubMed] [Google Scholar]
- 31.Bielby RC, Boccaccini AR, Polak JM, Buttery LD. In vitro differentiation and in vivo mineralization of osteogenic cells derived from human embryonic stem cells. Tissue Eng. 2004;10:1518–1525. doi: 10.1089/ten.2004.10.1518. [DOI] [PubMed] [Google Scholar]
- 32.Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 33.Yu J, Hu K, Smuga-Otto K, Tian S, Stewart R, Slukvin II, Thomson JA. Human induced pluripotent stem cells free of vector and transgene sequences. Science. 2009;324:797–801. doi: 10.1126/science.1172482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kaji K, Norrby K, Paca A, Mileikovsky M, Mohseni P, Woltjen K. Virus-free induction of pluripotency and subsequent excision of reprogramming factors. Nature. 2009;458:771–775. doi: 10.1038/nature07864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Woltjen K, Michael IP, Mohseni P, Desai R, Mileikovsky M, Hamalainen R, Cowling R, Wang W, Liu P, Gertsenstein M, Kaji K, Sung HK, Nagy A. PiggyBac transposition reprograms fibroblasts to induced pluripotent stem cells. Nature. 2009;458:766–770. doi: 10.1038/nature07863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yusa K, Rad R, Takeda J, Bradley A. Generation of transgene-free induced pluripotent mouse stem cells by the piggyBac transposon. Nat Methods. 2009;6:363–369. doi: 10.1038/nmeth.1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jia F, Wilson KD, Sun N, Gupta DM, Huang M, Li Z, Panetta NJ, Chen ZY, Robbins RC, Kay MA, Longaker MT, Wu JC. A nonviral minicircle vector for deriving human iPS cells. Nat Methods. 2010;7:197–199. doi: 10.1038/nmeth.1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim D, Kim CH, Moon JI, Chung YG, Chang MY, Han BS, Ko S, Yang E, Cha KY, Lanza R, Kim KS. Generation of human induced pluripotent stem cells by direct delivery of reprogramming proteins. Cell Stem Cell. 2009;4:472–476. doi: 10.1016/j.stem.2009.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhou H, Wu S, Joo JY, Zhu S, Han DW, Lin T, Trauger S, Bien G, Yao S, Zhu Y, Siuzdak G, Scholer HR, Duan L, Ding S. Generation of induced pluripotent stem cells using recombinant proteins. Cell Stem Cell. 2009;4:381–384. doi: 10.1016/j.stem.2009.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Warren L, Manos PD, Ahfeldt T, Loh YH, Li H, Lau F, Ebina W, Mandal PK, Smith ZD, Meissner A, Daley GQ, Brack AS, Collins JJ, Cowan C, Schlaeger TM, Rossi DJ. Highly efficient reprogramming to pluripotency and directed differentiation of human cells with synthetic modified mRNA. Cell Stem Cell. 2010;7:618–630. doi: 10.1016/j.stem.2010.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Huangfu D, Maehr R, Guo W, Eijkelenboom A, Snitow M, Chen AE, Melton DA. Induction of pluripotent stem cells by defined factors is greatly improved by small-molecule compounds. Nat Biotechnol. 2008;26:795–797. doi: 10.1038/nbt1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Huangfu D, Osafune K, Maehr R, Guo W, Eijkelenboom A, Chen S, Muhlestein W, Melton DA. Induction of pluripotent stem cells from primary human fibroblasts with only Oct4 and Sox2. Nat Biotechnol. 2008;26:1269–1275. doi: 10.1038/nbt.1502. [DOI] [PubMed] [Google Scholar]
- 43.Mikkelsen TS, Hanna J, Zhang X, Ku M, Wernig M, Schorderet P, Bernstein BE, Jaenisch R, Lander ES, Meissner A. Dissecting direct reprogramming through integrative genomic analysis. Nature. 2008;454:49–55. doi: 10.1038/nature07056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhu S, Li W, Zhou H, Wei W, Ambasudhan R, Lin T, Kim J, Zhang K, Ding S. Reprogramming of human primary somatic cells by OCT4 and chemical compounds. Cell Stem Cell. 2010;7:651–655. doi: 10.1016/j.stem.2010.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Miyoshi N, Ishii H, Nagano H, Haraguchi N, Dewi DL, Kano Y, Nishikawa S, Tanemura M, Mimori K, Tanaka F, Saito T, Nishimura J, Takemasa I, Mizushima T, Ikeda M, Yamamoto H, Sekimoto M, Doki Y, Mori M. Reprogramming of mouse and human cells to pluripotency using mature microRNAs. Cell Stem Cell. 2011;8:633–638. doi: 10.1016/j.stem.2011.05.001. [DOI] [PubMed] [Google Scholar]
- 46.Anokye-Danso F, Trivedi CM, Juhr D, Gupta M, Cui Z, Tian Y, Zhang Y, Yang W, Gruber PJ, Epstein JA, Morrisey EE. Highly efficient miRNA-mediated reprogramming of mouse and human somatic cells to pluripotency. Cell Stem Cell. 2011;8:376–388. doi: 10.1016/j.stem.2011.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Prescott C. The business of exploiting induced pluripotent stem cells. Philos Trans R Soc Lond B Biol Sci. 2011;366:2323–2328. doi: 10.1098/rstb.2011.0047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bahadur G, Morrison M. Patenting human pluripotent cells: balancing commercial, academic and ethical interests. Hum Reprod. 2010;25:14–21. doi: 10.1093/humrep/dep369. [DOI] [PubMed] [Google Scholar]
- 49.Baxter MA, Rowe C, Alder J, Harrison S, Hanley KP, Park BK, Kitteringham NR, Goldring CE, Hanley NA. Generating hepatic cell lineages from pluripotent stem cells for drug toxicity screening. Stem Cell Res. 2010;5:4–22. doi: 10.1016/j.scr.2010.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cai J, Zhao Y, Liu Y, Ye F, Song Z, Qin H, Meng S, Chen Y, Zhou R, Song X, Guo Y, Ding M, Deng H. Directed differentiation of human embryonic stem cells into functional hepatic cells. Hepatology. 2007;45:1229–1239. doi: 10.1002/hep.21582. [DOI] [PubMed] [Google Scholar]
- 51.Ek M, Soderdahl T, Kuppers-Munther B, Edsbagge J, Andersson TB, Bjorquist P, Cotgreave I, Jernstrom B, Ingelman-Sundberg M, Johansson I. Expression of drug metabolizing enzymes in hepatocyte-like cells derived from human embryonic stem cells. Biochem Pharmacol. 2007;74:496–503. doi: 10.1016/j.bcp.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 52.Soderdahl T, Kuppers-Munther B, Heins N, Edsbagge J, Bjorquist P, Cotgreave I, Jernstrom B. Glutathione transferases in hepatocyte-like cells derived from human embryonic stem cells. Toxicol In Vitro. 2007;21:929–937. doi: 10.1016/j.tiv.2007.01.021. [DOI] [PubMed] [Google Scholar]
- 53.Hay DC, Fletcher J, Payne C, Terrace JD, Gallagher RC, Snoeys J, Black JR, Wojtacha D, Samuel K, Hannoun Z, Pryde A, Filippi C, Currie IS, Forbes SJ, Ross JA, Newsome PN, Iredale JP. Highly efficient differentiation of hESCs to functional hepatic endoderm requires ActivinA and Wnt3a signaling. Proc Natl Acad Sci U S A. 2008;105:12301–12306. doi: 10.1073/pnas.0806522105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shiraki N, Umeda K, Sakashita N, Takeya M, Kume K, Kume S. Differentiation of mouse and human embryonic stem cells into hepatic lineages. Genes Cells. 2008;13:731–746. doi: 10.1111/j.1365-2443.2008.01201.x. [DOI] [PubMed] [Google Scholar]
- 55.Agarwal S, Holton KL, Lanza R. Efficient differentiation of functional hepatocytes from human embryonic stem cells. Stem Cells. 2008;26:1117–1127. doi: 10.1634/stemcells.2007-1102. [DOI] [PubMed] [Google Scholar]
- 56.Moore RN, Moghe PV. Expedited growth factor-mediated specification of human embryonic stem cells toward the hepatic lineage. Stem. Cell Res. 2009;3:51–62. doi: 10.1016/j.scr.2009.04.004. [DOI] [PubMed] [Google Scholar]
- 57.Basma H, Soto-Gutierrez A, Yannam GR, Liu L, Ito R, Yamamoto T, Ellis E, Carson SD, Sato S, Chen Y, Muirhead D, Navarro-Alvarez N, Wong RJ, Roy-Chowdhury J, Platt JL, Mercer DF, Miller JD, Strom SC, Kobayashi N, Fox IJ. Differentiation and transplantation of human embryonic stem cell-derived hepatocytes. Gastroenterology. 2009;136:990–999. doi: 10.1053/j.gastro.2008.10.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Song Z, Cai J, Liu Y, Zhao D, Yong J, Duo S, Song X, Guo Y, Zhao Y, Qin H, Yin X, Wu C, Che J, Lu S, Ding M, Deng H. Efficient generation of hepatocyte-like cells from human induced pluripotent stem cells. Cell Res. 2009;19:1233–1242. doi: 10.1038/cr.2009.107. [DOI] [PubMed] [Google Scholar]
- 59.Duan Y, Ma X, Zou WEI, Wang C, Bahbahan IS, Ahuja TP, Tolstikov V, Zern MA. Differentiation and characterization of metabolically functioning hepatocytes from human embryonic stem cells. Stem Cells. 2010;28:674–686. doi: 10.1002/stem.315. [DOI] [PubMed] [Google Scholar]
- 60.Synnergren J, Heins N, Brolen G, Eriksson G, Lindahl A, Hyllner J, Olsson B, Sartipy P, Bjorquist P. Transcriptional profiling of human embryonic stem cells differentiating to definitive and primitive endoderm and further toward the hepatic lineage. Stem Cells Dev. 2010;19:961–978. doi: 10.1089/scd.2009.0220. [DOI] [PubMed] [Google Scholar]
- 61.Touboul T, Hannan NR, Corbineau S, Martinez A, Martinet C, Branchereau S, Mainot S, Strick-Marchand H, Pedersen R, Di Santo J, Weber A, Vallier L. Generation of functional hepatocytes from human embryonic stem cells under chemically defined conditions that recapitulate liver development. Hepatology. 2010;51:1754–1765. doi: 10.1002/hep.23506. [DOI] [PubMed] [Google Scholar]
- 62.Brolen G, Sivertsson L, Bjorquist P, Eriksson G, Ek M, Semb H, Johansson I, Andersson TB, Ingelman-Sundberg M, Heins N. Hepatocyte-like cells derived from human embryonic stem cells specifically via definitive endoderm and a progenitor stage. J Biotechnol. 2010;145:284–294. doi: 10.1016/j.jbiotec.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 63.Liu H, Ye Z, Kim Y, Sharkis S, Jang YY. Generation of endoderm-derived human induced pluripotent stem cells from primary hepatocytes. Hepatology. 2010;51:1810–1819. doi: 10.1002/hep.23626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bone HK, Nelson AS, Goldring CE, Tosh D, Welham MJ. A novel chemically directed route for the generation of definitive endoderm from human embryonic stem cells based on inhibition of GSK-3. J Cell Sci. 2011;124:1992–2000. doi: 10.1242/jcs.081679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Si-Tayeb K, Noto FK, Nagaoka M, Li J, Battle MA, Duris C, North PE, Dalton S, Duncan SA. Highly efficient generation of human hepatocyte-like cells from induced pluripotent stem cells. Hepatology. 2010;51:297–305. doi: 10.1002/hep.23354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhang S, Chen S, Li W, Guo X, Zhao P, Xu J, Chen Y, Pan Q, Liu X, Zychlinski D, Lu H, Tortorella MD, Schambach A, Wang Y, Pei D, Esteban MA. Rescue of ATP7B function in hepatocyte-like cells from Wilson's disease induced pluripotent stem cells using gene therapy or the chaperone drug curcumin. Hum Mol Genet. 2011;20:3176–3187. doi: 10.1093/hmg/ddr223. [DOI] [PubMed] [Google Scholar]
- 67.Chen YF, Tseng CY, Wang HW, Kuo HC, Yang VW, Lee OK. Rapid generation of mature hepatocyte-like cells from human induced pluripotent stem cells by an efficient three-step protocol. Hepatology. 2012;55:1193–1203. doi: 10.1002/hep.24790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sancho-Bru P, Najimi M, Caruso M, Pauwelyn K, Cantz T, Forbes S, Roskams T, Ott M, Gehling U, Sokal E, Verfaillie CM, Muraca M. Stem and progenitor cells for liver repopulation: can we standardise the process from bench to bedside? Gut. 2009;58:594–603. doi: 10.1136/gut.2008.171116. [DOI] [PubMed] [Google Scholar]
- 69.Yildirimman R, Brolen G, Vilardell M, Eriksson G, Synnergren J, Gmuender H, Kamburov A, Ingelman-Sundberg M, Castell J, Lahoz A, Kleinjans J, van Delft J, Bjorquist P, Herwig R. Human embryonic stem cell derived hepatocyte-like cells as a tool for in vitro hazard assessment of chemical carcinogenicity. Toxicol Sci. 2011;124:278–290. doi: 10.1093/toxsci/kfr225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wobus AM, Loser P. Present state and future perspectives of using pluripotent stem cells in toxicology research. Arch Toxicol. 2011;85:79–117. doi: 10.1007/s00204-010-0641-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Robinton DA, Daley GQ. The promise of induced pluripotent stem cells in research and therapy. Nature. 2012;481:295–305. doi: 10.1038/nature10761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hewitt NJ, Lechon MJ, Houston JB, Hallifax D, Brown HS, Maurel P, Kenna JG, Gustavsson L, Lohmann C, Skonberg C, Guillouzo A, Tuschl G, Li AP, LeCluyse E, Groothuis GM, Hengstler JG. Primary hepatocytes: current understanding of the regulation of metabolic enzymes and transporter proteins, and pharmaceutical practice for the use of hepatocytes in metabolism, enzyme induction, transporter, clearance, and hepatotoxicity studies. Drug Metab Rev. 2007;39:159–234. doi: 10.1080/03602530601093489. [DOI] [PubMed] [Google Scholar]
- 73.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 74.Hobert O. Gene regulation by transcription factors and microRNAs. Science. 2008;319:1785–1786. doi: 10.1126/science.1151651. [DOI] [PubMed] [Google Scholar]
- 75.Lin SL, Chang DC, Lin CH, Ying SY, Leu D, Wu DT. Regulation of somatic cell reprogramming through inducible mir-302 expression. Nucleic Acids Res. 2011;39:1054–1065. doi: 10.1093/nar/gkq850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Pfaff N, Fiedler J, Holzmann A, Schambach A, Moritz T, Cantz T, Thum T. miRNA screening reveals a new miRNA family stimulating iPS cell generation via regulation of Meox2. EMBO Rep. 2011;12:1153–1159. doi: 10.1038/embor.2011.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Subramanyam D, Lamouille S, Judson RL, Liu JY, Bucay N, Derynck R, Blelloch R. Multiple targets of miR-302 and miR-372 promote reprogramming of human fibroblasts to induced pluripotent stem cells. Nat Biotechnol. 2011;29:443–448. doi: 10.1038/nbt.1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Barrilleaux B, Knoepfler PS. Inducing iPSCs to escape the dish. Cell Stem Cell. 2011;9:103–111. doi: 10.1016/j.stem.2011.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Liao B, Bao X, Liu L, Feng S, Zovoilis A, Liu W, Xue Y, Cai J, Guo X, Qin B, Zhang R, Wu J, Lai L, Teng M, Niu L, Zhang B, Esteban MA, Pei D. MicroRNA cluster 302–367 enhances somatic cell reprogramming by accelerating a mesenchymal-to-epithelial transition. J Biol Chem. 2011;286:17359–17364. doi: 10.1074/jbc.C111.235960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bar-Nur O, Russ HA, Efrat S, Benvenisty N. Epigenetic memory and preferential lineage-specific differentiation in induced pluripotent stem cells derived from human pancreatic islet beta cells. Cell Stem Cell. 2011;9:17–23. doi: 10.1016/j.stem.2011.06.007. [DOI] [PubMed] [Google Scholar]
- 81.Sullivan GJ, Hay DC, Park I-H, Fletcher J, Hannoun Z, Payne CM, Dalgetty D, Black JR, Ross JA, Samuel K, Wang G, Daley GQ, Lee J-H, Church GM, Forbes SJ, Iredale JP, Wilmut I. Generation of functional human hepatic endoderm from human induced pluripotent stem cells. Hepatology. 2010;51:329–335. doi: 10.1002/hep.23335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ghodsizadeh A, Taei A, Totonchi M, Seifinejad A, Gourabi H, Pournasr B, Aghdami N, Malekzadeh R, Almadani N, Salekdeh GH, Baharvand H. Generation of liver disease-specific induced pluripotent stem cells along with efficient differentiation to functional hepatocyte-like cells. Stem Cell Rev. 2010;6:622–632. doi: 10.1007/s12015-010-9189-3. [DOI] [PubMed] [Google Scholar]
- 83.Rashid ST, Corbineau S, Hannan N, Marciniak SJ, Miranda E, Alexander G, Huang-Doran I, Griffin J, Ahrlund-Richter L, Skepper J, Semple R, Weber A, Lomas DA, Vallier L. Modeling inherited metabolic disorders of the liver using human induced pluripotent stem cells. J Clin Invest. 2010;120:3127–3136. doi: 10.1172/JCI43122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Yusa K, Rashid ST, Strick-Marchand H, Varela I, Liu P-Q, Paschon DE, Miranda E, Ordonez A, Hannan NRF, Rouhani FJ, Darche S, Alexander G, Marciniak SJ, Fusaki N, Hasegawa M, Holmes MC, Di Santo JP, Lomas DA, Bradley A, Vallier L. Targeted gene correction of alpha1-antitrypsin deficiency in induced pluripotent stem cells. Nature. 2011;478:391–394. doi: 10.1038/nature10424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Jozefczuk J, Prigione A, Chavez L, Adjaye J. Comparative analysis of human embryonic stem cell and induced pluripotent stem cell-derived hepatocyte-like cells reveals current drawbacks and possible strategies for improved differentiation. Stem Cells Dev. 2011;20:1259–1275. doi: 10.1089/scd.2010.0361. [DOI] [PubMed] [Google Scholar]
- 86.Newman AM, Cooper JB. Lab-specific gene expression signatures in pluripotent stem cells. Cell Stem Cell. 2010;7:258–262. doi: 10.1016/j.stem.2010.06.016. [DOI] [PubMed] [Google Scholar]
- 87.Shirahashi H, Wu J, Yamamoto N, Catana A, Wege H, Wager B, Okita K, Zern MA. Differentiation of human and mouse embryonic stem cells along a hepatocyte lineage. Cell Transplant. 2004;13:197–211. doi: 10.3727/000000004783984016. [DOI] [PubMed] [Google Scholar]