

The need for a greater transparency in the process of how regulatory authorities (RAs) evaluate new medicines has been advocated by the public and the scientific communities [1–3]. Also transparency about the outcomes of marketing authorization procedures is important for the purpose of a better understanding of the reasons why certain procedures tend to result in either a successful or a failed application [4]. While the US Food and Drug Administration (FDA) is not obliged to disclose information on drug applications withdrawn prior to the conclusion of the evaluation process or refused at the end of it [3], the European Union (EU) legislation requires the European Medicines Agency (EMA) to do so [5, 6]. Indeed, the EMA publishes on its website assessment reports of withdrawn and refused applications.
We retrieved and evaluated such public assessment reports on withdrawals and refusals (i.e. a negative opinion given by the EMA) of all initial authorization applications published between 2003 (the date of publication of the first available refusal report) and 31 December 2010. A total of 86 drug applications could be identified with either a withdrawal (70 out of 86) or a refusal (16 out of 86). The main therapeutic categories in the failed submissions were the following: i) oncology/immunology (29 out of 86 drugs = 34%), ii) central nervous system (CNS) (15 out of 86 drugs = 17%), iii) cardiovascular/metabolic diseases (14 out of 86 drugs = 16%) and iv) infectious diseases (12 out of 86 drugs = 14%). The reasons for withdrawal or refusal were related to one or more of the three assessment criteria, i.e. quality, safety and efficacy.
Overall, 156 major objections on these three criteria were raised by the EMA: 106 (67.9%) objections due to efficacy deficiencies, while 27 (17.3%) due to safety and 23 (21.6%) due to quality. Within the scope of efficacy major objections, five main categories could be identified: i) lack of clinical relevance (44 out of 106, 41.5%), ii) methodological deficiencies (23 out of 106, 21.6%), iii) pharmacokinetic (PK) issues, including bioequivalence (20 out of 106, 18.8%), iv) lack of statistical significance (13 cases, 12.2%) and v) major Good Clinical Practice (GCP) issues (5 out of 106, 4.7%) (see Figure 1). Within the safety objections, clinical safety (e.g. increased risk of adverse reactions and potential risk identified) was present in 23 out of 27 cases (85.2%). The objections concerned non-clinical safety/toxicological issues in only four cases (14.8%), (see Figure S1). Within the quality objections, two categories could be identified: one related to drug substance and/or drug product (19 cases, 82.6%) and one related to Good Manufacturing Practice (GMP) issues (4 cases, 17.4%). Withdrawal was due solely to quality issues in only two cases (Docetaxel Mylan and Mycrograb). In none of the cases, was withdrawal solely due to safety (clinical or non-clinical). Interestingly, in four cases following an initial refusal (i.e. Cimzia, Valdoxan/Thymanax, Yondelis) a subsequent submission at a later stage led to a positive opinion by the EMA.
Figure 1.
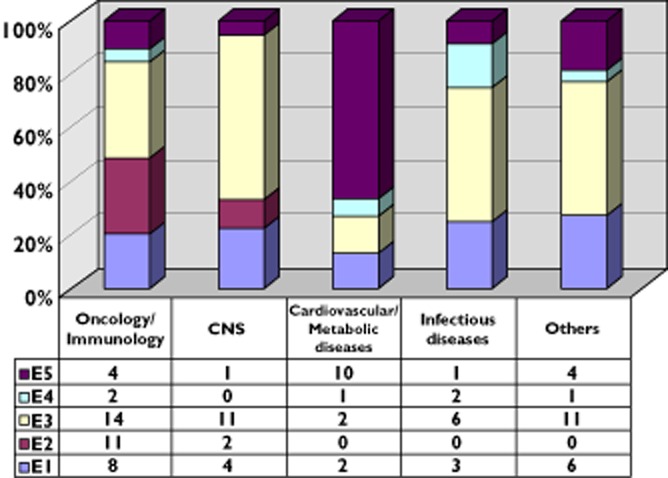
Types of efficacy deficiency per therapeutic area.: E1: methodological issues; E2: lack of statistical significance; E3: lack of clinical relevance; E4: GCP issues; E5: PK issues/bioequivalence
Disclosure of the grounds behind failed applications is a step forward on regulatory transparency and can be considered as a positive implementation of the EU legislation 726/2004. We queried several other RAs across the world in order to check whether they have similar transparency measures in place on failed drug applications. Apart from the EMA, only Australia has introduced such a system in 2009 (see Table S1). The process for an increased transparency in Europe has been further strengthened with regard to pharmacovigilance [7]. In July 2012 the EMA announced that it will systematically publish all of its committees’ agendas and minutes before the end of 2013 [8].
Our analysis confirms that failed justification of the clinical relevance by the applicant is a main predictor for withdrawal or refusal of an application for a marketing authorization of a new medicine, in line with previous publications [9]. A clear propensity of a positive EMA opinion seems to be a robust clinical trial programme, with a good rationale, and an efficient trial performance. Statistical significance alone is not sufficient enough to acquire an approval, but most importantly robust clinical benefit to the patients to be treated with the new medicine must be demonstrated.
Regulators have the legal task to evaluate all the available data and to come to an informed decision about the benefit-risk of the product under review. Withdrawn or refused applications provide an important insight into what may go wrong in bringing a product from bench to the clinic, and what could be improved in future applications. Over the last decade Europe has made important steps forwards in disclosing such withdrawal and refusal information. However, this should not be the end of improving transparency. Further studies are needed to gain better insight and understanding on failed drug development.
Competing Interests
‘All authors have completed the Unified Competing Interest form at http://www.icmje.org/coi_disclosure.pdf (available on request from the corresponding author) and declare no support from any organization for the submitted work, no financial relationships with any organizations that might have an interest in the submitted work in the previous 3 years and no other relationships or activities that could appear to have influenced the submitted work.’
Funding
None.
Supporting Information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site:
Figure S1
Rate of efficacy, safety and quality deficiencies per therapeutic area. Legend: S,Q and E are related to Safety, Quality and Efficacy deficiencies respectively.
{kind=link}
Table S1
Query on disclosure of withdrawal and/or refused drug applications directed to a sample of drug regulatory authorities in various countries.
References
- 1.Garattini S, Bertele V. Europe's opportunity to open up drug regulation. BMJ. 2010;340:c1578. doi: 10.1136/bmj.c1578. [DOI] [PubMed] [Google Scholar]
- 2.European Medicines Agency. More transparency needed. Lancet. 2010;375:1753. doi: 10.1016/S0140-6736(10)60785-4. [DOI] [PubMed] [Google Scholar]
- 3.Asamoah AK, Sharfstein JM. Transparency at the Food and Drug Administration. N Engl J Med. 2010;362:2341–2343. doi: 10.1056/NEJMp1005202. [DOI] [PubMed] [Google Scholar]
- 4.Eichler H, Aronsson B, Abadie E, Salmonson T. New drug approval success rate in Europe in 2009. Nat Rev Drug Discov. 2010;9:355–356. doi: 10.1038/nrd3169. [DOI] [PubMed] [Google Scholar]
- 5. Regulation (EC) No 726/2004 of the European Parliament and of the Council of 31 March 2004 laying down Community procedures for the authorisation and supervision of medicinal products for human and veterinary use and establishing a European Medicines Agency.
- 6. Reflection paper: publication of withdrawals of marketing authorisation applications for human medicinal products. London, 5 October 2006. Doc. Ref. EMEA/239350/2005 Rev.1.
- 7. Plan for implementation of the pharmacovigilance legislation by the European Medicines Agency Activities to protect and promote public health 2012 in partnership with European Member States. EMA/64750/2012. 2 February 2012.
- 8. European Medicines Agency announces plan to publish committee agendas and minutes. EMA/480386/2012 Press release, 18 July 2012.
- 9.Pignatti F, Aronsson B, Gate N, Vamvakas S, Wade G, Moulon I, Le Courtois P. The review of drug applications submitted to the European Medicines Evaluation Agency: frequently raised objections, and outcome. Eur J Clin Pharmacol. 2002;58:573–580. doi: 10.1007/s00228-002-0532-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.