

Abstract
Purpose
High-throughput chemosensitivity testing of low-passage cancer cell lines can be used to prioritize agents for personalized chemotherapy. However, generating cell lines from primary cancers is difficult, because contaminating stromal cells overgrow the malignant cells.
Experimental Design
We produced a series of hypoxanthine phosphoribosyl transferase (hprt)-null immunodeficient mice. During growth of human cancers in these mice, hprt-null murine stromal cells replace their human counterparts.
Results
Pancreatic and ovarian cancers explanted from these mice were grown in selection media to produce pure human cancer cell lines. We screened one cell line with a 3,131-drug panel and identified seventy-seven FDA approved drugs with activity, including two novel drugs to which the cell line was uniquely sensitive. Xenografts of this carcinoma were selectively responsive to both drugs.
Conclusion
Chemotherapy can be personalized using patient-specific cell lines derived in biochemically selectable mice.
Keywords: personalized chemotherapy, hprt, mice, cancer cell lines, drug screening
Introduction
Selecting the most appropriate chemotherapy for a given patient has historically been based on histopathology and past studies demonstrating that specific drugs are generally active against that malignancy. However, drugs deemed active against a particular type of cancer, are most commonly active in only a subset of patients with those cancers. Further, chemotherapeutic agents are generally more toxic and expensive than most other medications in widespread use. Accordingly, an alternative strategy is to test functionally whether a given patient’s cancer is likely or not to respond to specific drugs. In vitro chemotherapy sensitivity and resistance assays (CSRAs), a natural extrapolation of antimicrobial susceptibility testing, was first reported in 1957 (1). Some 50 years later, however, the official position of the American Society of Clinical Oncology has been that this type of testing is still not ready for routine clinical use (2). Problems with CSRA include the many different formats for this testing (2), and results from clinical trials, comparing CSRA guided therapy to conventional treatment, have varied from no significant difference to highly significant benefit (3). Finally, most groups test cancer cells immediately after resection, and these may be sick or dying from hypoxia, anesthetic drugs, or overnight shipping, and so any toxicity to these cells may reflect synergistic toxicity of the drugs tested with any of these effects.
We hypothesize that low-passage cell lines might better represent their respective tumors and therefore more accurately predict in vivo chemosensitivity. Isolating cancer cell lines, however, can, counterintuitively, be difficult, especially from solid primary tumors(4). To date, the success rate for the generation of cell lines is only 10–40% for many solid tumors(4–8). The most significant barrier to routine cancer cell line production is that when tumors are explanted into tissue culture, fibroblasts and other stromal cells proliferate, overgrow and eliminate the malignant cells.
We report production of nude-, SCID- (severe combined immunodeficiency) and NSG- (Non obese diabetic [NOD], SCID, Interleukin-2 receptor Gamma [IL2Rγ]), hprt-null mice. During growth of implanted human cancers in these mice, biochemically defective mouse stromal cells replace the human stromal cells from the original tumor(9, 10). After explanting these xenografts into culture, mouse cells can be eliminated by selection in hypoxanthine, aminopterin, and thymidine (HAT) containing media(11, 12) to isolate pure human cancer cells. Using this system, we isolated a total of 6 pancreatic ductal adenocarcinoma (PDA) cell lines and one ovarian cancer cell line. One of the PDA cell lines was isolated from the corresponding surgically resected sample and tested for in vitro chemosensitivity using a 3,131-drug panel(13). In comparison to other PDA cell lines, this cell line in vitro was differentially more sensitive to digitoxin and nogalamycin, which correlated with in vivo response in mice, where the same two drugs demonstrated selective activity against xenografted tumors. These data suggest a possible novel paradigm for functional personalized chemotherapeutic selection by isolating low-passage cancer cell lines and screening them with large drug panels.
Methods
Patient samples and xenografting
Primary tumors from patient-derived resection specimens or xenografts from standard nude mice were harvested with informed consent and IRB approval. They were implanted in anesthetized standard B6 nude and hprt-null B6 nude mice. Following a small skin incision, a 5mm3 sample coated with Matrigel Matrix (BD Biosciences, Bedford, MA) was implanted subcutaneously in each upper flank, the incision was closed using staples, and the site was disinfected with Betadine (Purdue Frederick, Stamford, CT). Among the 6 pancreatic cancers, 3 were selected because they were familial, and 3 because they have undergone full exomic DNA sequencing as xenografted samples(14).
Tumor harvest and culture
When tumors reached approximately 1 cm in diameter, mice were sacrificed, and tumors harvested. In a laminar flow biosafety cabinet, tumors were finely minced and incubated in DMEM containing 750 units/ml of type IV collagenase (Invitrogen) and 500 units/ml of hyaluronidase (Sigma-Aldrich Inc., St. Louis, MO) for 1 hour at 37°C, and pipetting up and down 20 times to mechanically fragment the tumor. The cell solution was plated at 0.5, 1 and 2 mls per well on 6-well plates coated with or without type I rat tail collagen (BD Biosciences) in MEM with 20% fetal bovine serum, supplemented with standard concentrations of penicillin/streptomycin, L-glutamine, 5 ng/ml of human recombinant EGF (all Invitrogen) and 0.2 U/ml of human insulin (Sigma-Aldrich). Following two days recovery (designated day 0), 1X HAT (100uM sodium hypoxanthine, 400nM aminopterin, 16nM thymidine, Invitrogen) media was fed to half of the cultures. Cells are fed with or without HAT media every 2–3 days. All cell lines were confirmed as patient in origin by DNA fingerprinting with the powerplex 1.2 kit per manufacturers instructions (Promega, Madison, WI). They were also stained with anti-cytokeratin (Ventana, Tucson, AZ, 760-2595) and anti-smooth muscle actin (Ventana, 760-2835) by immunohistochemistry and demonstrated to be tumorigenic in nude mice.
Chemosensitivity testing-Initial library screening and confirmation
Cancer cells were screened for chemosensitivity using the Johns Hopkins Drug Library (JHDL) consisting of 3,131 drugs arrayed in 96-well plates. This includes 1,907 United States Food and Drug Administration (FDA) approved drugs, 570 drugs approved in other countries, 654 other drugs in various phases of development(13). All drugs are maintained at minus 70 at a stock concentration of 200 microMolar in PBS containing 2% DMSO and 10% FBS.
Tumor cells were subcultured, counted, and inoculated into 96-well plates at 5,000 cells/well in complete media and allowed to adhere overnight. Drugs were then added to the plates in duplicate wells at a final concentration of 10 μM for a total of 48 hours. After the first 24 hours in drug, 1 microCi [3H]-thymidine was added, and incubated for an additional 24 hours (in both drug and [3H]-thymidine). Cells were then trypsinized using 1x trypsin-EDTA (Invitrogen). The suspended cells were transferred onto FilterMat-A glass fiber filters (Wallac, Turku, Finland) using the Harvester-96 cell harvester (Tomtec, Hamden, CT). The glass fiber filters were washed five times with water to wash away unincorporated free [3H]-thymidine. On each plate, the first and last columns were used as no-drug controls. Incorporated thymidine was read on a MicroBeta plate reader (Perkin Elmer), and the mean % inhibition was calculated relative to no-drug control wells. A histogram of the % inhibition for the drugs in the JHDL was established using 2.5% size bins, and normal distribution curve fitting was performed by the least squares method using GraphPad ver. 5.02 (GraphPad Software Inc., LaJolla, CA). The mean +/− 3 SD encompasses 3,014 drugs. Using Gaussian statistics and 3 SD as the cutoff (40.4% inhibition), then we expect ~4 drugs to be false positives among the drugs demonstrating 40–100% inhibition (3014 × 0.13=3.9).
IC50s of selected drugs were determined from % growth inhibition of ten different concentrations of the drugs obtained in the same experimental design described above. IC50s of each drug for each cell line were calculated using four-parameter logistic equation with three replicates results by GraphPad ver. 5.02.
Growth inhibition of subcutaneous xenografted tumors
Tumor xenografts were generated by subcutaneous injection of 10 × 106 cells bilaterally into 6 weeks athymic nude-Foxn1nu mice (Harlan, Indianapolis, IN). Tumor volumes were measured and when the tumors reached approximately 100 mm3 in size, the mice were stratified (day 0) to treatment and non-treatment groups with 5 mice per group so that each group was equivalent based on tumor volume. Tumor volume is obtained by the formula: length x (width)2 × 0.5, where length is the longest diameter and width is the shortest diameter perpendicular to the length. The mice received treatment with daily intraperitoneal injection of nogalamycin (0.2 and 1 mg/kg in vehicle), digitoxin (0.4 and 2 mg/kg in vehicle), or vehicle (0.9 % sodium chloride with 1 % DMSO) control for 30 days (day 1-day 30). Tumor volumes were measured twice a week. At the completion of the study, mice were euthanized and tumors were measured, harvested and weighed. The tumor volume index (TVI) was determined from a ratio of the tumor volume on a given day divided by the tumor volume of day 0. The harvested tumors were then weighed and means and standard deviations calculated. The normalized tumor weight of treatment group was calculated by dividing the treatment values by the control group for each cell line (i.e. the mean tumor weight of control group for each cell line is 100). Statistical analysis was performed using the unpaired Student’s t test on Graph Pad Prism ver. 5.02.
Results
Production of hprt-null immunodeficient mice
A series of hprt-null immunodeficient mice, including nude hprt-null (Supplemental Fig. S1), SCID hprt-null (Supplemental Fig. S2) and NOD-SCID Interleukin2γ(NSG) hprt-null, were produced by breeding immunocompetent hprt-null mice with the appropriate immunodeficient mice (see Supplemental Information). Mice that are homozygous or hemizygous for the hprt-null allele and homozygous nu/nu appear to have no phenotypic variation from standard Foxn1nu/nu mice (Fig. 1A). To confirm genotyping (Fig. 1B, Supplementary Fig. S3), we harvested tail cuttings from mice II and III (Fig. 1A), trypsinized them and grew the cells in tissue culture. These samples were grown in the presence of HAT or 6-thioguanine (6TG) to select for or against HPRT function, respectively (Fig. 1C). As predicted from their genotype (Fig. 1B), cells from the hprt-null mouse died in HAT media and survived in 6TG containing media. The HPRT proficient heterozygous hprt-null/wt mouse cells gave the opposite result, as expected.
Fig. 1. Athymic hprt-null mice.

A. Third cross Foxn1nu/nu mice. The nude hprt-null mouse (II) and heterozygous littermate controls (I, III, IV) show similar phenotypic characteristics in size, hair growth cycles, and skin pigmentation. B. hprt genotyping: hprt PCR products are separated on a 1% agarose gel. Wild type (+) and null (Δ) alleles are assayed in separate PCR reactions. Wild type (WT) mice result in a 746 bp band in the wild type reaction, and only the control 500 bp band in the Δ reaction. Homozygous null females (II, female Null) and hemizygous null males (male Null) result in a 628 bp band in the Δ reaction, but no band in the wild type reaction. Heterozygous females (I, III, IV, HET) result in a 746 bp band in the wild type reaction, and a 628 bp band in the Δ reaction. Note, a 500 bp control band is in the Δ reaction in all lanes. C. Tail cell phenotyping. Homozygous hprt-null (II) and heterozygous (III) tail snips were harvested from female mice, trypsinized and plated in the presence of HAT or 6TG containing media. Brightfield microscopy of crystal violet stained cells.
Patient-specific cancer cell line production
A PDA xenograft (Panc410) was implanted into these mice and the tumors were harvested for cell culture. After plating and 2 days of recovery, cultures of Panc410 (Day 0) showed a mixture of cancer cells clustering together to form “islands”, and scattered fibroblasts and other stromal cells (Fig. 2A and 2D). Cultures explanted from the hprt-null mouse and treated with HAT selection media typically yielded fibroblast free cancer cell lines after 2–3 weeks (Fig. 2A–C). During this time, HAT selection eliminated the fibroblasts and other stromal cells, allowing for the small cancer cell “islands” to expand and eventually coalesce. In contrast, a sister culture, grown in the absence of HAT, was heavily contaminated with fibroblasts (Fig. 2D–F). These stromal cells surrounded cancer cell islands (Fig. 2E), overgrew them (Fig. 2F), and precluded producing cancer cell lines from these cultures. The resultant cell line matched the patient’s microsatelllites and was tumorigenic in nude mice. To confirm its epithelial origin, the cell line was stained for cytokeratin and smooth muscle actin, which stained positive and negative respectively, as expected (data not shown).
Fig. 2. Rapid pancreatic cancer cell line production.
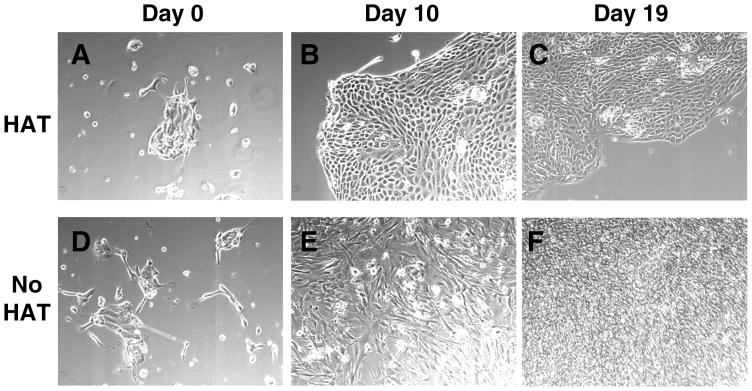
Samples explanted from Panc410 tumors grown in a nude hprt-null mouse (A–F) in the presence (A–C) or absence (D–F) of HAT media, and photographed using phase microscopy at T=0, 10 and 19 days in culture. Note that after 19 days in the absence of HAT media (F), fibroblasts have overgrown the culture.
Chemosensitivity-Initial Screen
One potential application of rapid cell line isolation is personalized chemosensitivity testing. To demonstrate utility of the mice for this purpose, we first implanted SCID hprt-null mice with a surgically resected PDA and isolated the cell line Panc502 (Supplementary Fig. S4). We then screened this cell line against the JHDL (13) consisting of 3,131 drugs (Supplementary Fig. S5). The library includes 1,907 US FDA approved drugs, 570 drugs approved for clinical use in other countries and 654 other drugs in various phases of development. A histogram of the number of drugs, graphed as a function of the percent growth inhibition (Fig. 3A), showed that most drugs fit under a Gaussian distribution (mean 5.0%, SD 11.8%), which we interpreted as inactive drugs for this cancer cell line. The mean plus or minus 3 standard deviations encompasses 2817 drugs, or ~90% of those tested. More importantly, 314 drugs (~10%) showed activity above the normal distribution, and these were considered potentially active drugs (Supplementary Table 1). These 314 drugs included 77 drugs that are currently FDA approved and 12 drugs previously approved, but currently discontinued (combined ~2.8% of drugs in the library).
Fig. 3. Chemosensitivity of a low-passage familial pancreatic cancer from surgery.

Histogram of the number of drugs (frequency) as a function of % growth inhibition (A). Curve-fitting of Gaussian distribution onto the histogram (black line) distinguishes the distribution of drugs with little or no activity from those which demonstrate some level of activity above this distribution. Arrowhead indicates 3 standard deviations (+3SD) above the mean. Arrows indicate the % growth inhibition for nogalamycin (N) and digitoxin (D). Cell line specific sensitivity of nogalamycin and digitoxin in the cell lines Panc502, Panc486 and Panc410 (B). Values shown are the mean IC50 values of 3 replicates and error bars are the 95% confidence intervals. In vivo growth curves of subcutaneous mouse xenografted tumors raised from the Panc410 and Panc502 cell lines after treatment with nogalamycin, digitoxin or control (C). Control (empty ovals), nogalamycin 0.2 mg/kg (empty squares), nogalamycin 1.0 mg/kg (filled squares), digitoxin 0.4 mg/kg (empty triangles), digitoxin 2.0 mg (filled triangles). Normalized weight of tumors explanted from mice after 30 days of treatment (D). Normalized tumor weight of Panc410 and Panc502 in white columns or grey columns, respectively. Error bars are standard deviations. Fold changes between Panc410 and Panc502 are noted.
The FDA approved drug, digoxin, showed 95% growth inhibition, consistent with its recent discovery as a potential anti-tumor agent(15). We also identified two terpenoid drugs, pristimerin and triptonide, isolated from Chinese medicinal plants, as potent inhibitors of growth(16–18). Among the 125 antineoplastic drugs tested (Supplementary Table 2), only a third (49 drugs, 39.2%) showed activity above the bell-shaped curve. Nine drugs demonstrated greater than 99% growth inhibition, including 7 nucleic acid-targeting chemotherapeutics (actinomycin C, actinomycin D, chromomycin A3, camptothecin, doxorubicin, gemcitabine, and mitoxantrone) and 2 terpenoid natural products (pristimerin and triptonide).
Novel drugs with activity against PDA
From the drugs demonstrating 75% or higher inhibition at 10μM final concentration, we selected 10 drugs to study further (Table 1), after excluding drugs with well-documented activity against pancreatic cancer, and those with relatively high toxicities (i.e. low LD50 values in mice). Only one of these drugs is FDA approved. We generated dose-response curves to confirm activity and establish IC50 values for Panc502, Panc410 and another low-passage PDA cell line, Panc486 described below. From these, we selected two drugs because they showed significant variability in their IC50s among the three low passage PDA cell lines (Fig. 3B). The anthracycline, nogalamycin, was approximately 11x more effective against Panc502 compared to either Panc410 or the non-transformed pancreatic duct cell line, HPDE (data not shown). Finally, the cardiac glycoside digitoxin was substantially more toxic to Panc502 than Panc410, while Panc486 had an intermediate sensitivity. We also generated dose response curves using a panel of topoisomerase inhibitors, showing that topoisomerase I inhibitors were generally more effective than topoisomerase II inhibitors (Supplementary Fig. S6).
Table 1.
Ten drugs with 75% or greater inhibition
| Drug | % growth inhibition at 10uM on Panc502 | IC50 (nM)
|
||
|---|---|---|---|---|
| Panc502 | Panc486 | Panc410 | ||
| Triptolide (for Triptonide) | 98.1 | 7.3 | 10.2 | 8.3 |
| Nogalamycin | 75.4 | 23.3 | 265.2 | 281.9 |
| Ouabain | 95.6 | 39.7 | 57.9 | 81 |
| Digitoxin | 95.1 | 62.7 | 199.7 | 572.2 |
| Strophanthin K | 98.9 | 69.3 | 96.8 | 132.5 |
| Digoxin | 94.7 | 70.8 | 101.6 | 193.2 |
| Dithiazanine iodide | 99.2 | 234.1 | 156.4 | 283.3 |
| Strophanthidin | 98 | 430.1 | 549 | 988.9 |
| Quinacrine | 100 | 657.7 | 756.6 | 961.2 |
| Celastrol (for Pristimerin) | 100 | 753.6 | 1229 | 1076 |
Does in vitro chemosensivity predict in vivo response?
To address the hypothesis that in vitro response could predict in vivo response, we then raised xenografted tumors from Panc410 and Panc502 cell lines, and treated the mice harboring these xenografts with nogalamycin, digitoxin or control for 30 days. We measured the size of tumors twice a week during this time (Fig. 3C). Both nogalamycin and digitoxin demonstrated more activity against Panc502 than Panc410, supporting the notion that in vitro sensitivity does predict in vivo response, at least with these 2 drugs in these cell lines, as judged by tumor size (Fig. 3C), weight of the tumors after completing the treatment (Fig. 3D), and by visual inspection of the residual tumors after treatment (Supplementary Fig. S7).
Isolation of additional cell lines
To test whether we could use this system to routinely generate cell lines from solid cancers, we also isolated a cell line from an ovarian cancer, another highly lethal cancer. The ovarian cancer cell line, FM108, was established from an existing xenograft (Supplementary Fig. S8). We also isolated an additional cell line from a surgically resected PDA. Panc486 was isolated from a xenograft from a patient with a family history of pancreatic cancer (Supplementary Fig. S9). In each case, cell lines were documented to be tumorigenic in athymic mice, sequenced to document oncogenic mutations in the Kras2 gene, and DNA fingerprinted to confirm their patient origin (information available on request). During this time, we isolated 3 additional cell lines from other PDAs, but were unable to produce cell lines from two other PDA xenografts using similar approaches (total 7 cell lines, out of 9 attempts, 78%). This suggests that the in vitro propagation of the 2 failed cell lines is dependent on one of a number of factors such as factors intrinsic to the carcinoma, factors related to the non-neoplastic stromal cells, putative paracrine growth factors, or critical media components that were missing and remain to be identified.
Discussion
We report successful production of nude, SCID, and NSG immunodeficient mice whose hprt-null cells are biochemically selectable in vitro. Using this system, we were able to isolate 3 PDA and 1 ovarian cancer cell line. The Panc502 familial PDA cell line was isolated directly from the tumor surgically resected from a patient, and we screened it against a panel of 3,131 drugs. Approximately 314 drugs demonstrated potential activity, where the vast majority of them (265, 84%) were not designated as antineoplastics. Of 125 anti-neoplastic drugs, only 49 (39%) demonstrated activity. Two novel drugs demonstrated selective activity in vitro, and this correlated with in vivo response. We also identified 2 terpenoid drugs with >99% growth inhibition.
In this paper, we describe an alternative strategy to traditional CSRA. In contrast to traditional CSRA, the approach described herein uses: i) pure low-passage human cancer cells, ii) cells that are viable and well-adapted to in vitro growth, and iii) larger drug panels. The use of pure cancer cells, means that any signal detected is due to the drug’s effect on the malignant cells rather than stromal cell toxicity. Because the cancer cells are growing and healthy, measured effects are a combined effect of the drug and the cancer cells and not due to synergistic effects of the drug with anoxia, starvation, overnight shipping, etc. Finally, because the cells are expanded into a low-passage cell line, drug panels with thousands of drugs can be screened, rather than the 5–10 typically tested in traditional CSRA. This permits one to test all FDA approved anti-neoplastics, FDA approved drugs for other indications, late phase clinical trial drugs, herbal drugs and drugs approved in other countries for clinical use. The idea of screening drugs using cancer cell lines is well established (reviewed in (19)), and the personalized approach has recently been validated for one patient with a rare type of pancreatic cancer, acincar-cell carcinoma (20).
The approach describe herein differs from the xenograft-based approach demonstrated by Hidalgo’s group (21, 22). The advantage of the low-passage cell line approach is that more drugs can be screened under highly controlled conditions. A possible advantage of the xenograft approach is that xenografted tumor response may be more biologically predictive of response in patients. We are eager to test whether low-passage personalized cell line responses can predict patient response. This may be best tested with a malignancy for which several alternative first-line chemotherapeutic choices are available.
Drugs with activity in vivo may not be limited to those that demonstrate >95% growth inhibition. While digitoxin demonstrated 95.1% growth inhibition in vitro, nogalamycin was chosen in part because it only had 75.4% growth inhibition in vitro. Finally, it is possible that the drugs identified could be synergistic with those currently in clinical use such as gemcitabine, although this remains to be demonstrated. In the future, the combination of the patient’s germline SNPs, somatic mutations, mRNA expression and epigenetic changes in the cancer, may play an increasing role in the choice of chemotherapy. Despite this, one can also imagine a role for functional CSRAs. This is currently being attempted for various cancers(23–26), although it is controversial whether this testing is truly predictive of in vivo response(2, 3). This initial sample took about 8 months from surgical resection to full drug panel testing, however we estimate that, with additional optimization, this testing could be accomplished much shorter time. The shorter time could be achieved by implanting more mice, with additional experience, and most importantly by using robotics for the drug screening. Certain clinical settings, such as identifying drugs after a Whipple procedure for PDA, might be especially amenable to this time frame. Applying a similar approach to a cohort of genetically defined cell lines, could define a standardized core set of drugs to be tested on all resected PDAs.
Cell lines are useful for many other studies of cancer biology since they: i) can be expanded indefinitely, ii) contain all of the gene mutations present in the patient’s primary cancer, iii) can be manipulated in vitro by adding or eliminating genes, iv) can be implanted in mice to test the effects of these manipulations on the ability to form tumors(21, 27), and v) carry few if any additional genetic changes from the primary cancer(28). Cancer cell lines are also important for other in vitro functional studies, molecular imaging, and possibly isogenic cancer vaccine production. With increasingly powerful tools such as whole genome DNA sequencing, RNAi libraries, and high-throughput drug screens, one can anticipate that the need for cell lines from human cancers will likely increase(13, 29–31).
We envision several applications of this system for cancer cell line production. First, there are no cell lines representing some malignancies, such as oligodendrogliomas and pheochromocytomas, and in others, such as esophageal, thyroid, and salivary gland tumors, many cell lines are contaminants (32). For others, such as prostate, ovarian and breast cancers, the number of available cell lines cannot fully represent the full spectrum of disease (e.g. most existing prostate cancer cell lines are hormone refractory). Additionally, some cancers contain unique genetic defects, and successful cell line production is essential to identify pathway members, and perhaps most importantly, for high-throughput drug screening.
There are some limitations of this system. For some cancers, stromal cells from the tumor microenvironment may provide paracrine growth stimuli to the malignant cells, and this may explain why we were unable to isolate two of the cell lines. It is generally believed that all of the human stromal cells are replaced by mouse stromal cells in a single passage in immunodefective mice. While we generally agree with this assessment based on phase microscopy of HAT treated cultures (unpublished data), this has never been formally quantified. The replacement of stromal cells may be slightly less than 100% based on the extremely faint residual PCR product from DNA isolated from xenografts of cancers containing homozygous deletions(33). In this regard, it is possible that some tumors may need to be passaged through additional mice to get complete replacement of the human stromal cells. It is also possible that some cancers require unique cytokines that are not currently components of tissue culture medium.
In addition to cell line production, one can envision other situations where an animal model is required to study a biological process, but where recovery of the exogenous cells is subsequently required. For example, the mouse may be useful for isolation of organ-specific spontaneous metastases or small numbers of dormant malignant cells refractory to chemotherapeutic agents. Finally, high-throughput functional chemosensitivity profiling may be especially warranted for those patients who have failed conventional chemotherapeutic options.
Supplementary Material
Statement of Translational Relevance.
Cancer cell lines are essential for functional studies of cancer. When explanted tumors are grown in vitro, however, non-cancer fibroblasts paradoxically overgrow the malignant cells. Here we report production of a series of immunodeficient hprt-null mice. When human tumors are expanded in these mice, the resultant tumor is composed of human cancer cells and biochemically defective mouse stromal cells. When these tumors are explanted into culture and grown in HAT media, human cancer cell lines can be readily isolated. We demonstrate proof-of-principle of the mice for personalized cell line isolation and chemosensitivity testing. This approach may be used to guide chemotherapy selection in the future.
Acknowledgments
We dedicate this work to the memory of Dr. Dawn Audi. We thank Dr. Scott Kern for the suggestion to use hprt as the selectable marker (PCT/US01/31219), and Dr. Ming-Sound Tsao for generously providing the HPDE cell line. We acknowledge Drs. Yoshihisa Matsushita, Bert Vogelstein, Cynthia Zahnow, Julie Watson, Curt Civin, Cory Brayton, Don Price, Phil Wong, Craig Henke, Mehtab Khan and Rajni Sharma for helpful discussions. This work was funded in part from NIH grants CA130938 (JRE), CA62924 (Drs. Scott Kern, RHH, AM, JRE), CA122581 (RBSR), The Sol Goldman Pancreatic Cancer Research Center, The Stewart Trust Fund, The Lustgarten Foundation, the Mary Lou Wootton Pancreatic Cancer Research Fund, The Michael Rolfe Pancreatic Cancer Foundation and the HERA Foundation (RBSR).
Abbreviations
- HAT
hypoxanthine, aminopterin, thymidine
- HPRT
hypoxanthine phosphoribosyl transferase
- IL2Rγ
interleukin-2 receptor gamma
- JHDL
Johns Hopkins Drug Library
- NOD
non obese diabetic
- NSG
triple immunodeficient, SCID, NOD, IL2Rγ
- PDA
pancreatic ductal adenocarcinoma
- SCID
severe combined immunodeficiency
- 6TG
6-thioguanine
Footnotes
Notes: The hprt-null NSG mice are available from Jackson Labs (Bar Harbor, Maine) as mouse #012480 (NOD.Cg-Prkdc<scid> IL2Rγ<tm1Wjl> Hprt<b-m3>/EshJ).
Author Contributions: AM and JRE conceived the project. HK, SR, JSS, CAK, GF, LH, MK, FWS, MTL, RMB, BK, HL, MEM, GM conducted the experiments. MH, EJ, RHH, HAJ, RBSR, AJ, JOL provided essential reagents and critical oversight.
Potential Conflict of Interest: Drs. Rauenzahn, Maitra and Eshleman may receive royalty payments if the mice are licensed; a patent licensed to Myriad Genetics (Drs. Hruban and Eshleman); and Advisory Board membership in Roche Molecular Diagnostics (Dr. Eshleman). No potential conflicts of interest were declared by other authors.
LITERATURE CITED
- 1.Wright JC, Cobb JP, Gumport SL, Golomb FM, Safadi D. Investigation of the relation between clinical and tissue-culture response to chemotherapeutic agents on human cancer. N Engl J Med. 1957;257:1207–11. doi: 10.1056/NEJM195712192572502. [DOI] [PubMed] [Google Scholar]
- 2.Schrag D, Garewal HS, Burstein HJ, Samson DJ, Von Hoff DD, Somerfield MR. American Society of Clinical Oncology Technology Assessment: chemotherapy sensitivity and resistance assays. J Clin Oncol. 2004;22:3631–8. doi: 10.1200/JCO.2004.05.065. [DOI] [PubMed] [Google Scholar]
- 3.Samson DJ, Seidenfeld J, Ziegler K, Aronson N. Chemotherapy sensitivity and resistance assays: a systematic review. J Clin Oncol. 2004;22:3618–30. doi: 10.1200/JCO.2004.04.077. [DOI] [PubMed] [Google Scholar]
- 4.Dangles-Marie V, Pocard M, Richon S, Weiswald LB, Assayag F, Saulnier P, et al. Establishment of human colon cancer cell lines from fresh tumors versus xenografts: comparison of success rate and cell line features. Cancer Res. 2007;67:398–407. doi: 10.1158/0008-5472.CAN-06-0594. [DOI] [PubMed] [Google Scholar]
- 5.Gazdar AF, Kurvari V, Virmani A, Gollahon L, Sakaguchi M, Westerfield M, et al. Characterization of paired tumor and non-tumor cell lines established from patients with breast cancer. Int J Cancer. 1998;78:766–74. doi: 10.1002/(sici)1097-0215(19981209)78:6<766::aid-ijc15>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 6.Jaffee EM, Schutte M, Gossett J, Morsberger LA, Adler AJ, Thomas M, et al. Development and characterization of a cytokine-secreting pancreatic adenocarcinoma vaccine from primary tumors for use in clinical trials. Cancer J Sci Am. 1998;4:194–203. [PubMed] [Google Scholar]
- 7.Phelps RM, Johnson BE, Ihde DC, Gazdar AF, Carbone DP, McClintock PR, et al. NCI-Navy Medical Oncology Branch cell line data base. J Cell Biochem Suppl. 1996;24:32–91. doi: 10.1002/jcb.240630505. [DOI] [PubMed] [Google Scholar]
- 8.McBain JA, Weese JL, Meisner LF, Wolberg WH, Willson JK. Establishment and characterization of human colorectal cancer cell lines. Cancer Res. 1984;44:5813–21. [PubMed] [Google Scholar]
- 9.Hooper M, Hardy K, Handyside A, Hunter S, Monk M. HPRT-deficient (Lesch-Nyhan) mouse embryos derived from germline colonization by cultured cells. Nature. 1987;326:292–5. doi: 10.1038/326292a0. [DOI] [PubMed] [Google Scholar]
- 10.Jinnah HA, Page T, Friedmann T. Brain purines in a genetic mouse model of Lesch-Nyhan disease. J Neurochem. 1993;60:2036–45. doi: 10.1111/j.1471-4159.1993.tb03488.x. [DOI] [PubMed] [Google Scholar]
- 11.Littlefield JW. Selection of Hybrids from Matings of Fibroblasts in Vitro and Their Presumed Recombinants. Science. 1964;145:709–10. doi: 10.1126/science.145.3633.709. [DOI] [PubMed] [Google Scholar]
- 12.Kohler G, Milstein C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature. 1975;256:495–7. doi: 10.1038/256495a0. [DOI] [PubMed] [Google Scholar]
- 13.Chong CR, Xu J, Lu J, Bhat S, Sullivan DJ, Jr, Liu JO. Inhibition of angiogenesis by the antifungal drug itraconazole. ACS Chem Biol. 2007;2:263–70. doi: 10.1021/cb600362d. [DOI] [PubMed] [Google Scholar]
- 14.Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Angenendt P, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321:1801–6. doi: 10.1126/science.1164368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang H, Qian DZ, Tan YS, Lee K, Gao P, Ren YR, et al. Digoxin and other cardiac glycosides inhibit HIF-1alpha synthesis and block tumor growth. Proc Natl Acad Sci U S A. 2008;105:19579–86. doi: 10.1073/pnas.0809763105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chang FR, Hayashi K, Chen IH, Liaw CC, Bastow KF, Nakanishi Y, et al. Antitumor agents. 228. five new agarofurans, Reissantins A–E, and cytotoxic principles from Reissantia buchananii. J Nat Prod. 2003;66:1416–20. doi: 10.1021/np030241v. [DOI] [PubMed] [Google Scholar]
- 17.Phillips PA, Dudeja V, McCarroll JA, Borja-Cacho D, Dawra RK, Grizzle WE, et al. Triptolide induces pancreatic cancer cell death via inhibition of heat shock protein 70. Cancer Res. 2007;67:9407–16. doi: 10.1158/0008-5472.CAN-07-1077. [DOI] [PubMed] [Google Scholar]
- 18.Titov DV, Gilman B, He QL, Bhat S, Low WK, Dang Y, et al. XPB, a subunit of TFIIH, is a target of the natural product triptolide. Nat Chem Biol. 2011;7:182–8. doi: 10.1038/nchembio.522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sharma SV, Haber DA, Settleman J. Cell line-based platforms to evaluate the therapeutic efficacy of candidate anticancer agents. Nat Rev Cancer. 2010;10:241–53. doi: 10.1038/nrc2820. [DOI] [PubMed] [Google Scholar]
- 20.Armstrong MD, Von Hoff D, Barber B, Marlow LA, von Roemeling C, Cooper SJ, et al. An effective personalized approach to a rare tumor: prolonged survival in metastatic pancreatic acinar cell carcinoma based on genetic analysis and cell line development. J Cancer. 2011;2:142–52. doi: 10.7150/jca.2.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rubio-Viqueira B, Jimeno A, Cusatis G, Zhang X, Iacobuzio-Donahue C, Karikari C, et al. An in vivo platform for translational drug development in pancreatic cancer. Clin Cancer Res. 2006;12:4652–61. doi: 10.1158/1078-0432.CCR-06-0113. [DOI] [PubMed] [Google Scholar]
- 22.Hidalgo M, Bruckheimer E, Rajeshkumar NV, Garrido-Laguna I, De Oliveira E, Rubio-Viqueira B, et al. A pilot clinical study of treatment guided by personalized tumorgrafts in patients with advanced cancer. Molecular cancer therapeutics. 2011;10:1311–6. doi: 10.1158/1535-7163.MCT-11-0233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Eltabbakh GH, Piver MS, Hempling RE, Recio FO, Lele SB, Marchetti DL, et al. Correlation between extreme drug resistance assay and response to primary paclitaxel and cisplatin in patients with epithelial ovarian cancer. Gynecol Oncol. 1998;70:392–7. doi: 10.1006/gyno.1998.5109. [DOI] [PubMed] [Google Scholar]
- 24.Fruehauf JP, Alberts DS. Assay-assisted treatment selection for women with breast or ovarian cancer. Recent Results Cancer Res. 2003;161:126–45. doi: 10.1007/978-3-642-19022-3_12. [DOI] [PubMed] [Google Scholar]
- 25.Furukawa T, Kubota T, Hoffman RM. Clinical applications of the histoculture drug response assay. Clin Cancer Res. 1995;1:305–11. [PubMed] [Google Scholar]
- 26.Ugurel S, Schadendorf D, Pfohler C, Neuber K, Thoelke A, Ulrich J, et al. In vitro drug sensitivity predicts response and survival after individualized sensitivity-directed chemotherapy in metastatic melanoma: a multicenter phase II trial of the Dermatologic Cooperative Oncology Group. Clin Cancer Res. 2006;12:5454–63. doi: 10.1158/1078-0432.CCR-05-2763. [DOI] [PubMed] [Google Scholar]
- 27.Jimeno A, Amador ML, Kulesza P, Wang X, Rubio-Viqueira B, Zhang X, et al. Assessment of celecoxib pharmacodynamics in pancreatic cancer. Molecular cancer therapeutics. 2006;5:3240–7. doi: 10.1158/1535-7163.MCT-06-0565. [DOI] [PubMed] [Google Scholar]
- 28.Jones S, Chen WD, Parmigiani G, Diehl F, Beerenwinkel N, Antal T, et al. Comparative lesion sequencing provides insights into tumor evolution. Proc Natl Acad Sci U S A. 2008;105:4283–8. doi: 10.1073/pnas.0712345105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wood LD, Parsons DW, Jones S, Lin J, Sjoblom T, Leary RJ, et al. The genomic landscapes of human breast and colorectal cancers. Science. 2007;318:1108–13. doi: 10.1126/science.1145720. [DOI] [PubMed] [Google Scholar]
- 30.Hannon GJ. RNA interference. Nature. 2002;418:244–51. doi: 10.1038/418244a. [DOI] [PubMed] [Google Scholar]
- 31.Hudson TJ, Anderson W, Artez A, Barker AD, Bell C, Bernabe RR, et al. International network of cancer genome projects. Nature. 2010;464:993–8. doi: 10.1038/nature08987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Boonstra JJ, van Marion R, Beer DG, Lin L, Chaves P, Ribeiro C, et al. Verification and Unmasking of Widely Used Human Esophageal Adenocarcinoma Cell Lines. J Natl Cancer Inst. 2010 doi: 10.1093/jnci/djp499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hahn SA, Seymour AB, Hoque AT, Schutte M, da Costa LT, Redston MS, et al. Allelotype of pancreatic adenocarcinoma using xenograft enrichment. Cancer Res. 1995;55:4670–5. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.