

Abstract
Aim
Total imatinib concentrations are currently measured for the therapeutic drug monitoring of imatinib, whereas only free drug equilibrates with cells for pharmacological action. Due to technical and cost limitations, routine measurement of free concentrations is generally not performed. In this study, free and total imatinib concentrations were measured to establish a model allowing the confident prediction of imatinib free concentrations based on total concentrations and plasma proteins measurements.
Methods
One hundred and fifty total and free plasma concentrations of imatinib were measured in 49 patients with gastrointestinal stromal tumours. A population pharmacokinetic model was built up to characterize mean total and free concentrations with inter-patient and intrapatient variability, while taking into account α1-acid glycoprotein (AGP) and human serum albumin (HSA) concentrations, in addition to other demographic and environmental covariates.
Results
A one compartment model with first order absorption was used to characterize total and free imatinib concentrations. Only AGP influenced imatinib total clearance. Imatinib free concentrations were best predicted using a non-linear binding model to AGP, with a dissociation constant Kd of 319 ng ml−1, assuming a 1:1 molar binding ratio. The addition of HSA in the equation did not improve the prediction of imatinib unbound concentrations.
Conclusion
Although free concentration monitoring is probably more appropriate than total concentrations, it requires an additional ultrafiltration step and sensitive analytical technology, not always available in clinical laboratories. The model proposed might represent a convenient approach to estimate imatinib free concentrations. However, therapeutic ranges for free imatinib concentrations remain to be established before it enters into routine practice.
Keywords: cytochrome P450 enzyme, drug monitoring, protein binding, pharmacokinetics, proton pump inhibitors, tyrosine kinase inhibitor
What is Already Known about This Subject
Variability in binding of imatinib to α1-acid glycoprotein can influence its activity since only the free drug is likely to equilibrate with the intracellular milieu to exert its pharmacological action.
While the therapeutic drug monitoring of total imatinib concentration is currently proposed, routine measurement of free concentrations is generally not performed due to cost and technical issues.
What this Study Adds
This study allowed for the first time the characterization of the population pharmacokinetic profile of total and unbound imatinib concentrations and the confident prediction of imatinib free concentrations based on total concentrations and α1-acid glycoprotein levels.
Introduction
Imatinib is a selective inhibitor of tyrosine kinases, comprising Bcr-Abl fusion protein in chronic myeloid leukaemia (CML) and the c-kit proto-oncogene in gastrointestinal stromal tumour (GIST). It has demonstrated an impressive clinical efficacy in both malignancies. Inducing durable responses and achieving prolonged survival, imatinib has become the standard of care for the treatment of these diseases. However, imatinib treatment is not devoid of toxicity and resistance occurs in some instances. Besides cellular mechanisms of resistance (gene amplification and mutation), variability in binding to α1-acid glycoprotein (AGP) can modulate its activity [1, 2–5]. Indeed, only the free drug is likely to equilibrate with the intracellular milieu to exert its pharmacological action. Moreover, a small change in the extent of protein binding may result in a significant impact on imatinib free fraction and on its concentration–effect relationships [5, 6].
Although there are many circulating proteins in plasma capable of binding drugs, the majority of drugs bind to human serum albumin (HSA) and AGP [7]. HSA is the most abundant protein in plasma, whereas the normal AGP concentrations are much lower, resulting in a lower capacity to bind drugs and a more rapid saturation of the protein [8]. Both are capable of binding a broad variety of drugs with sufficient affinity to impact on the pharmacologically active free fraction. HSA is the primary binding protein for acidic drugs, while binding to AGP is more commonly observed with basic lipophilic agents. Changes in the concentration, conformation, and/or other physicochemical characteristics of these proteins may result in significant changes in the drug free fraction [9]. Alterations in albumin concentrations in plasma occur as a result of altered synthesis, loss, or a shift of fluids between body compartments. The most common alteration, hypoalbuminaemia, is associated with a wide variety of pathologic and physiologic conditions, such as inflammation, nephrotic syndrome, burn injury and cancer. Similarly, several disease states (inflammation, renal and hepatic disease), physiological conditions (age, pregnancy, obesity), genetic factors or co-administration of some drugs can markedly alter AGP concentrations (see [8] for review).
Binding studies of imatinib in plasma, in particular to HSA and AGP, showed that imatinib binds mainly to AGP, resulting in a mean free fraction of about 4% [4, 10]. An in vitro binding study performed by Kretz et al. [4], with radioactive imatinib incubated in conditions reflecting the clinical situation, reported free fractions of 3.1% and 20% in AGP and HSA solutions, respectively, and of 4.3% in healthy human plasma. Several studies showed that imatinib has about 50 to 60-fold higher affinity for AGP than for HSA [2, 11]. AGP of most individuals is a mixture of two or three genetic variants (F1 and/or S and A), which are encoded by two different genes [12, 13]. The F1 and S are encoded by alleles of the same gene, while the A variant is the product of the other gene [12, 13]. Moreover, it has been shown that imatinib binds with a stronger selectivity to the F1-S genetic variant of AGP, while its binding on the A variant is weaker and less specific [12, 13].
For drugs of intermediate to low hepatic extraction and high protein binding such as imatinib, a change in protein concentrations or in binding affinity alters total plasma concentrations, while free drug concentrations are expected to remain mostly unchanged [14]. However, an altered free fraction modifies the apparent total concentration−effect relationships, and may thus compromise the correct interpretation of therapeutic drug monitoring results based on total plasma concentrations, especially for strongly protein bound drugs such as imatinib. Moreover, under certain pathophysiological conditions competing with normal binding, such as uraemia, liver disease, hypoproteinaemia or drug interactions, free drug concentration may be significantly elevated despite total concentrations within the therapeutic range [15].
The relationship between unbound drug exposure and efficacy and toxicity endpoints has been recently investigated. Studies have shown that unbound drug exposure was correlated with haematological toxicity (absolute neutrophil count), whereas no significant association with treatment efficacy in GIST patients could be detected [16, 17]. Widmer et al. [18] confirmed that both total (in GIST) and free imatinib exposure (in CML and GIST) were correlated with the occurrence and number of side effects, and that higher free drug exposure also predicted a higher probability of therapeutic response in GIST when taking into account tumour KIT genotype [18]. All these results were however obtained using extrapolated free concentrations rather than real measurements. A formal confirmation of them is therefore still warranted.
The aim of the present study was to extend our previously proposed model enabling the prediction of free imatinib concentrations based on total imatinib concentrations [17]. The objectives were therefore (i) to characterize the population pharmacokinetics of total and free imatinib plasma concentrations, (ii) to evaluate the influence of both AGP and HSA concentrations in addition to demographic variables and co-medications on total and free imatinib pharmacokinetics and (iii) to refine our model for the prediction of imatinib free concentrations based on total concentrations along with other potential influencing factors.
Methods
Study population
Data from 49 GIST patients, providing a total of 150 plasma samples, were collected over 2 years (of these patients, three participated in our previous study [17], but no concentrations overlapped between the latter and this study). Patients received imatinib at daily oral doses ranging from 200 to 800 mg and all patients were pooled regardless of their medical history. Most peripheral blood samples were drawn at 1–6 month intervals on follow-up visits as part of an observational clinical pharmacokinetic study and of routine TDM laboratory tests for medical purposes. The median number of measurements for each patient was 3 (range 1–11) and were obtained under steady-state conditions. The following data were recorded for each patient: body weight, gender, age, HSA and AGP concentrations (g l−1), as well as other administered drugs. α1-acid glycoprotein concentrations measured twice in one patient during the same study day resulted in very different concentrations of AGP (0.7 and 1.99 mg l−1); these results were considered unreliable because of a possible degradation due to inappropriate storage and time interval until analysis, and were thus excluded from the covariate analysis). Concomitant medications were categorized into inducers or inhibitors of the cytochromes (CYP) P450 3A4, mainly responsible for the metabolism of imatinib [10, 16]. In addition, proton pump inhibitors were recorded separately to test for a potential effect on imatinib absorption or relative bioavailability through an effect on gastric pH. The study was approved by the Ethics Committee of the Lausanne Faculty of Biology and Medicine. Informed written consent was obtained from all participants.
Analytical equipment
The high performance liquid chromatography system involved a Rheos 2200 binary pump (Flux Instruments, Basel, Switzerland) equipped with an online degasser and a temperature-controlled 324 vial autosampler maintained at +10°C (CTC Analytics AG, Zwingen, Switzerland). The chromatographic system was coupled to a triple stage quadrupole mass spectrometer (TSQ Quantum Discovery; Thermo Electron Corporation, Waltham, MA, USA) equipped with an electrospray ionization interface operated in positive ion mode and controlled with the Xcalibur 1.1 software (Thermo Electron Corporation, San Jose, CA, USA).
Free and total drug analysis
Peripheral blood samples (5 ml) were collected from patients into EDTA Monovette® syringes (Sarstedt, Nümbrecht, Germany). Plasma was isolated by centrifugation at 1850 g for 10 min at +4°C (Beckman model J-6B centrifuge; Beckman Coulter, Fullerton, CA, USA) and stored at −20°C until analysis. Total plasma concentrations (Ctot) of imatinib were measured by reverse phase liquid chromatography coupled with tandem mass spectrometry (LC-MS/MS) after plasma protein precipitation with acetonitrile, using an adaptation of our previously reported method [19]. The selected mass transitions for imatinib and its internal standard imatinib-D8 were m/z 494.3→ 394.1 and m/z 502.3→ 394.1, respectively. The method was validated according to the recommendations published on-line by the Food and Drugs Administration (FDA) [20]. The method was precise and accurate within the range of calibration (1–10 000 ng ml−1) with inter-assay precision (CV%) and accuracy (bias%) for the low, medium and high quality control plasma samples (3, 2000, 8000 ng ml−1, respectively) ranging between 3.2 to 14.1% and −3.1 to 5.6%, respectively. The lower limit of quantification (LLOQ) for total plasma concentration determination was 1 ng ml−1. Our laboratory participates to an External Quality Control program for imatinib, organized initially within the frame of the European Treatment and Outcome Study (EUTOS) of European Leukaemia Net (http://www.leukemia-net.org/).
For determination of free plasma concentrations, ultrafiltration Amicon Centrifree® Filter Systems (cutoff 30 kDa; Millipore Corporation, Bedford, MA, USA) were used to separate the free (unbound) fraction from the total plasma concentration based on a methodology developed and validated in our laboratory [21]. In brief, Amicon Centrifree® filters were first conditioned prior to use by subjecting them to ultrafiltration (2000 g, 30 min, 26°C) with 500 μl of ultrapure water in a fixed-edge, temperature-controlled centrifuge (Avanti® J-30I High Performance Centrifuge System, Beckman Coulter). Free imatinib concentrations were measured in patient plasma samples as follows: plasma aliquots (500 μl) were thawed and allowed to equilibrate at room temperature before being subjected to ultrafiltration in pre-washed Centrifree® filters for 30 min at 2000 g at 26°C in the Avanti fixed-edge centrifuge and the ultrafiltrate was collected in plastic cups. The 30 min ultrafiltrate collection was diluted 1:1 with MeOH without delay, to avoid the adsorption of the free imatinib species from the aqueous ultrafiltrate medium onto the cup's plastic wall. After the addition of 100 μl of internal standard solution (imatinib D8 20 ng ml−1) to 100 μl aliquot of each ultrafiltrate/MeOH 1:1 mixture, they were injected into the LC-MS/MS for the determination of free imatinib concentrations (Cu). The LC-MS/MS method for free concentration measurements was calibrated using matrix-matched standards (prepared in blank plasma ultrafiltrate diluted 1:1 with imatinib solution in MeOH, to yield calibration concentrations ranging from 1 to 1000 ng ml−1). The LOQ for the assay was 1 ng ml−1. The blank ultrafiltrate pool used for the preparation of calibration and quality control ultrafiltrate samples was obtained from blank plasma subjected to ultrafiltration (1850 g, 30 min, +4°C), onto an Amicon Centricon® Plus-20 Filter System (cutoff 30 kDa; Millipore Corporation) and distributed as 100 μl aliquots stored at −20°C until use.
Of importance, the potential loss of drug onto the filter membrane because of adsorption especially during the early step of ultrafiltration (a phenomenon that has been observed with other therapeutic classes, namely some antiretroviral drugs [21]), was carefully ascertained. Our experiments performed with plasma spiked with imatinib at clinically relevant total plasma concentrations (500, 1000 and 4000 ng ml−1) have shown that the free concentrations of imatinib determined in the ultrafiltrate collected in four fractions (0–8, 8–16, 16–24 and 24–30 min), and the corresponding fu values, remained constant throughout the entire 30 min duration of the ultrafiltration process. Notably, no significant drop of imatinib free concentrations could be noticed in the early (0–8 min) ultrafiltration fraction collection, indicating that no loss of imatinib was to be expected due to membrane adsorption in the Centrifree® filters. These results depart somewhat from those reported by Streit et al. [22], possibly because spiked ultrafiltrate and phosphate buffer matrices were used in their adsorption experiments. These aqueous media, in which imatinib is probably less soluble, could be more prone to adsorption than the whole plasma we used. Finally, subjecting spiked control plasma samples to a single freezing-thawing cycle had no significant influence on the measured free plasma concentrations.
HSA and AGP concentrations were measured using commercially available assays from Roche Diagnostics based on colorimetric and immunoturbidimetric methods, respectively, carried out on a Roche Cobas® Integra® 400 apparatus (Roche Diagnostics, Rotkreuz, Switzerland). The inter-assay precision (CV%) of the assay for HSA, determined in the clinically relevant range of concentrations with control plasma samples at 23.5 and 48.8 g l−1 of albumin is 1.1% and 1.3%, respectively. The inter-assay precision of the assay of AGP is 2.4 and 1.5%, at AGP plasma concentrations of 0.62 and 2.22 g l−1, respectively.
Pharmacokinetic modelling
The analysis was performed using the nonmem® software (version VI ICON Development Solutions, Ellicott city, MO, USA, with NM-TRAN version II and a gfortran compiler). The program uses mixed (fixed and random) effects regression to estimate population means and variances of the pharmacokinetic parameters and to identify factors that may affect them. Equations used for the description of the protein binding were in part derived using Mathematica (Version 6.0, Wolfram, Champaign, IL, USA, for Sun Solaris SPARC).
Total and free imatinib pharmacokinetic models
Structural model
Total and free imatinib concentrations were analyzed first separately, using a one compartment model with first order absorption according to our previously published model [17]. Estimated parameters were total imatinib clearance CLtot, total volume of distribution Vd,tot, first order absorption rate katot for total concentrations, and unbound clearance CLu, unbound volume of distribution Vd,u and kau for the description of free imatinib concentrations. In the absence of intravenous data, bioavailability (F) was fixed to 1, in accordance with the almost complete absorption reported for imatinib [23, 24].
Covariate model
At first, individual Bayesian estimates of CL, Vd and ka were derived and plotted against demographic covariates (body weight, gender, age, AGP and HSA concentrations, CYP3A4 inhibitors or inducers and proton pump inhibitors) to identify possible influences and to evaluate the shape of the relationship. Available covariates were then sequentially incorporated in the model and tested for significance on CLtot, CLu and Vd,tot, Vd,u or katot. The influence of body weight (BW), age (AGE), AGP and HSA concentrations expressed as the relative deviation of the individual BW, AGE, AGP and HSA concentrations from the population mean (BWmean = 70 kg, AGEmean = 50 years, AGPmean = 0.9 g l−1 and HSA mean = 34 g l−1, respectively) were tested using linear relationships, allometric or power functions as appropriate. Dichotomous variables were used for gender and concomitant medications use. The influence of antacids was also tested using a relative bioavailability factor, where F was fixed to 1 for individuals without any treatment and estimated for those under treatment with proton pump inhibitors.
Prediction of free imatinib concentrations
Basic equations
Several models were tested for the simultaneous analysis of imatinib Cu and Ctot data, in order to characterize their relationship. The first baseline model used a simple ratio of both moieties, assuming a constant, non-saturable free fraction (Equation 1]). Further models included protein concentrations in the relationships, testing for either linear binding kinetics (Equation 2]) or non-linear binding equilibrium (Equation 3]) as previously proposed [17, 25–28]. The equations are as follows:
| (1) |
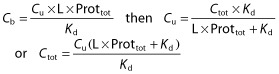 |
(2) |
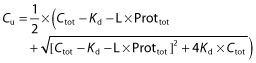 |
(3) |
In these equations, Cb is the bound concentration, calculated as Ctot − Cu. L is a scaling factor that accounts for the difference in concentration unit between Ctot (ng ml−1) and total protein concentration (AGP or HSA g l−1), Prottot corresponds to the total protein concentration (AGP or HSA), and Kd is the equilibrium dissociation constant. As L and Kd are correlated and cannot be independently estimated, L was fixed to 11 700, assuming a 1:1 molar binding ratio [2] and considering a molar mass of 493.6 g mol−1 for imatinib and a mean molar mass of 42 000 g mol−1 for AGP. For albumin, a wide variety of drugs have appreciable affinity for one or more binding sites of HSA, but no study has determined the number of binding sites for imatinib. Therefore, considering a molar mass of 493.6 g mol−1 for imatinib, and a mean molar mass of 68 000 g mol−1 for HSA, different L values were tried, assuming a 1:1 molar binding ratio leading to an L of 7300, a 1:2, 1:3, 1:4 and 1:5 molar binding ratio with L values of respectively 3600, 2400 and 1800 and 1400.
Statistical analyses and model building
At first, non-linear regression analyses of Cu as a function of Ctot and AGP or HSA concentrations were performed for an initial estimation of the Kd values. Then, Ctot data were fitted using a one compartment model described above and Cu estimated using linear and non-linear binding kinetic with the proteins (Equation 2] and Equation 3]). In these models, Kd was either fixed to 90 ng ml−1 according to our previous model [17] or estimated. Finally, the contribution and the potential interaction between AGP and HSA on the prediction of Cu were tested using those relationships (see Appendix 1] for the derived equations describing the interaction models).
Since unbound pharmacokinetic (PK) parameters (CLu and Vu) were thought to represent more closely the physiologic elimination process, we also fitted Cu data and predicted Ctot based on the above equations. Both approaches provided very close results and similar estimations of the Kd value (Table 3).
Table 3.
Population pharmacokinetic parameters of the final models using unbound and total concentrations of imatinib for the prediction of Cu (Model A) or Ctot (Model B)(simultaneous analysis)
| Total PK Parameters | Total PK parameters | Unbound PK parameters | Unbound PK parameters | ||
|---|---|---|---|---|---|
| Model A: Cu = f (Ctot)* | Model B: Ctot = f (Cu)† | ||||
| Estimate | SE (%)‡ | Estimate | SE (%)§ | ||
| CLtot (l h−1) | 13.8 | 5 | CLu (l h−1) | 386 | 6 |
| Vd,tot (l) | 409 | 17 | Vd,u (l) | 9 580 | 20 |
| katot (h−1) | 0.80 | 73 | kau (h−1) | 0.93 | 70 |
| Kd (ng ml−1) | 319 | 2 | Kd (ng ml−1) | 316 | 3 |
| L | 11 700¶ | – | L | 11 700¶ | – |
| Interindividual variability | Interindividual variability | ||||
| ωCL,tot (CV%)** | 27 | 53§ | ωCLu (CV%)** | 25 | 66§ |
| ωVd,tot (CV%)** | 48 | 75§ | ωVd,u (CV%)** | 64 | 81§ |
| Correlation CL-Vd | 0.59 | Correlation CL-Vd | 0.65 | ||
| Residual variability | Residual variability | ||||
| σ Ctot (CV%)†† | 14 | 40§ | σ Ctot (CV%)†† | 15 | 41%§ |
| σ Cu (CV%)†† | 17 | 41§ | σ Cu (CV%)†† | 14 | 41%§ |
| Correlation Cu-Ctot | 0.72 | Correlation Cu-Ctot | 0.66 | ||
In this model, total PK parameters CLtot, Vd,tot and katot are estimated and Cu are predicted using:
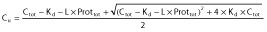 (see text).
(see text).
in this model, total unbound PK parameters are estimated CLu, Vd,u and kau, and Ctot are predicted using:
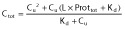 (see text).
(see text).
SE, standard error of the estimates, expressed as CV%.
SE, standard error of the variance components, taken as square (SEestimate/estimate), expressed as a percentage.
Fixed value that represent the scaling factor that accounts for the difference in concentration unit between Ctot (ng ml−1) and total protein concentration AGP (g l−1), assuming a one-to-one molar binding ratio.
Estimates of variability expressed as coefficient of variation (CV%).
Residual variability of the plasma concentration, expressed as CV%.
Statistical model
A hierarchical model was used to account for individual and residual variability. The individual PK parameters θj were modelled assuming a log-normal distribution of the general form 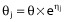 where θ is the population mean, and ηj are independent normally distributed random effects with mean zero and variance Ω. A proportional model was used to describe the residual variability of imatinib. For the generic response Ŷ and the corresponding prediction Y, the ith measurement for the jth individual takes the form
where θ is the population mean, and ηj are independent normally distributed random effects with mean zero and variance Ω. A proportional model was used to describe the residual variability of imatinib. For the generic response Ŷ and the corresponding prediction Y, the ith measurement for the jth individual takes the form 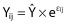 where εij is independent normally distributed with mean zero and a variance Σ. Separate error models were allowed for total and free concentrations and correlation between measures were tested, using the L2 function in nonmem®.
where εij is independent normally distributed with mean zero and a variance Σ. Separate error models were allowed for total and free concentrations and correlation between measures were tested, using the L2 function in nonmem®.
Model estimation
The regression analyses of Cu as a function of Ctot using non-linear binding kinetics (Equation 3]) were performed in nonmem®. The PK data were fitted using the first order conditional method (FOCE INTERACTION) with the subroutine ADVAN 6. To determine the statistical significance between models, different selection criteria were used. A decrease in the nonmem® objective function (OF), which corresponds to minus twice the logarithm of the maximum likelihood of the model and is approximately χ2 distributed, has been used to choose between nested models and the Akaike criterion was used for non-hierarchical models. A decrease >6.64 points for one additional parameter (P = 0.01) was considered statistically significant. In addition, goodness-of-fit plots and visual predictive checks (VPCs) were used to select models. Regression diagnostic plots were generated with R (version 2.10.1; The R Foundation for Statistical Computing, Vienna, Austria). The predictive performance of the developed models was also evaluated on the basis of the bias and the precision of the individual predicted concentrations vs. actual (total or free) observations. Bias (i.e. [predicted free concentration – measured free concentration]/measured free concentration) and precision with 95% confidence interval (95% CI) were assessed using percent mean predictive error (MPE) and percent root mean-squared predictive error (RMSE), as previously described [29].
Results
The 150 imatinib plasma total concentration values measured in the 49 patients ranged between 355 and 4440 μg l−1, and between 11 and 166 μg l−1 for free concentrations. The characteristics of the population are summarized in Table 1.
Table 1.
Summary of the characteristics of the patients
| Characteristic | Patients | |
|---|---|---|
| Gender (n) | Men | 24 |
| Woman | 25 | |
| Age (years) | Median | 66 |
| Range | 25–88 | |
| Body weight (kg) | Median | 72 |
| Range | 48–105 | |
| AGP plasma concentration (g l−1) | Median | 0.9 |
| Range | 0.4–2.0 | |
| HSA plasma concentration (g l−1) | Median | 34.4 |
| Range | 8.0–43.8 | |
| CYP3A4 inhibitors co-administered | Levothyroxine, valproic acid | 3 |
| CYP3A4 inducers co-administered | Dexamethasone | 1 |
| Proton pump inhibitors co-administrered | Omeprazole, esomeprazole, pantoprazole, rabeprazole, lansoprazole | 14 |
Total and free imatinib concentrations model
The independent analyses of total and unbound imatinib concentration data showed that the disposition of both moieties could adequately be described by a one compartment model with first order absorption from the gastrointestinal tract. An intersubject variability was observed on CL and Vd for both total and free concentrations, with a significant covariance between both parameters. Among the covariates tested, only AGP concentrations showed a statistically significant influence on imatinib CLtot (ΔOF = −35.4, P = 2.7 × 10−9) and Vd,tot (ΔOF = −7.8, P = 5.2 × 10−3), but the influence of this variable on Vd,tot did not remain statistically significant in the multivariate analysis. No influence of this protein was observed on CLu and Vd,u, and HSA concentrations had no impact on unbound or bound PK parameters either. A small increase of 10% and 14% on CLtot and CLu, respectively, and of 18% and 28% on both Vd,tot and Vd,u was observed on body weight doubling, which however did not reach statistical significance using either linear or allometric functions. A small 12% decrease in relative bioavailability was detected in patients under treatment with proton pump inhibitors, but did not reach statistical significance either, and no effect of these drugs on katot was observed. Neither did the other demographic factors, HSA concentrations, CYP3A inhibitors or inducers show any significant effect on imatinib total or unbound kinetics. The final population estimates are presented in Table 2 for total and unbound concentration data. Figure 1 illustrates the observed free and total plasma concentrations for the patients receiving 400 mg once daily imatinib, along with the population mean and 95% prediction interval.
Table 2.
Final population pharmacokinetic parameters of total and free imatinib concentrations (independent analyses)
| Model | Parameters | Population mean | Inter-individual variability | ||
|---|---|---|---|---|---|
| Estimate | SE† | Estimate* | SE‡ | ||
| Total imatinib concentrations | CLtot (l h−1) | 13.5 | 5% | 23% | 34% |
| Vd,tot (l) | 314 | 22% | 31% | 150% | |
| katot (h−1) | 0.45 | 71% | – | – | |
| θAGP CL,tot (l h−1) | −5.8 | 13% | |||
| Correlation CLtot-Vd,tot | 0.81 | ||||
| Residual variability σ (CV%)§ | 28% | 39%‡ | |||
| Free imatinib concentrations | CLu (l h−1) | 389 | 6% | 27% | 58% |
| Vd,u (l) | 8410 | 19% | 36% | 84% | |
| kau (h−1) | 0.48 | 32% | - | ||
| Correlation CLu-Vd,u | 0.80 | ||||
| Residual variability σ (CV%)§ | 30% | 39%‡ | |||
CL: clearance, Vd: volume of distribution, ka: absorption rate constant; tot: total PK parameters, u: unbound PK parameters.
Estimates of variability expressed as coefficient of variation (CV%).
SE, standard error of the estimates, expressed as CV%.
SE, standard error of the variance components, taken as square root (SEestimate/estimate), expressed as a percentage.
Residual variability of the plasma concentration, expressed as CV%.
Figure 1.
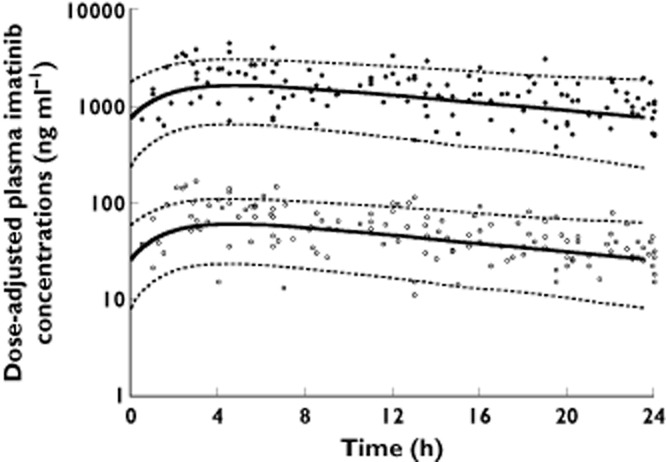
Total (black circles) and free (white circles) imatinib observed concentrations in patients scaled for 400 mg once daily imatinib, with the mean population prediction (solid line) and 95% prediction interval (dashed lines)
Prediction of free imatinib concentrations
Statistical analyses
The regression of Cu as a function of Ctot and AGP concentrations estimated a Kd value of 327.0 ± 7.9 ng ml−1 (Equations 2] and 3]), significantly better than the baseline model (Equation 1]) (ΔOF > −33, P = 9.2 × 10−9). No influence of HSA using either linear or non-linear binding was observed (ΔOF> +26 compared with the baseline model) and Kd was much higher (4580 ± 144 ng ml−1). The interaction model (Equation 6] Appendix 1]) improved significantly the prediction of Cu compared with the model including non-linear binding to AGP solely (ΔOF = −7) with a Kd for AGP and HSA of, respectively, 421 and 23 300 ng ml−1.
Pharmacokinetic modelling
Several models were then sequentially elaborated to describe the relationship between Cu and Ctot. The baseline model (Equation 1]) predicted a free fraction (Equation 1]) of 3.5% with a small non-significant inter-patient variability of 11%. Allowing for a correlation between the residual errors for Cu and Ctot resulted in a marked improvement of the model fit (ΔOF = −68.7, P = 1.1 × 10−16),
The predictions of Cu based on our previous non-linear model (Equation 3]) [17], using a fixed Kd of 90 ng ml−1, are presented in Figure 2A. This model was not able to predict imatinib free concentrations observed in our population, yielding a major bias of −70% (MPE range −66/−71%), and a poor precision of 250%. A difference of about three-fold was apparent between measured and predicted free imatinib concentrations. Refinement of the model by allowing Kd to be estimated markedly improved the fit (ΔOF = −166, P = 5.5 × 10−38), finally yielding a Kd of 319 ng ml−1. This model described the data much better that the one assuming a constant free fraction (Equation 1], ΔOF = −37.7, P = 8.3 × 10−10). This approach drastically decreased the bias (from −70 to 4%, with 95% CI −2, 10%) and improved the precision (43%) of the model, showing a good correlation between measured and estimated free imatinib concentrations (Figure 2B). A reduced model using a linear (Equation 2]) function of AGP described the data worse than the non-linear model (ΔOF = +12.9). The hyperbolic dependency of imatinib observed free fractions on plasma AGP concentrations depicted in Figure 3 is in accordance with this finding.
Figure 2.
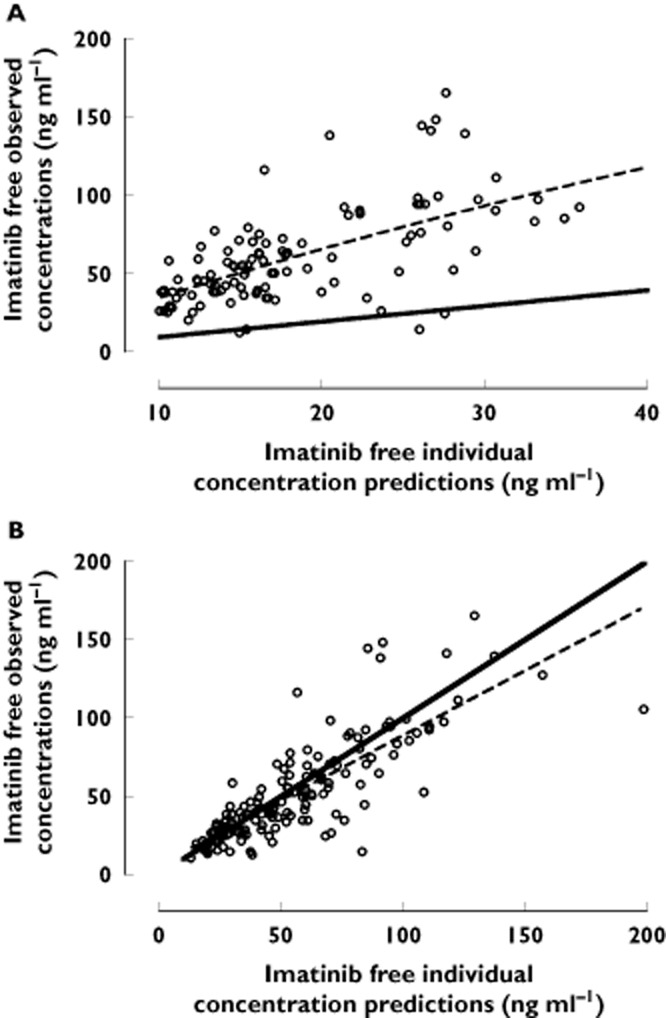
(A)Individual predicted (predicted by Widmer et al. equation [17]) vs. measured free concentrations of imatinib. (B) Individual predictions (predicted by our final model, see text) vs. measured free imatinib concentrations. The dotted line represents a non-parametric local regression function while the black dotted line represents the identity line
Figure 3.
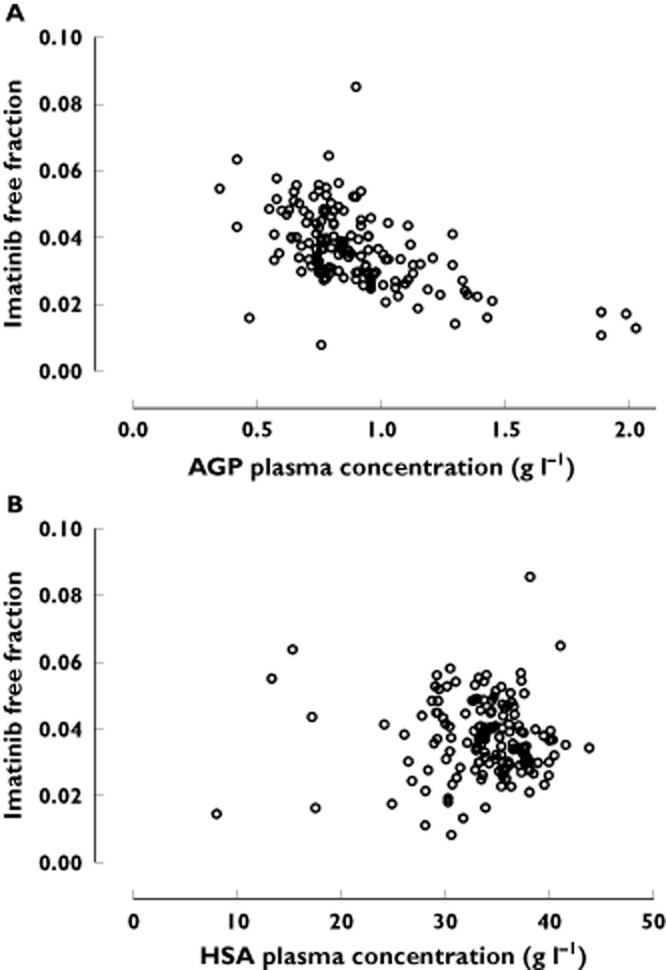
Imatinibfree fractions vs. A) AGP and B) HSA plasma concentrations
For albumin, the linear model did not fit the data better than the baseline model (Equation 1]) or the model integrating AGP, and no further improvement was observed using non-linear binding kinetics to this protein (ΔOF >61.2). The Kd value was 4370 ng ml−1 (assuming two binding sites). The fit did not change assuming one, two, three or four binding sites to this protein. No clear relationship was apparent between observed imatinib free fractions and plasma HSA concentrations (Figure 3).
Interaction models assessing the simultaneous influence of linear binding to AGP and linear or non-linear binding to HSA (Equation 4] or Equation 5], Appendix 1]), did not fit the data as well as the non-linear AGP model solely (ΔOF < −3.5, P = 0.06), whatever the number of binding sites assumed for HSA. Conversely, by using Equation 6] (non-linear AGP and linear HSA relationship) a statistical improvement of the model was observed (ΔOF < −9.8, P = 1.7 × 10−3). The Kd value for AGP was 411 ± 39.1 ng ml−1 and for HSA was 22 300 ± 7850 ng ml−1 for two binding sites (no difference was observed considering one, two or three binding sites). The goodness-of-fit plots were, however, not better than the ones considering AGP only and showed similar non-significant bias (MPE 4%) and precision (MRSE 42%). The influence of HSA on the prediction of Cu was thus considered marginal and was not retained. The final results of the analyses of Ctot as a function of Cu and conversely are presented in Table 3. The model-based relationship between imatinib total and free concentrations based on the final relationship (Equation 3]) using a Kd of 319 ng ml−1 and a L of 11 700, stratified according to several levels of AGP concentrations, is presented in Figure 4. Figure 5 provides goodness-of-fit plots of observed vs. predicted and individual predicted free imatinib concentrations from the final model.
Figure 4.
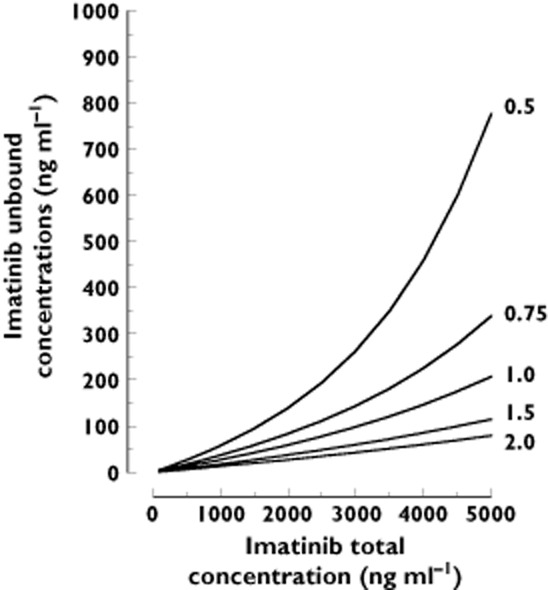
Imatinib total concentrations Ctot vs. predicted free concentrations based on the final model determined in this study, integrating AGP values of 0.5, 0.75, 1, 1.5 g l−1 and 2 g l−1
Figure 5.

Goodness-of-fitplots of imatinib observed total concentrations (A and B) and unbound concentrations (C and D) vs. population predictions (A and C) and individual predictions (B and D) for the simultaneous fit
Discussion
This study allowed for the first time the full characterization of the pharmacokinetic profile of total and unbound imatinib concentrations and the description of the relationships governing the equilibrium between total and free imatinib concentrations.
The population pharmacokinetics of total imatinib concentrations could be adequately described using a one compartment model for total and unbound concentrations. The estimated values of CLtot and Vd,tot are in close accordance with our previous results and in good agreement with previously reported studies [10, 30–32]. Intersubject variabilities on CL and Vd were of the same magnitude for total and free concentrations, although poorly estimated. Among the tested covariates, only AGP had a significant influence on CLtot. It is expected that an increase in AGP concentrations induces a reduction in the free fraction of imatinib, therefore decreasing total clearance, whereas CLu remains unchanged. The resulting effect is an increase in total plasma concentrations despite constant unbound concentrations. No effect of HSA concentrations was observed in univariate analyses. The interaction model revealed a small influence of this protein, which could indicate that HSA, a carrier with lower affinity but higher capacity, might have a small residual influence once accounting for the predominant effect of AGP. The prediction of Cu was, however, not improved using this more complicated model. In addition, owing to the much larger amount of HSA in blood and its lower affinity to imatinib, it is not expected that changes in HSA concentrations could alter imatinib concentrations to a significant extent at therapeutic concentrations. Furthermore, although the model including non-linear binding to both AGP and HSA could not be tested due to the complexity of the relationship, it is very unlikely that saturation of HSA occurs at therapeutic concentrations.
Among the demographic covariates tested, only body weight was associated with a small increase in CLtot and CLu, and Vd,tot and Vd,u in our population, which did not reach statistical significance. Menon-Andersen et al. showed that total body weight was the only covariate found to affect CLtot and Vd,tot and reported an increased clearance by 23% and Vd by 32% on body weight doubling [32]. The study of Schmidli et al. [30] revealed a small and similar 12% increase in CLtot on doubling body weight and a 32% increase in Vd,tot. A much more important effect of body weight was found in our previous study [17], which increased CLtot and CLu by 99% and 91%, respectively. The lack of correlation might be related to power issues, the range of body weight being relatively restricted in our population (SD ± 15). Gender and age were not shown to affect imatinib pharmacokinetics, in accordance with several studies having reported that both factors are unlikely to be clinically significant in GIST and CML patients [10, 30, 33–35]. Whether these demographic parameters have an influence on imatinib free concentrations or not should be confirmed. No influence of comedications was found, owing probably to the limited number of patients with cytochromes P450 3A4 inducers or inhibitors. No effect of proton pump inhibitors was found either [36]. Previous studies with Mg2+/Al3+-based antacids had shown negative results as well [37].
We could challenge our previous theoretical model [17] using experimental measurements of unbound imatinib concentrations actually determined in GIST patients. The average imatinib free fraction estimated in our study (3.5%) is in close agreement with free fractions reported of 4% [10], 3.1% [4] and 5% [22] in the literature. HSA and AGP are the most important drug-binding proteins in plasma. In healthy subjects, the concentration of AGP in plasma varies in the range of 0.55–1.4 g l−1 [38]. HSA, the major protein component of plasma [38], is present in the plasma of healthy individuals at concentrations ranging from 35–52 g l−1 [39]. The modular structural organization of HSA provides a variety of ligand binding sites, although two appear to predominate [38].
AGP plasma concentrations proved to have a marked influence on imatinib bound pharmacokinetics, whereas HSA did not show any relevant effect. The reported association binding constant (Ka) values of imatinib to AGP are of 4.9 × 106 m−1, 2.4 × 106 m−1, 1.7 × 106 m−1 and 1.4 × 106 m−1 [2, 3, 11], corresponding to Kd values of respectively 100, 210, 290 and 350 ng ml−1. These values are in good agreement with the Kd of 319 ng ml−1 estimated in this study, using non-linear binding and assuming a 1:1 binding ratio 4]. Ka values of 2.3 × 105 m−1, 3 × 104 m−1 and 7 × 103 m−1 [2–4, 11], have been reported for imatinib binding to HSA, suggesting very different Kd values (2100, 16 500 and 70 500 ng ml−1) and a much weaker affinity for HSA. These higher values are in accordance with the estimated Kd for albumin in our study. The limited additional effect of HSA on imatinib kinetics observed in our study is thus compatible with its approximately 60 times higher affinity for AGP than for HSA [2]. The major bias observed by applying our previous model was solely due to the inaccurate estimation of the Kd value determined without actual free imatinib concentrations measurements, which underestimated unbound concentrations by a factor of three approximately. It must be recalled that this study used a fairly indirect method to estimate Kd only from total concentration values [17]. The small residual and non-significant bias might be related to a few data points measured during the absorption phase that were under-estimated by the model.
Despite the validity of this approach in our population, it must be acknowledged that the prediction of Cu had some limitations. Indeed, some pathophysiological conditions or concomitant medications could alter the binding affinity or capacity to AGP, resulting in changes in unbound concentrations that are not directly proportional to changes in protein levels. Confounding factors have thus to be considered while using this approach for the prediction of unbound concentrations.
In conclusion, the prediction of free imatinib concentrations can be based on measurement of total concentrations and AGP concentrations using the following relationship:
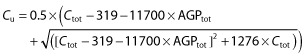 |
where Cu and Ctot are expressed in ng ml−1 and AGPtot in g l−1.
Individualization of imatinib therapy based on free imatinib plasma concentration extrapolated with our model could be advised, in particular when changes in plasma binding are expected under specific pathophysiological conditions. Either the determination of free concentrations of imatinib or its model-derived estimation in a larger population of patients might also help to understand better the relationship between free concentrations and efficacy or toxicity [22]. In routine TDM practice, this approach could represent a more practical and affordable method to derive free concentrations of imatinib from total concentrations, having only to take into account AGP plasma concentration measurements.
Acknowledgments
This project was carried out at the initiative of the late Dr Ulrich Schnorf, former president of the Swiss association of patients suffering from GIST (GIST-Gruppe Schweiz) with a research grant from Novartis Schweiz AG (as part of the ‘European Treatment and Outcome Study’ [EUTOS] initiative of the European Leukemia Net [ELN]). The Swiss National Science Foundation (Switzerland) has also supported in part the salary of AH. TB, have received support from the Nano-Tera initiative, funded by the Swiss National Science Foundation to develop novel tools for the monitoring of drug concentrations. Finally, the authors are grateful to the Vital-it platform (http://www.vital-it.ch) of the Swiss Institute of Bioinformatics for providing the computational resources for the pharmacokinetic modelling.
Appendix 1
Linear relationship between Cu and Cb:
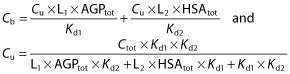 |
(4) |
Linear relationship with AGP and non-linear relationship with HSA:
| (5) |
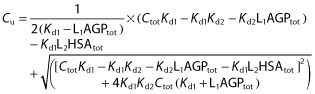 |
Non-linear relationship with AGP and linear relationship with HSA:
| (6) |
Non-linear relationship with AGP and HSA
| (7) |
The cubic equation describing Cu as a function of Ctot is not shown due to the complexity of the relationship, but can be provided upon request.
Competing Interests
L. Decosterd has received a grant-in-aid from Novartis for the development of the LC-MS/MS assay for free plasma concentrations of imatinib. L. Decosterd and T. Buclin have received unrestricted research grants from Novartis. A. Haouala, N. Widmer, T. Buclin and L. Decosterd have received travel grants from Novartis for participating to International meetings on CML and on Therapeutic Drug Monitoring of TKIs.
References
- 1.Ceciliani F, Pocacqua V. The acute phase protein alpha1-acid glycoprotein: a model for altered glycosylation during diseases. Curr Protein Pept Sci. 2007;8:91–108. doi: 10.2174/138920307779941497. [DOI] [PubMed] [Google Scholar]
- 2.Fitos I, Visy J, Zsila F, Mady G, Simonyi M. Selective binding of imatinib to the genetic variants of human alpha1-acid glycoprotein. Biochim Biophys Acta. 2006;1760:1704–1712. doi: 10.1016/j.bbagen.2006.08.015. [DOI] [PubMed] [Google Scholar]
- 3.Gambacorti-Passerini C, Barni R, le Cortre CP, Zucchetti M, Cabrita G, Cleris L, Rossi F, Gianazza E, Brueggen J, Cozens R, Pioltelli P, Pogliani E, Corneo G, Formelli F, D'Incalci M. Role of alpha1 acid glycoprotein in the in vivo resistance of human BCR-ABL(+) leukemic cells to the abl inhibitor STI571. J Natl Cancer Inst. 2000;92:1641–1650. doi: 10.1093/jnci/92.20.1641. [DOI] [PubMed] [Google Scholar]
- 4.Kretz O, Weiss HM, Schumacher MM, Gross G. In vitro blood distribution and plasma protein binding of the tyrosine kinase inhibitor imatinib and its active metabolite, CGP74588, in rat, mouse, dog, monkey, healthy humans and patients with acute lymphatic leukaemia. Br J Clin Pharmacol. 2004;58:212–216. doi: 10.1111/j.1365-2125.2004.02117.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Smith KD, Paterson S. Binding of alpha-1-acid glycoprotein to imatinib following increased dosage of drug. Haematologica. 2005;90(Suppl):ELT01. [PubMed] [Google Scholar]
- 6.Kwong TC. Free drug measurements: methodology and clinical significance. Clin Chim Acta. 1985;151:193–216. doi: 10.1016/0009-8981(85)90082-8. [DOI] [PubMed] [Google Scholar]
- 7.Bailey DN, Briggs JR. The binding of selected therapeutic drugs to human serum alpha-1 acid glycoprotein and to human serum albumin in vitro. Ther Drug Monit. 2004;26:40–43. doi: 10.1097/00007691-200402000-00009. [DOI] [PubMed] [Google Scholar]
- 8.Kremer JM, Wilting J, Janssen LH. Drug binding to human alpha-1-acid glycoprotein in health and disease. Pharmacol Rev. 1988;40:1–47. [PubMed] [Google Scholar]
- 9.Svensson CK, Woodruff MN, Baxter JG, Lalka D. Free drug concentration monitoring in clinical practice. Rationale and current status. Clin Pharmacokinet. 1986;11:450–469. doi: 10.2165/00003088-198611060-00003. [DOI] [PubMed] [Google Scholar]
- 10.Petain A, Kattygnarath D, Azard J, Chatelut E, Delbaldo C, Geoerger B, Barrois M, Seronie-Vivien S, LeCesne A, Vassal G. Population pharmacokinetics and pharmacogenetics of imatinib in children and adults. Clin Cancer Res. 2008;14:7102–7109. doi: 10.1158/1078-0432.CCR-08-0950. [DOI] [PubMed] [Google Scholar]
- 11.Zsila F, Fitos I, Bencze G, Keri G, Orfi L. Determination of human serum alpha1-acid glycoprotein and albumin binding of various marketed and preclinical kinase inhibitors. Curr Med Chem. 2009;16:1964–1977. doi: 10.2174/092986709788682191. [DOI] [PubMed] [Google Scholar]
- 12.Duche JC, Herve F, Tillement JP. Study of the expression of the genetic variants of human alpha1-acid glycoprotein in healthy subjects using isoelectric focusing and immunoblotting. J Chromatogr B Biomed Sci Appl. 1998;715:103–109. doi: 10.1016/s0378-4347(98)00084-x. [DOI] [PubMed] [Google Scholar]
- 13.Herve F, Duche JC, Jaurand MC. Changes in expression and microheterogeneity of the genetic variants of human alpha1-acid glycoprotein in malignant mesothelioma. J Chromatogr B Biomed Sci Appl. 1998;715:111–123. doi: 10.1016/s0378-4347(98)00085-1. [DOI] [PubMed] [Google Scholar]
- 14.Rowland M. Protein binding and drug clearance. Clin Pharmacokinet. 1984;9(Suppl. 1):10–17. doi: 10.2165/00003088-198400091-00002. [DOI] [PubMed] [Google Scholar]
- 15.Dasgupta A. Clinical utility of free drug monitoring. Clin Chem Lab Med. 2002;40:986–993. doi: 10.1515/CCLM.2002.172. [DOI] [PubMed] [Google Scholar]
- 16.Delbaldo C, Chatelut E, Re M, Deroussent A, Seronie-Vivien S, Jambu A, Berthaud P, Le CA, Blay JY, Vassal G. Pharmacokinetic-pharmacodynamic relationships of imatinib and its main metabolite in patients with advanced gastrointestinal stromal tumors. Clin Cancer Res. 2006;12(20 Pt 1):6073–6078. doi: 10.1158/1078-0432.CCR-05-2596. [DOI] [PubMed] [Google Scholar]
- 17.Widmer N, Decosterd LA, Csajka C, Leyvraz S, Duchosal MA, Rosselet A, Rochat B, Eap CB, Henry H, Biollaz J, Buclin T. Population pharmacokinetics of imatinib and the role of alpha-acid glycoprotein. Br J Clin Pharmacol. 2006;62:97–112. doi: 10.1111/j.1365-2125.2006.02719.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Widmer N, Decosterd LA, Leyvraz S, Duchosal MA, Rosselet A, biec-Rychter M, Csajka C, Biollaz J, Buclin T. Relationship of imatinib-free plasma levels and target genotype with efficacy and tolerability. Br J Cancer. 2008;98:1633–1640. doi: 10.1038/sj.bjc.6604355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Haouala A, Zanolari B, Rochat B, Montemurro M, Zaman K, Duchosal MA, Ris HB, Leyvraz S, Widmer N, Decosterd LA. Therapeutic drug monitoring of the new targeted anticancer agents imatinib, nilotinib, dasatinib, sunitinib, sorafenib and lapatinib by LC tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 2009;877:1982–1996. doi: 10.1016/j.jchromb.2009.04.045. [DOI] [PubMed] [Google Scholar]
- 20.FDA. 2001. Homepage. Guidance for industry: bioanalytical method validation [online]. Available at http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm070107.pdf (last accessed 14 May 2012)
- 21.Fayet A, Beguin A, de Tejada BM, Colombo S, Cavassini M, Gerber S, Eap CB, Telenti A, Buclin T, Biollaz J, Decosterd LA. Determination of unbound antiretroviral drug concentrations by a modified ultrafiltration method reveals high variability in the free fraction. Ther Drug Monit. 2008;30:511–522. doi: 10.1097/FTD.0b013e3181817318. [DOI] [PubMed] [Google Scholar]
- 22.Streit F, Binder L, Hafke A, Brandhorst G, Braulke F, Haase D, Armbrust T, Cameron S, Ramadori G, Oellerich M, Walson P. Use of total and unbound imatinib and metabolite LC-MS/MS assay to understand individual responses in CML and GIST patients. Ther Drug Monit. 2011;33:632–643. doi: 10.1097/FTD.0b013e3182263ac4. [DOI] [PubMed] [Google Scholar]
- 23.Cohen MH, Williams G, Johnson JR, Duan J, Gobburu J, Rahman A, Benson K, Leighton J, Kim SK, Wood R, Rothmann M, Chen G, U KM, Staten AM, Pazdur R. Approval summary for imatinib mesylate capsules in the treatment of chronic myelogenous leukemia. Clin Cancer Res. 2002;8:935–942. [PubMed] [Google Scholar]
- 24.Peng B, Dutreix C, Mehring G, Hayes MJ, Ben-Am M, Seiberling M, Pokorny R, Capdeville R, Lloyd P. Absolute bioavailability of imatinib (Glivec) orally versus intravenous infusion. J Clin Pharmacol. 2004;44:158–162. doi: 10.1177/0091270003262101. [DOI] [PubMed] [Google Scholar]
- 25.Belpaire FM, Braeckman RA, Bogaert MG. Binding of oxprenolol and propranolol to serum, albumin and alpha 1-acid glycoprotein in man and other species. Biochem Pharmacol. 1984;33:2065–2069. doi: 10.1016/0006-2952(84)90575-6. [DOI] [PubMed] [Google Scholar]
- 26.MacKichan J. Influence of protein binding and use of unbound (free) drug concentrations. In: Burton M, Shaw L, Schentag JJ, Evans W, Wilkins LW, editors. Applied Pharmacokinetics and Pharmacodynamics: Principles of Therapeutic Drug Monitoring. Baltimore, MD: Lippincott, Williams and Wilkins; 2006. pp. 82–113. [Google Scholar]
- 27.Lagneau F, Marty J, Beyne P, Tod M. Physiological modeling for indirect evaluation of drug tissular pharmacokinetics under non-steady-state conditions: an example of antimicrobial prophylaxis during liver surgery. J Pharmacokinet Pharmacodyn. 2005;32:1–32. doi: 10.1007/s10928-005-2101-0. [DOI] [PubMed] [Google Scholar]
- 28.Son DS, Hariya S, Shimoda M, Kokue E. Contribution of alpha 1-acid glycoprotein to plasma protein binding of some basic antimicrobials in pigs. J Vet Pharmacol Ther. 1996;19:176–183. doi: 10.1111/j.1365-2885.1996.tb00036.x. [DOI] [PubMed] [Google Scholar]
- 29.Sheiner LB, Beal SL. Some suggestions for measuring predictive performance. J Pharmacokinet Biopharm. 1981;9:503–512. doi: 10.1007/BF01060893. [DOI] [PubMed] [Google Scholar]
- 30.Schmidli H, Peng B, Riviere GJ, Capdeville R, Hensley M, Gathmann I, Bolton AE, Racine-Poon A. Population pharmacokinetics of imatinib mesylate in patients with chronic-phase chronic myeloid leukaemia: results of a phase III study. Br J Clin Pharmacol. 2005;60:35–44. doi: 10.1111/j.1365-2125.2005.02372.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Judson I, Ma P, Peng B, Verweij J, Racine A, di Paola ED, van Glabbeke M, Dimitrijevic S, Scurr M, Dumez H, van Oosterom A. Imatinib pharmacokinetics in patients with gastrointestinal stromal tumour: a retrospective population pharmacokinetic study over time. EORTC Soft Tissue and Bone Sarcoma Group. Cancer Chemother Pharmacol. 2005;55:379–386. doi: 10.1007/s00280-004-0876-0. [DOI] [PubMed] [Google Scholar]
- 32.Menon-Andersen D, Mondick JT, Jayaraman B, Thompson PA, Blaney SM, Bernstein M, Bond M, Champagne M, Fossler MJ, Barrett JS. Population pharmacokinetics of imatinib mesylate and its metabolite in children and young adults. Cancer Chemother Pharmacol. 2009;63:229–238. doi: 10.1007/s00280-008-0730-x. [DOI] [PubMed] [Google Scholar]
- 33.Cortes JE, Egorin MJ, Guilhot F, Molimard M, Mahon FX. Pharmacokinetic/pharmacodynamic correlation and blood-level testing in imatinib therapy for chronic myeloid leukemia. Leukemia. 2009;23:1537–1544. doi: 10.1038/leu.2009.88. [DOI] [PubMed] [Google Scholar]
- 34.Larson RA, Druker BJ, Guilhot F, O'Brien SG, Riviere GJ, Krahnke T, Gathmann I, Wang Y. Imatinib pharmacokinetics and its correlation with response and safety in chronic-phase chronic myeloid leukemia: a subanalysis of the IRIS study. Blood. 2008;111:4022–4028. doi: 10.1182/blood-2007-10-116475. [DOI] [PubMed] [Google Scholar]
- 35.Yoo C, Ryu MH, Kang BW, Yoon SK, Ryoo BY, Chang HM, Lee JL, Beck MY, Kim TW, Kang YK. Cross-sectional study of imatinib plasma trough levels in patients with advanced gastrointestinal stromal tumors: impact of gastrointestinal resection on exposure to imatinib. J Clin Oncol. 2010;28:1554–1559. doi: 10.1200/JCO.2009.26.5785. [DOI] [PubMed] [Google Scholar]
- 36.Ogawa R, Echizen H. Clinically significant drug interactions with antacids: an update. Drugs. 2011;71:1839–1864. doi: 10.2165/11593990-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 37.Sparano BA, Egorin MJ, Parise RA, Walters J, Komazec KA, Redner RL, Beumer JH. Effect of antacid on imatinib absorption. Cancer Chemother Pharmacol. 2009;63:525–528. doi: 10.1007/s00280-008-0778-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.L.Trainor G. The importance of plasma protein binding in drug discovery. Exp Opin Drug Discov. 2007;2:51–64. doi: 10.1517/17460441.2.1.51. [DOI] [PubMed] [Google Scholar]
- 39.Denko CW, Gabriel P. Age and sex related levels of albumin, ceruloplasmin, alpha 1 antitrypsin, alpha 1 acid glycoprotein, and transferrin. Ann Clin Lab Sci. 1981;11:63–68. [PubMed] [Google Scholar]