

Abstract
Elevated intraocular pressure (IOP) is a causative risk factor for the development and progression of glaucoma. Glaucomatous mutations in myocilin (MYOC) damage the trabecular meshwork and elevate IOP in humans and in mice. Animal models of glaucoma are important to discover and better understand molecular pathogenic pathways and to test new glaucoma therapeutics. Although a number of different animal models of glaucoma have been developed and characterized, there are no true models of human primary open angle glaucoma (POAG). The overall goal of this work is to develop the first inducible mouse model of POAG using a human POAG relevant transgene (i.e. mutant MYOC) expression in mouse eyes to elevate IOP and cause pressure induced damage to the optic nerve. Four mouse strains (A/J, BALB/cJ, C57BL/6J, and C3H/HeJ) were used in this study. Ad5.MYOC.Y437H (5 × 107 pfu) was injected intravitreally into one eye, with the uninjected contralateral eye serving as the control eye. Conscious IOP measurements were taken using a TonoLab rebound tonometer. Optic nerve damage was determined by scoring PPD stained optic nerve cross sections. Retinal ganglion cell and superior colliculus damage was assessed by Nissl stain cell counts. Intravitreal administration of viral vector Ad5.MYOC.Y437H caused a prolonged, reproducible, and statistically significant IOP elevation in BALB/cJ, A/J, and C57BL/6J mice. IOPs increased to approximately 25 mm Hg for 8 weeks (p<0.0001). In contrast, the C3H/HeJ mouse strain was resistant to Ad5.MYOC.Y437H induced IOP elevation for the 8-week time period. IOPs were stable (12–15 mm Hg) in the uninjected control eyes. We also determined whether there were any strain differences in pressure-induced optic nerve damage. Even though IOP was similarly elevated in three of the strains tested (BALB/cJ, C57BL/6J, and A/J ) only the A/J strain had considerable and significant optic nerve damage at the end of 8 weeks with optic nerve damage score of 2.64 +/− 0.19 (n=18, p<0.001) in the injected eye. There was no statistical difference in retinal ganglion cell death or superior colliculus damage at the 8-week time point in any of the strains tested. These results demonstrate strain dependent responses to Ad5.MYOC.Y437H-induced ocular hypertension and pressure-induced optic nerve damage.
Keywords: glaucoma, mouse model, optic nerve, intraocular pressure
1. Introduction
Elevated IOP is the most important causative risk factor for the development and progression of primary open angle glaucoma (POAG). The elevated IOP is due to increased aqueous humor outflow resistance and is associated with morphological and biochemical changes in the trabecular meshwork (TM) (Clark and Wordinger, 2009; Wordinger and Clark, 1999). A number of different mouse models of glaucoma have been developed (Pang and Clark, 2007), but these models do not mimic many aspects of human POAG.
Several risk factors have been identified for the development of POAG. Much of this data has been assessed by longitudinal population-based studies such as the Baltimore Eye Study (Sommer, 1996), the Barbados Incidence Study of Eye Diseases (Leske et al., 2008), the Melbourne Visual Impairment Project (Le et al., 2003), the Rotterdam Eye Study (de Voogd et al., 2006), and the Ocular Hypertension Treatment Study (Gordon et al., 2002). Each of these studies identified a combination of physiological and hereditary risk factors for glaucoma, including IOP, age, race, and family history. The common risk factor identified in all studies was high IOP. It was found that approximately 1 mm Hg increase in IOP from baseline was a strong predictor for development of glaucoma with a 10–14% increased risk (de Voogd et al., 2006; Gordon et al., 2002; Le et al., 2003; Leske et al., 2008). There are several hypotheses on what types of insults may lead to IOP elevation, such as extensive malformations of drainage structures, focal malformations of drainage structures, aging, lack of gene function, and cellular abnormalities (Libby et al., 2005b). However, some of these deficits could be subtle and below clinical thresholds for detection. Therefore, animal models are crucial to begin to understand the pathogenic processes of the disease.
Although it is difficult to determine the exact influence of heredity in a slowly progressive, adult onset, multifactorial disease, the prevalence of glaucoma is higher in first degree relatives of glaucoma patients (Mackey, 2008), and numerous glaucoma loci have been mapped and several glaucoma genes identified (Allingham et al., 2009; Mackey, 2008). Using linkage studies, mutations in myocilin were found to be associated with juvenile open angle glaucoma and a small subset (3–5%) of POAG (Alward et al., 1998; Stone et al., 1997). To date, mutations in the myocilin gene are the most common genetic cause of glaucoma, but the function of mutant myocilin in the development of glaucoma is largely unknown.
The molecular pathogenesis of myocilin glaucoma is poorly understood, although several new pathogenic pathways have recently been implicated. It has been shown that myocilin mutations cause a gain of function phenotype (Jacobson et al., 2001; Kim et al., 2001; Shepard et al., 2007). In addition, glaucomatous mutant myocilin is not secreted from TM cells (Jacobson et al., 2001; Liu and Vollrath, 2004), and is thought to instead accumulate and induces endoplasmic reticulum stress (Joe et al., 2003). Mutant myocilin also appears to expose a cryptic carboxy terminal PTS1 site that abnormally localizes myocilin to peroxisomes (Shepard et al., 2007). We have previously published that over-expression of mutant human myocilin (particularly Y437H) in mouse eyes elevated IOP, and IOP elevation required both the glaucoma mutation and the PTS1 carboxy terminal signal (Shepard et al., 2007).
Here, we have developed a novel approach to generate mouse models of glaucoma using a viral vector harboring human mutant myocilin. In this study, we demonstrate that intravitreal injections of Ad5.MYOC.Y437H in A/J mice cause elevated IOP and glaucomatous optic neuropathy; thus providing a new mouse model of human POAG. We also report strain differences in the response to Ad5.MYOC.Y437H. These data provide an excellent resource for future studies on the development and progression of POAG.
2. Materials and methods
2.1. Animal
All experiments were conducted in compliance with the ARVO Statement of the Use of Animals in Ophthalmic and Vision Research and the University of North Texas Health Science Center (UNTHSC) Animal Care and Use Committee regulations. A/J, BALB/cJ, C57BL/6J, and C3H/HeJ mice were obtained from The Jackson Laboratory (Bar Harbor, ME) and subsequently aged at UNTHSC. All mice were 6–7 month old females unless otherwise noted.
2.2. Adenoviruses
We have previously reported the preparation and use of the Ad5 vectors: Ad5.RSV-MYOC.Y437H, Ad5.RSV-MYOC.WT, and Ad5.CMV-null (Shepard et al., 2007). The Ad5 vectors were prepared by the Gene Transfer Core Facility, University of Iowa, Iowa City, IA. Intraocular injections with Ad5.MYOC.WT (wild-type human myocilin, n=6 mice), Ad5.null (n=5 mice/strain), and uninjected eyes were used as negative controls. Mice were injected intravitreally with one of the Ad5 expression vectors (5 ×107 pfu) into one eye of each animal. Intravitreal injections in mice have been shown to produce a more pronounced and consistent response compared to intracameral injections. These data have been previously shown and speculated to be due to the vitreous serving as a depot for the vector resulting in a slow release and longer duration of transduction (Millar et al., 2008; Shepard et al., 2007; Shepard et al., 2010). Experiments using the Ad5.MYOC.Y437H vector were conducted using two independent cohorts of mice. In the first cohort, 10–15 mice/strain were injected. All animals were clinically evaluated throughout the 8 week time course using a direct ophthalmoscope. As previously shown in adenoviral injections, the adenovirus itself can cause increased overall corneal thickness, modest anterior chamber inflammation, and a propensity toward juxtaposition of the corneal endothelium to the iris (Shepard et al., 2010), yet the majority of eyes in our study showed no signs of decreased lenticular or corneal transparency or IOP changes due to these phenotypes as compared with controls. Two mice from the A/J strain, one mouse from the BALB/cJ strain, and two mice from the C57BL/6J strain were removed from the study due to injection-induced cataract formation or severe inflammation. The final sample size for the first cohort of mice was as follows: n=14, BALB/cJ; n=8, A/J; n=10, C57BL/6J; and n=10, C3H/HeJ. The sample size for the second cohort of mice was as follows: n=8, BALB/cJ; n=6, A/J; n=7, C57BL/6J; and n=10, C3H/HeJ.
2.3. Immunohistochemistry
Mice from four different mouse strains (A/J, BALB/cJ, C57BL/6J, and C3H/HeJ) were injected intravitreally with Ad5.MYOC.Y437H or Ad5.null (n=5 mice per strain), and at either 7 days or 8 weeks post-injection, whole eyes were removed and processed for immunohistochemistry to detect myocilin overexpression. Eyes were fixed in 4% PFA for 24 hours, processed and embedded in paraffin. 5-μm sections were cut and sections were transferred to glass slides. Paraffin sections were dewaxed 2 times in xylene, 100% ethanol, and 95% ethanol for 2 minutes each. Slides were then soaked in PBS for 5 minutes. Goat anti-myocilin antibody (N-15; Santa Cruz sc-21243) was used at a 1:100 dilution, followed by biotinylated secondary anti-goat antibody. Direct ABC immunohistochemistry (Vectastain ABC Kit, Vector Laboratories, Burlingame, CA) was performed with 3, 3′-diaminobenzidine tetrahydrochloride (DAB chromogen, DAKO, Carpinteria, CA) as the substrate and is visible as brown in images. Hematoxylin staining was performed on additional sections from each eye and is visible as purple stain in the images.
2.5. Intraocular pressure measurements
IOP was measured with a rebound tonometer following previously described methodology (Millar et al., 2011; Shepard et al., 2010; Wang et al., 2008; Wang et al., 2005). To summarize, mice were acclimated to the procedure room, and IOP was measured using a non-invasive method in conscious mice with the TonoLab tonometer (Colonial Medical Supply, Franconia, NH). All measurements were made during the same 3-hour period of the lights-on phase, 1–3 times per week for 8 weeks post-injection. All IOP’s are represented as means (+/− SD) at each time point with p values calculated by two-tailed paired Student’s t-test. Area under the curve (AUC) was calculated for each individual mouse and then averaged for each mouse strain. IOP exposure was calculated by subtracting the AUC of control eyes from the AUC of the Ad5.MYOC.Y437H injected eyes.
2.6. Assessment of optic nerve damage
Retro-orbital optic nerves were fixed, processed, and embedded in plastic as previously reported (Libby et al., 2005a; Libby et al., 2007; Libby et al., 2005c). Optic nerve cross-sections were stained with paraphenylenediamine (PPD) that stains myelin sheaths and more darkly stains the axoplasm of sick or dying axons. Optic nerve damage has previously been successfully evaluated using semi-quantitative optic nerve grading schemes (Chauhan et al., 2006; Fortune et al., 2004; Libby et al., 2005a; Pang and Clark, 2007). Here, we used the semi- quantitative five-point optic nerve grading scheme, optic nerve damage score (ONDS) (Pang and Clark, 2007). Two to three investigators performed masked evaluations to grade each optic nerve, and optic nerve damage scores for each sample were averaged. ONDS’s are represented as means (+/− SEM) from two independent studies conducted in an identical manner.
2.7. Assessment of retinal ganglion cell loss
To quantify cell loss from the retinal ganglion cell (RGC) layer, retinas from fixed eyes were carefully dissected, flat-mounted, and Nissl-stained with cresyl violet as previously described (Dietz et al., 2008; Li et al., 2007; Libby et al., 2005c). The retinas were dissected and “mounted” RGC layer side up on “Plus” glass slides (Fischer Scientific), dried, and flattened under a weighed coverslip. The retinas were then stained with 1% cresyl violet in 0.25% acetic acid for 30–45 seconds. The stained retinas were dehydrated in 95% ethanol, 100% ethanol, and cleared in xylene. The retinas were then coverslipped for microscopic evaluation. Digital images of 400X magnification were captured from peripheral and midperipheral regions from each of 4 quadrants (8 images per retina), processed and counted using Adobe Photoshop software. Four equal sized regions were counted from each image, resulting in a total of about 2% of all retina ganglion cells counted. Cell loss was calculated by comparing average cell counts in the Ad5 vector injected eyes with average cell counts in the uninjected contralateral control eyes, +/− SD with p values calculated by two-tailed paired Student’s t-test.
2.8. Assessment of superior colliculus damage
To map the superficial layers of the superior colliculus (SC), 2 μl of Cholera Toxin B-subunit (CTB) conjugated with AlexaFluor 488 was injected intravitreally into BALB/cJ mice (n=3). The CTB conjugate was taken up by RGCs and anterograde transported to the SC. The mice were sacrificed 48 hours after the intravitreal injection, and the brain coronal sections were imaged to identify the regions in the SC innervated by RGC axons. Templates were prepared to highlight this region. These templates mapped the region of the SC that was used to count the neurons in our experimental mice. For cresyl violet staining, the brain was fixed in NBF for 24 hours, paraffin embedded, sectioned coronally, and stained with 0.04% cresyl violet in 0.3% acetic acid for 1 hour. The stained sections were dehydrated in 95% ethanol, 100% ethanol, and cleared in xylene. Neurons in the template regions from 5 SC sections of each brain were counted.
3. Results
3.1. Ad5.MYOC.Y437H is expressed in TM after injection
Because myocilin is expressed in human TM cells and Adenovirus 5 has previously been reported to have tropism for TM cells (Millar et al., 2008), we chose Adenovirus 5 as our gene delivery method. To confirm that intravitreal injection of the virus transduced the TM and that mutant myocilin protein was expressed, we evaluated the transduction profile of Ad5.MYOC.Y437H. Ad5.MYOC.Y437H was injected intravitreally, and the eyes were harvested 7 days post-injection for immunohistochemistry. Myocilin was expressed in the TM of each of the four different mouse strains tested (A/J, BALB/cJ, C576BL/6J, and C3H/HeJ; n=5 mice per strain) (Figures 1B, 1F, 1J, 1N respectively; arrows indicate myocilin overexpression in the TM). We also saw some expression in the iris, ciliary body, and corneal endothelium. This confirmed that intravitreal injection of Ad5 viral vectors transduces tissues in the anterior segment of the eye, including the TM as previously reported (Millar et al., 2008; Shepard et al., 2007; Shepard et al., 2010). Small amounts of endogenous mouse myocilin expression are evident in the uninjected control eyes (Figures 1A, 1E, 1I, 1M). In addition, in each of the four strains tested the irideocorneal angle was open and free of any gross abnormalities in both the Ad5.MYOC.Y437H injected eye and the contralateral uninjected control eye (Figure 1, stars indicate open angles). Similarly, it was recently found that overexpression of mutant human myocilin in a transgenic mouse also yielded expression in the TM as well as open irideocorneal angles (Zode et al., 2011). These data show that Ad5 viral vectors have tropism for the TM, and that the human myocilin gene is expressed in the TM after transduction while maintaining open irideocorneal angles.
Figure 1. Myocilin is overexpressed in the trabecular meshwork after adenovirus transduction.

All mice were injected in one eye with 5×107 pfu Ad5.MYOC.Y437H. The contralateral uninjected eye served as a control. All samples were collected 7 days post-injection. The first two columns are labeled with anti-myocilin antibody and myocilin expression is represented by the brown labeling (A–B, E–F, I–J, M–N; n= 5 mice per strain). Myocilin expression in the TM is indicated by black arrows (B, F, J, N). The last two columns are sections from the same eye used in the first two columns and are stained with hematoxylin. The stars represent an open iridiocorneal angle (C–D, G–H, K–L, O–P; n=5 mice per strain).
3.2. Mutant myocilin adenovirus causes strain dependent ocular hypertension in mice
Genetic background can dramatically alter the response to specific transgenes; therefore, we tested our viral transgenes in several different mouse strains to identify the most susceptible strain, and best candidate for a model of POAG. We tested four genetically distinct mouse strains, A/J, BALB/cJ, C57BL/6J, and C3H/HeJ, to determine their susceptibility to mutant myocilin-induced ocular hypertension. We evaluated the IOP of each individual mouse 1–3 times a week post-injection of mutant MYOCY437H in two independent studies, represented as a combined data set. Each individual mouse was not necessarily measured at each time point (A/J n= 8–18 mice/time point, BALB/cJ n=10–22 mice/time point, C57BL/6J n=9–17 mice/time point, C3H/HeJ n=10–20 mice/time point). Intravitreal injection of Ad5.MYOC.Y437H significantly elevated IOP in A/J, BALB/cJ, and C57BL/6J mice for 8 weeks without affecting the IOP of the uninjected contralateral eye (Figure 2A, 2B, 2C). In the A/J, BALB/cJ, and C57BL/6J mouse strains the IOP was significantly increased by 5 days post-injection (A/J n=8, injected eye 18.4 +/− 3.0 mmHg, uninjected eye 11.4 +/− 1.9 mmHg, p=0.0002; BALB/cJ n=18, injected eye 20.2 +/− 5.8 mmHg, uninjected eye 12.7 +/− 2.9 mmHg, p<0.0001; C57BL/6J n=9, injected eye 17.1 +/− 3.9 mmHg, uninjected eye 12.8 +/− 2.4 mmHg, p=0.0393). IOP remained significantly elevated in these mouse strains throughout the 8-week time course in comparison to the contralateral uninjected eyes (A/J, BALB/cJ, and C57BL/6J, days 7–56, p< 0.001). IOP exposure throughout the 8-week time course was calculated by the AUC (Table 1). Both the A/J and BALB/cJ strains had a similar IOP exposure, while the C57BL/6J mice had a significantly smaller IOP exposure. Interestingly, despite expression of the transgene in the TM (Figure 1), the C3H/HeJ mouse strain was resistant to Ad5.MYOC.Y437H induction of ocular hypertension throughout the 8-week time course (Figure 2D). There was a small, but statistically significant, difference in IOP in the C3H/HeJ mice at day 5 (injected eye 15.8 +/− 2.9 mmHg, uninjected eye 12.1 +/− 2.3 mmHg, p=0.0018, n=20), day 7 (injected eye 15.0 +/− 3.5 mmHg, uninjected eye 12.1 +/− 1.5 mmHg, p=0.0042, n=20), and day 14 (injected eye 15.0 +/− 2.7 mmHg, uninjected eye 12.4 +/− 1.9 mmHg (p=0.0105, n=20) (Figure 2D), but not at any other time point. These data show that mutant human myocilin induces IOP phenotypes in mice and that there is strain differences in the response to the mutant transgene, suggesting genetic modifiers exist. The ocular hypertension responsive strains, A/J, BALB/cJ, and C57BL/6J, are potential models of human POAG. With only a minimal response to changes in IOP, the C3H/HeJ strain provides an excellent resource to genetically map and study the biology of IOP regulation in mice.
Figure 2. Ad5.MYOC.Y437H induces strain dependent ocular hypertension in mice.

All mice were injected in one eye with 5×107 pfu Ad5.MYOC.Y437H and IOP was measured for 8 weeks post injection. The contralateral uninjected eye served as a control. (A) A/J mice had significant IOP elevation beginning at 5 days post-injection (p=0.0002). The IOP elevation remained significant for at least 56 days (p<0.0001, Days 7–56). (B) BALB/cJ mice had significant IOP elevation beginning at 5 days post-injection (p<0.0001). The IOP elevation remained significant for at least 56 days (p<0.0001, Days 7–56). (C) C57BL/6J mice had significant IOP elevation beginning at 5 days post-injection (p=0.0393). The IOP elevation remained significant for at least 56 days (p<0.0001, Days 7–56). (D) C3H/HeJ mice had significant IOP elevation at Day 5 (p=0.0018), Day 7 (p=0.0042) and Day 14 (p=0.0104). At all other time points there was no significant elevation in IOP. Ad5.MYOC.Y437H injected eyes represented in grey, uninjected control eye represented in black. Data reported as mean +/− SD.
Table 1.
Ad5.MYOC.Y437H induced IOP exposure in mice.
| AUC (mmHg*day) injected eyes | AUC (mmHg*day) contro eyes | IOP exposure (mmHg*day) | |||||
|---|---|---|---|---|---|---|---|
| Average | Std. Dev | Average | Std. Dev | Injected-Control | Std. Dev. | p value (compared to A/J) | |
| A/J | 1452.50 | 232.69 | 671.65 | 61.51 | 780.84 | 240.69 | - |
| BALB/cJ | 1419.07 | 185.02 | 699.82 | 103.95 | 719.26 | 212.22 | 0.3954 |
| C57BL/6J | 1219.46 | 132.38 | 741.78 | 89.32 | 477.67 | 159.70 | <0.0001 |
| C3H/HeJ | 726.03 | 56.53 | 697.68 | 80.53 | 28.35 | 98.39 | 0.0001 |
AUC (area under the curve) was calculate for each individual mouse and then averaged for each mouse strain. IOP exposure was calculated by subtracting the AUC of control eyes from the AUC of the Ad5. MYOC. Y437H injected eyes.
The elevated IOP in the 3 responsive strains were correlated with increased myocilin expression in the TM assessed by immunohistochemical staining (Figure 1). Only minimal endogenous mouse myocilin expression was seen in the uninjected eyes. It is very difficult to quantify mutant myocilin expression levels in the TM of single mouse eyes by western immunoblotting. All of the mouse eyes within a given strain injected with Ad5.MYOC.Y437H developed comparable levels of elevated IOP. For these reasons, we could not obtain a graded correlation between differences in myocilin expression and IOP elevation.
3.3. Wild-type myocilin has no effect on intraocular pressure
As previously shown, injection of Ad5.MYOC.WT has no effect on mouse IOP (Shepard et al., 2007). Here we validate those results in BALB/cJ mice over an 8-week time course (Figure 3). There was no difference in IOP in the injected eye versus uninjected control eye. These data confirm that the IOP phenotype is due to a gain of function response in mutant myocilin and is not an artifact of gene delivery.
Figure 3. Ad5.MYOC.WT has no effect on IOP.
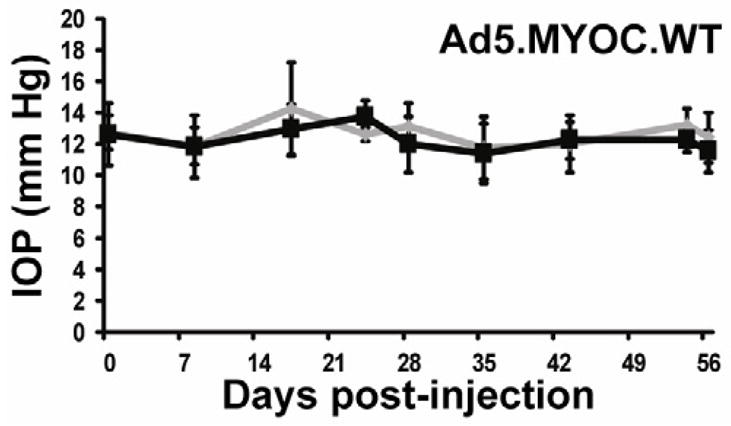
BALB/cJ mice were injected in one eye with 5×107 pfu Ad5.MYOC.WT and IOP was measured for 8 weeks post injection. The contralateral uninjected eye served as a control. There was no statistical difference in IOP between the injected and uninjected eye at any time point throughout the 8-week time period. Ad5.MYOC.WT injected eyes represented in grey, uninjected control eye represented in black. Data reported as mean +/− SD.
3.4. Mutant myocilin adenovirus causes optic nerve damage in A/J mice
Since we identified strain differences in responsiveness to mutant myocilin induced ocular hypertension, we tested whether these strains also harbored glaucomatous optic nerve damage. Even though IOP was elevated in three of the strains tested (A/J, BALB/cJ, and C57BL/6J) (Table 1, Figure 2), only the A/J strain had considerable and significant optic nerve damage at the end of 8 weeks (injected eye ONDS=2.64 +/− 0.19, n=18; uninjected eye ONDS=1.40 +/− 0.14, n=18, p<0.001) (Figure 4). These data suggest that at 8 weeks post-injection, the A/J mice harbor early signs of the glaucoma phenotype of optic nerve damage. Since the C57BL/6J mice had a lower IOP exposure than the A/J mice (Table 1, Figure 2), it is possible that with a prolonged higher IOP these animals could also develop glaucomatous optic nerve damage. It has previously been hypothesized that the initial neurobiological insult in glaucoma is at the optic nerve head (Howell et al., 2007; Libby et al., 2005c; Schlamp et al., 2006; Soto et al., 2011). Here, we confirm these data and provide a model to study the timing and onset of damage. Our data suggest that the A/J mouse strain is the most susceptible to mutant human myocilin induced ocular hypertension and optic nerve damage, identifying a novel mouse model of human POAG.
Figure 4. Ad5.MYOC.Y437H damages the optic nerve in A/J mice at 8 weeks post injection.
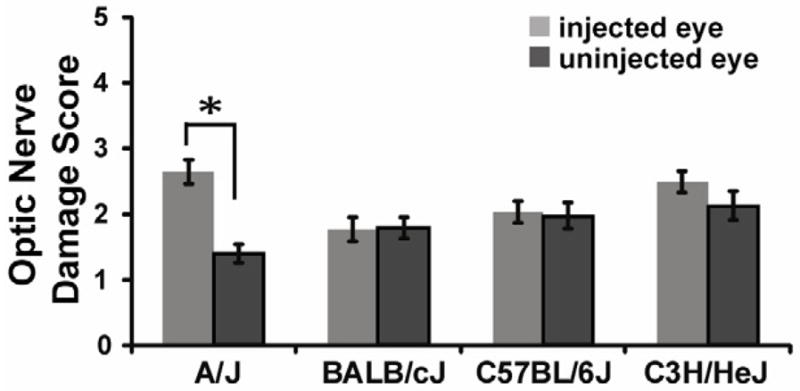
Optic nerve damage was assessed by PPD stain and quantified by using the optic nerve damage score (ONDS) system, which clinically grades optic nerve damage from 1 (no damage) to 5 (severe damage). At 8 weeks post-injection there was a significant difference in ONDS in A/J mice (injected eye ONDS=2.64 +/− 0.19, n=18; uninjected eye ONDS=1.40 +/− 0.14, n=18, p<0.001). There was no significant difference in ONDS in BALB/cJ, C57BL/6J or C3H/HeJ mice at 8 weeks. Data represented as mean +/− SEM.
3.5. Mutant myocilin adenovirus does not cause measurable RGC or SC damage in mice after 8 weeks
Since we identified damage to the optic nerve, we also tested whether damage had progressed to the RGC layer and SC in the brain. At 8-weeks post-injection of Ad5.MYOC.Y437H, there was no biologically significant loss of cells in the RGC layer (Figure 5A) or SC damage (Figure 5B) in the four strains tested. Since we saw only moderate damage to the optic nerve of A/J at the 8-week time point, it was not surprising that these mice had not progressed to more extensive damage to the RGCs and SC. Further studies are necessary to determine the onset and extent of damage in these tissues.
Figure 5. Ad5.MYOC.Y437H has no effect on RGC loss or SC damage at 8 weeks post injection.
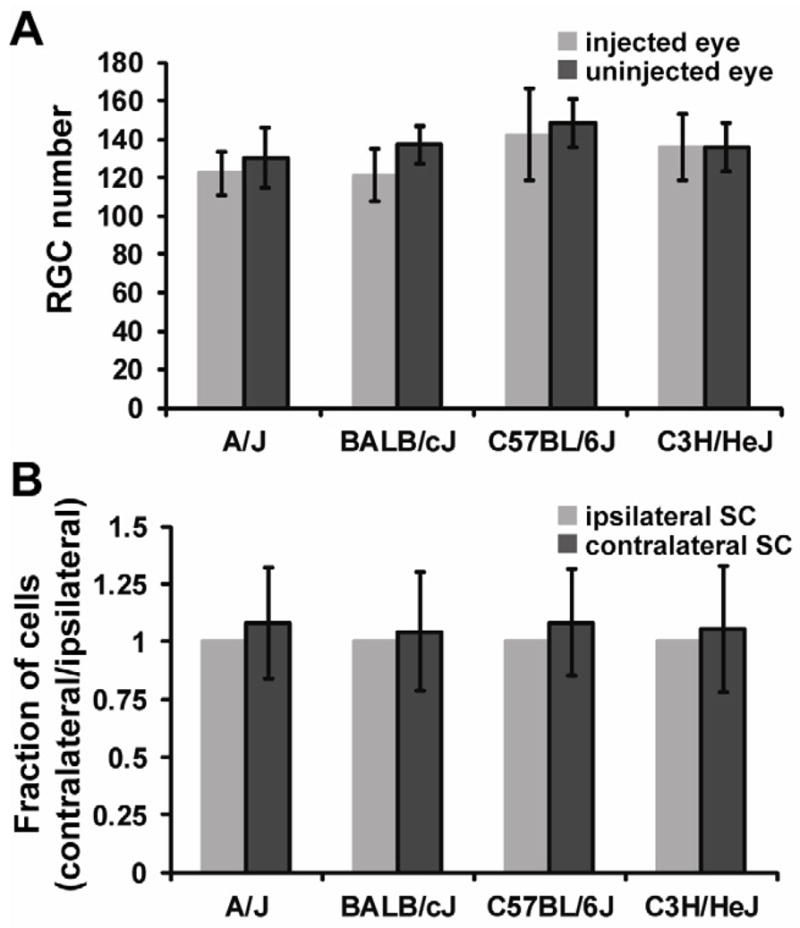
(A) Nissl stained retina flat mounts were used to quantify RGC loss 8 weeks after Ad5.MYOC.Y437H injection. At 8 weeks post-injection there was no significant difference in RGC number in A/J (n=8), BALB/cJ (n=10), C57BL/6J (n=10), or C3H/HeJ (n=10) mice. Two images (400X) were taken from each quadrant of the retina flat mount (8 images total) and four regions within each image were counted, representing about 2% of total RGC’s. (B) Nissl stained superior colliculus sections were used to quantitate damage 8 weeks after Ad5.MYOC.Y437H injection. At 8 weeks post-injection there was no significant difference in cell number in the superior colliculus in A/J (n=10), BALB/cJ (n=10), C57BL/6J (n=10), or C3H/HeJ (n=8) mice. Data represented as mean +/− SD.
3.6. Mutant myocilin is expressed in the TM 8 weeks post-injection in A/J mice
Given the IOP and optic nerve damage response to mutant myocilin in A/J mice, we also evaluated the anterior chamber morphology and myocilin expression at 8 weeks post-injection. Mice were injected with Ad5.MYOC.Y437H in one eye and the contralateral eye served as the uninjected control. The uninjected control eye showed minimal myocilin expression in the TM and the irideocorenal angles were open (Figures 6A, 6B; n=5 mice). The Ad5.MYOC.Y437H injected eye showed myocilin expression and the irideocorneal angles were open 8 weeks post-injection (Figure 6C, 6D; n=5 mice). Ad5.null injected eyes and contralateral control eyes showed minimal myocilin expression and the irideocorneal angles were open (Figure 6E, 6F, 6G, 6H; n=5 mice). These data suggest that the elevated IOP response in mutant myocilin injected A/J mice is due to changes in the TM and not to defects in the irideocorneal angle. Further studies are necessary to determine the molecular and cellular changes occurring within the TM cells.
Figure 6. Mutant myocilin is expressed in the TM 8 weeks post-injection in A/J mice.
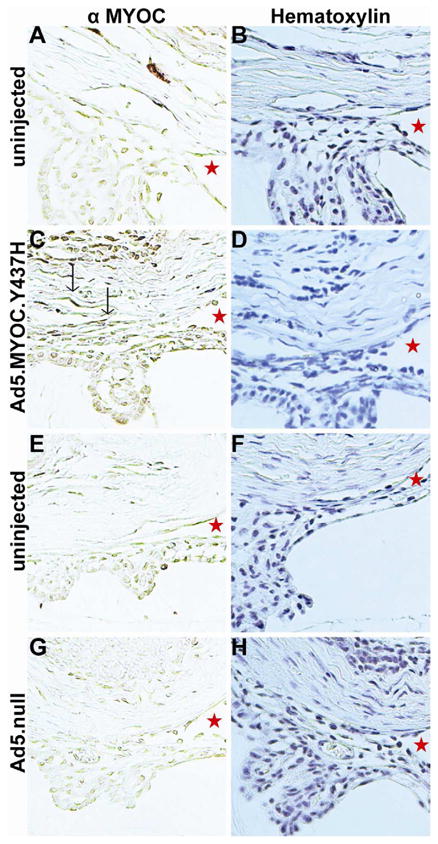
All mice were injected in one eye with 5×107 pfu Ad5.MYOC.Y437H. The contralateral uninjected eye served as a control. All samples were collected 8 weeks post-injection. The left column is labeled with anti-myocilin antibody and myocilin expression is represented by the brown labeling (A, C, E, G; n= 5 mice per strain). Myocilin expression in the TM is indicated by black arrows. The right column is sections from the same eye used in the left column and are stained with hematoxylin. The stars represent open iridiocorneal angles (B, D, F, H; n=5 mice per strain).
4. Discussion
We report strain differences in the responsiveness to transduction of mouse eyes with mutant human myocilin. These data show two important, tractable and genetically distinct differences in the response to a known human glaucoma causing gene. First, intravitreal injection of Ad5.MYOC.Y437H induced ocular hypertension in A/J, BALB/cJ, and C57BL/6J mice but had minimal effect on C3H/HeJ mice. Second, intravitreal injection of Ad5.MYOC.Y437H induced optic nerve damage in A/J mice but had no significant damaging effect on optic nerves of BALB/cJ and C57BL/6J mice, even though all 3 strains had ocular hypertension phenotypes. As expected, given the lack of ocular hypertension, the C3H/HeJ mice had no optic nerve damage. These data provide an excellent resource to study the onset and progression of glaucoma phenotypes in mice, to elucidate genetic pathways involved in IOP regulation and optic nerve damage through genetic mapping experiments, as well as provide a novel model system that can be applied to any mouse strain.
The genetic and molecular pathways involved in IOP regulation are not well known. In addition, changes in aqueous humor dynamics and IOP are major risk factors for developing glaucoma. IOP measurement is also the primary clinical tool for monitoring the potential onset and development of other glaucoma phenotypes. Here we show strain differences in IOP elevation in response to human mutant myocilin. The exact mechanism of how mutant myocilin affects IOP has not been completely elucidated. Wild-type myocilin is secreted from TM and is expressed in other eye tissues (Jacobson et al., 2001; Nguyen et al., 1998), but its function is not known. Previously it has been shown that mutations in human myocilin induce exposure of a cryptic peroxisomal targeting sequence, whose interaction with the PTS1R is necessary for IOP elevation in mice (Shepard et al., 2007). Based on these data it has been hypothesized that instead of being secreted, mutant myocilin is misfolded and shuttled to peroxisomes, leading to cellular and endoplasmic reticulum stress and resulting in deleterious TM function. The human mutant myocilin adenovirus used in our studies presented here is presumably acting in a similar fashion, resulting in peroxisome dysfunction. However, ocular hypertension was only induced in three of the strains tested. C3H/HeJ mice did not develop a pronounced mutant myocilin induced ocular hypertension even though the human myocilin transgene was expressed in the TM (Figure 1). Interestingly, C3H/HeJ mice have altered levels of enzymes associated with peroxisome proliferation as well as polymorphisms in PPARG (peroxisome proliferator activated receptor γ) (Ackert-Bicknell et al., 2008; Butler et al., 1988). Therefore, it is possible that C3H/HeJ mice may have altered peroxisome function affecting how the TM cells process mutant myocilin, resulting in maintenance of IOP versus the increased IOP seen in other mouse strains. Further experiments are needed to test this hypothesis. By demonstrating dramatic differences in genetically distinct mouse strains, we can now utilize the power of mouse genetics to begin to tease out the genes and pathways responsible for these differences.
We have shown that A/J mice are susceptible to mutant myocilin induced ocular hypertension and optic nerve damage, whereas BALB/cJ and C57BL/6J mice displayed only the ocular hypertension phenotype. These data suggest that there are strain differences in the ability to progress from increased IOP to axonal damage in the optic nerve. Previously, glaucoma related strain differences have been identified between the DBA/2J and C57BL/6J mouse strains (Anderson et al., 2006). However, in that particular case when the disease causing genes were transferred from the DBA/2J background to the C57BL/6J background, both the IOP and optic nerve damage phenotypes were lost. Here, we were able to separate the IOP phenotype from the optic nerve damage phenotype. Interestingly, the BALB/cJ strain was not sensitive to optic nerve damage or RGC loss in our model, but this strain is most susceptible to RGC loss in the optic nerve crush model (Li et al., 2007). Expansion of the strain survey and/or backcross studies could identify the modifier genes associated with this response, which will lead to a better understanding of the molecular mechanisms responsible for glaucomatous optic nerve and RGC damage. This model would also be ideally suited to look at the roles of individual genes in glaucomatous damage and optic neuropathy neuroprotection. Thus, our model may provide an invaluable resource to begin to dissect the genetic and molecular components involved in the progression from increased IOP to axon and RGC damage.
It has been shown in other models of glaucoma that damage to the optic nerve precedes RGC damage and visual center damage in the brain (Howell et al., 2007; Libby et al., 2005c; Schlamp et al., 2006; Soto et al., 2011). Here, we recapitulate those results in our model. The A/J mice were susceptible to both ocular hypertension and optic nerve damage. However, there was no evidence of RGC damage or damage to the SC after 8-weeks post-injection of the human mutant myocilin adenovirus. This suggests that the initial insult is first occurring at the optic nerve. Further time course analysis is necessary to evaluate the temporal sequence of RGC and SC damage after the initial optic nerve insult.
Recently, there have been several mouse models of glaucoma developed using mutant myocilin transgenic mice (Senatorov et al., 2006; Zhou et al., 2008; Zode et al., 2011). These model systems are beneficial in that they have TM damage with open irideocorneal angles, elevated IOP, RGC damage, and optic nerve damage. Zode et al have also demonstrated that therapeutic intervention can protect against glaucomatous damage in their transgenic mouse (Zode et al., 2011). The model system we used in this study also has several benefits compared to many of the glaucoma mouse models developed to date. Using human mutant myocilin adenovirus is unique in that it is inducible and can be applied to any mouse strain, without having to extensively breed animals to transfer mutant genes from one background to another. The insult also mimics human glaucoma like the transgenic mice, by using a similar pathogenic pathway (mutant myocilin) to raise IOP. The response within each mouse strain was also homogeneous, yielding similar phenotypes in all animals. IOP elevation was achieved within a week post-injection of the adenovirus and optic nerve damage was shown by 8 weeks, providing a reasonable and cost effective time frame. Since we have identified strain differences in response to human mutant myocilin, we can use the model to exploit the power of mouse genetics to molecularly dissect pathogenic mechanisms.
5. Conclusions
We have identified mouse strain differences in ocular hypertension and optic nerve damage in response to mutant human myocilin in mice. These data provide an excellent resource to identify the initiating factors in glaucoma pathogenesis as well as the pathways involved in the disease progressing from the onset of elevated IOP to later stages of glaucomatous retinopathy and neuropathy.
A mouse model of human POAG using mutant myocilin transgene expression in mice.
Mouse strain differences in myocilin induced ocular hypertension.
Mouse strain differences in myocilin induced optic nerve damage.
Acknowledgments
The authors acknowledge Holly E. Tebow, Terri A. Beckwith, Sandra K. Neubauer, Gareth R. Howell and The Jackson Laboratory Services for assistance with processing and analyzing histological sections. This work was supported by NEI/NIH grant R21 EY019977 (AFC) with partial support from EY11721 (SWMJ). SWMJ is an Investigator of the Howard Hughes Medical Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ackert-Bicknell CL, Demissie S, Marin de Evsikova C, Hsu YH, DeMambro VE, Karasik D, Cupples LA, Ordovas JM, Tucker KL, Cho K, Canalis E, Paigen B, Churchill GA, Forejt J, Beamer WG, Ferrari S, Bouxsein ML, Kiel DP, Rosen CJ. PPARG by dietary fat interaction influences bone mass in mice and humans. J Bone Miner Res. 2008;23:1398–1408. doi: 10.1359/JBMR.080419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allingham RR, Liu Y, Rhee DJ. The genetics of primary open-angle glaucoma: a review. Exp Eye Res. 2009;88:837–844. doi: 10.1016/j.exer.2008.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alward WL, Fingert JH, Coote MA, Johnson AT, Lerner SF, Junqua D, Durcan FJ, McCartney PJ, Mackey DA, Sheffield VC, Stone EM. Clinical features associated with mutations in the chromosome 1 open-angle glaucoma gene (GLC1A) N Engl J Med. 1998;338:1022–1027. doi: 10.1056/NEJM199804093381503. [DOI] [PubMed] [Google Scholar]
- Anderson MG, Libby RT, Mao M, Cosma IM, Wilson LA, Smith RS, John SW. Genetic context determines susceptibility to intraocular pressure elevation in a mouse pigmentary glaucoma. BMC Biol. 2006;4:20. doi: 10.1186/1741-7007-4-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler EG, England PJ, Williams GM. Genetic differences in enzymes associated with peroxisome proliferation and hydrogen peroxide metabolism in inbred mouse strains. Carcinogenesis. 1988;9:1459–1463. doi: 10.1093/carcin/9.8.1459. [DOI] [PubMed] [Google Scholar]
- Chauhan BC, Levatte TL, Garnier KL, Tremblay F, Pang IH, Clark AF, Archibald ML. Semiquantitative optic nerve grading scheme for determining axonal loss in experimental optic neuropathy. Invest Ophthalmol Vis Sci. 2006;47:634–640. doi: 10.1167/iovs.05-1206. [DOI] [PubMed] [Google Scholar]
- Clark AF, Wordinger RJ. The role of steroids in outflow resistance. Exp Eye Res. 2009;88:752–759. doi: 10.1016/j.exer.2008.10.004. [DOI] [PubMed] [Google Scholar]
- de Voogd S, Ikram MK, Wolfs RC, Jansonius NM, Witteman JC, Hofman A, de Jong PT. Is diabetes mellitus a risk factor for open-angle glaucoma? The Rotterdam Study. Ophthalmology. 2006;113:1827–1831. doi: 10.1016/j.ophtha.2006.03.063. [DOI] [PubMed] [Google Scholar]
- Dietz JA, Li Y, Chung LM, Yandell BS, Schlamp CL, Nickells RW. Rgcs1, a dominant QTL that affects retinal ganglion cell death after optic nerve crush in mice. BMC Neurosci. 2008;9:74. doi: 10.1186/1471-2202-9-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortune B, Bui BV, Morrison JC, Johnson EC, Dong J, Cepurna WO, Jia L, Barber S, Cioffi GA. Selective ganglion cell functional loss in rats with experimental glaucoma. Invest Ophthalmol Vis Sci. 2004;45:1854–1862. doi: 10.1167/iovs.03-1411. [DOI] [PubMed] [Google Scholar]
- Gordon MO, Beiser JA, Brandt JD, Heuer DK, Higginbotham EJ, Johnson CA, Keltner JL, Miller JP, Parrish RK, 2nd, Wilson MR, Kass MA. The Ocular Hypertension Treatment Study: baseline factors that predict the onset of primary open-angle glaucoma. Arch Ophthalmol. 2002;120:714–720. doi: 10.1001/archopht.120.6.714. discussion 829–730. [DOI] [PubMed] [Google Scholar]
- Howell GR, Libby RT, Jakobs TC, Smith RS, Phalan FC, Barter JW, Barbay JM, Marchant JK, Mahesh N, Porciatti V, Whitmore AV, Masland RH, John SW. Axons of retinal ganglion cells are insulted in the optic nerve early in DBA/2J glaucoma. J Cell Biol. 2007;179:1523–1537. doi: 10.1083/jcb.200706181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson N, Andrews M, Shepard AR, Nishimura D, Searby C, Fingert JH, Hageman G, Mullins R, Davidson BL, Kwon YH, Alward WL, Stone EM, Clark AF, Sheffield VC. Non-secretion of mutant proteins of the glaucoma gene myocilin in cultured trabecular meshwork cells and in aqueous humor. Hum Mol Genet. 2001;10:117–125. doi: 10.1093/hmg/10.2.117. [DOI] [PubMed] [Google Scholar]
- Joe MK, Sohn S, Hur W, Moon Y, Choi YR, Kee C. Accumulation of mutant myocilins in ER leads to ER stress and potential cytotoxicity in human trabecular meshwork cells. Biochem Biophys Res Commun. 2003;312:592–600. doi: 10.1016/j.bbrc.2003.10.162. [DOI] [PubMed] [Google Scholar]
- Kim BS, Savinova OV, Reedy MV, Martin J, Lun Y, Gan L, Smith RS, Tomarev SI, John SW, Johnson RL. Targeted Disruption of the Myocilin Gene (Myoc) Suggests that Human Glaucoma- Causing Mutations Are Gain of Function. Mol Cell Biol. 2001;21:7707–7713. doi: 10.1128/MCB.21.22.7707-7713.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le A, Mukesh BN, McCarty CA, Taylor HR. Risk factors associated with the incidence of open-angle glaucoma: the visual impairment project. Invest Ophthalmol Vis Sci. 2003;44:3783–3789. doi: 10.1167/iovs.03-0077. [DOI] [PubMed] [Google Scholar]
- Leske MC, Wu SY, Hennis A, Honkanen R, Nemesure B. Risk factors for incident open-angle glaucoma: the Barbados Eye Studies. Ophthalmology. 2008;115:85–93. doi: 10.1016/j.ophtha.2007.03.017. [DOI] [PubMed] [Google Scholar]
- Li Y, Semaan SJ, Schlamp CL, Nickells RW. Dominant inheritance of retinal ganglion cell resistance to optic nerve crush in mice. BMC Neurosci. 2007;8:19. doi: 10.1186/1471-2202-8-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libby RT, Anderson MG, Pang IH, Robinson ZH, Savinova OV, Cosma IM, Snow A, Wilson LA, Smith RS, Clark AF, John SW. Inherited glaucoma in DBA/2J mice: pertinent disease features for studying the neurodegeneration. Vis Neurosci. 2005a;22:637–648. doi: 10.1017/S0952523805225130. [DOI] [PubMed] [Google Scholar]
- Libby RT, Gould DB, Anderson MG, John SW. Complex genetics of glaucoma susceptibility. Annu Rev Genomics Hum Genet. 2005b;6:15–44. doi: 10.1146/annurev.genom.6.080604.162209. [DOI] [PubMed] [Google Scholar]
- Libby RT, Howell GR, Pang IH, Savinova OV, Mehalow AK, Barter JW, Smith RS, Clark AF, John SW. Inducible nitric oxide synthase, Nos2, does not mediate optic neuropathy and retinopathy in the DBA/2J glaucoma model. BMC Neurosci. 2007;8:108. doi: 10.1186/1471-2202-8-108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libby RT, Li Y, Savinova OV, Barter J, Smith RS, Nickells RW, John SW. Susceptibility to neurodegeneration in a glaucoma is modified by Bax gene dosage. PLoS Genet. 2005c;1:17–26. doi: 10.1371/journal.pgen.0010004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Vollrath D. Reversal of mutant myocilin non-secretion and cell killing: implications for glaucoma. Hum Mol Genet. 2004;13:1193–1204. doi: 10.1093/hmg/ddh128. [DOI] [PubMed] [Google Scholar]
- Mackey DA. Gillies lecture: dissecting glaucoma: understanding the molecular risk factors. Clin Experiment Ophthalmol. 2008;36:403–409. doi: 10.1111/j.1442-9071.2008.001798.x. [DOI] [PubMed] [Google Scholar]
- Millar JC, Clark AF, Pang IH. Assessment of aqueous humor dynamics in the mouse by a novel method of constant-flow infusion. Invest Ophthalmol Vis Sci. 2011;52:685–694. doi: 10.1167/iovs.10-6069. [DOI] [PubMed] [Google Scholar]
- Millar JC, Pang IH, Wang WH, Wang Y, Clark AF. Effect of immunomodulation with anti-CD40L antibody on adenoviral-mediated transgene expression in mouse anterior segment. Mol Vis. 2008;14:10–19. [PMC free article] [PubMed] [Google Scholar]
- Nguyen TD, Chen P, Huang WD, Chen H, Johnson D, Polansky JR. Gene structure and properties of TIGR, an olfactomedin-related glycoprotein cloned from glucocorticoid-induced trabecular meshwork cells. J Biol Chem. 1998;273:6341–6350. doi: 10.1074/jbc.273.11.6341. [DOI] [PubMed] [Google Scholar]
- Pang IH, Clark AF. Rodent models for glaucoma retinopathy and optic neuropathy. J Glaucoma. 2007;16:483–505. doi: 10.1097/IJG.0b013e3181405d4f. [DOI] [PubMed] [Google Scholar]
- Schlamp CL, Li Y, Dietz JA, Janssen KT, Nickells RW. Progressive ganglion cell loss and optic nerve degeneration in DBA/2J mice is variable and asymmetric. BMC Neurosci. 2006;7:66. doi: 10.1186/1471-2202-7-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senatorov V, Malyukova I, Fariss R, Wawrousek EF, Swaminathan S, Sharan SK, Tomarev S. Expression of mutated mouse myocilin induces open-angle glaucoma in transgenic mice. J Neurosci. 2006;26:11903–11914. doi: 10.1523/JNEUROSCI.3020-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepard AR, Jacobson N, Millar JC, Pang IH, Steely HT, Searby CC, Sheffield VC, Stone EM, Clark AF. Glaucoma-causing myocilin mutants require the Peroxisomal targeting signal-1 receptor (PTS1R) to elevate intraocular pressure. Hum Mol Genet. 2007;16:609–617. doi: 10.1093/hmg/ddm001. [DOI] [PubMed] [Google Scholar]
- Shepard AR, Millar JC, Pang IH, Jacobson N, Wang WH, Clark AF. Adenoviral gene transfer of active human transforming growth factor-{beta}2 elevates intraocular pressure and reduces outflow facility in rodent eyes. Invest Ophthalmol Vis Sci. 2010;51:2067–2076. doi: 10.1167/iovs.09-4567. [DOI] [PubMed] [Google Scholar]
- Sommer A. Glaucoma risk factors observed in the Baltimore Eye Survey. Curr Opin Ophthalmol. 1996;7:93–98. doi: 10.1097/00055735-199604000-00016. [DOI] [PubMed] [Google Scholar]
- Soto I, Pease ME, Son JL, Shi X, Quigley HA, Marsh-Armstrong N. Retinal ganglion cell loss in a rat ocular hypertension model is sectorial and involves early optic nerve axon loss. Invest Ophthalmol Vis Sci. 2011;52:434–441. doi: 10.1167/iovs.10-5856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone EM, Fingert JH, Alward WL, Nguyen TD, Polansky JR, Sunden SL, Nishimura D, Clark AF, Nystuen A, Nichols BE, Mackey DA, Ritch R, Kalenak JW, Craven ER, Sheffield VC. Identification of a gene that causes primary open angle glaucoma. Science. 1997;275:668–670. doi: 10.1126/science.275.5300.668. [DOI] [PubMed] [Google Scholar]
- Wang WH, McNatt LG, Pang IH, Millar JC, Hellberg PE, Hellberg MH, Steely HT, Rubin JS, Fingert JH, Sheffield VC, Stone EM, Clark AF. Increased expression of the WNT antagonist sFRP-1 in glaucoma elevates intraocular pressure. J Clin Invest. 2008;118:1056–1064. doi: 10.1172/JCI33871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang WH, Millar JC, Pang IH, Wax MB, Clark AF. Noninvasive measurement of rodent intraocular pressure with a rebound tonometer. Invest Ophthalmol Vis Sci. 2005;46:4617–4621. doi: 10.1167/iovs.05-0781. [DOI] [PubMed] [Google Scholar]
- Wordinger RJ, Clark AF. Effects of glucocorticoids on the trabecular meshwork: towards a better understanding of glaucoma. Prog Retin Eye Res. 1999;18:629–667. doi: 10.1016/s1350-9462(98)00035-4. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Grinchuk O, Tomarev SI. Transgenic mice expressing the Tyr437His mutant of human myocilin protein develop glaucoma. Invest Ophthalmol Vis Sci. 2008;49:1932–1939. doi: 10.1167/iovs.07-1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zode GS, Kuehn MH, Nishimura DY, Searby CC, Mohan K, Grozdanic SD, Bugge K, Anderson MG, Clark AF, Stone EM, Sheffield VC. Reduction of ER stress via a chemical chaperone prevents disease phenotypes in a mouse model of primary open angle glaucoma. J Clin Invest. 2011;121:3542–3553. doi: 10.1172/JCI58183. [DOI] [PMC free article] [PubMed] [Google Scholar]