

Abstract
Smoking is a leading global cause of disease and mortality1. We performed a genomewide meta-analytic association study of smoking-related behavioral traits in a total sample of 41,150 individuals drawn from 20 disease, population, and control cohorts. Our analysis confirmed an effect on smoking quantity (SQ) at a locus on 15q25 (P=9.45e-19) that includes three genes encoding neuronal nicotinic acetylcholine receptor subunits (CHRNA5, CHRNA3, CHRNB4). We used data from the 1000 Genomes project to investigate the region using imputation, which allowed analysis of virtually all common variants in the region and offered a five-fold increase in coverage over the HapMap. This increased the spectrum of potentially causal single nucleotide polymorphisms (SNPs), which included a novel SNP that showed the highest significance, rs55853698, located within the promoter region of CHRNA5. Conditional analysis also identified a secondary locus (rs6495308) in CHRNA3.
Smoking behavior and Nicotine Dependence (ND) are multifactorial traits with substantial genetic influences2. There is an urgent need to better understand the molecular neurobiology of ND, in order to design targeted, more effective therapies3. Recently, genome-wide association scans (GWAS) have established one locus in ND and Smoking Quantity (SQ), which implicates a cluster of three genes encoding neuronal nicotinic acetylcholine receptor subunits, CHRNA5, CHRNA3, and CHRNB4, on chromosome 15q254–8. The locus is also associated with lung cancer7,9,10, peripheral arterial disease7, and chronic obstructive pulmonary disease and lung function11.
We initially performed a GWAS meta-analytic study of smoking-related traits in a total sample of 41,150 individuals of white European descent, sourced from multiple disease, population and control cohorts (Table 1, Supplementary Table 1, Online Methods). As the cohorts were genotyped on a variety of different genome-wide SNP arrays (Table 1, Supplementary Table 1), we first imputed genotypes for all datasets12, for all SNPs in the HapMap version release 2213.
Table 1.
Summary information for the cohorts used in meta-analysis. Further details are given in Online Methods and Supplementary Table 1.
| Label | Description | Genotyping | Sample Sizes | |||||
|---|---|---|---|---|---|---|---|---|
| All | CPD>0 | Ever | Never | Current | Non- current |
|||
| WTCCC-RA | Rheumatoid Arthritis cases | Affymetrix 500K | 1860 | n/a | n/a | n/a | 262 | 558 |
| EPIC | Obesity case-control | Affymetrix 500K | 3516 | n/a | 1927 | 1589 | 353 | 1574 |
| WTCCC-HT | Hypertension cases | Affymetrix 500K | 1952 | 830 | n/a | n/a | 1274 | 672 |
| GEMS | Dyslipidemia case-control | Affymetrix 500K | 1847 | 862 | 910 | 793 | 268 | 642 |
| GSK-COPD | COPD case-control | Illumina 550 | 1633 | 1632 | n/a | n/a | 725 | 905 |
| GSK-BIPOLAR | Bipolar depression case-control | Illumina 550 | 1805 | 944 | 1008 | 790 | 498 | 510 |
| GSK-UPD | Unipolar depression case-control | Illumina 550 | 1792 | 899 | 935 | 856 | 503 | 432 |
| WTCCC-IBD | Crohn's disease cases | Affymetrix 500K | 1748 | n/a | 713 | 540 | 713 | 420 |
| KORA | Population-based | Affymetrix 500K | 1644 | 253 | 811 | 831 | 217 | 1425 |
| KORCULA | Population-based | Illumina 300 | 827 | n/a | 376 | 451 | 179 | 654 |
| LOLIPOP | Population-based | Affymetrix 500K | 1288 | 650 | 653 | 635 | 258 | 395 |
| MEDSTAR | Coronary Artery Disease case-control | Affymetrix 6.0 | 1322 | 820 | 853 | 469 | 300 | 553 |
| ORCADES | Population-based | Illumina 300 | 692 | n/a | 288 | 404 | 60 | 632 |
| PENNCATH | Coronary Artery Disease case-control | Affymetrix 6.0 | 1401 | n/a | n/a | n/a | 464 | 612 |
| POPGEN | Population-based | Affymetrix 6.0 | 1107 | 573 | 495 | 608 | n/a | n/a |
| CoLaus | Population-based | Affymetrix 500K | 5636 | 3132 | 3357 | 2275 | 1485 | 1872 |
| SardiNIA | Population-based | Affymetrix 500+10K | 4,305 | 1731 | 1743 | 2562 | 873 | 3432 |
| SHIP | Population-based | Affymetrix 6.0 | 4080 | 2011 | 2631 | 1449 | 1240 | 2840 |
| VIS | Population-based | Illumina 300 | 769 | n/a | 441 | 328 | 212 | 557 |
| WTCCC-CAD | Coronary Artery Disease cases | Affymetrix 500K | 1926 | 1237 | 1457 | 461 | 239 | 1218 |
| TOTALS | 41150 | 15574 | 18598 | 15041 | 10123 | 19903 | ||
The main focus of our analysis was on SQ within current or past smokers, as a semi-quantitative trait based on the self-reported variable of Cigarettes-per-Day (CPD)7. We performed association analysis separately within each cohort under an additive model, using covariate effects for age and sex, disease case/control status where applicable, and other cohort-specific covariates (Supplementary Table 1). The meta-analysis was then carried out by combining study-specific β- estimates using a fixed effects model14. In total, 15,574 subjects reported CPD values >0 and were used for meta-analysis of SQ (Table 1, Supplementary Table 1). We followed up our most promising association findings by comparing them with results from two concurrent GWAS meta-analyses of smoking; the ENGAGE study of 46,481 subjects15, and the TAG study of 74,035 subjects16. We also made our meta-analysis results available to the authors of those studies to check their top findings for replication.
Our meta-analysis of SQ identified the CHRNA5/CHRNA3 locus on 15q25 as the single outstandingly significant locus in the genome (Figure 1, Table 2, Supplementary Table 2), with a minimum P=9.45e-19 for rs1051730, which has been a SNP commonly reported4–8, and very low P values for many other SNPs in the region (Supplementary Figure 1, Supplementary Table 2). All cohorts in the analysis contributed at least somewhat to the 15q25 association (Supplementary Figure 1). Each copy of the ‘high-smoking’ A allele (34% frequency) had a quantitative effect size on SQ of 0.079 (95% CI 0.070–0.088) which is inline with previous estimates7. Joint analysis of our total dataset together with TAG and ENGAGE, for rs1051730, yielded P=1.71E-66 (Table 2).
Figure 1.
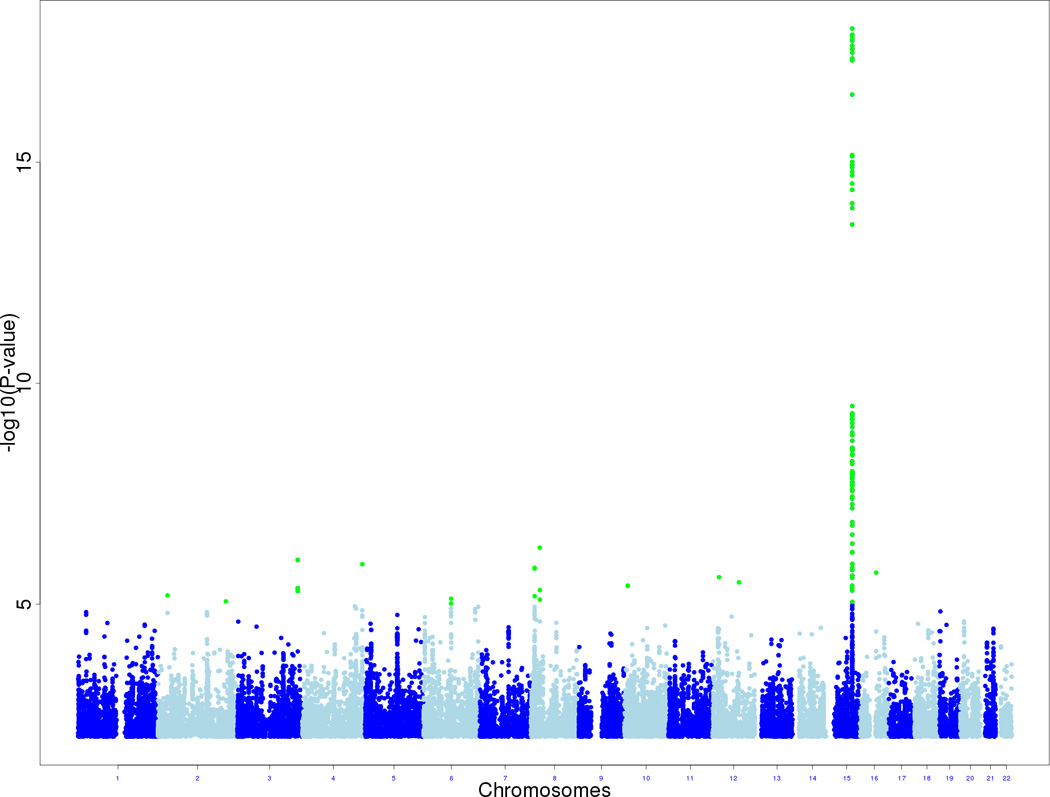
Manhattan plot showing the significance of association of all SNPs in genome-wide SQ meta-analysis. SNPs are plotted on the x-axis according to their positions on each chromosome, against association with SQ on the y-axis (−log10 P-value). SNPs with p-values less than 1.0E-05 are highlighted in green.
Table 2.
Summary information for selected SNPs at 15q25 from meta-analysis of association with the Smoking Quantity (SQ) phenotype. Our study is referred to as OX-GSK. Information for all SNPs spanning the 15q25 locus in our genomewide analysis is given in Supplementary Table 2.
| SNP (rs ID) | Chr | Position | Coded Allele |
Coded Allele MAF |
OX-GSK | TAG | ENGAGE | Combined | |||
|---|---|---|---|---|---|---|---|---|---|---|---|
| P-value | heterozygosity P-value |
P-value | P-value | P-value | Beta | SE | |||||
| rs588765 | 15 | 76652480 | T | 0.43 | 1.74×10−3 | 0.50 | NA | NA | NA | NA | NA |
| rs16969968 | 15 | 76669980 | G | 0.65 | 1.64×10−18 | 0.86 | 1.85×10−27 | 1.53×10−23 | 4.29×10−65 | −0.078 | 0.0046 |
| rs1051730 | 15 | 76681394 | G | 0.66 | 9.45×10−19 | 0.68 | 3.62×10−27 | 9.98×10−25 | 1.71×10−66 | −0.079 | 0.0046 |
| rs6495308 | 15 | 76694711 | T | 0.77 | 3.30×10−10 | 0.10 | 7.99×10−24 | 1.60×10−13 | 5.82×10−44 | 0.073 | 0.0052 |
Multiple variants at the 15q25 locus have been suggested to underlie its effect, including a non-synonymous SNP in CHRNA5, together with variants that affect mRNA expression levels17–19. We decided to use our very large sample, in combination with data from the 1000 Genomes Project (see URL below), to perform fine mapping and modeling of the 15q25 locus in relation to SQ. We reasoned that, with the near complete information on common variants derived from 1000 Genomes, it might be possible to pinpoint a variant, or combination of variants, that can explain all the signal of association at 15q25. We used data from 108 estimated CEU haplotypes from the April 2009 release of the 1000 Genomes Pilot 1 data. This contained 2189 SNPs in our region of interest (See Online Methods), approximately a five-fold increase in coverage compared to 437 SNPs in release 22 of the HapMap. By imputing genotypes for all SNPs across this locus from 1000 Genomes, and repeating the meta-analysis, we found that the most significant association was with a novel and previously untested SNP, not in the HapMap, located within the 5' untranslated region (UTR) of CHRNA5, which makes it a candidate for affecting mRNA transcription (rs55853698, P = 1.31E-16; Figure 2). The p-value for the commonly reported SNP rs1051730 in this analysis was similar but a little higher, P=1.47E-15. (P values for our 1000 Genomes analysis are generally higher than our HapMap-based analysis because not all cohorts were included in the 1000 Genomes imputation - see Online Methods.) SNP rs55853698 is a G/T substitution where the G allele has a frequency ranging from 0.313 to 0.378 across the various cohorts.
Figure 2.
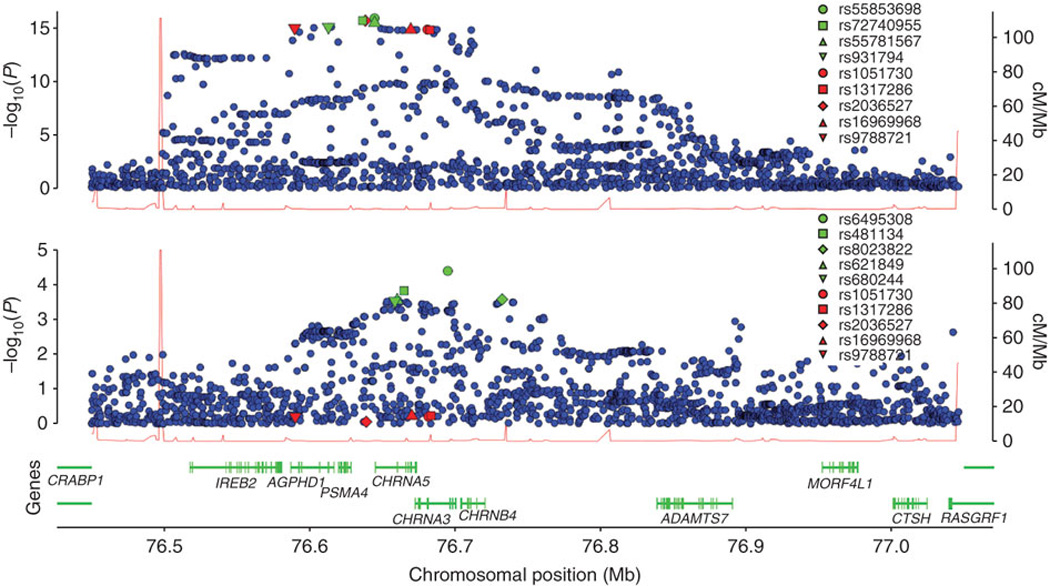
Chromosome 15q25 signal plots. Top: Signal plot based on 1000 Genomes imputation and meta-analysis of SQ association. SNPs are plotted by their positions on the chromosome, against association with SQ (−log10 p-value) on the left Y-axis. The five SNPs with the lowest p-values from the HapMap imputation are highlighted in red. The five SNPs with the lowest p-values from the 1000 Genomes imputation are highlighted in green (unless already coloured red). The rs identities of highlighted SNPs are given in the box. Recombination rates across the region are shown by the red line plotted against the right y-axis. Middle: Chromosome 15q25 signal plot based on 1000 Genomes imputation and meta-analysis of SQ association, conditional on the SNP rs55853698. The five SNPs with the lowest p-values from the conditional analysis are highlighted in green. The five SNPs with the lowest p-values from the unconditioned HapMap-imputation analysis are highlighted in red. Bottom: Genes and the positions of exons (using data from the UCSC genome browser; URL is given below).
To investigate whether the association at 15q25 can be explained completely by rs55853698, we carried out tests of association for all SNPs spanning the CHRNA5/CHRNA3 locus conditional upon this SNP (Figure 2). Residual association was still detected at many SNPs in the region, with the most significant signal occurring at rs6495308 (P= 3.96E-05), located within an intron of CHRNA3 (Figure 2). In unconditioned analysis rs6495308 has a marginal association in the meta-analysis of P=3.30E-10. Further conditioning on rs6495308, after already conditioning on rs55853698, leaves no obvious signal of association in the region (Supplementary Figure 2), suggesting that these two SNPs together could be sufficient to explain this genetic effect.
Wang et al.18 suggested that a non-synonymous SNP rs16969968, in CHRNA5, is functional for ND risk (and lung cancer risk), but also that variants that cause high expression of CHRNA5 mRNA, tagged by SNP rs588765, increase the risk for ND independently. The marginal p-values of rs16969968 and rs588765 in our meta-analysis were P=1.64E-18 and P=1.74E-03. Conditional analysis on rs16969968 within our cohorts still left residual association within the region (Supplementary Figure 2), with the most significant signal again occurring at rs6495308 (P=1.54E-05). Conditioning on both rs16969968 and rs588765, i.e. the proposed combination of Wang et al.18, leaves no obvious signal of association (Supplementary Figure 2). To further investigate which pair of SNPs best explains the signal of association we used the Bayesian Information Criteria (BIC) measure of model fit 20. For the model of Wang et al.18, i.e. conditioning on both rs16969968 and rs588765, we obtained BIC = 22719.87, posterior probability 0.15. For the model conditioning on the novel promoter SNP rs55853698, and rs6495308, we obtained BIC = 22716.49, posterior probability 0.85, which indicates a better model fit.
Examination of the LD structure between the SNPs that we have considered shows that rs1051730, rs16969968, and rs55853698 are all close tagging proxies of each other (all pairwise R2 > 0.96). These variants tag, or cause, the principal risk for high SQ attributable to the 15q25 locus, but the high LD makes it difficult to assign causality. The ‘residual association’ SNPs rs588765 and rs6495308 are in low LD with each other (R2 = 0.21), and are both only in modest LD with the principal SNPs (maximum R2 = 0.47). It is not therefore clear that this locus can be completely understood in the way proposed by Wang et al.18. While the non-synonymous SNP in CHRNA5, rs16969968, may be important, we have identified a novel and potentially functional SNP in the 5' UTR of this gene that is a close proxy to the non-synonymous SNP in terms of LD, but shows a slightly more significant association in our meta-analysis. Then, while rs588765 can explain much of the secondary or residual association at this locus, we find that a largely independent variant within CHRNA3, rs6495308, is the best tagger of the residually associated variation, while also contributing to a better fitting 2-SNP model, and having a much stronger marginal significance in unconditioned analysis (P=3.30E-10 for rs6495308 compared to P=1.74E-03 for rs588765).
Our analysis has, for the first time, surveyed virtually all of the common variants in the 15q25 region, and provides one of the first examples of how data from the 1000 Genomes Project can contribute new information to mapping and characterizing loci for complex traits. We recommend that further analysis of this locus should not be limited in focus to CHRNA5, nor particularly to the common, non-synonymous SNP rs16969968. It is notoriously difficult to distinguish functional variation in the context of high LD across a region21. There are numerous ways in which variants can be functional, including expression regulatory changes that affect close or distant genes, epigenetic changes, splicing effects, alterations to microRNA binding sites, or non-coding RNAs21. It is also conceivable that association with common variants can arise through the effects of multiple rarer variants that happen to be relatively restricted to specific haplotype backgrounds.
The second strongest association within the genome in our meta-analysis, for SQ, was at a locus on 8p21 that received modest support from the TAG and ENGAGE studies (Supplementary Table 2, Supplementary Figure 3; P=5.26E-07 for rs11782673). This locus would not survive correcting for genome-wide multiple testing, although it is noteworthy that the locus spans another neuronal nicotinic acetylcholine receptor subunit gene, CHRNA2.
In addition to our analysis of SQ, we also tested genome-wide for allelic differences between those who reported currently smoking, or smoking in the past, versus those who said they had never been smokers (the EVER/NEVER phenotype; sample sizes in Table 1, Supplementary Table 1). This was in order to identify genetic effects on the establishment of a smoking habit. No locus achieved genome-wide significance, and none of the top 15 loci showed evidence for replication (Supplementary Table 2, Supplementary Figure 4). Likewise, no consistent results emerged when we tested for allelic differences between those who reported currently smoking versus those who had smoked in the past but had stopped at the time of interview (Supplementary Table 2, Supplementary Figure 4). When age-adjusted, this is a rough measure of smoking cessation.
Our study identified association at some loci which, while not reaching genomewide significance in our own meta-analysis, supported findings from the concurrent TAG and ENGAGE studies15,16. These include novel loci on chromosomes 8 and 19 for SQ, 11 for EVER/NEVER, and 9 for Current/Non-Current15,16. These findings have provided further novel insights into the biology of smoking behavior.
ONLINE METHODS
Study samples
Study collections and their basic characteristics are listed in Table 1 and Supplementary Table 1. Subjects used in our analysis were adults of white European descent. Summary descriptions of the collections are given below, together with primary citations that describe the collections fully. Data were used in accordance with the ethical permissions and consents relating to each collection.
GEMS22: The Genetic Epidemiology of Metabolic Syndrome (GEMS) study consists of dyslipidaemic cases (age 20–65 years) matched with normolipidaemic controls by sex and recruitment site, drawn from non-Mediterranean subjects of the Genetic Epidemiology of Metabolic Syndrome study (Finland, Switzerland, Canada, Australia, USA).
CoLaus23: The Cohorte Lausannoise (CoLaus) is a single-center, cross-sectional population-based study, including individuals aged 35 to 75 years randomly selected from the list of residents of the city of Lausanne (Switzerland).
GSK COPD11: This collection includes cases with chronic obstructive pulmonary disease diagnosed according to Global Initiative of Chronic Obstructive Lung Disease (GOLD) criteria, and unaffected controls recruited from Bergen, Norway.
GSK UPD24: This collection includes cases with recurrent major depression according to DSM-IV criteria and age- and gender-matched non-affected controls, recruited at the Max-Planck Institute of Psychiatry in Munich, Germany; patients were also recruited at two satellite recruiting hospitals (BKH Augsburg and Klinikum Ingolstadt) in the Munich area.
GSK Bipolar25: The Bipolar collection includes DSM-IV Bipolar cases and controls from subjects recruited at 3 study sites: the Institute of Psychiatry (IOP) in London, U.K.; the Centre for Addiction and Mental Health in Toronto, Canada; and the University of Dundee, U.K.
GSK Lolipop26: The London Life Sciences Prospective Population (LOLIPOP) is a population based study including Indian Asian and European white men and women recruited from the lists of 58 General Practitioners in West London.
GSK Medstar27: The MedStar cohort includes cases with acute coronary syndrome or chronic coronary artery disease from Washington DC, and unaffected controls.
PennCath27: The Penn-CATH cohort is a University of Pennsylvania Medical Center based angiographic study, from which cases with coronary artery disease (CAD) and controls with no evidence of CAD at the coronary angiography were derived.
EPIC28: The EPIC-Obesity cohort is a case-control cohort for obesity drawn from the EPIC-Norfolk cohort, which includes white European men and women aged 39–79 years recruited in Norfolk, UK.
KORA29: The Co-operative Health Research in the Region of Augsburg (KORA) study is an epidemiological survey of the general population living in the city of Augsburg, Southern Germany, and two adjacent counties.
WTCCC HT30: The WTCCC-HT collection comprises severely hypertensive probands ascertained from families with multiple affected members in the UK as part of the BRIGHT study.
WTCCC CAD, WTCCC CD, WTCCC RA30: include patients with Coronary Artery Disease, Chrohn’s disease and Rheumatoid Arthritis from the Wellcome Trust Case Control Consortium Study.
POPGEN study31: The Population Genetic Cohort (POPGEN) is a cross sectional epidemiological surveys of regional German populations from Schleswig-Holstein, northern Germany.
SHIP Study32: The Study of Health in Pomerania (SHIP) is a longitudinal, population-based survey from West Pomerania, Germany. Data from the baseline cohort were used for this study.
VIS Study33: This study includes unselected Croatians, aged 18–93 years, recruited from the villages of Vis and Komiza on the Dalmatian island of Vis.
ORCADES Study34: The Orkney Complex Disease Study is a family-based, cross-sectional study that seeks to identify genetic factors influencing cardiovascular and other disease risk in the population isolate of the Orkney Isles in northern Scotland.
KORCULA Study35: The KORCULA study includes healthy volunteers aged 18 and over from the villages of Lumbarda, Žrnovo, and Račišće on the Island of Korcula, Croatia.
SardiNIA Study36: The SardiNIA is a population-based longitudinal cohort study that includes male and female related individuals, aged 14 years and above, from a cluster of four towns in the Ogliastra province of Sardinia, Italy.
Genotyping, quality control and imputation
Supplementary Table 1 lists the various genotype platforms used for each cohort, genotype calling algorithms, SNP and sample quality control, and details of the imputation and association analysis software used. The quality control measures from previous analyses of each cohort were adopted for this study and are detailed in the table. We used NCBI Build 36 co-ordinates for SNP base-pair positions so that all the cohorts could be combined seamlessly.
We imputed all SNPs reported in the CEU sample in HapMap Phase II using various imputation algorithms12,37 (see the URL section for a link to the software ProbABEL). Imputations were performed after excluding samples and SNPs that did not meet the study-specific quality control criteria. Genotypes were imputed for SNPs not present in the genome-wide arrays or for those where genotyping had failed to meet the QC criteria.
Only imputed SNPs with good imputation quality were included in the meta-analysis. This was defined as proper_info≥0.5 (for studies analysed with IMPUTE/SNPTEST12) or rsq-hat≥0.5 (for studies analysed using MACH37) and Imp_info≥0.5 (for studies analysed using ProbABEL).
Derivation of smoking phenotypes
We used the categorical SQ levels defined by Thorgeirsson et al.7. The SQ levels were 0 (1–10 cigarettes per day), 1, (11–20), 2 (21–30) and 3 (31 or more). Each increment represents an increase in SQ of 10 cigarettes per day. Most of the cohorts in our study have maximal CPD recorded on each sample but a few have collected average CPD (Supplementary Table 1). We examined the distributions of CPD across cohorts and found no large differences between those cohorts with average or maximal CPD. The mean and standard deviation of the CPD measurements in each cohort are given in Supplementary Table 1. The Ever/Never and Current/Non-current phenotypes used were those collected by the individual cohorts. Not all cohorts had all three phenotypes collected. Precise details of the phenotypes collected in each cohort are given in Supplementary Table 1. An assessment would typically be questionnaire-based, following a structure such as:
-
Tick the option that best describes you:
-
-
I smoke now
-
-
I don’t smoke now. I have stopped for … years.
-
-
I have never smoked
-
-
About how many cigarettes do you or did you smoke per day?
Put the number of years you have smoked.
Statistical Analysis and Meta-analysis
Each cohort was analyzed separately for each of the 3 phenotypes considered. The majority of the analysis was carried out on the raw genotype data in Oxford but some cohorts (SardiNIA, VIS, KORCULA, ORCADES, SHIP) carried out their own analysis and submitted results for the meta-analysis. For the binary traits (Ever/Never, Current/Non-Current) tests for additive genetic effects on the logodds scale were carried out using logistic regression. For the categorical SQ phenotype, tests for additive genetic effects were carried out on a linear scale using linear regression. The programs SNPTEST, probABEL and MERLIN were used on the various cohorts to fit these models taking account of the genotype uncertainty at imputed SNPs. All tests conditioned on Sex and Age and for some cohorts other covariates of self-reported ancestry, country of origin or PCA-derived covariates were included (a complete list is given in Supplementary Table 1). A Genomic Control (GC) lambda estimate was calculated for each phenotype and each cohort (Supplementary Table 3).
The meta-analysis was carried out by combining study-specific β-estimates using a fixed effects model14 using the inverse of the variance of the study-specific β-estimates to weight the contribution of each study. The variance of each cohort’s β-estimate was multiplied by the GC lambda estimate to correct for observed inflation38. Specifically,
where βi and λi are the β-estimate, β-estimate variance and GC lambda estimate for the ith cohort. This method is appropriate when the same phenotype and measurement scale are used in each cohort and has the advantage that measures of effect size (eβ is an estimate of the Odds Ratio of the risk allele) and its standard error can be calculated. We also repeated the analysis of SQ by combining Z-scores from each cohort weighted by their sample size38 and obtained almost identical results. All meta-analysis was carried out using the SNPMETA program (see URL list). After performing each meta-analysis the overall lambda estimate for each phenotype was: SQ 1.0145, Ever/Never 1.002, Current/Non-Current 0.998. For each SNP we also calculated a p-value for the heterogeneity across the studies38.
SNP selection for replication
In collaboration with two other groups carrying out similar meta-analysis of smoking related traits (ENGAGE15 and TAG16) we agreed to an in-silico replication strategy in which for each phenotype (SQ, EVER/NEVER, CURRENT/NON-CURRENT) each group would select 15 regions of the genome showing evidence for association, and summary data (p-values, β-estimate, β-estimate variances, sample sizes, GC-lambda estimates and sample sizes) would be shared across groups to facilitate replication. We selected the top 15 regions for each phenotype based on the p-values we obtained in our own meta-analysis. We excluded regions in which only a small number of cohorts contributed to the study because the information measure at the SNPs in the excluded cohorts were below our thresholds, or where the heterogeneity between the studies was high. Each selected region consisted of several SNPs showing evidence of association in our meta-analysis with p-values below 1e-5. For each of the three phenotypes the results from all the cohorts in all three concurrent studies were combined together using the same GC-corrected inverse-variance meta-analysis method described above. A full list of the selected regions and the summary information from all 3 phenotypes is given in Supplementary Table 2.
1000 Genomes imputation analysis of the 15q22 associated region for SQ
We used 108 estimated CEU haplotypes from the April 2009 release of the 1000 Genomes Pilot 1 data to carry out our fine-mapping experiments at the 15q25 locus (see the URL list for a link to the data source). We used these haplotypes to carry out imputation in the interval 76.4–77.0Mb on chr15 in 12 of the cohorts (GSK-Bipolar, GSK-Unipolar, GSK-COPD, KORA, POPGEN, Lausanne, GSKLolipop, GSK-GEMS, Medstar, SHIP, WTCCC-CAD and WTCCC-HT) using the program IMPUTE12. This release contains 2189 SNPs in this interval compared to 437 SNPs in release 22 of the HapMap data. Meta-analysis of the imputed data was then carried out in the same way as described above. An important technical detail when carrying out imputation using the 1000 Genomes haplotype data is how to align it with the genotype data from genome-wide studies. The program IMPUTE aligns SNPs between the haplotype and genotype data based on base-pair position (and not using SNP identifiers such as rs IDs) so as long as the same co-ordinate system is used for both the haplotype and genotype data the alignment is automatic.
Conditional analysis and modeling
The analysis conditional upon SNPs was carried out using all of the centrally analyzed cohorts (Bipolar, Unipolar, COPD, KORA, POPGEN, Lausanne, LOLIPOP, GEMS, MEDSTAR, SHIP, WTCCC-CAD and WTCCC-HT). At the SNP being conditioned upon we used expected genotype counts as this allowed us to combine data from cohorts which had imputed the SNP and cohorts which had genotyed the SNP. These expected counts where included into the baseline null model as an additional covariate along with the other covariates such as Age, Sex and covariates coding for population structure. The same method was used when conditioning upon two SNPs. The model selection analysis of the two pairs of SNPs in the 15q25 region was carried out using the expected genotype counts. Analysis was carried out using the R statistical package.
Supplementary Material
ACKNOWLEDGEMENTS
GlaxoSmithKline plc (GSK), a pharmaceuticals company that is interested to develop novel cessation therapies for smoking, funded a postdoctoral fellowship for JL at Oxford University. GSK also funded the collection, characterization, and in some cases the genotyping and genotype data preparation for several of the cohorts used in this study. Allen Roses and Paul Matthews played crucial roles in establishing and funding the Medical Genetics activities at GSK. Acknowledgements that are specific to individual cohorts are given in the Supplementary Note.
Footnotes
AUTHOR CONTRIBUTIONS
JZL carried out most of the analysis for this study. JM and CF conceived and directed this study and wrote the manuscript. FT, DMW and VM were involved in study design and helped to coordinate the inclusion of many of the GSK cohorts. SGP, PM, LM, WB, CWK, XY, GW, PV, MP, NJW, JHZ, RJFL, IB, KK, SG, PB, RM, AK, RM, JBV, JS, JLK, AF, PM, RD, KM, PB, AG, SL, MI, TB, SH, HEW, RR, ND, CL, OP, LZ, JH, SC, JK, JCC, MSB, JMD, ADP, KMK. LS, JML, RW, SE, JFW, SHW, HC, VV, MPR, ML, LQ, RW, WM, HHH, DJR, AF, MW, AS, MU, AT, XX, FB, PS, DS, DStC, DR, GRA, HJG, AT, HV, AP, UJ, IR, CH, AFW, IK. BJW, JRT, AJB, ASH, NJS, CAA, TA, CGM, MP, JS, MC, PBM, MF, AD, JW, WT, SE, AB, & WTCCC prepared and shared datasets and, in some cases, cohort-specific results from their own primary analysis.
COMPETING INTERESTS STATEMENT
FT, CF, DMW, VM, PM, SGP, CWK are/were full time employees of the company GlaxoSmithKline (GSK). GSK also funded several aspects of the study as detailed in ACKNOWLEDGEMENTS. There were no competing interests arising from GSK’s involvement in this study.
URLs
ProbABEL software: http://mga.bionet.nsc.ru/yurii/ABEL/
SNPMETA software: http://www.stats.ox.ac.uk/~marchini/software/gwas/snpmeta.html
1000 Genomes Project: http://www.1000genomes.org/
April 2009 release of the 1000 Genomes Pilot 1 data: ftp://ftp-trace.ncbi.nih.gov/1000genomes/ftp/pilot_data/release/2009_04/
UCSC Genome Browser: http://genome.ucsc.edu/
References
- 1.Ezzati M, Lopez AD, Rodgers A, Vander Hoorn S, Murray CJ. Selected major risk factors and global and regional burden of disease. Lancet. 2002;360:1347–1360. doi: 10.1016/S0140-6736(02)11403-6. [DOI] [PubMed] [Google Scholar]
- 2.Li MD. The genetics of nicotine dependence. Curr.Psychiatry Rep. 2006;8:158–164. doi: 10.1007/s11920-006-0016-0. [DOI] [PubMed] [Google Scholar]
- 3.Benowitz NL. Neurobiology of nicotine addiction: implications for smoking cessation treatment. Am J Med. 2008;121:S3–S10. doi: 10.1016/j.amjmed.2008.01.015. [DOI] [PubMed] [Google Scholar]
- 4.Berrettini W, et al. [alpha]-5//[alpha]-3 nicotinic receptor subunit alleles increase risk for heavy smoking. Mol Psychiatry. 2008;13:368–373. doi: 10.1038/sj.mp.4002154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bierut LJ, et al. Novel genes identified in a high-density genome wide association study for nicotine dependence. Human Molecular Genetics. 2007;16:24–35. doi: 10.1093/hmg/ddl441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li MD. Identifying susceptibility loci for nicotine dependence: 2008 update based on recent genome-wide linkage analyses. Hum Genet. 2008;123:119–131. doi: 10.1007/s00439-008-0473-0. [DOI] [PubMed] [Google Scholar]
- 7.Thorgeirsson TE, et al. A variant associated with nicotine dependence, lung cancer and peripheral arterial disease. Nature. 2008;452:638–642. doi: 10.1038/nature06846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Caporaso N, et al. Genome-wide and candidate gene association study of cigarette smoking behaviors. PLoS One. 2009;4:e4653. doi: 10.1371/journal.pone.0004653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Amos CI, et al. Genome-wide association scan of tag SNPs identifies a susceptibility locus for lung cancer at 15q25.1. Nat Genet. 2008;40:616–622. doi: 10.1038/ng.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hung RJ, et al. A susceptibility locus for lung cancer maps to nicotinic acetylcholine receptor subunit genes on 15q25. Nature. 2008;452:633–637. doi: 10.1038/nature06885. [DOI] [PubMed] [Google Scholar]
- 11.Pillai SG, et al. A genome-wide association study in chronic obstructive pulmonary disease (COPD): identification of two major susceptibility loci. PLoS Genet. 2009;5:e1000421. doi: 10.1371/journal.pgen.1000421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Marchini J, Howie B, Myers S, McVean G, Donnelly P. A new multipoint method for genome-wide association studies by imputation of genotypes. Nat Genet. 2007;39:906–913. doi: 10.1038/ng2088. [DOI] [PubMed] [Google Scholar]
- 13.Frazer KA, et al. A second generation human haplotype map of over 3.1 million SNPs. Nature. 2007;449:851–861. doi: 10.1038/nature06258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Normand SL. Meta-analysis: formulating, evaluating, combining, and reporting. Stat Med. 1999;18:321–359. doi: 10.1002/(sici)1097-0258(19990215)18:3<321::aid-sim28>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 15.ENGAGE Smoking Consortium. Sequence variants at CHRNB3-CHRNA6 and CYP2A6 influence smoking behavior. (Submitted). [Google Scholar]
- 16.Tobacco and Genetics Consortium. Meta-analyses of genomewide association studies implicate loci on chromosomes 9, 11 and 15 for smoking behavior. (Submitted). [Google Scholar]
- 17.Falvella FS, et al. Transcription deregulation at the 15q25 locus in association with lung adenocarcinoma risk. Clin Cancer Res. 2009;15:1837–1842. doi: 10.1158/1078-0432.CCR-08-2107. [DOI] [PubMed] [Google Scholar]
- 18.Wang JC, et al. Risk for nicotine dependence and lung cancer is conferred by mRNA expression levels and amino acid change in CHRNA5. Hum. Mol. Genet. 2009;18:3125–3135. doi: 10.1093/hmg/ddp231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang JC, et al. Genetic variation in the CHRNA5 gene affects mRNA levels and is associated with risk for alcohol dependence. Mol Psychiatry. 2008 doi: 10.1038/mp.2008.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schwarz G. Estimating the dimension of a model. Annals of Statistics. 1978;6:461–464. [Google Scholar]
- 21.Ioannidis JP, Thomas G, Daly MJ. Validating, augmenting and refining genome-wide association signals. Nat Rev Genet. 2009;10:318–329. doi: 10.1038/nrg2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stirnadel H, et al. Genetic and phenotypic architecture of metabolic syndrome-associated components in dyslipidemic and normolipidemic subjects: the GEMS Study. Atherosclerosis. 2008;197:868–876. doi: 10.1016/j.atherosclerosis.2007.07.038. [DOI] [PubMed] [Google Scholar]
- 23.Firmann M, et al. The CoLaus study: a population-based study to investigate the epidemiology and genetic determinants of cardiovascular risk factors and metabolic syndrome. BMC Cardiovasc Disord. 2008;8:6. doi: 10.1186/1471-2261-8-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Muglia P, et al. Genome-wide association study of recurrent major depressive disorder in two European case-control cohorts. Mol Psychiatry. 2008 doi: 10.1038/mp.2008.131. [DOI] [PubMed] [Google Scholar]
- 25.Scott LJ, et al. Genome-wide association and meta-analysis of bipolar disorder in individuals of European ancestry. Proc Natl Acad Sci U S A. 2009;106:7501–7506. doi: 10.1073/pnas.0813386106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chahal NS, et al. Ethnicity-Related Differences in Left Ventricular Function, Structure and Geometry: A Population Study of UK Indian Asians and European Whites. Heart. 2009 doi: 10.1136/hrt.2009.173153. [DOI] [PubMed] [Google Scholar]
- 27.Kathiresan S, et al. Genome-wide association of early-onset myocardial infarction with single nucleotide polymorphisms and copy number variants. Nat Genet. 2009;41:334–341. doi: 10.1038/ng.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Day N, et al. EPIC-Norfolk: study design and characteristics of the cohort. European Prospective Investigation of Cancer. Br J Cancer. 1999;80(Suppl 1):95–103. [PubMed] [Google Scholar]
- 29.Wichmann HE, Gieger C, Illig T. KORA-gen--resource for population genetics, controls and a broad spectrum of disease phenotypes. Gesundheitswesen. 2005;67:S26–S30. doi: 10.1055/s-2005-858226. [DOI] [PubMed] [Google Scholar]
- 30.Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Krawczak M, et al. PopGen: population-based recruitment of patients and controls for the analysis of complex genotype-phenotype relationships. Community Genet. 2006;9:55–61. doi: 10.1159/000090694. [DOI] [PubMed] [Google Scholar]
- 32.John U GB, Hensel E, Lüdemann J, Piek M, Sauer S, Adam C, Born G, Alte D GE, Haertel U, Hense H-W, Haerting J, Willich S, Kessler C. Study of Health in Pomerania (SHIP): a health examination survey in an east German region: objectives and design. Soz-Präventivmed. 2001;46:186–194. doi: 10.1007/BF01324255. [DOI] [PubMed] [Google Scholar]
- 33.Vitart V, et al. SLC2A9 is a newly identified urate transporter influencing serum urate concentration, urate excretion and gout. Nat Genet. 2008;40:437–442. doi: 10.1038/ng.106. [DOI] [PubMed] [Google Scholar]
- 34.McQuillan R, et al. Runs of homozygosity in European populations. Am J Hum Genet. 2008;83:359–372. doi: 10.1016/j.ajhg.2008.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zemunik T, et al. Genome-wide association study of biochemical traits in Korcula Island, Croatia. Croat Med J. 2009;50:23–33. doi: 10.3325/cmj.2009.50.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pilia G, et al. Heritability of cardiovascular and personality traits in 6,148 Sardinians. PLoS Genet. 2006;2:e132. doi: 10.1371/journal.pgen.0020132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li Y, Abecasis GR. Mach 1.0: Rapid Haplotype Reconstruction and Missing Genotype Inference. Am J Hum Genet. 2006;S79:2290. [Google Scholar]
- 38.de Bakker PI, et al. Practical aspects of imputation-driven meta-analysis of genome-wide association studies. Hum Mol Genet. 2008;17:R122–R128. doi: 10.1093/hmg/ddn288. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.