

Key Points
Our study demonstrates aberrant genome-wide deposition of histone 3 lysine 79 dimethylation on MLL-target genes in MLL-AF6–driven leukemia cells.
We provide evidence that leukemia cells bearing the MLL-AF6 fusion are sensitive to genetic and pharmacologic DOT1L inhibition.
Abstract
The t(6;11)(q27;q23) is a recurrent chromosomal rearrangement that encodes the MLLAF6 fusion oncoprotein and is observed in patients with diverse hematologic malignancies. The presence of the t(6;11)(q27;q23) has been linked to poor overall survival in patients with AML. In this study, we demonstrate that MLL-AF6 requires continued activity of the histone-methyltransferase DOT1L to maintain expression of the MLL-AF6-driven oncogenic gene-expression program. Using gene-expression analysis and genome-wide chromatin immunoprecipitation studies followed by next generation sequencing, we found that MLL-fusion target genes display markedly high levels of histone 3 at lysine 79 (H3K79) dimethylation in murine MLL-AF6 leukemias as well as in ML2, a human myelomonocytic leukemia cell line bearing the t(6;11)(q27;q23) translocation. Targeted disruption of Dot1l using a conditional knockout mouse model inhibited leukemogenesis mediated by the MLL-AF6 fusion oncogene. Moreover, both murine MLL-AF6–transformed cells as well as the human MLL-AF6–positive ML2 leukemia cell line displayed specific sensitivity to EPZ0004777, a recently described, selective, small-molecule inhibitor of Dot1l. Dot1l inhibition resulted in significantly decreased proliferation, decreased expression of MLL-AF6 target genes, and cell cycle arrest of MLL-AF6–transformed cells. These results indicate that patients bearing the t(6;11)(q27;q23) translocation may benefit from therapeutic agents targeting aberrant H3K79 methylation.
Introduction
Genomic rearrangements of the human 11q23 chromosomal band, involving the mixed lineage leukemia (MLL) gene, are observed in a diverse range of myeloid as well as lymphoblastic leukemia subtypes, with a strong association with pediatric and therapy-related leukemias.1-3 In these leukemias, the MLL gene is fused to one of more than 60 different partner genes, resulting in the formation of dominantly acting MLL fusion-oncoproteins.3-5 The partners of MLL are nuclear-, cytoplasmic-, or membrane-associated proteins involved in diverse functional processes ranging from chromatin modification and transcriptional elongation to cellular adhesion, endocytosis, cytoskeleton organization, and signal transduction (reviewed in Meyer et al4). A number of MLL fusion partners, especially nuclear proteins such as AF4, AF9, ENL, ELL, and AF10, fusions of which together account for the vast majority of MLL patients, are components of large, multi-subunit, protein complexes that control gene expression. Several such complexes have been identified, including the family of elongation-assisting proteins, the super-elongation complex,6 the related AF4/ENL/p-Tefb (AEP) complex,7 and the chromatin-modifying Dot1l complex.8 In MLL-rearranged leukemias, constitutive recruitment of one or more of these complexes by chimeric MLL fusion proteins is believed to facilitate sustained expression of MLL-target genes, resulting in leukemic transformation. These complexes represent possible targets for pharmacologic inhibition, with several studies demonstrating the importance of different components of these complexes to MLL fusion-mediated transformation (reviewed in Muntean et al3). One such promising candidate for therapeutic intervention is the histone methyltransferase Dot1l, the central component of the Dot1l complex. Dot1l is the only known enzyme catalyzing the methylation of histone 3 at lysine 79 (H3K79). Studies using various human MLL-rearranged leukemia cells demonstrate high levels of H3K79 methylation on MLL-fusion target genes, suggesting that DOT1L may play an important role in leukemia driven by a variety of MLL-fusion proteins.9-15 Moreover, a specific small-molecule inhibitor of DOT1L has been shown to have selective activity against MLL-rearranged human leukemia cell lines,16 raising hopes for therapeutic inhibition of Dot1l as a novel strategy for patients with MLL-rearranged leukemias.
Leukemias with t(6;11)(q27;q23), which encodes the MLL-AF6 fusion protein, constitute the largest subgroup of MLL-rearranged leukemias in which MLL is fused with a predominantly cytoplasmic protein.5 Retrospective studies have shown that presence of the t(6;11)(q27;q23) translocation predicts a particularly poor prognosis,17,18 and thus new therapeutic approaches are clearly needed. Given the recent studies demonstrating an important role for Dot1l in leukemias driven by various MLL-fusion proteins and the development of small-molecule Dot1l inhibitors, we wondered if the MLL-AF6 fusion protein, where the MLL fusion partner is normally cytoplasmic and thus unlikely to be associated with transcriptional complexes, might also require Dot1l to maintain the oncogenic gene expression program. MLL-AF6 leukemias have been modeled in mice, where it was recently shown that the MLL-AF6 fusion can transform hematopoietic progenitors in vitro and in vivo, a process dependent on the dimerizing activity of the Ras-interacting domain of AF6.19 We sought to analyze a potential role for H3K79 methylation by conducting a genome-wide analysis of H3K79 dimethylation in MLL-AF6 leukemia cells. We also assessed whether MLL-AF6 fusion-mediated transformation was dependent on aberrant H3K79 methylation by genetic or pharmacologic inhibition of the Dot1l histone methyltransferase.
Materials and methods
Generation of transformed murine cells and leukemia
The MSCV-neo MLL-AF6 plasmid consisting of amino acids 35 to 347 comprising the AF6 N-terminal conserved region cloned in the MSCV-neo 5′ MLL construct has been described before19 and was a kind gift from Ruud Delwel (Erasumus, Rotterdam). The Mi-Tomato plasmid and the CRE-Mi-Tomato plasmids have been described before.15 Sorted Lin-Sca-1+cKit+ (LSK) cells from mouse bone marrow cells were transduced with the MLL-AF6 retrovirus and expanded for 2 weeks in methylcellulose M3234 (Stem Cell Technologies) supplemented with cytokines (6 ng/mL interleukin [IL]-3, 10 ng/mL IL6, and 20 ng/mL stem cell factor) and 1 mg/mL of G418. After 2 weeks of selection, MLL-AF6–transformed cells were either injected into syngenic recipients to generate leukemias or transduced with either Cre-Mi-Tomato or the empty Mi-Tomato control vector. At 48 hours after transduction with Mi-Tomato or Cre-Mi-Tomato, tdTomato-positive cells were sorted and used for colony-forming assays. For leukemia maintenance experiments, bone marrow cells harvested from primary leukemic mice were transduced with Mi-Tomato or Cre-Mi-Tomato and 72 hours later, 200 000 sorted tdTomato-positive cells were injected into sublethally (650 Rad) irradiated BL6 × 129 recipient mice. All mice used in this study were housed in the Animal Research Facility at Children’s Hospital Boston. Animal experiments and protocols were approved by the Internal Animal Care and Use Committee.
Mutant mice
Dot1l conditional knockout mice in which the active site of Dot1l (exon5) is flanked by LoxP sites have been previously described 12. Bone marrow cells from 7- to 10-week-old mice in Dot1l +/+ or Dot1lf/f backgrounds were used for transformation assays and subsequent biochemical experiments.
Colony-forming assays
Colony-forming cell assays were performed by plating 1000 tdTomato-positive cells/mL of cytokine-supplemented methylcellulose M3234 (6 ng/mL IL3, 10 ng/mL IL6, and 20 ng/mL stem cell factor). On day 6 to 7 after plating, colonies were scored using the Nikon Eclipse TS100 inverted microscope (Nikon, Tokyo, Japan). Because almost all colonies were either compact or hypercellular (blast-like) or small and diffuse (consistent with differentiation), colonies were classified into these 2 categories.
Microarray and real-time polymerase chain reaction
Total RNA was isolated using Trizol (Invitrogen, Carlsbad, CA). RNA from 3 independently derived murine MLL-AF6 leukemic bone marrow cells and from the human MLL-AF6–positive cell line ML2 was used for gene expression profiling using Affymetrix 430 2.A and U133 HG-U133_Plus_2 microarrays, respectively (accession no. GSE43069). Data were analyzed using Genepattern.20 Complementary DNA was generated using the tetro cDNA synthesis kit (Bioline, Taunton, MA) using oligo-dT primers. Real-time polymerase chain reaction (PCR) was performed using prevalidated Taqman probes (Applied Biosystems) on the ABI 7700 Sequence Detection System (Applied Biosystems). Primer and probe information will be provided upon request. Average Ct values were normalized to the housekeeping gene GAPDH.
Histone extraction and chromatin IP-sequencing
Histones were extracted by overnight acid extraction using the following protocol outlined on the Abcam website (www.abcam.com). Western blotting was done with standard procedures using a 10% Bis-Tris Gel (Invitrogen) and transferred onto polyvinylidene difluoride membranes. Chromatin IP-sequencing (ChIP-seq) was performed as previously described.9,12
More details regarding reagents and methods for cell proliferation assays, cytospins, western blotting, ChIP-seq and analysis, cell cycle and apoptosis, etc. can be found in the supplemental Methods section.
Results
Elevated H3K79 dimethylation on MLL-fusion target genes in murine MLL-AF6 leukemias
The presence of abnormally high levels of H3K79 dimethylation (H3K79me2) at MLL-fusion target genes has been shown to be a characteristic of cells bearing MLL fusions with predominantly nuclear proteins such as AF9 and AF4. In 2011, Yokoyama et al7 demonstrated the co-occurrence of the AEP complex, H3K79me2, as well as the MLL-AF6 fusion protein at the chromatin of select MLL-target genes in the MLL-AF6–positive cell line ML2. We decided to probe genome-wide H3K79me2 in MLL-AF6–transformed leukemia cells in order to assess whether H3K79 methylation might be involved in driving MLL-target gene expression more broadly in MLL-AF6 leukemias. We established MLL-AF6–driven leukemias using a bone marrow transplantation model in which lineage LSK cells were transformed with the MLL-AF6 fusion gene. MLL-AF6 expression was confirmed by western blot following overexpression in 293-T cells (supplemental Figure 1). All mice that developed leukemia were found to have acute myelogenous leukemia (AML), with >90% of cells expressing the Gr-1 and Mac-1 myeloid markers in the bone marrow and spleen (supplemental Figure 2). We performed gene expression profiling of 3 independently derived MLL-AF6 leukemias and conducted a genome-wide analysis of H3K79me2 by ChIP-seq using H3K79me2-specific antibodies on the same leukemic bone marrow cells. We observed high levels of H3K79me2 at well-characterized MLL-target genes in all the MLL-AF6 leukemias studied (Figure 1A). Expectedly, genes showing high expression levels in the MLL-AF6 leukemias as assessed by microarray also exhibited high levels of H3K79me2 (red line) in contrast to nonexpressed genes that had little H3K79 dimethylation (blue line). To analyze whether MLL-target loci possessed higher relative levels of H3K79me2 than other highly expressed genes, we compared the average distribution of H3K79me2 on a set of previously defined MLL-core target genes12 with 3 randomly chosen sets of size- and expression-matched genes as control (gray lines). We observed a consistently higher deposition of H3K79me2 associated with MLL-fusion core target genes (cyan line) compared with controls (gray lines) (Figure 1B).
Figure 1.
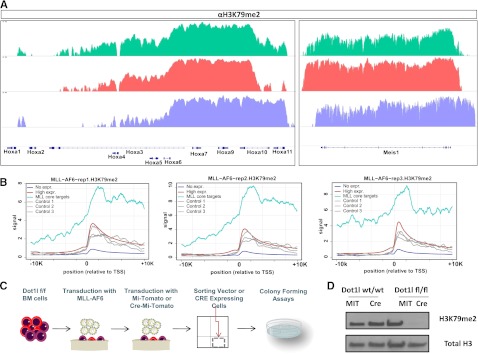
H3K79 methylation in MLL-AF6–transformed cells. (A) H3K79me2 profiles of select MLL-AF9 target genes: Hoxa cluster genes (left) Hox co-factor Meis1 (right). (B) Level and distribution of H3K79me2 profiles around the transcription start site (TSS) of MLL core targets (cyan lines) compared with 3 sets of size-matched, randomly chosen, highly expressed genes based on microarray data from MLL-AF6 leukemic bone marrow cells (gray lines). H3K79 methylation profiles of the highly expressed and nonexpressed control genes are depicted with red and blue lines respectively. Each panel represents data from an independently derived, MLL-AF6–driven murine leukemia. (C) Schematic depiction of experimental design. (D) Western blot showing a marked decrease in H3K79me2 methylation 6 days after transduction with the Cre recombinase compared with control vector (Mi-Tomato)-transduced cells.
To assess whether H3K79 methylation is required for MLL-AF6–mediated transformation, we transduced LSK cells from mice harboring homozygous floxed Dot1l alleles (Dot1lfl/fl) or Dot1l wild-type controls (Dot1l+/+) with the retrovirus encoding MLL-AF6. Subsequently, the MLL-AF6–transformed cells were selected with neomycin and transduced with an MSCV-based retrovirus that expresses Cre recombinase and tdTomato (Cre-Mi-Tomato). tdTomato-positive cells were sorted and plated in methylcellulose-based media supplemented with cytokines and replated every week for up to 3 weeks (schematic in Figure 1C). Controls were MLL-AF6–transformed Dot1l+/+ LSKs transduced with Cre-Mi-Tomato or MLL-AF6–transformed Dot1lfl/fl LSK cells transduced with the MSCV-based retrovirus without the Cre-recombinase (Mi-Tomato). Immunoblot analysis demonstrated that H3K79me2 levels were significantly reduced in MLL-AF6–transformed Dot1lfl/fl bone marrow cells after Cre expression compared with controls (Figure 1D).
Dot1l deletion inhibits MLL-AF6–mediated transformation
To characterize the effects of Dot1l loss on MLL-AF6 transformation, we first performed colony-forming assays. We found that Dot1l excision significantly diminished the clonogenic capacity of MLL-AF6–transformed cells in the first week (Figure 2A). In contrast to the dense and compact “blast-like” colonies observed in control vector-transduced cells, colonies generated upon Dot1l deletion were mostly small and diffuse, and Wright Giemsa-stained cytospins of these colonies showed features characteristic of monocytic differentiation (Figure 2B). Genotyping PCR of the Cre-transduced colonies at week 1, 2, and 3 of replating showed a progressive emergence of the unexcised Dot1l allele, demonstrating eventual outgrowth of cells that had escaped deletion of Dot1l (supplemental Figure 3). These results indicate a strong selective pressure against Dot1l-deleted cells. As we and others have previously published, we noted the lack of such strong selection in HoxA9-Meis1–transformed LSK cells, and individual HoxA9-Meis1–transformed Dot1l-excised colonies could be picked and propagated for several weeks (data not shown). These results demonstrate a selective requirement for Dot1l in MLL-AF6–transformed cells, but not all transformed cells. Because aberrant H3K79 methylation at target chromatin is believed to result in inappropriate expression of MLL-fusion target genes, we sought to assess the effects of Dot1l deletion on H3K79me2 at select MLL-fusion target genes. Consistent with the ChIP-seq data from MLL-AF6 leukemias, we found high enrichment of H3K79me2 on the promoter proximal regions of the Hoxa cluster genes compared with input chromatin (Figure 2C dark bars). Cre-mediated excision of Dot1l from these MLL-AF6–transformed cells decreased H3K79me2 levels at the promoters of MLL-target genes and β-actin (Figure 2C). Reduction in the levels of H3K79me2 resulted in a specific reduction in the transcript levels of MLL-target genes Hoxa9, Hoxa10, and Meis1, but not the housekeeping gene β-actin (Figure 2D). This result demonstrates that similar to other MLL-fusion–driven leukemias, continued expression of MLL-fusion target genes in MLL-AF6 leukemias requires activity of the Dot1l methyltransferase.
Figure 2.
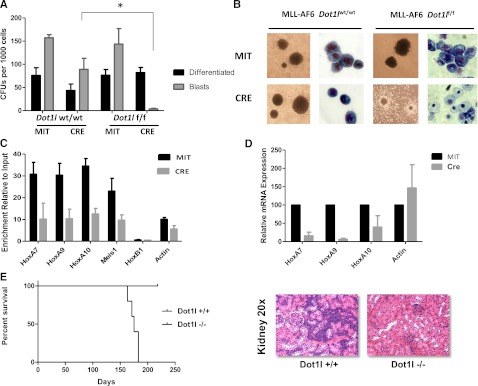
Dot1l deletion impairs the transforming capacity of MLL-AF6–transformed bone marrow cells. (A) Differential colony counts from Dot1l-excised, MLL-AF6–transformed cells 7 days after Cre transduction compared with vector (Mi-Tomato)-expressing controls (n = 3 independent experiments). Light gray bars represent dense and compact blast-like colonies, whereas dark gray bars represent small and diffuse colonies showing marked features of differentiation. Colony-forming units (CFU)/1000 cells averaged from 3 independent experiments are plotted with standard error of the mean (SEM) values. *P value = 0.002. (B) Morphological changes in colony and cell types upon Dot1l deletion in bone marrow cells immortalized by MLL-AF6 (colonies, ×10; cell morphology, ×40). (C) Enrichment of H3K79me2 normalized to input DNA on the chromatin locus of Hoxa/Meis1 gene promoters as assessed by quantitative reverse-transcription (qRT)-PCR is shown 6-9 days after Cre transduction compared with Mi-Tomato–transduced cells (n = 2 independent experiments). (D). RT and qRT-PCR 5 days after transduction with Cre-Mi-Tomato or Mi-Tomato. Expression levels normalized to Gapdh and expressed relative to Mi-Tomato–transduced cells (set to 100%) are shown. Error bars represent the SEM (n = 2 independent experiments). (E) Survival curves for mice injected with 2 × 105 wild-type or Dot1l-excised MLL-AF6 primary leukemia cells (left). Hematoxylin and eosin staining showing blast infiltration in the kidney of a representative leukemic mouse injected with Dot1l +/+ MLL-AF6 leukemias is presented (right). Staining from a disease-free mouse injected with Dot1l −/− cells and sacrificed at an identical time point after injection is shown for comparison.
We then investigated the requirement of Dot1l for the maintenance of established murine MLL-AF6 leukemias. We observed that mice injected with leukemia cells harboring wild-type Dot1l alleles succumbed to acute myeloid leukemia in an average of 175 d after injection. The leukemic mice showed highly elevated white blood cell counts and anemia accompanied by multi-organ blast infiltration and splenomegaly (Figure 2E; supplemental Figure 4; data not shown). In stark contrast, mice injected with Dot1l-deleted leukemic blasts failed to show any signs of leukemic engraftment and retained completely normal white blood cell and red blood cell counts for up to 210 days of observation (Figure 2E; supplemental Figure 4).
The Dotl1 inhibitor EPZ004777 selectively impairs proliferation of murine MLL-AF6–transformed cells
Because genetic inactivation of Dot1l severely impaired MLL-AF6 transformation, we sought to assess whether a specific small-molecule inhibitor of Dot1l would similarly affect MLL-AF6–transformed cells. The specific, small-molecule Dot1l inhibitor EPZ004777 has been recently described to selectively impair the proliferation of several MLL-rearranged human cell lines, although its impact on MLL-AF6–transformed cells has not yet been tested. We incubated MLL-AF6–transformed LSK cells with increasing concentrations (0.1-10 µM) of EPZ004777 or dimethylsulfoxide vehicle control and assessed cell number over several days. MLL-AF9–transformed LSKs and HoxA9/Meis1–transformed LSKs served as positive and negative controls, respectively. A dose-dependent reduction of H3K79me2 was seen in all the transformed cell populations by western blotting after incubation with EPZ004777 (Figure 3A). We used 10 µM of the EPZ004777 inhibitor for the proliferation assays, because this is a dose that consistently affects the proliferation of MLL-AF9–transformed cells (used as a positive control) but is still much lower than the >50-µM dose that is needed for off-target effects. As shown in Figure 3B, the expansion of MLL-AF6 as well as MLL-AF9 cells was dramatically impaired by EPZ004777, whereas the growth of HoxA9/Meis1–transformed cells was not significantly altered. Consistent with previous findings with human MLL-AF9 and MLL-AF4 leukemia cell lines, the effects on MLL-AF6–transformed primary murine hematopoietic progenitor cells became apparent only after 7 days. Nevertheless, when exposed to EPZ004777 for a longer time, MLL-AF6–transformed cells showed a dramatic progressive decrease in cell number. In contrast, expansion of the Hoxa9/Meis1–transformed cells was not significantly affected in the presence of the EPZ004777 inhibitor, excluding a drop in viable cell numbers at day 10, which was seen in some but not all experiments (Figure 3B). We then used quantitative reverse-transcription-PCR to quantify expression levels of Hoxa9, Hoxa10, and Meis1 in cells exposed to EPZ004777. A decrease in Hoxa9, Hoxa10, and Meis1 transcript levels was observed in 3 days, with a significant drop at day 7 and 10 after drug exposure (Figure 3C).
Figure 3.
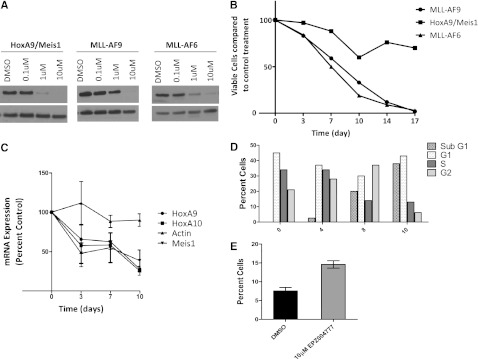
Selective inhibition of MLL-AF6–transformed cells by EPZ004777. (A) Inhibition of cellular H3K79me2 levels in MLL-AF6, MLL-AF9, or Hoxa9/Meis1a transformed hematopoietic progenitor cells following 7 days of treatment with the indicated concentrations of EPZ004777 as measured by immunoblot analysis of extracted histones with an anti-H3K79me2 antibody. (B) Representative results from the growth kinetics of MLL-AF6, MLL-AF9, and HoxA9/Meis1a transformed murine bone marrow cells exposed to 10 µM of EPZ004777. Viable cells were counted and replated at equal cell numbers in fresh media with fresh compound every 3-4 days. Results were plotted as percentage of split-adjusted viable cells in the presence of 10 µM EPZ004777 compared with dimethylsulfoxide (DMSO) vehicle control. Results are representative of 3 independent experiments. (C) Time course of HoxA9 and Meis1 mRNA expression in MLL-AF6–transformed cells over 10 days of incubation with 10 µM EPZ004777 as measured by quantitative real-time PCR. Expression levels were normalized to Gapdh and expressed relative to those at d 0 (set to 100%). Error bars represent the standard error of the mean (n = 3 independent experiments). (D) Representative graph of cell cycle changes (BrdU/7-AAD flow cytometry) in MLL-AF6–transformed bone marrow cells after being treated with 10 µM EPZ004777 for 0, 4, 8, or 10 days. Similar results were obtained in 2 independent experiments. (E) Annexin V staining in MLL-AF6–transformed bone marrow cells 10 days after treatment with 10 µM EPZ004777 or DMSO control (n = 2 independent experiments). Error bars represent standard error of the mean.
To assess the effect of EPZ004777 on the MLL-AF6–transformed cells in more detail, we analyzed changes in cell cycle and apoptosis upon exposure of the MLL-AF6–transformed cells to EPZ004777 by flow cytometry for DNA content and Annexin V staining. Dot1l inhibition reduced the number of actively proliferating MLL-AF6 cells, with a progressive increase in the percentage of cells in the sub-G1 fraction and a concomitant decrease in the S-phase fraction after 10 days of incubation with 10 µM EPZ004777 (Figure 3D). These changes in the cell cycle were accompanied by an increase in the percentage of Annexin V-positive cells, consistent with apoptotic cell death after EPZ004777 treatment (compared with dimethylsulfoxide control) (Figure 3E). These findings demonstrate that MLL-AF6–driven leukemias are selectively sensitive to pharmacologic inhibition of Dot1l.
Detection of the elevated H3K79me2 at MLL-fusion target genes in a human MLL-AF6–positive cell line
To determine whether human leukemia cells bearing the MLL-AF6 fusion gene also display high H3K79me2 on MLL-fusion target genes, we performed microarray analysis coupled with a genome-wide ChIP-seq for H3K79me2 on the human MLL-AF6–positive cell line ML2. ChIP-seq for H3K79me2 in the MLL-germline cell line HL60 and the MLL-AF9–positive cell line MOLM-13 served as negative and positive controls, respectively. ML2 cells showed very high levels of H3K79 dimethylation on the HOXA gene cluster, similar to the MLL-AF9–positive MOLM-13 cell line, whereas HL60 cells displayed low to undetectable levels of enrichment of this modification at the same locus (Figure 4A). H3K79me2 levels at other MLL-target genes such as MEF2C were similarly high in ML2 and MOLM-13 compared with HL60 (Figure 4A). Of note, unlike MOLM-13 cells, the ML2 cell line did not display high H3K79 methylation on the MEIS1 locus, consistent with a previous report.7 On a more global scale, MLL-fusion core targets showed significantly high levels of mean H3K79me2 signal in the ML2 cell line (Figure 4B). A recent study identified a subset of direct binding targets of the MLL-AF6 fusion protein using ChIP-Chip in the human t(6;11)(q27;q23)-positive ML2 cell line.21 We observed that mean H3K79me2 levels at these 24 MLL-AF6 target genes were markedly higher than even the MLL-fusion core target genes (Figure 4B). These findings demonstrate that similar to murine leukemias, the human MLL-AF6–positive cell line ML2 harbors elevated H3K79me2 on MLL-fusion target genes. We then sought to assess whether the expression of MLL-target genes in human MLL-AF6 patients also correlates with high H3K79 dimethylation. For this, we performed an integrative analysis of gene expression from published MLL-AF6–positive human AML patient samples18 with our ChIP-seq data from the ML2 cell line. Expectedly, when analyzed on a global scale, the levels of H3K79 dimethylation in ML2 positively correlated with the level of transcript expression in these patients (supplemental Figure 5, left panel). Also, mean levels of H3K79 dimethylation on both MLL-core targets as well as MLL-AF6 targets were significantly higher compared with 3 randomly selected size- and expression-matched sets of highly expressed genes (supplemental Figure 5, right panel), suggesting a correlation of H3K79me2 and MLL-target gene expression also in human MLL-AF6–positive leukemia.
Figure 4.
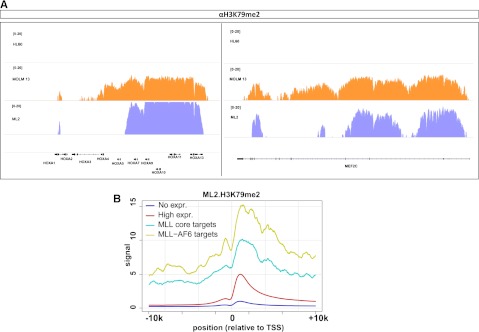
Abnormal H3K79me2 on MLL targets in the ML2 cell line. (A) H3K79me2 profiles of select MLL-target genes in MLL nonrearranged HL60 cells, MLL-AF9–positive MOLM-13 cells, and the MLL-AF6–positive ML2 cell line. (B) Level and distribution of H3K79me2 profiles around the transcription start site (TSS) of MLL core targets (cyan line) and MLL-AF6 direct targets (gold line) in the ML2 cell line. Genes with high levels of expression are depicted with the red line and genes with no expression are marked with the blue line.
The Dotl1 inhibitor EPZ004777 impairs proliferation of the t(6;11)-positive cell line ML2
To determine the effects of small-molecule inhibition of DOT1L in human leukemia cells, we assessed whether the DOT1L inhibitor EPZ0004777 affected the proliferation, differentiation, or survival of the ML2 cell line with HL60 and MOLM13 as negative and positive controls, respectively. The ML2 cell line showed sensitivity to EPZ004777, with a significant reduction in cell numbers starting from d 10 after drug exposure (Figure 5A). Despite the fact that H3K79me2 was significantly diminished upon exposure to EPZ004777 in all 3 tested cell lines (Figure 5B), only MOLM-13 and ML2 showed significant sensitivity to EPZ004777, further supporting the selectivity of this small molecule for MLL-rearranged cell lines. Exposure of ML2 cells to 10 µM EPZ004777 resulted in a progressive reduction in cells entering the S-phase starting from 3 days after drug exposure with an accumulation in the subG1 phase (Figure 5C left). EPZ004777 treatment also led to a modest but statistically insignificant increase in the percentage of Annexin V-positive cells, indicating that in contrast to cell cycle changes, the effects of EPZ00477 on apoptosis of ML2 cells was not as profound (Figure 5C right). Moreover, the expression of MLL-fusion target genes such as HOXA9 and HOXA10 was significantly down-regulated starting from d 7 after exposure of the ML2 cells to EPZ004777 (Figure 5D). Taken together, the results from the murine and human MLL-AF6–positive leukemia cells indicate that MLL-AF6–driven leukemias may show sensitivity to pharmacologic DOT1L inhibition.
Figure 5.
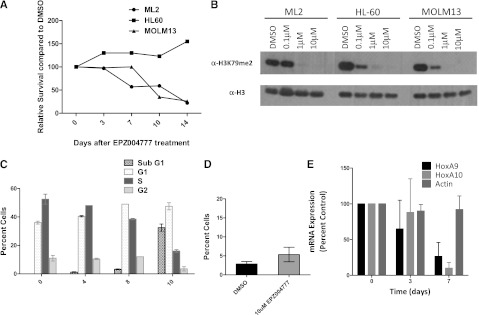
Selective inhibition of the ML2 cell line by EPZ004777. (A) Effect of EPZ004777 on the proliferation of leukemia cell lines bearing an MLL-AF6 (ML2) or MLL-AF9 (MOLM-13) fusion or a cell line lacking an MLL rearrangement (HL60). Cell lines were maintained in the presence of increasing concentrations of EPZ004777: 0.1, 1, and 10 μM. (B) Immunoblots showing the levels of H3K79me2 upon exposure of the different cell lines to indicated concentrations of EPZ004777 or dimethylsulfoxide (DMSO) carrier controls. (C) Cell cycle changes (Hoechst staining) in ML2 cell line upon treatment with 10 μM EPZ004777 for 0, 4, 8, or 10 days. Similar results were obtained in 2 independent experiments, each having 2 technical replicates. (D) Percentage of Annexin V-positive cells plotted 10 d after treatment with 10 μM of EPZ004777 or DMSO control. (E) Time course of HOXA9 and HOXA10 mRNA expression in the ML2 cell line for 7 days of incubation with 10 µM EPZ004777 as measured by quantitative real-time PCR. Expression levels were normalized to GAPDH and expressed relative to those at d 0 (set to 100%). Error bars represent the standard error of the mean (n = 3 independent experiments).
Discussion
MLL-AF6 fusions comprise a significant proportion of MLL-rearranged AMLs.5 Still, barring notable exceptions, there have been few studies assessing mechanisms of oncogenic transformation in MLL-AF6–driven leukemia. In 2 recent studies on pediatric leukemia, MLL-AF6 fusions were shown to be predictive of a significantly poor prognosis compared with most other common MLL-fusions.17,18 The identification of molecular mechanisms of transformation in the MLL-AF6 leukemias, therefore, is of significant clinical relevance. In this study, we demonstrate that both mouse and human MLL-AF6 leukemia cells harbor elevated H3K79 methylation on MLL-target genes, indicating aberrant DOT1L activity in these leukemias. We further show that loss of Dot1l function via conditional gene inactivation or inhibition of Dot1l using specific, small-molecule inhibitors reduces H3K79 on MLL-target genes and dramatically impairs the transforming ability of the MLL-AF6 fusion gene. The human MLL-AF6–positive cell line ML2 also reflects our findings in the murine models, suggesting that DOT1L plays a critical role in human MLL-AF6 leukemia.
Several MLL fusion proteins recruit transcriptional elongation complexes such as the AEP complex and the DOT1L complex to chromatin near MLL-fusion target genes, resulting in their constitutive and aberrant activation in MLL-rearranged leukemia. A number of MLL fusion partners such as AF9, ENL, and AF10 are normally components of these multi-protein complexes and thus when fused to MLL may directly recruit DOT1L, resulting in the pathological activation of downstream MLL-fusion target genes.12,22,23 This explanation suggests that MLL-fusion proteins consisting of a partner gene that is not normally part of a chromatin-associated transcriptional complex (such as MLL-AF6) might not be critically dependent on Dot1l and H3K79 methylation. The AF6 protein in its nonrearranged form is localized primarily at adherens junctions and binds to F-actin.24,25 Even though a shorter spliced form of AF6 has been reported to shuttle between the nucleus and the cytoplasm26 and therefore could potentially be involved in DOT1L and/or AEP recruitment, AF6 has not been co-purified in any of the described transcriptional elongation complexes that include DOT1L (reviewed in Deshpande27). A recent study showed that in the MLL-AF6–positive human cell line ML-2, the AEP complex co-localizes, along with MLL-AF6, on MLL-fusion target genes even though the MLL-AF6 fusion cannot directly recruit this complex.7 The ML2 cell line also showed H3K79 methylation on select MLL-fusion targets, but in the absence of genome-wide data it was not possible to determine if this level was above what would be expected for other highly expressed genes.7 Thus, although both the AEP and DOT1L complexes co-localize along with the MLL-AF6 fusion on MLL-target loci through a hitherto unknown mechanism, it has remained unclear if MLL-AF6 would depend on DOT1L and H3K79 for continued proliferation. Our data using genetic as well as pharmacological Dot1l inhibition strategies on mouse and human MLL-AF6 leukemia cells clearly demonstrate a critical role for DOT1L in MLL-AF6–mediated leukemia.
The requirement of DOT1L for transformation by cytoplasmic MLL-fusion partners such as MLL-AF6 suggests that direct biochemical interaction with an MLL-fusion partner may not be the only mechanism by which DOT1L contributes to MLL-leukemogenesis. Indeed, it cannot be ruled out that different mechanisms of DOT1L recruitment operate in nuclear vs cytoplasmic fusion partners of MLL. Alternatively, it is also possible that the dysregulation of DOT1L stability, enzymatic activity, or interactions between different transcriptional complexes may play an important role in MLL-fusion–mediated transformation, perhaps in addition to DOT1L recruitment. In this regard, it is conceivable that recruitment of the polymerase-associated factor complex by the N-terminal part of MLL is an alternative, indirect way of deregulating H3K79 methylation as previously suggested,28 thereby highlighting the role of the N-terminal MLL part of the fusion as opposed to the fusion partner in establishment of the aberrant H3K79 “lesion” at MLL-target genes. Even as the exact mechanisms of DOT1L recruitment by MLL fusion proteins as well as the role of transcriptional complexes in MLL-leukemogenesis warrant further investigation, there is mounting evidence indicating that DOT1L is an attractive target for therapy in the MLL leukemias. The FDA approval of small molecules targeting epigenetic enzymes such as the DNA methyltransferase inhibitors 5-azacytidine and decitabine and the HDAC inhibitors vorinostat and romidepsin have raised hopes for a new wave of targeted epigenetic therapies against human malignancies. Several efforts are being made to discover small-molecule inhibitors of histone methyltransferases such as DOT1L,16,29 and clinical trials with some of these inhibitors have begun. We and others have previously demonstrated that transformation mediated by other oncogenes such as Hoxa9 and Meis1, E2A-PBX, as well as E2A-HLF is insensitive to Dot1l inhibition.11-13 Also, a number of human cell lines that do not harbor MLL fusions are refractory to DOT1L inactivation using short hairpin RNAs or small-molecule inhibition.12,16 These results strongly suggest that aberrant H3K79 methylation is a molecular feature characteristic to certain, but not all, hematological malignancies. Against this backdrop, our observation that MLL-AF6 leukemias, which make up a substantial proportion of MLL-rearranged leukemia, are dependent on aberrant DOT1L activity is therefore of high clinical relevance.
In summary, the studies discussed above indicate that several MLL fusions, independent of the subcellular localization of the C-terminal partner, may directly or indirectly recruit common transcriptional effector complexes for constitutive activation of the MLL transcriptional program. This potentially shared mechanism of transformation could prove to be an attractive therapeutic target in the MLL leukemias. The findings of this study showing that MLL-AF6–mediated transformation is dependent upon Dot1l and can be inhibited using small-molecule inhibitors could help inform future clinical trials with clinically effective DOT1Ll inhibitors.
Supplementary Material
Acknowledgments
The MLL-AF6 plasmid used in this study was a kind gift from the laboratory of Ruud Delwel (Erasmus MC, Rotterdam).The authors thank Ronald Mathieu and Mahnaz Pakhtinat for their excellent technical assistance with flow cytometry.
This work was supported by grants (to S.A.A.) from the National Institutes of Health (National Cancer Institute grants CA66996 and CA140575; and National Institute of Diabetes and Digestive and Kidney Diseases grant DK049216), and grants from the Harvard Stem Cell Institute, and the Leukemia and Lymphoma Society. A.J.D. is supported by the NCI Howard Temin Pathway to Independence Award. M.F. was supported by a summer fellowship from the Giovanni Armenise-Harvard foundation.
Footnotes
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: A.J.D., L.C., K.M.B., and S.A.A. designed the study; A.J.D., L.C., M.F., D.B., J.C., and S.D. performed the mouse experiments; A.U.S. analyzed the genomics data and performed the statistical and bioinformatics analyses; A.J.D., L.C., and S.A.A. wrote the manuscript; E.J.O., S.R.D., V.M.R., and R.M.P. participated in analyzing and interpreting the DOT1L inhibitor data; and all authors reviewed and approved the final version of the manuscript.
Conflict-of-interest disclosure: E.J.O., S.R.D., V.M.R., and R.M.P. are employees of Epizyme Inc. and S.A.A. is a consultant for Epizyme Inc. The remaining authors declare no competing financial interests.
Correspondence: Scott Armstrong, 1275 York Ave, Box 20, New York, NY 10065; e-mail: armstros@mskcc.org.
References
- 1.Rowley JD. The critical role of chromosome translocations in human leukemias. Annu Rev Genet. 1998;32:495–519. doi: 10.1146/annurev.genet.32.1.495. [DOI] [PubMed] [Google Scholar]
- 2.Daser A, Rabbitts TH. The versatile mixed lineage leukaemia gene MLL and its many associations in leukaemogenesis. Semin Cancer Biol. 2005;15(3):175–188. doi: 10.1016/j.semcancer.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 3.Muntean AG, Hess JL. The pathogenesis of mixed-lineage leukemia. Annual Review of Pathology: Mechanisms of Disease. 2012;7(1):283–301. doi: 10.1146/annurev-pathol-011811-132434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Meyer C, Kowarz E, Hofmann J, et al. New insights to the MLL recombinome of acute leukemias. Leukemia. 2009;23(8):1490–1499. doi: 10.1038/leu.2009.33. [DOI] [PubMed] [Google Scholar]
- 5.Krivtsov AV, Armstrong SA. MLL translocations, histone modifications and leukaemia stem-cell development. Nat Rev Cancer. 2007;7(11):823–833. doi: 10.1038/nrc2253. [DOI] [PubMed] [Google Scholar]
- 6.Lin C, Smith ER, Takahashi H, et al. AFF4, a component of the ELL/P-TEFb elongation complex and a shared subunit of MLL chimeras, can link transcription elongation to leukemia. Mol Cell. 2010;37(3):429–437. doi: 10.1016/j.molcel.2010.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yokoyama A, Lin M, Naresh A, Kitabayashi I, Cleary ML. A higher-order complex containing AF4 and ENL family proteins with P-TEFb facilitates oncogenic and physiologic MLL-dependent transcription. Cancer Cell. 2010;17(2):198–212. doi: 10.1016/j.ccr.2009.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mohan M, Herz HM, Takahashi YH, et al. Linking H3K79 trimethylation to Wnt signaling through a novel Dot1-containing complex (DotCom). Genes Dev. 2010;24(6):574–589. doi: 10.1101/gad.1898410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Krivtsov AV, Feng Z, Lemieux ME, et al. H3K79 methylation profiles define murine and human MLL-AF4 leukemias. Cancer Cell. 2008;14(5):355–368. doi: 10.1016/j.ccr.2008.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Guenther MG, Lawton LN, Rozovskaia T, et al. Aberrant chromatin at genes encoding stem cell regulators in human mixed-lineage leukemia. Genes Dev. 2008;22(24):3403–3408. doi: 10.1101/gad.1741408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chang MJ, Wu H, Achille NJ, et al. Histone H3 lysine 79 methyltransferase Dot1 is required for immortalization by MLL oncogenes. Cancer Res. 2010;70(24):10234–10242. doi: 10.1158/0008-5472.CAN-10-3294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bernt KM, Zhu N, Sinha AU, et al. MLL-rearranged leukemia is dependent on aberrant H3K79 methylation by DOT1L. Cancer Cell. 2011;20(1):66–78. doi: 10.1016/j.ccr.2011.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jo SY, Granowicz EM, Maillard I, et al. Requirement for Dot1l in murine postnatal hematopoiesis and leukemogenesis by MLL translocation. Blood. 2011;117(18):4759–4768. doi: 10.1182/blood-2010-12-327668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nguyen AT, Taranova O, He J, et al. DOT1L, the H3K79 methyltransferase, is required for MLL-AF9-mediated leukemogenesis. Blood. 2011;117(25):6912–6922. doi: 10.1182/blood-2011-02-334359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen L, Deshpande AJ, Banka D, et al. Abrogation of MLL-AF10 and CALM-AF10-mediated transformation through genetic inactivation or pharmacological inhibition of the H3K79 methyltransferase Dot1 [published online ahead of print Nov 9, 2012]. Leukemia. 2012 doi: 10.1038/leu.2012.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Daigle SR, Olhava EJ, Therkelsen CA, et al. Selective killing of mixed lineage leukemia cells by a potent small-molecule DOT1L inhibitor. Cancer Cell. 2011;20(1):53–65. doi: 10.1016/j.ccr.2011.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Balgobind BV, Raimondi SC, Harbott J, et al. Novel prognostic subgroups in childhood 11q23/MLL-rearranged acute myeloid leukemia: results of an international retrospective study. Blood. 2009;114(12):2489–2496. doi: 10.1182/blood-2009-04-215152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pigazzi M, Masetti R, Bresolin S, et al. MLL partner genes drive distinct gene expression profiles and genomic alterations in pediatric acute myeloid leukemia: an AIEOP study. Leukemia. 2011;25(3):560–563. doi: 10.1038/leu.2010.316. [DOI] [PubMed] [Google Scholar]
- 19.Liedtke M, Ayton PM, Somervaille TC, et al. Self-association mediated by the Ras association 1 domain of AF6 activates the oncogenic potential of MLL-AF6. Blood. 2010;116(1):63–70. doi: 10.1182/blood-2009-09-243386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Reich M, Liefeld T, Gould J, et al. GenePattern 2.0. Nat Genet. 2006;38(5):500–501. doi: 10.1038/ng0506-500. [DOI] [PubMed] [Google Scholar]
- 21.Wang QF, Wu G, Mi S, et al. MLL fusion proteins preferentially regulate a subset of wild-type MLL target genes in the leukemic genome. Blood. 2011;117(25):6895–6905. doi: 10.1182/blood-2010-12-324699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mueller D, Bach C, Zeisig D, et al. A role for the MLL fusion partner ENL in transcriptional elongation and chromatin modification. Blood. 2007;110(13):4445–4454. doi: 10.1182/blood-2007-05-090514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Okada Y, Feng Q, Lin Y, et al. hDOT1L links histone methylation to leukemogenesis. Cell. 2005;121(2):167–178. doi: 10.1016/j.cell.2005.02.020. [DOI] [PubMed] [Google Scholar]
- 24.Yamamoto T, Harada N, Kano K, et al. The Ras target AF-6 interacts with ZO-1 and serves as a peripheral component of tight junctions in epithelial cells. J Cell Biol. 1997;139(3):785–795. doi: 10.1083/jcb.139.3.785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mandai K, Nakanishi H, Satoh A, et al. Afadin: a novel actin filament-binding protein with one PDZ domain localized at cadherin-based cell-to-cell adherens junction. J Cell Biol. 1997;139(2):517–528. doi: 10.1083/jcb.139.2.517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Buchert M, Poon C, King JA, et al. AF6/s-afadin is a dual residency protein and localizes to a novel subnuclear compartment. J Cell Physiol. 2007;210(1):212–223. doi: 10.1002/jcp.20853. [DOI] [PubMed] [Google Scholar]
- 27.Deshpande AJ, Bradner J, Armstrong SA. Chromatin modifications as therapeutic targets in MLL-rearranged leukemia. Trends Immunol. 2012;33(11):563–570. doi: 10.1016/j.it.2012.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tan J, Muntean AG, Hess JL. PAFc, a key player in MLL-rearranged leukemogenesis. Oncotarget. 2010;1(6):461–465. doi: 10.18632/oncotarget.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yao Y, Chen P, Diao J, et al. Selective inhibitors of histone methyltransferase DOT1L: design, synthesis, and crystallographic studies. J Am Chem Soc. 2011;133(42):16746–16749. doi: 10.1021/ja206312b. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.