

Abstract
This article reviews the latest developments in understanding the pathogenesis, detection and treatment of small intestinal damage and bleeding caused by nonsteroidal anti-inflammatory drugs (NSAIDs). With improvements in the detection of NSAID-induced damage in the small intestine, it is now clear that this injury and the associated bleeding occurs more frequently than that occurring in the stomach and duodenum, and can also be regarded as more dangerous. However, there are no proven-effective therapies for NSAID-enteropathy, and detection remains a challenge, particularly because of the poor correlation between tissue injury and symptoms. Moreover, recent studies suggest that commonly used drugs for protecting the upper gastrointestinal tract (i.e., proton pump inhibitors) can significantly worsen NSAID-induced damage in the small intestine. The pathogenesis of NSAID-enteropathy is complex, but studies in animal models are shedding light on the key factors that contribute to ulceration and bleeding, and are providing clues to the development of effective therapies and prevention strategies. Novel NSAIDs that do not cause small intestinal damage in animal models offer hope for a solution to this serious adverse effect of one of the most widely used classes of drugs.
Keywords: Anti-inflammatory, Ulcer, Prostaglandin, Non-steroidal, Bleeding, Intestinal, Bile, Enterohepatic, Bacteria, Hydrogen sulfide, Aspirin, Hemorrhage
INTRODUCTION
Nonsteroidal anti-inflammatory drugs (NSAIDs) are among the widely used prescription and over-the-counter medications. They are used to treat the symptoms of a variety of inflammatory conditions, most notably osteoarthritis, rheumatoid arthritis, ankylosing spondylitis and gout. In such conditions, NSAIDs are used chronically, and the affected patients frequently have co-morbidities such as hypertension, diabetes and obesity, as well as often also taking glucocorticoids or anti-coagulants.
By inhibiting the activity of cyclo-oxygenase (COX), NSAIDs prevent the formation of prostaglandin (PG) H2, which is the precursor for the production of all other PG and thromboxane subtypes. Most NSAIDs inhibit COX activity in a competitive fashion, whereas aspirin is an irreversible inhibitor of the enzyme. Indeed, the ability of aspirin to irreversibly inhibit thromboxane synthesis by platelets, and the lack of capacity of platelets to synthesize more COX, underlie the utility of chronic, low-dose aspirin as an anti-thrombotic drug, reducing the incidence of several adverse cardiovascular events (e.g., stroke, myocardial infarction).
Inhibition of COX is central to the major anti-inflammatory actions of NSAIDs. By inhibiting the production of PGs (particularly PGE2 and PGI2), NSAIDs reduce two key elements of inflammation: vasodilation and pain.By reducing blood flow to a damaged and inflamed site, NSAIDs also contribute to a reduction of edema.
Unfortunately, PGs do not only contribute to the cardinal signs of inflammation. They also play important roles in many physiological processes. In the gastrointestinal (GI) tract, PGs are very important mediators of mucosal defence and repair[1]. Inhibition of their synthesis renders GI tissues much more susceptible to damage induced by luminal irritants (including gastric acid and bile), and less able to restore mucosal structure and function after injury[1]. Suppression of PG synthesis is the key effect of NSAIDs that leads to gastro-duodenal ulceration and bleeding. However, several other effects of NSAIDs appear to be central to the ability of these drugs to cause damage in the small intestine.
OVERVIEW OF THE CLINICAL PROBLEM
For several decades, the ability of NSAIDs to induce significant damage to the small intestine was largely unappreciated, being over-shadowed by the attention paid to damage induced by these agents in the stomach and proximal duodenum. The prevalence and clinical significance of NSAID-enteropathy continues to be greatly under-recognized. NSAID-induced enteropathy and bleeding occur more frequently that NSAID-induced gastropathy[2,3]. Significant small intestinal damage and bleeding can be observed in about 70% of chronic NSAID users[4,5], and in the majority of patients the injury is sub-clinical[6].
Unlike the case for NSAID-gastropathy, there are no proven-effective preventative therapies for NSAID-enteropathy, and the pathogenesis is poorly understood[7]. Iron-deficient anemia is a common first presentation of NSAID-enteropathy, and serious complications can include massive bleeding, perforation and strictures, sometimes leading to death[2,6,8].
Aspirin is the most commonly used NSAID, and it is a very frequent cause of small intestinal bleeding. In the United States and Europe, in over 50% of cases, aspirin has been identified as the precipitator of GI bleeding leading to hospital admissions[3,9,10]. Aspirin-induced small intestinal damage appears to occur more frequently when the aspirin is enteric-coated[8,11]. There is a lack of recognition of the frequency and potential severity of aspirin-induced lower GI injury, particularly when the aspirin is given at low doses for cardiovascular protection. In a recent clinical trial that involved over 1200 patients taking aspirin and another anti-platelet therapy for cardiovascular protection, lower GI bleeding was found to occur 3-times more frequently than upper GI bleeding[12]. Zhu et al[13] reported that only about 3.5% of patients prescribed low-dose aspirin also received a prescription for a proton pump inhibitor (PPI), histamine H2 receptor antagonist (H2RA) or muco-protective drug, suggesting that the prescribing physicians did not recognize the potential for GI adverse effects of low-dose aspirin. The pathogenesis of aspirin-induced small intestinal damage differs in several respects to that of the ulceration caused by other NSAIDs (discussed in more detail below).
Selective COX-2 inhibitors were introduced to the marketplace at the beginning of this century with a promise of GI safety[14,15]. While some selective COX-2 inhibitors produce less gastroduodenal damage in some circumstances, the promise of these drugs has been largely unfulfilled[16,17]. Selective COX-2 inhibitors cause small intestinal damage and bleeding (the latter effect is somewhat surprising given the minimal inhibitory effects these drugs of these drugs on platelet function). McCarthy[3] noted that in the VIGOR study, the majority of the GI bleeds originated from lesions in the small intestine (distal to the ligament of Treitz): 58% of the GI bleeds in patients taking rofecoxib and 52% of the GI bleeds in patients taking naproxen[13].
There are several reasons for the lack of recognition of the prevalence and seriousness of NSAID-enteropathy. First, it is more difficult to detect small bowel damage than that induced by NSAIDs in the stomach and proximal duodenum: “The single most important reason for underestimating the clinical importance of NSAID enteropathy is the difficulty in making a diagnosis”[2]. Second, there is a poor correlation between NSAID-induced small intestinal damage and clinical symptoms.The vast majority of NSAID-enteropathy is sub-clinical[6], and when there are symptoms, they are largely non-specific (including iron deficiency anemia, occult blood, diarrhea, hypoalbuminemia, and malabsorption of vitamin B12 and/or bile acids). Thirdly, some researchers have argued that the focus of large pharmaceutical companies on the development of “gastroprotective” drugs, such as H2RA, PPI, and putative gastric-sparing drugs (selective COX-2 inhibitors, NSAID pro-drugs) has led to a preoccupation of physicians and researchers with the stomach and proximal duodenum, at the expense of consideration of the detrimental effects of NSAIDs in the small (and large) intestine. The fact that there are no proven-effective treatments for NSAID-enteropathy likely also contributes to the lack of recognition of this serious condition[7].
DETECTION OF NSAID-ENTEROPATHY
Until recently, detection of NSAID-enteropathy has been very difficult, with most of the evidence for its occurrence coming from post-mortem studies or through indirect measures of intestinal bleeding[4,18,19]. Several indirect methods for detecting and measuring the severity of NSAID-enteropathy were developed, prior to improved endoscopic techniques for viewing the small intestine becoming widely available. These included measuring small intestinal permeability with sugars[20,21] or small molecular weight radioactive probes[22], measuring bleeding (presumed of intestinal origin) with radiolabelled red blood cells[23], and measuring leukocyte markers in the small intestine (radiographically)[24] or in feces[25]. All of these methods provide useful information, but none have become recognized as a “gold standard” for detecting and quantifying enteropathy, because of lack of specificity and/or sensitivity. However, with video capsule endoscopy (VCE) and double-balloon enteroscopy, it is now possible to directly visualize of NSAID-induced damage and bleeding throughout the small intestine. Using these methods, it has become clear that NSAID-enteropathy occurs frequently, even in low-risk subjects (healthy, young volunteers) with low-risk treatment protocols (short-term ingestion of NSAIDs, sometimes together with a “gastro-protective” agent). For example, using VCE, Graham et al[5] found a high prevalence of ulcers in long-term NSAID users. More than 70% of these patients (taking NSAID for more than 3 mo) had intestinal inflammation accompanied by bleeding and protein loss.Symptoms persisted after stopping the therapy (by as long as 16 mo in some patients). Maiden et al[25] reported gross damage in 68% of healthy volunteers taking diclofenac plus omeprazole for 2 wk. Even low-dose aspirin was found to cause significant small intestinal damage with short-term administration; thus, Endo et al[26] reported that 80% of patients taking low-dose aspirin for 2 wk had intestinal damage.
POLYPHARMACY CONUNDRUM: SHIFTING GI INJURY MORE DISTALLY
Animal studies of NSAID injury to the GI tract usually involve the use of healthy animals. Of course, the people most commonly taking NSAIDs on a chronic basis are those with chronic illnesses, and more often than not, they are affected by more than one disease. It is also the case that disorders such as rheumatoid arthritis, obesity and diabetes can increase the susceptibility of the patient to the GI (and other) adverse effects of NSAIDs[27-29]. Moreover, these patients are often taking a number of different drugs, which can also affect susceptibility to NSAID-induce GI injury and bleeding. Polypharmacy is now commonplace, even in patients that do not have disorders other than the one for which NSAID therapy is indicated. Consider a disorder like osteoarthritis, which is more common in the elderly. Cardiovascular diseases are common in this group of patients, often leading to co-prescription of low-dose aspirin and sometimes of other anticoagulants. Low-dose aspirin is also frequently co-prescribed with selective COX-2 inhibitors and conventional NSAIDs because of concerns about the elevated risk of serious cardiovascular events in patients taking those drugs[30]. Of course, co-administration of low-dose aspirin together with a selective COX-2 inhibitor essentially eliminates any advantage, in terms of upper GI safety, of the selective COX-2 inhibitor as compared to a conventional NSAID[15,31-33]. To reduce the expected upper GI toxicity of the combination of an NSAID and low-dose aspirin, PPIs are typically prescribed as well. Indeed, there are now fixed-dose, enteric-coated, combination tablets available that contain an NSAID and a PPI[34]. While there is strong evidence for PPIs reducing the severity of damage and bleeding in the stomach and duodenum, where the role of acid in the production of damage has been clearly demonstrated[1,35], there is no evidence to suggest that a PPI (or other anti-secretory drug) would reduce the severity of NSAID-induced enteropathy. Indeed, antisecretory drugs have been described as “useless either in preventing or treating mucosal lesions” induced by NSAIDs in the intestine[36]. It is worth repeating that the majority of damage and bleeding caused by NSAIDs occurs in the small intestine, distal to the ligament of Treitz[3,13].
Using a rat model, we attempted to replicate common clinical scenarios of polypharmacy to determine the effects on the small intestine[37]. Groups of rats were treated with combinations of anti-inflammatory doses of NSAIDs (naproxen, celecoxib or a novel hydrogen sulfide-releasing NSAID, ATB-346)[38], a PPI (omeprazole or lanzoprazole) and an anti-thrombotic dose of aspirin. In rats that received only the NSAID, the levels of small intestinal damage and bleeding were very low (Figures 1 and 2). However, when co-administered with a PPI or with low-dose aspirin, the levels of small intestinal damage and bleeding in rats treated with naproxen or celecoxib increased significantly (Figures 1 and 2). This effect has been confirmed in a recent study by Satoh et al[39]. The combination of an NSAID with both a PPI and low-dose aspirin resulted in extensive damage and bleeding (the latter was evident post-mortem and also by marked decreases in hematocrit). ATB-346 did not produce small intestinal damage alone or in combination with a PPI and/or low-dose aspirin (Figure 2).
Figure 1.
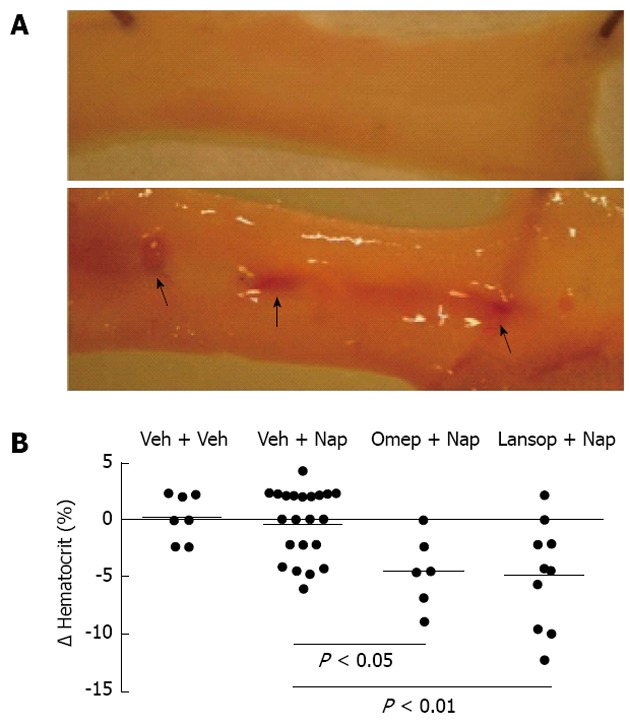
Proton pump inhibitors exacerbate naproxen-induced ulceration and bleeding. In panel A, the top image is of the jejunum of a rat treated with naproxen for 4.5 d (10 mg/kg twice-daily). There are no ulcers present. The bottom image is of a jejunum of a rat receiving the same naproxen treatment, but also treated with omeprazole at a dose that suppressed gastric acid secretion[37]. The arrows indicate the numerous hemorrhagic ulcers that form with this combination of treatments; Panel B shows the change in hematocrit of rats treated with naproxen (Nap) plus vehicle (Veh), omeprazole (Omep) or lansoprazole (Lansop)[37]. The two proton pump inhibitors significantly enhanced the decrease in hematocrit when co-administered with naproxen (no decrease in hematocrit was observed in rats treated with a proton pump inhibitors alone).
Figure 2.
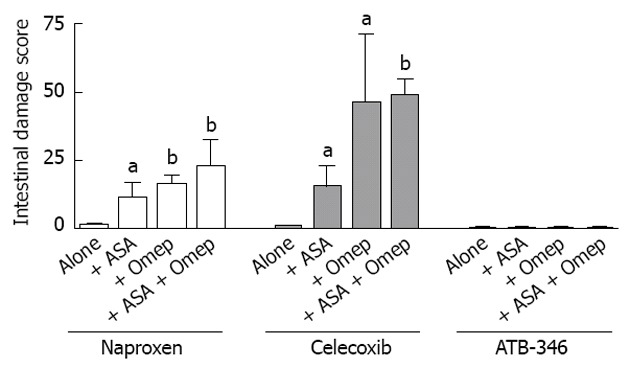
Proton pump inhibitors and low-dose aspirin significantly exacerbate nonsteroidal anti-inflammatory drug-induced small intestinal ulceration. Rats were treated orally, twice-daily for 4.5 d with equi-effective anti-inflammatory doses of naproxen (10 mg/kg), celecoxib (10 mg/kg) or ATB-346 (14.5 mg/kg). ATB-346 is a hydrogen sulfide-releasing derivative of naproxen[38]. Starting 5 d before the nonsteroidal anti-inflammatory drugs (NSAIDs), the rats began receiving twice-daily treatments with omeprazole (Omep) (10 mg/kg) or vehicle. Starting 3 d before the NSAIDs, the rats began receiving daily doses of low-dose aspirin (10 mg/kg) or vehicle. The results are shown as the mean ± SE of at least 6 rats per group. aP < 0.05, bP < 0.01 vs the corresponding group treated with the NSAID alone. No intestinal damage was observed in rats treated with aspirin (ASA) alone. The exacerbation of small intestinal ulceration with omeprazole was also observed with another proton pump inhibitor, lanzoprazole[37]. This figure was constructed using data from Blackler et al[175].
We then performed experiments to try to determine the mechanisms underlying the exacerbation of small intestinal damage by the PPIs. As discussed in more detail below, there is evidence that the bacteria residing in the small intestine play a significant role in the pathogenesis of NSAID-enteropathy. Given the evidence that marked suppression of gastric acid secretion by PPIs can alter the numbers of bacteria in the small intestine[40-42], we focused our investigation on potential changes in intestinal microbiota. Treatment of rats with omeprazole resulted in a dramatic shift in the types of bacteria in the small intestine (dysbiosis). In particular, there was a marked reduction of the Actinobacteria, particularly of Bifidobacteria spp. (> 80% reduction in the jejunum). This diminution of Bifidobacteria was an important factor in the PPI-induced increase in NSAID-induced intestinal damage: replenishment of intestinal Bifidobacteria in PPI-treated rats reduced levels of naproxen-induced intestinal damage those seen in rats not receiving a PPI. Further evidence that it was the dysbiosis induced by the PPI that resulted in elevated susceptibility to NSAID-enteropathy came from studies of germ-free mice[37]. Groups of germ-free mice were colonized with intestinal contents from rats that had been treated with a PPI or vehicle. Beginning one week later, the mice were treated with naproxen for 4 d, and the severity of intestinal damage was then blindly evaluated. Mice that had been colonized with bacteria from PPI-treated rats developed significantly worse intestinal damage than those colonized with bacteria from vehicle-treated rats.
While no clinical studies have been published that directly tested the hypothesis that treatment with PPIs could cause dysbiosis and thereby exacerbate NSAID-induced intestinal damage, there are several reports with data that are consistent with our hypothesis, as summarized by Daniell[43]. In addition to numerous studies documenting that PPIs altering the gut microbiota, resulting in diarrhea[40-42,44], there is evidence from two studies for the presence of intestinal inflammation (detected by elevated fecal calprotectin levels) in patients taking PPIs[45,46], and evidence for microscopic colitis in patients taking NSAIDs or PPIs[47-49], and particularly in patients taking both types of drugs concurrently[49]. In addition, two studies reported greater small intestinal damage in healthy volunteers taking an NSAID plus a PPI as compared to a group taking only a selective COX-2 inhibitor[50,51], and it is now clear that the ability of selective COX-2 inhibitors to damage the small intestine is comparable to that of non-selective NSAIDs[17].
PATHOGENESIS
The key to development of treatments and prevention strategies for NSAID-enteropathy lies in better understanding of the pathogenesis of this injury. Fortunately, the animal models of NSAID enteropathy are very good, reproducible and simple, and can serve as useful tools for gaining a better understanding of the pathogenesis of this disorder and for testing potential therapeutic/preventative agents. Administration of NSAIDs to rats, for example, results in ulceration predominantly in the distal jejunum and ileum[52], the same regions where ulcers are concentrated humans[53,54]. While there will undoubtedly be some differences between rodent models and humans, the existing data suggest that the animal models will be predictive in terms of treatment and prevention strategies. Figure 3 shows some of the key mechanisms suggested to be involved in NSAID-enteropathy, which are discussed in more detail below.
Figure 3.
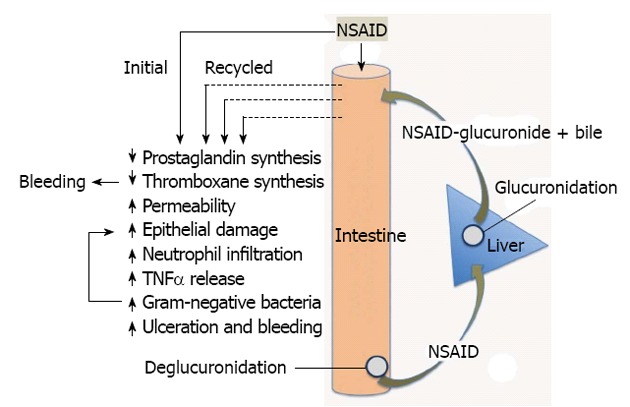
Pathogenesis of nonsteroidal anti-inflammatory drugs-Induced enteropathy. Nonsteroidal anti-inflammatory drugs (NSAIDs) produce effects during their initial exposure to the small intestine, and when secreted back into the proximal small intestine, along with bile, following their absorption in the distal intestine, and glucuronidation in the liver. Suppression of thromboxane synthesis likely plays an important role in promoting bleeding (especially with aspirin, an irreversible inhibitor of platelet thromboxane synthesis). Repeated exposure of the intestinal epithelium to the combination of NSAIDs and bile will promote damage, and the damage is likely exacerbated by the shift in intestinal bacteria stimulated by the NSAID (elevated gram-negative bacteria). These effects appear to be mediated by endotoxin, acting at least in part through toll-like receptor-4. The interplay among bile, bacteria and recirculation of the NSAID is complex. For example, bacterial enzymes convert primary bile acids to secondary bile acids (which may be more damaging) and bacterial enzymes are necessary for deglucuronidation, which permits reabsorption and enterohepatic recirculation of NSAIDs. TNFα: Tumor necrosis factor-alpha.
Inhibition of cyclooxygenase activity
Flower et al[55] first suggested the existence of more than one isoform of COX in 1972. It was almost 20 years later that the two isoforms, now known as COX-1 and COX-2, were sequenced[56,57]. In the decade that followed, a tremendous amount of research was focused on understanding the physiology and pharmacology of these enzymes, largely fueled by the interest of several large pharmaceutical companies in the notion that selective inhibitors of COX-2 would provide all of the anti-inflammatory activities of NSAIDs without the major adverse effects. However, as the science of COX-2 caught up with the marketing of COX-2, it became evident that the delineation of functions of the two COX isoforms was not so clear-cut as had been proposed and heavily promoted. COX-1 contributes significantly to inflammation while COX-2 contributes significantly to many physiological functions, including mucosal defence[58]. This was shown clearly both by studies of mice lacking the gene for one of the COX isoforms and pharmacological studies[59-63]. A striking finding from our laboratory was that injection of carrageenan into the hind-paw of COX-2-deficient mice resulted in inflammation that did not resolve, as it would in a normal mouse[60], suggesting an important role for COX-2-derived prostanoids in resolution of inflammation and healing. Gilroy et al[61] provided compelling evidence from animal models of pleurisy showing the same, and identifying specific COX-2-derived prostanoids that contributed significantly to down-regulating inflammation. Serhan et al[64] described a family of previously unrecognized lipid mediators (lipoxins, resolvins, protectins), some of which were derived from COX-2, that act at several levels of the inflammatory cascade to “turn off” inflammation and allow for a coordinated restoration of tissue homeostasis[65].
The same was true in the GI tract, as COX-2-derived prostanoids were found to contribute significantly to maintenance of the integrity of the tissue, to repair of mucosal injury and to resolution of inflammation[58]. Thus, COX-2 is the isoform that produces PGs at the margins of gastric ulcers, which contribute significantly to the healing of those ulcers[66,67]. In the colon, prostaglandins derived from COX-2 play a very important role in down-regulating inflammation and promoting repair of mucosal injury[52,68,69]. Suppression of COX-2 activity has been shown to exacerbate experimental colitis[52,69]. Indeed, COX-2 is up-regulated throughout the GI tract when the tissue is injured or when there is insufficient PG production via COX-1[52,63,70]. For example, COX-2 is rapidly induced in the stomach in response to suppression of COX-1 by aspirin[70], and it helps to enhance mucosal defence in such circumstances. One of the mechanisms through which this is achieved is via the production, via COX-2, of a potent gastroprotective and anti-inflammatory substance, 15-epi-lipoxin A4[71,72]. Induction of damage in the stomach, in the absence of any other toxic challenge, requires suppression of both COX-1 and COX-2[62], and this also appears to be the case in the small intestine[63].
Clinical studies generally show that selective COX-2 inhibitors produce less gastroduodenal injury and bleeding than conventional NSAIDs, but the small intestinal damage may not differ substantially between the two sub-classes of NSAIDs. For example, Maiden et al[73] performed a VCE study comparing the enteropathy produced in patients on long-term NSAID or selective COX-2 inhibitor therapy, and the key finding was that NSAIDs and selective COX-2 inhibitors produced comparable levels of small bowel damage (small intestinal injury was observed in 50% of the patients treated with a selective COX-2 inhibitor vs 62% of patents treated with a conventional NSAID; not significantly different).
While suppression of COX activity undoubtedly contributes to the pathogenesis of NSAID-enteropathy, it is clear that other factors probably play a more significant role. Suppression of COX activity likely contributes to this disorder mainly through the impairment of repair processes, such as angiogenesis[74], and through inhibition of platelet aggregation, leading to bleeding. The latter effect, however, is most apparent with aspirin, which irreversibly inhibits platelet COX-1, and with NSAIDs that have a long half-life.
Mitochondrial injury
One of the earliest changes that can be detected after NSAID administration, in addition to inhibition of COX activity, is mitochondrial injury[75]. Morphological evidence of mitochondrial damage can be detected within 1 h of administration of an NSAID to rats, and in vitro studies of liver showed that the NSAID could rapidly cause uncoupling of oxidative phosphorylation[75]. This provides a mechanistic explanation for the ability of NSAIDs to damage intestinal epithelial cells and to increase epithelial permeability, as have been demonstrated by several groups[22,52,76]. On the other hand, this mechanism does not explain the localization of ulcers in the jejunum and ileum in animal models and in humans. In their endoscopic study of diclofenac-induced small intestinal injury, Fujimori et al[54] observed denuded regions throughout the small intestine (perhaps indicative of a topical erosive effect), but ulcers were concentrated in the distal jejunum and ileum.
Role of bile and enterohepatic circulation
Several observations suggest important roles for bile and for enterohepatic circulation of NSAIDs in the pathogenesis of NSAID-enteropathy (Figure 3). Ligation of the bile duct in rats prevents NSAID-induced intestinal damage[75-78]. There have also been reports that NSAIDs that do not re-circulate enterohepatically do not cause small intestinal damage[52,75], although aspirin is a notable exception, at least when administered intraduodenally or in an enteric-coated formulation[11,78]. Also, in rats lacking the hepatocanalicular conjugate export pump, which is required for excretion of conjugated NSAIDs into bile, but not for the flow of bile itself, intestinal damage induced by an NSAID (diclofenac) was prevented[79]. On the other hand, induction of higher expression of the export pump aggravated NSAID-induced intestinal damage[79]. A number of studies have demonstrated that a combination of an NSAID and bile is damaging to intestinal epithelial cells[80,81] and non-GI cells[82]. It is noteworthy that in all of these studies, it was secondary bile acids that were found, in combination with NSAIDs, to be effective in damaging cells. Moreover, it has been shown that administration of an NSAID to rats results in increased concentrations of secondary bile acids in bile[83]. Thus, when an NSAID recirculates enterohepatically, the intestinal epithelium is repeatedly exposed to a damaging combination of the NSAID and bile. If this were the primary mechanism of injury in NSAID-enteropathy, however, one would expect to see ulcers produced where the highest concentrations of NSAID and bile would be found (i.e., near the Sphincter of Oddi), whereas the most severe tissue injury is concentrated in the more distal parts of the small intestine[54]. It has been suggested that the sites of ulceration correspond to the sites of NSAID re-absorption, and related to the deconjugation of the NSAIDs at those sites by bacterial β-glucuronidases[79,84-86].
Role of bacteria
There is an abundance of evidence that intestinal bacteria contribute to the pathogenesis of NSAID-enteropathy, but it remains unclear if there is a primary role, initiating the tissue damage, or just a secondary role, exacerbating tissue injury and impeding repair. One of the key observations leading some to propose a primary role of bacteria in NSAID-enteropathy is that germ-free rats and mice develop little or no intestinal damage when given an NSAID, but when colonized by gram-negative bacteria, these animals become susceptible to NSAID-enteropathy[87,88]. Several studies have documented dramatic shifts in the types of bacteria in the small intestine following NSAID administration, with increases in gram-negative bacteria generally being observed, and a concomitant reduction in gram-positive bacteria[89-93]. In some studies, there appeared to be an enrichment of specific bacteria, such as Enterococcus faecalis, Clostridium, Bacteroides and Escherichia coli (E. coli)[89-91]. A number of studies reported protective effects of antibiotics against NSAID-enteropathy, particularly when the antibiotics were effective in reducing number of gram-negative bacteria[88,89,93,94]. Similarly, some probiotics have been reported to reduce the severity of NSAID-enteropathy, especially when they prevent increases in the number of gram-negative bacteria in the intestine[93,95,96]. Despite a considerable number of studies examining the potential contribution of bacteria to NSAID-enteropathy, there remains a lack of clear evidence for a primary role of bacteria in initiation of tissue injury. Bacteria rapidly colonize sites of ulceration and can interfere with ulcer healing[97,98]. In one of the earliest papers on the pathogenesis of NSAID-enteropathy, Kent et al[89] remarked “since the antibiotics do not prevent completely the ulceration, we think that these agents reduce the severity of the lesion by allowing healing to start sooner”. A similar conclusion was drawn by Yamada et al[99].
The apparent importance of gram-negative bacteria in the pathogenesis of NSAID-enteropathy is consistent with reports of a role for lipopolysaccharide (LPS) in driving tissue inflammation and impairment of ulcer healing. Hagiwara et al[91] showed that heat-killed E. coli and their purified LPS caused “deterioration” of NSAID-induced ileal ulcers, but could not cause ulcers themselves in the absence of the NSAID). Koga et al[94] reported that systemic administration of LPS reversed the beneficial effects of an antibiotic in reducing the severity of NSAID-enteropathy in rats, and further demonstrated that T cell function was not required for NSAIDs to induce intestinal ulceration. Watanabe et al[93] demonstrated that mice lacking the endotoxin receptor, toll-like receptor-4, developed much less (about 80%) intestinal damage when given an NSAID than the normal counterparts. These data are once again consistent with the notion that bacteria play a secondary role in NSAID-enteropathy, exacerbating tissue injury and interfering with ulcer healing. These effects may be in part attributable to activation of neutrophils in the mucosal microcirculation, which has been shown to contribute significantly to ulceration[100-104], and local generation of tumor necrosis factor-alpha may be one of the main triggers leading to neutrophil recruitment and/or activation[93,105-107].
As mentioned above, one of the key observations supporting an important role of bacteria in the pathogenesis of NSAID-enteropathy was that germ-free animals do not develop significant small intestinal damage following NSAID administration[75-78]. However, one must bear in mind that ligation of the bile duct blocks the secretion of bile and the enterohepatic circulation of NSAIDs, both of which have been implicated in intestinal injury by these drugs (Figure 3). The conversion of primary bile acids to secondary bile acids is dependent on intestinal bacterial enzymes. Thus, germ-free animals lack secondary bile acids. As mentioned above, most studies that have shown that bile acids (alone or in combination with an NSAID) can cause damage to intestinal epithelial cells have used secondary, rather than primary bile acids[80,81]. Moreover, the re-absorption of NSAIDs in the distal small intestine is largely dependent on bacterial β-glucuronidase activity, which de-conjugates NSAID-glucuronides, allowing the NSAID to be transported across the epithelium[84]. Enterohepatic circulation of NSAIDs is negligible in animals that lack intestinal bacteria, resulting in decreased exposure of the intestine to the NSAID, and therefore decreased tissue injury. Recently, LoGuidice et al[85] demonstrated that an inhibitor of bacterial β-glucuronidase could significantly reduce the severity of diclofenac-induced small intestinal injury in mice. β-glucuronidase has been shown to be expressed in Clostridium, Peptostreptococcus, Staphylococcus and E. coli[85,108].
TREATING AND PREVENTING NSAID-ENTEROPATHY
In sharp contrast to NSAID-induced gastroduodenal damage, where several options are available to provide protection to a patient, no treatments or prevention strategies for NSAID-enteropathy have been convincingly shown to be effective.As outlined above, PPIs provide upper GI protection against NSAIDs but worsen NSAID-enteropathy in animals, and there is emerging evidence that the same is the case in humans. There are novel NSAIDs in development that do not cause enteropathy in animals (discussed below).
Misoprostol, metronidazole and sulfasalazine have all been suggested to be beneficial in treatment or prevention of NSAID-enteropathy in humans, but the studies suggesting this had significant limitations (open-label, not controlled, and/or small sample sizes)[22,24,109-111]. Misoprostol, H2RA and sucralfate were found to be ineffective in reducing NSAID-induced intestinal permeability in humans[112,113], though in one, open-label study misoprostol reduced the elevated intestinal permeability induced by indomethacin[114]. Based on the animal data showing beneficial effects of metronidazole in reducing NSAID-enteropathy[99], Bjarnason et al[24] performed an open-label human study of chronic NSAID users. The patients took metronidazole for 2-12 wk while continuing their NSAID treatment. The endpoints were fecal excretion of 51Cr-labeled erythrocytes and 111In-labelled neutrophils. Both markers declined significantly during metronidazole treatment, leading the authors to conclude that “these results suggest that the neutrophil is the main damaging effector cell in NSAID induced enteropathy” and that the main chemoattractant “may be a metronidazole sensitive microbe”.
The observations from animal studies that NSAID-enteropathy was accompanied by dramatic shifts in numbers and types of intestinal bacteria led to a number of studies of the potential value of probiotics for treatment or prevention of NSAID-enteropathy. In studies in rats, Kinouchi et al[95] demonstrated that Lactobacillus acidophilus and Bifidobacteria adolescentis administration markedly reduced the severity of NSAID-induced ileal ulceration. Syer et al[96] also showed a marked protective effect of Bifidobacteria adolescentis in a rat model of NSAID-enteropathy. Only two clinical trials of a probiotic for NSAID-enteropathy have been reported to date. Montalto et al[115] performed a randomized, double-bind, placebo-controlled trial of VSL#3, a probiotic formulation consisting of 8 different live bacteria. Volunteers received indomethacin daily for 4 d, and fecal calprotectin levels were the endpoint. The placebo-treated volunteers exhibited markedly elevated fecal calprotectin levels during the period of indomethacin treatment, while during treatment with VSL#3 the fecal calprotectin levels remained within the normal range. In a study by Endo et al[116], 25 patients with unexplained iron deficiency anemia who had been taking low-dose enteric-coated aspirin plus omeprazole for more than 3 mo were given either Lactobacillus casei (L. casei) or placebo for 3 mo while continuing the aspirin and omeprazole therapy. VCE at the end of the treatment period showed a significant reduction of mucosal breaks and “capsule endoscopy score” in the group receiving L. casei. The results of this small clinical study are consistent with a study of L. casei (strain Shirota) in a rat model of indomethacin-induced enteropathy[117].
Lactoferrin has been shown to prevent NSAID-induced bleeding in rodents[118] and this effect may be related to its ability to promote the growth of Bifidobacteria in the small intestine[119]. Oral treatment of healthy volunteers with recombinant lactoferrin was shown to reduce indomethacin-induced changes in small intestinal permeability[120]. However, in this short-term study, only a very modest increase in intestinal permeability was seen, with only a single administration of lactoferrin that would have been unlikely to have significantly affected the intestinal microbiome.
Rebamipide is a quinolinone derivative that is used to promote the healing of GI ulcers and for mucosal protection. Its mechanism of action is not fully understood, though it appears to stimulate mucus secretion and PG synthesis[121] and to scavenge oxygen-derived free radicals[6]. It has been shown to significantly reduce the severity of NSAID-induced enteropathy in rats[122]. Niwa et al[123] performed a pilot study in healthy humans to examine the effectiveness of rebamipride in preventing NSAID-enteropathy. The volunteers received placebo or rebamipride together with diclofenac for 7 d. The small intestine was examined at the end of the study by VCE. Damage was observed in 8 of the 10 of placebo-treated group (2 ulcers, 1 bleed), but in only 2 of the 10 of rebamipide-treated group (no ulcers or bleeding). However, a larger study of healthy volunteers treated for 14 d with an NSAID (diclofenac), a PPI (omeprazole) and either rebamipide or placebo, failed to a detect a significant benefit of rebamipide in terms of reducing the incidence of intestinal mucosal injury[124]. Larger studies of rebamipide, ideally in patients receiving NSAID therapy for an inflammatory disorder, are needed to clarify if this drug will have benefit in reducing the incidence and/or severity of NSAID-enteropathy.
Studies in animal models have suggested other possible approaches to prevention of NSAID-enteropathy, but have not yet been assessed in humans. For example, in a mouse model of acute indomethacin-induced intestinal damage, Yasuda et al[125] found that dopamine D2 receptor antagonists reduced the severity of damage, and these effects were mediated through the activation of endogenous anti-inflammatory pathways mediated by via α7-nicotinic acetylcholine receptors, as had been observed previously[126]. Using the same model, Kato et al[127] demonstrated that certain 5-HT receptors could modulate susceptibility to NSAID-enteropathy. They reported that antagonists of the 5-HT3 receptor (ondansetron and ramosetron) dose-dependently reduced intestinal damage, while a 5-HT4 antagonist (GR113808) aggravated damage. A 5-HT4 agonist (mosapride) significantly reduced damage. As in the case of protection with dopamine D2 receptor antagonists, the authors suggested that the beneficial effects the 5HT4 agonist may be mediated through activation of α7-nicotinic acetylcholine receptors. There have also been studies demonstrating a significant increase in intestinal motor activity after administration of NSAIDs, and have suggested that this contributes to the generation of injury, but pharmacological approaches targeting this 5-HT/α7-nicotinic acetylcholine receptor axis have not yet been evaluated in human NSAID-enteropathy.
NSAID pro-drugs: The enteropathy remains
Pro-drugs have been defined as “bioreversible derivatives of drug molecules that undergo an enzymatic and/or chemical transformation in vivo to release the active parent drug, which can then exert the desired pharmacological effect”[128,129]. A number of NSAID pro-drugs have been developed, based on the premise that if the drug can pass through the stomach in an inactive form, it will not inhibit PG synthesis in the stomach, and therefore will not be ulcerogenic. In essence, an NSAID pro-drug of this design does not differ significantly from an enteric coated NSAID, and the problems associated with the latter are well documented[8,36]. Moreover, there are several problems with the premise upon which NSAID pro-drugs are based. First, once the drug is absorbed and transformed to release the parent drug, that drug will produce “the desired pharmacological effect”. That effect, reduction of pain and inflammation, is attributable to systemic inhibition of COX activity. In the absence of any “protective” intervention, systemic inhibition of COX activity will result in damage and bleeding in the upper GI tract. Thus, NSAIDs administered systemically induce significant gastrointestinal ulceration and bleeding[130-132]. If a pro-drug is formulated such that it produces a marked delay in the release of the parent drug, there will be a similar delay in the onset of the desired activity. Second, the pro-drug approach is focused entirely on sparing the upper GI tract of injury, ignoring the potential of the drug to cause small intestinal injury, particularly if it undergoes enterohepatic recirculation. Once the pro-drug is metabolized to release the parent drug, the parent drug will behave, pharmacokinetically and pharmacodynamically, in the same way as if the parent drug itself had been administered. These are points that have been acknowledged on the website of a company that is developing a naproxen pro-drug: “the pro-drug approach will not address the GI damage associated with the systemic inhibition of COX after release of the parent drug, nor will it address the toxic effects of metabolites delivered into the gut lumen with bile”[133].
Clinical trials of pro-drugs have often produced data that are very favourable to the pro-drug. However, this is largely because such studies have typically focused on acute gastric or gastroduodenal damage (erosions and “endoscopic ulcers”)[134] that are of questionable clinical significance, since they do not necessarily predict the incidence of true ulcers[135]. Indeed, the same is true for most of the trials of selective COX-2 inhibitors and of PPIs, which gave a false signal of the GI safety of those classes of drugs because of reliance on inappropriate endpoints for upper GI damage and lack of consideration of the potential damaging effects of these drugs on the small intestine. When examined in “real world” scenarios, using clinically meaningful endpoints[135], there is little, if any, evidence of significant benefit of NSAID pro-drugs over the parent drugs or over other NSAIDs. This topic has been very well reviewed by Graham[135]. Thus, while the pro-drug sulindac rarely caused erosions or “endoscopic ulcers” in short-term studies of human volunteers[136,137], longer term studies in at-risk patients showed this drug to offer no upper GI safety benefit as compared to other NSAIDs[138]. Likewise, nabumetone was purported to be a GI-safe pro-drug, and acute upper GI studies suggested that this was the case[139], but in at-risk patients the drug did not offer any benefit over other NSAIDs[140]. Neither sulindac nor nabumetone have been specifically examined with respect to their propensity to cause small intestinal ulceration and bleeding. Moreover, there are suggestions in the literature[132,141], supported by animal studies[130,131], that systemic administration of NSAIDs, which completely avoids contact of the drug with the lining of the stomach and duodenum, does not offer significant benefit in terms of reducing the incidence of significant GI ulceration and bleeding.
Novel intestinal-sparing NSAIDs
The advances that have been made in understanding the pathogenesis of NSAID-enteropathy provide important clues for designing novel NSAIDs that will not damage in the small intestine (or the stomach). Several approaches have been taken that show promise, mainly using the “co-drug” model of drug design[142]. Co-drugs are somewhat like pro-drugs, with the key difference being that the pro-moiety is not inert; rather, it exerts important pharmacological effects[129]. Two such classes of co-drugs are the nitric oxide (NO)-releasing NSAIDs and the hydrogen sulfide (H2S)-releasing NSAIDs[143-145]. In each case, the NSAID portion of the co-drug behaves the same as expected (inhibition of COX-1 and COX-2, leading to anti-inflammatory and analgesic effects), while the gaseous mediator portion of the co-drug exerts mucosal protective effects, very similar to the effects of endogenous prostaglandins[146,147]. Both of these gaseous mediators are vasodilators and can inhibit leukocyte adherence to the vascular endothelium[148,149]. Suppression of mucosal synthesis of NO or H2S reduces the resistance of the stomach to the damaging effects of NSAIDs and other irritants, and impairs the healing of pre-existing damage[148,150-156]. NO and H2S donors can increase the resistance of the gastric mucosa to injury induced by NSAIDs and other noxious substances[148,151,156,157] and can accelerate healing of ulcers in rodent models[37,150,153,154,158]. Some of the other actions of NO-NSAIDs and H2S-NSAIDs and their underlying mechanisms have been reviewed previously[143,145,152].
NO-NSAIDs were shown to cause significantly less intestinal damage than the parent drugs[159,160], and to be well tolerated in rats with pre-existing colitis[159]. In a small, short-term clinical trial, an NO-NSAID produced significantly less of an increase in small intestinal permeability than was produced by an equivalent dose of the parent drug (naproxen)[161]. Despite very promising results from clinical trials that demonstrated efficacy and safety in osteoarthritis[162-166], NO-NSAIDs have not obtained regulatory approval because the safety advantages over the parent drug (naproxen) have not been sufficiently demonstrated. One key GI safety clinical trial fell just short of showing a significant benefit as compared to naproxen (P = 0.066)[167].
H2S-releasing NSAIDs exhibit enhanced anti-inflammatory activity relative to the parent drugs[37,151,155,168,169], presumably attributable to the anti-inflammatory and pro-resolution effects of the H2S released from these drugs[149,158,170-174]. In addition to sparing the gastric mucosa of damage in several circumstances of impaired mucosal defence[38,175], H2S-releasing NSAIDs have been shown not to cause damage in the small intestine of rats[38,155,175]. Moreover, when tested in co-morbidity and polypharmacy models, with repeated administration over several days, an H2S-releasing derivative of naproxen (ATB-346) did not cause small intestinal damage[175] (Figures 2 and 4). For example, obese rats that exhibited markedly greater naproxen-induced enteropathy than was observed in lean rats, but ATB-346 did not elicit damage in lean or obese rats[175]. Interestingly, Zucker obese rats have a microbiota distinct from that of their lean littermates, with a marked reduction in intestinal levels of Bifidobacteria[176]. Recall that we observed that PPIs increased the severity of NSAID-enteropathy in rats, and found that this was largely attributable to a decrease in intestinal Bifidobacteria levels[37]. ATB-346 retained its favourable profile in the intestine even when co-administered with a PPI and or low-dose aspirin[175] (Figure 2).
Figure 4.
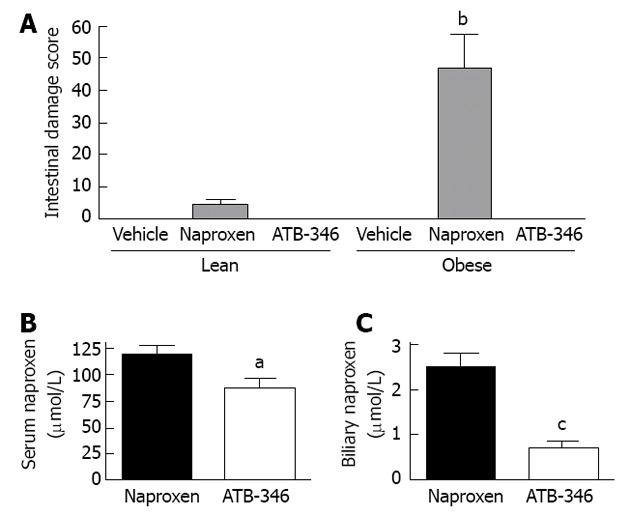
Intestinal safety and altered biliary excretion of ATB-346. A: ATB-346 is a hydrogen sulfide-releasing derivative of naproxen[38].When administered to obese Zucker rats, twice-daily for 4.5 d at 10 mg/kg, naproxen induced small intestinal damage that was significantly more severe in the obese rats (bP < 0.01). However, at an equimolar dose, ATB-346 did not induce intestinal damage in lean or obese rats[175]; B: The serum levels of naproxen in normal rats after 4.5 d of twice-daily administration of ATB-346 were marginally, but significantly (aP < 0.05) lower than those in rats treated with an equimolar dose of naproxen; C: Biliary levels of naproxen in rats treated with ATB-346 (as above) were markedly reduced compared to those in rats treated with an equimolar dose of naproxen (cP < 0.001).The data shown in this graph are from Blackler et al[175].
A particularly important feature of ATB-346 that may be very important in terms of its lack of damaging effects in the small intestine is that, though metabolized to release naproxen, there are relatively low levels of naproxen in bile after administration of this compound[175] (Figure 4). Moreover, the biliary levels of naproxen-glucuronide were reduced by 72% in the ATB-346 group as compared to the naproxen group[175]. These altered pharmacokinetics of ATB-346 vs naproxen did not alter the anti-inflammatory activity of the drug[37], but could contribute significantly to the intestinal-sparing properties of ATB-346.
Recently, a class of drugs was described that consists of an NSAID attached to moieties releasing both NO and H2S[177]. These compounds show comparable actions as the parent drugs in terms of inhibiting COX activity, but there ares no available data on their GI toxicity.
NSAIDs pre-associated with phospholipids are a unique type of “co-drug”.
Surface-active phospholipids have been proposed to constitute an important component of the epithelial “barrier” to acid back-diffusion, and NSAIDs can to disrupt this barrier[178,179]. Lichtenberger et al[180] demonstrated that pre-associating an NSAID with a zwitterionic phospholipid prevents the NSAID from disrupting the barrier function of the epithelium. Thus, covalently linking phosphatidylcholine to aspirin, ibuprofen and other NSAIDs results in compounds with equivalent anti-inflammatory properties to the parent drug, but with markedly reduced gastric toxicity[181]. This has been demonstrated in endoscopic clinical trials for an aspirin derivative[181], and also with an ibuprofen derivative[182], though in the latter trial, statistical significance was only seen in an older subset of the patients studied. Recently, Lichtenberger et al[78] demonstrated that pre-associating aspirin with phosphatidylcholine greatly reduced the small intestinal damage produced by intraduodenal administration of this compound, as compared to aspirin alone.
CONCLUSION
NSAID-enteropathy has largely been ignored for decades as a result of the focus on NSAID-gastropathy, driven largely by the development of several commercially successful drugs targeting that disorder (H2RA, PPIs, selective COX-2 inhibitors). Moreover, the difficulty in detecting NSAID-enteropathy and the lack of any proven-effective preventative or treatment options has contributed to an under-appreciation of the magnitude of this significant adverse reaction to a very widely used class of drugs. With the development of video capsule endoscopy, the frequency and severity of NSAID-enteropathy has become more evident. Techniques such as VCE also permit more conclusive studies of the safety of novel NSAIDs and of potential prevention or treatment strategies.
The animal models for NSAID-enteropathy are very good, and they have provided a great deal of information on the pathogenesis of this disorder. Moreover, the animal studies have given some direction as to viable strategies for preventing NSAID-enteropathy, and the models are useful for testing novel therapeutics agents.
As is the case with NSAID-induced injury in the upper GI tract, it is important that studies of NSAID-enteropathy focus on animal models that are most similar to the patients that use these drugs and most often develop serious adverse effects. Thus, future studies should focus on the use of animal models with relevant co-morbidities that display increased susceptibility to NSAID-enteropathy, and on patients most at risk of developing intestinal damage and bleeding.
ACKNOWLEDGMENTS
Dr. Wallace is a founder and shareholder of Antibe Therapeutics Inc., a company developing novel NSAIDs.
Footnotes
Supported by A grant from the Canadian Institutes of Health Research
P- Reviewer Tarnawski AS S- Editor Wen LL L- Editor A E- Editor Li JY
References
- 1.Wallace JL. Prostaglandins, NSAIDs, and gastric mucosal protection: why doesn’t the stomach digest itself? Physiol Rev. 2008;88:1547–1565. doi: 10.1152/physrev.00004.2008. [DOI] [PubMed] [Google Scholar]
- 2.Adebayo D, Bjarnason I. Is non-steroidal anti-inflammaory drug (NSAID) enteropathy clinically more important than NSAID gastropathy? Postgrad Med J. 2006;82:186–191. doi: 10.1136/pgmj.2005.039586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McCarthy DM. GI bleeding: problems that persist. Gastrointest Endosc. 2009;70:225–228. doi: 10.1016/j.gie.2008.12.247. [DOI] [PubMed] [Google Scholar]
- 4.Bjarnason I, Hayllar J, MacPherson AJ, Russell AS. Side effects of nonsteroidal anti-inflammatory drugs on the small and large intestine in humans. Gastroenterology. 1993;104:1832–1847. doi: 10.1016/0016-5085(93)90667-2. [DOI] [PubMed] [Google Scholar]
- 5.Graham DY, Opekun AR, Willingham FF, Qureshi WA. Visible small-intestinal mucosal injury in chronic NSAID users. Clin Gastroenterol Hepatol. 2005;3:55–59. doi: 10.1016/s1542-3565(04)00603-2. [DOI] [PubMed] [Google Scholar]
- 6.Park SC, Chun HJ, Kang CD, Sul D. Prevention and management of non-steroidal anti-inflammatory drugs-induced small intestinal injury. World J Gastroenterol. 2011;17:4647–4653. doi: 10.3748/wjg.v17.i42.4647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wallace JL. NSAID gastropathy and enteropathy: distinct pathogenesis likely necessitates distinct prevention strategies. Br J Pharmacol. 2012;165:67–74. doi: 10.1111/j.1476-5381.2011.01509.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Davies NM. Sustained release and enteric coated NSAIDs: are they really GI safe? J Pharm Pharm Sci. 1999;2:5–14. [PubMed] [Google Scholar]
- 9.Lanas A, Sekar MC, Hirschowitz BI. Objective evidence of aspirin use in both ulcer and nonulcer upper and lower gastrointestinal bleeding. Gastroenterology. 1992;103:862–869. doi: 10.1016/0016-5085(92)90018-t. [DOI] [PubMed] [Google Scholar]
- 10.Stack WA, Atherton JC, Hawkey GM, Logan RF, Hawkey CJ. Interactions between Helicobacter pylori and other risk factors for peptic ulcer bleeding. Aliment Pharmacol Ther. 2002;16:497–506. doi: 10.1046/j.1365-2036.2002.01197.x. [DOI] [PubMed] [Google Scholar]
- 11.Endo H, Sakai E, Higurashi T, Yamada E, Ohkubo H, Iida H, Koide T, Yoneda M, Abe Y, Inamori M, et al. Differences in the severity of small bowel mucosal injury based on the type of aspirin as evaluated by capsule endoscopy. Dig Liver Dis. 2012;44:833–838. doi: 10.1016/j.dld.2012.05.016. [DOI] [PubMed] [Google Scholar]
- 12.Casado Arroyo R, Polo-Tomas M, Roncalés MP, Scheiman J, Lanas A. Lower GI bleeding is more common than upper among patients on dual antiplatelet therapy: long-term follow-up of a cohort of patients commonly using PPI co-therapy. Heart. 2012;98:718–723. doi: 10.1136/heartjnl-2012-301632. [DOI] [PubMed] [Google Scholar]
- 13.Zhu LL, Xu LC, Chen Y, Zhou Q, Zeng S. Poor awareness of preventing aspirin-induced gastrointestinal injury with combined protective medications. World J Gastroenterol. 2012;18:3167–3172. doi: 10.3748/wjg.v18.i24.3167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bombardier C, Laine L, Reicin A, Shapiro D, Burgos-Vargas R, Davis B, Day R, Ferraz MB, Hawkey CJ, Hochberg MC, et al. Comparison of upper gastrointestinal toxicity of rofecoxib and naproxen in patients with rheumatoid arthritis. VIGOR Study Group. N Engl J Med. 2000;343:1520–1528. doi: 10.1056/NEJM200011233432103. [DOI] [PubMed] [Google Scholar]
- 15.Silverstein FE, Faich G, Goldstein JL, Simon LS, Pincus T, Whelton A, Makuch R, Eisen G, Agrawal NM, Stenson WF, et al. Gastrointestinal toxicity with celecoxib vs nonsteroidal anti-inflammatory drugs for osteoarthritis and rheumatoid arthritis: the CLASS study: A randomized controlled trial. Celecoxib Long-term Arthritis Safety Study. JAMA. 2000;284:1247–1255. doi: 10.1001/jama.284.10.1247. [DOI] [PubMed] [Google Scholar]
- 16.Wallace JL. Selective COX-2 inhibitors: is the water becoming muddy? Trends Pharmacol Sci. 1999;20:4–6. doi: 10.1016/s0165-6147(98)01283-8. [DOI] [PubMed] [Google Scholar]
- 17.Graham DY, Jewell NP, Chan FK. Rofecoxib and clinically significant upper and lower gastrointestinal events revisited based on documents from recent litigation. Am J Med Sci. 2011;342:356–364. doi: 10.1097/MAJ.0b013e3182113658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bjarnason I, Price AB, Zanelli G, Smethurst P, Burke M, Gumpel JM, Levi AJ. Clinicopathological features of nonsteroidal antiinflammatory drug-induced small intestinal strictures. Gastroenterology. 1988;94:1070–1074. doi: 10.1016/0016-5085(88)90568-9. [DOI] [PubMed] [Google Scholar]
- 19.Chan FK, Lanas A, Scheiman J, Berger MF, Nguyen H, Goldstein JL. Celecoxib versus omeprazole and diclofenac in patients with osteoarthritis and rheumatoid arthritis (CONDOR): a randomised trial. Lancet. 2010;376:173–179. doi: 10.1016/S0140-6736(10)60673-3. [DOI] [PubMed] [Google Scholar]
- 20.Sutherland LR, Verhoef M, Wallace JL, Van Rosendaal G, Crutcher R, Meddings JB. A simple, non-invasive marker of gastric damage: sucrose permeability. Lancet. 1994;343:998–1000. doi: 10.1016/s0140-6736(94)90125-2. [DOI] [PubMed] [Google Scholar]
- 21.Bjarnason I, Peters TJ. Intestinal permeability, non-steroidal anti-inflammatory drug enteropathy and inflammatory bowel disease: an overview. Gut. 1989;30 Spec No:22–28. doi: 10.1136/gut.30.spec_no.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bjarnason I, Smethurst P, Clark P, Menzies I, Levi J, Peters T. Effect of prostaglandin on indomethacin-induced increased intestinal permeability in man. Scand J Gastroenterol Suppl. 1989;164:97–102; discussion 102-103. doi: 10.3109/00365528909091195. [DOI] [PubMed] [Google Scholar]
- 23.Hunt RH, Bowen B, Mortensen ER, Simon TJ, James C, Cagliola A, Quan H, Bolognese JA. A randomized trial measuring fecal blood loss after treatment with rofecoxib, ibuprofen, or placebo in healthy subjects. Am J Med. 2000;109:201–206. doi: 10.1016/s0002-9343(00)00470-8. [DOI] [PubMed] [Google Scholar]
- 24.Bjarnason I, Hayllar J, Smethurst P, Price A, Gumpel MJ. Metronidazole reduces intestinal inflammation and blood loss in non-steroidal anti-inflammatory drug induced enteropathy. Gut. 1992;33:1204–1208. doi: 10.1136/gut.33.9.1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Maiden L, Thjodleifsson B, Theodors A, Gonzalez J, Bjarnason I. A quantitative analysis of NSAID-induced small bowel pathology by capsule enteroscopy. Gastroenterology. 2005;128:1172–1178. doi: 10.1053/j.gastro.2005.03.020. [DOI] [PubMed] [Google Scholar]
- 26.Endo H, Hosono K, Inamori M, Kato S, Nozaki Y, Yoneda K, Akiyama T, Fujita K, Takahashi H, Yoneda M, et al. Incidence of small bowel injury induced by low-dose aspirin: a crossover study using capsule endoscopy in healthy volunteers. Digestion. 2009;79:44–51. doi: 10.1159/000204465. [DOI] [PubMed] [Google Scholar]
- 27.Solomon DH, Gurwitz JH. Toxicity of nonsteroidal anti-inflammatory drugs in the elderly: is advanced age a risk factor? Am J Med. 1997;102:208–215. doi: 10.1016/s0002-9343(96)00380-4. [DOI] [PubMed] [Google Scholar]
- 28.Hernández-Díaz S, Rodríguez LA. Incidence of serious upper gastrointestinal bleeding/perforation in the general population: review of epidemiologic studies. J Clin Epidemiol. 2002;55:157–163. doi: 10.1016/s0895-4356(01)00461-9. [DOI] [PubMed] [Google Scholar]
- 29.Aro P, Storskrubb T, Ronkainen J, Bolling-Sternevald E, Engstrand L, Vieth M, Stolte M, Talley NJ, Agréus L. Peptic ulcer disease in a general adult population: the Kalixanda study: a random population-based study. Am J Epidemiol. 2006;163:1025–1034. doi: 10.1093/aje/kwj129. [DOI] [PubMed] [Google Scholar]
- 30.Kearney PM, Baigent C, Godwin J, Halls H, Emberson JR, Patrono C. Do selective cyclo-oxygenase-2 inhibitors and traditional non-steroidal anti-inflammatory drugs increase the risk of atherothrombosis? Meta-analysis of randomised trials. BMJ. 2006;332:1302–1308. doi: 10.1136/bmj.332.7553.1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Laine L, Maller ES, Yu C, Quan H, Simon T. Ulcer formation with low-dose enteric-coated aspirin and the effect of COX-2 selective inhibition: a double-blind trial. Gastroenterology. 2004;127:395–402. doi: 10.1053/j.gastro.2004.05.001. [DOI] [PubMed] [Google Scholar]
- 32.Jüni P, Rutjes AW, Dieppe PA. Are selective COX 2 inhibitors superior to traditional non steroidal anti-inflammatory drugs? BMJ. 2002;324:1287–1288. doi: 10.1136/bmj.324.7349.1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schnitzer TJ, Burmester GR, Mysler E, Hochberg MC, Doherty M, Ehrsam E, Gitton X, Krammer G, Mellein B, Matchaba P, et al. Comparison of lumiracoxib with naproxen and ibuprofen in the Therapeutic Arthritis Research and Gastrointestinal Event Trial (TARGET), reduction in ulcer complications: randomised controlled trial. Lancet. 2004;364:665–674. doi: 10.1016/S0140-6736(04)16893-1. [DOI] [PubMed] [Google Scholar]
- 34.Sostek MB, Fort JG, Estborn L, Vikman K. Long-term safety of naproxen and esomeprazole magnesium fixed-dose combination: phase III study in patients at risk for NSAID-associated gastric ulcers. Curr Med Res Opin. 2011;27:847–854. doi: 10.1185/03007995.2011.555756. [DOI] [PubMed] [Google Scholar]
- 35.Wallace JL. Nonsteroidal anti-inflammatory drugs and gastroenteropathy: the second hundred years. Gastroenterology. 1997;112:1000–1016. doi: 10.1053/gast.1997.v112.pm9041264. [DOI] [PubMed] [Google Scholar]
- 36.Lanas A, Scarpignato C. Microbial flora in NSAID-induced intestinal damage: a role for antibiotics? Digestion. 2006;73 Suppl 1:136–150. doi: 10.1159/000089789. [DOI] [PubMed] [Google Scholar]
- 37.Wallace JL, Syer S, Denou E, de Palma G, Vong L, McKnight W, Jury J, Bolla M, Bercik P, Collins SM, et al. Proton pump inhibitors exacerbate NSAID-induced small intestinal injury by inducing dysbiosis. Gastroenterology. 2011;141:1314–1322, 1322.e1-5. doi: 10.1053/j.gastro.2011.06.075. [DOI] [PubMed] [Google Scholar]
- 38.Wallace JL, Caliendo G, Santagada V, Cirino G. Markedly reduced toxicity of a hydrogen sulphide-releasing derivative of naproxen (ATB-346) Br J Pharmacol. 2010;159:1236–1246. doi: 10.1111/j.1476-5381.2009.00611.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Satoh H, Amagase K, Takeuchi K. Exacerbation of nonsteroidal anti-inflammatory drug-induced small intestinal lesions by antisecretory drugs in rats: the role of intestinal motility. J Pharmacol Exp Ther. 2012;343:270–277. doi: 10.1124/jpet.112.197475. [DOI] [PubMed] [Google Scholar]
- 40.Verdu E, Viani F, Armstrong D, Fraser R, Siegrist HH, Pignatelli B, Idström JP, Cederberg C, Blum AL, Fried M. Effect of omeprazole on intragastric bacterial counts, nitrates, nitrites, and N-nitroso compounds. Gut. 1994;35:455–460. doi: 10.1136/gut.35.4.455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Williams C, McColl KE. Review article: proton pump inhibitors and bacterial overgrowth. Aliment Pharmacol Ther. 2006;23:3–10. doi: 10.1111/j.1365-2036.2006.02707.x. [DOI] [PubMed] [Google Scholar]
- 42.Lombardo L, Foti M, Ruggia O, Chiecchio A. Increased incidence of small intestinal bacterial overgrowth during proton pump inhibitor therapy. Clin Gastroenterol Hepatol. 2010;8:504–508. doi: 10.1016/j.cgh.2009.12.022. [DOI] [PubMed] [Google Scholar]
- 43.Daniell HW. NSAID-PPI enteropathy in humans. Gastroenterology. 2012;142:e20; author reply e20–e21. doi: 10.1053/j.gastro.2012.02.004. [DOI] [PubMed] [Google Scholar]
- 44.Compare D, Pica L, Rocco A, De Giorgi F, Cuomo R, Sarnelli G, Romano M, Nardone G. Effects of long-term PPI treatment on producing bowel symptoms and SIBO. Eur J Clin Invest. 2011;41:380–386. doi: 10.1111/j.1365-2362.2010.02419.x. [DOI] [PubMed] [Google Scholar]
- 45.Poullis A, Foster R, Mendall MA, Shreeve D, Wiener K. Proton pump inhibitors are associated with elevation of faecal calprotectin and may affect specificity. Eur J Gastroenterol Hepatol. 2003;15:573–574; author reply 574. doi: 10.1097/00042737-200305000-00021. [DOI] [PubMed] [Google Scholar]
- 46.Andréasson K, Scheja A, Saxne T, Ohlsson B, Hesselstrand R. Faecal calprotectin: a biomarker of gastrointestinal disease in systemic sclerosis. J Intern Med. 2011;270:50–57. doi: 10.1111/j.1365-2796.2010.02340.x. [DOI] [PubMed] [Google Scholar]
- 47.Wilcox GM, Mattia AR. Microscopic colitis associated with omeprazole and esomeprazole exposure. J Clin Gastroenterol. 2009;43:551–553. doi: 10.1097/MCG.0b013e31817d3fa1. [DOI] [PubMed] [Google Scholar]
- 48.Keszthelyi D, Jansen SV, Schouten GA, de Kort S, Scholtes B, Engels LG, Masclee AA. Proton pump inhibitor use is associated with an increased risk for microscopic colitis: a case-control study. Aliment Pharmacol Ther. 2010;32:1124–1128. doi: 10.1111/j.1365-2036.2010.04453.x. [DOI] [PubMed] [Google Scholar]
- 49.Pardi DS, Kelly CP. Microscopic colitis. Gastroenterology. 2011;140:1155–1165. doi: 10.1053/j.gastro.2011.02.003. [DOI] [PubMed] [Google Scholar]
- 50.Goldstein JL, Eisen GM, Lewis B, Gralnek IM, Zlotnick S, Fort JG. Video capsule endoscopy to prospectively assess small bowel injury with celecoxib, naproxen plus omeprazole, and placebo. Clin Gastroenterol Hepatol. 2005;3:133–141. doi: 10.1016/s1542-3565(04)00619-6. [DOI] [PubMed] [Google Scholar]
- 51.Hawkey CJ, Ell C, Simon B, Albert J, Keuchel M, McAlindon M, Fortun P, Schumann S, Bolten W, Shonde A, et al. Less small-bowel injury with lumiracoxib compared with naproxen plus omeprazole. Clin Gastroenterol Hepatol. 2008;6:536–544. doi: 10.1016/j.cgh.2007.12.023. [DOI] [PubMed] [Google Scholar]
- 52.Reuter BK, Davies NM, Wallace JL. Nonsteroidal anti-inflammatory drug enteropathy in rats: role of permeability, bacteria, and enterohepatic circulation. Gastroenterology. 1997;112:109–117. doi: 10.1016/s0016-5085(97)70225-7. [DOI] [PubMed] [Google Scholar]
- 53.Morris AJ, Madhok R, Sturrock RD, Capell HA, MacKenzie JF. Enteroscopic diagnosis of small bowel ulceration in patients receiving non-steroidal anti-inflammatory drugs. Lancet. 1991;337:520. doi: 10.1016/0140-6736(91)91300-j. [DOI] [PubMed] [Google Scholar]
- 54.Fujimori S, Gudis K, Takahashi Y, Seo T, Yamada Y, Ehara A, Kobayashi T, Mitsui K, Yonezawa M, Tanaka S, et al. Distribution of small intestinal mucosal injuries as a result of NSAID administration. Eur J Clin Invest. 2010;40:504–510. doi: 10.1111/j.1365-2362.2010.02290.x. [DOI] [PubMed] [Google Scholar]
- 55.Flower RJ, Vane JR. Inhibition of prostaglandin synthetase in brain explains the anti-pyretic activity of paracetamol (4-acetamidophenol) Nature. 1972;240:410–411. doi: 10.1038/240410a0. [DOI] [PubMed] [Google Scholar]
- 56.Xie WL, Chipman JG, Robertson DL, Erikson RL, Simmons DL. Expression of a mitogen-responsive gene encoding prostaglandin synthase is regulated by mRNA splicing. Proc Natl Acad Sci USA. 1991;88:2692–2696. doi: 10.1073/pnas.88.7.2692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kujubu DA, Fletcher BS, Varnum BC, Lim RW, Herschman HR. TIS10, a phorbol ester tumor promoter-inducible mRNA from Swiss 3T3 cells, encodes a novel prostaglandin synthase/cyclooxygenase homologue. J Biol Chem. 1991;266:12866–12872. [PubMed] [Google Scholar]
- 58.Wallace JL, Devchand PR. Emerging roles for cyclooxygenase-2 in gastrointestinal mucosal defense. Br J Pharmacol. 2005;145:275–282. doi: 10.1038/sj.bjp.0706201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sigthorsson G, Simpson RJ, Walley M, Anthony A, Foster R, Hotz-Behoftsitz C, Palizban A, Pombo J, Watts J, Morham SG, et al. COX-1 and 2, intestinal integrity, and pathogenesis of nonsteroidal anti-inflammatory drug enteropathy in mice. Gastroenterology. 2002;122:1913–1923. doi: 10.1053/gast.2002.33647. [DOI] [PubMed] [Google Scholar]
- 60.Wallace JL, Bak A, McKnight W, Asfaha S, Sharkey KA, MacNaughton WK. Cyclooxygenase 1 contributes to inflammatory responses in rats and mice: implications for gastrointestinal toxicity. Gastroenterology. 1998;115:101–109. doi: 10.1016/s0016-5085(98)70370-1. [DOI] [PubMed] [Google Scholar]
- 61.Gilroy DW, Colville-Nash PR, Willis D, Chivers J, Paul-Clark MJ, Willoughby DA. Inducible cyclooxygenase may have anti-inflammatory properties. Nat Med. 1999;5:698–701. doi: 10.1038/9550. [DOI] [PubMed] [Google Scholar]
- 62.Wallace JL, McKnight W, Reuter BK, Vergnolle N. NSAID-induced gastric damage in rats: requirement for inhibition of both cyclooxygenase 1 and 2. Gastroenterology. 2000;119:706–714. doi: 10.1053/gast.2000.16510. [DOI] [PubMed] [Google Scholar]
- 63.Tanaka A, Hase S, Miyazawa T, Takeuchi K. Up-regulation of cyclooxygenase-2 by inhibition of cyclooxygenase-1: a key to nonsteroidal anti-inflammatory drug-induced intestinal damage. J Pharmacol Exp Ther. 2002;300:754–761. doi: 10.1124/jpet.300.3.754. [DOI] [PubMed] [Google Scholar]
- 64.Serhan CN, Oliw E. Unorthodox routes to prostanoid formation: new twists in cyclooxygenase-initiated pathways. J Clin Invest. 2001;107:1481–1489. doi: 10.1172/JCI13375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Serhan CN, Brain SD, Buckley CD, Gilroy DW, Haslett C, O’Neill LA, Perretti M, Rossi AG, Wallace JL. Resolution of inflammation: state of the art, definitions and terms. FASEB J. 2007;21:325–332. doi: 10.1096/fj.06-7227rev. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mizuno H, Sakamoto C, Matsuda K, Wada K, Uchida T, Noguchi H, Akamatsu T, Kasuga M. Induction of cyclooxygenase 2 in gastric mucosal lesions and its inhibition by the specific antagonist delays healing in mice. Gastroenterology. 1997;112:387–397. doi: 10.1053/gast.1997.v112.pm9024292. [DOI] [PubMed] [Google Scholar]
- 67.Ma L, del Soldato P, Wallace JL. Divergent effects of new cyclooxygenase inhibitors on gastric ulcer healing: Shifting the angiogenic balance. Proc Natl Acad Sci USA. 2002;99:13243–13247. doi: 10.1073/pnas.202392199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Schmassmann A, Peskar BM, Stettler C, Netzer P, Stroff T, Flogerzi B, Halter F. Effects of inhibition of prostaglandin endoperoxide synthase-2 in chronic gastro-intestinal ulcer models in rats. Br J Pharmacol. 1998;123:795–804. doi: 10.1038/sj.bjp.0701672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Okayama M, Hayashi S, Aoi Y, Nishio H, Kato S, Takeuchi K. Aggravation by selective COX-1 and COX-2 inhibitors of dextran sulfate sodium (DSS)-induced colon lesions in rats. Dig Dis Sci. 2007;52:2095–2103. doi: 10.1007/s10620-006-9597-z. [DOI] [PubMed] [Google Scholar]
- 70.Davies NM, Sharkey KA, Asfaha S, Macnaughton WK, Wallace JL. Aspirin causes rapid up-regulation of cyclo-oxygenase-2 expression in the stomach of rats. Aliment Pharmacol Ther. 1997;11:1101–1108. doi: 10.1046/j.1365-2036.1997.00247.x. [DOI] [PubMed] [Google Scholar]
- 71.Fiorucci S, de Lima OM, Mencarelli A, Palazzetti B, Distrutti E, McKnight W, Dicay M, Ma L, Romano M, Morelli A, et al. Cyclooxygenase-2-derived lipoxin A4 increases gastric resistance to aspirin-induced damage. Gastroenterology. 2002;123:1598–1606. doi: 10.1053/gast.2002.36558. [DOI] [PubMed] [Google Scholar]
- 72.Souza MH, de Lima OM, Zamuner SR, Fiorucci S, Wallace JL. Gastritis increases resistance to aspirin-induced mucosal injury via COX-2-mediated lipoxin synthesis. Am J Physiol Gastrointest Liver Physiol. 2003;285:G54–G61. doi: 10.1152/ajpgi.00525.2002. [DOI] [PubMed] [Google Scholar]
- 73.Maiden L, Thjodleifsson B, Seigal A, Bjarnason II, Scott D, Birgisson S, Bjarnason I. Long-term effects of nonsteroidal anti-inflammatory drugs and cyclooxygenase-2 selective agents on the small bowel: a cross-sectional capsule enteroscopy study. Clin Gastroenterol Hepatol. 2007;5:1040–1045. doi: 10.1016/j.cgh.2007.04.031. [DOI] [PubMed] [Google Scholar]
- 74.Jones MK, Wang H, Peskar BM, Levin E, Itani RM, Sarfeh IJ, Tarnawski AS. Inhibition of angiogenesis by nonsteroidal anti-inflammatory drugs: insight into mechanisms and implications for cancer growth and ulcer healing. Nat Med. 1999;5:1418–1423. doi: 10.1038/70995. [DOI] [PubMed] [Google Scholar]
- 75.Somasundaram S, Rafi S, Hayllar J, Sigthorsson G, Jacob M, Price AB, Macpherson A, Mahmod T, Scott D, Wrigglesworth JM, et al. Mitochondrial damage: a possible mechanism of the “topical” phase of NSAID induced injury to the rat intestine. Gut. 1997;41:344–353. doi: 10.1136/gut.41.3.344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wax J, Clinger WA, Varner P, Bass P, Winder CV. Relationship of the enterohepatic cycle to ulcerogenesis in the rat small bowel with flufenamic acid. Gastroenterology. 1970;58:772–780. [PubMed] [Google Scholar]
- 77.Jacob M, Foster R, Sigthorsson G, Simpson R, Bjarnason I. Role of bile in pathogenesis of indomethacin-induced enteropathy. Arch Toxicol. 2007;81:291–298. doi: 10.1007/s00204-006-0149-2. [DOI] [PubMed] [Google Scholar]
- 78.Lichtenberger LM, Phan T, Okabe S. Aspirin’s ability to induce intestinal injury in rats is dependent on bile and can be reversed if pre-associated with phosphatidylcholine. J Physiol Pharmacol. 2011;62:491–496. [PubMed] [Google Scholar]
- 79.Seitz S, Boelsterli UA. Diclofenac acyl glucuronide, a major biliary metabolite, is directly involved in small intestinal injury in rats. Gastroenterology. 1998;115:1476–1482. doi: 10.1016/s0016-5085(98)70026-5. [DOI] [PubMed] [Google Scholar]
- 80.Uchida A, Yamada T, Hayakawa T, Hoshino M. Taurochenodeoxycholic acid ameliorates and ursodeoxycholic acid exacerbates small intestinal inflammation. Am J Physiol. 1997;272:G1249–G1257. doi: 10.1152/ajpgi.1997.272.5.G1249. [DOI] [PubMed] [Google Scholar]
- 81.Zhou Y, Dial EJ, Doyen R, Lichtenberger LM. Effect of indomethacin on bile acid-phospholipid interactions: implication for small intestinal injury induced by nonsteroidal anti-inflammatory drugs. Am J Physiol Gastrointest Liver Physiol. 2010;298:G722–G731. doi: 10.1152/ajpgi.00387.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Dial EJ, Darling RL, Lichtenberger LM. Importance of biliary excretion of indomethacin in gastrointestinal and hepatic injury. J Gastroenterol Hepatol. 2008;23:e384–e389. doi: 10.1111/j.1440-1746.2007.05266.x. [DOI] [PubMed] [Google Scholar]
- 83.Yamada T, Hoshino M, Hayakawa T, Kamiya Y, Ohhara H, Mizuno K, Yamada H, Nakazawa T, Inagaki T, Uchida A, et al. Bile secretion in rats with indomethacin-induced intestinal inflammation. Am J Physiol. 1996;270:G804–G812. doi: 10.1152/ajpgi.1996.270.5.G804. [DOI] [PubMed] [Google Scholar]
- 84.Boelsterli UA, Ramirez-Alcantara V. NSAID acyl glucuronides and enteropathy. Curr Drug Metab. 2011;12:245–252. doi: 10.2174/138920011795101877. [DOI] [PubMed] [Google Scholar]
- 85.LoGuidice A, Wallace BD, Bendel L, Redinbo MR, Boelsterli UA. Pharmacologic targeting of bacterial β-glucuronidase alleviates nonsteroidal anti-inflammatory drug-induced enteropathy in mice. J Pharmacol Exp Ther. 2012;341:447–454. doi: 10.1124/jpet.111.191122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Atchison CR, West AB, Balakumaran A, Hargus SJ, Pohl LR, Daiker DH, Aronson JF, Hoffmann WE, Shipp BK, Treinen-Moslen M. Drug enterocyte adducts: possible causal factor for diclofenac enteropathy in rats. Gastroenterology. 2000;119:1537–1547. doi: 10.1053/gast.2000.20186. [DOI] [PubMed] [Google Scholar]
- 87.Robert A, Asano T. Resistance of germfree rats to indomethacin-induced intestinal lesions. Prostaglandins. 1977;14:333–341. doi: 10.1016/0090-6980(77)90178-2. [DOI] [PubMed] [Google Scholar]
- 88.Uejima M, Kinouchi T, Kataoka K, Hiraoka I, Ohnishi Y. Role of intestinal bacteria in ileal ulcer formation in rats treated with a nonsteroidal antiinflammatory drug. Microbiol Immunol. 1996;40:553–560. doi: 10.1111/j.1348-0421.1996.tb01108.x. [DOI] [PubMed] [Google Scholar]
- 89.Kent TH, Cardelli RM, Stamler FW. Small intestinal ulcers and intestinal flora in rats given indomethacin. Am J Pathol. 1969;54:237–249. [PMC free article] [PubMed] [Google Scholar]
- 90.Dalby AB, Frank DN, St Amand AL, Bendele AM, Pace NR. Culture-independent analysis of indomethacin-induced alterations in the rat gastrointestinal microbiota. Appl Environ Microbiol. 2006;72:6707–6715. doi: 10.1128/AEM.00378-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hagiwara M, Kataoka K, Arimochi H, Kuwahara T, Ohnishi Y. Role of unbalanced growth of gram-negative bacteria in ileal ulcer formation in rats treated with a nonsteroidal anti-inflammatory drug. J Med Invest. 2004;51:43–51. doi: 10.2152/jmi.51.43. [DOI] [PubMed] [Google Scholar]
- 92.Kato N, Mashita Y, Kato S, Mitsufuji S, Yoshikawa T, Takeuchi K. Sildenafil, an inhibitor of phosphodiesterase subtype 5, prevents indomethacin-induced small-intestinal ulceration in rats via a NO/cGMP-dependent mechanism. Dig Dis Sci. 2009;54:2346–2356. doi: 10.1007/s10620-008-0646-7. [DOI] [PubMed] [Google Scholar]
- 93.Watanabe T, Higuchi K, Kobata A, Nishio H, Tanigawa T, Shiba M, Tominaga K, Fujiwara Y, Oshitani N, Asahara T, et al. Non-steroidal anti-inflammatory drug-induced small intestinal damage is Toll-like receptor 4 dependent. Gut. 2008;57:181–187. doi: 10.1136/gut.2007.125963. [DOI] [PubMed] [Google Scholar]
- 94.Koga H, Aoyagi K, Matsumoto T, Iida M, Fujishima M. Experimental enteropathy in athymic and euthymic rats: synergistic role of lipopolysaccharide and indomethacin. Am J Physiol. 1999;276:G576–G582. doi: 10.1152/ajpgi.1999.276.3.G576. [DOI] [PubMed] [Google Scholar]
- 95.Kinouchi T, Kataoka K, Bing SR, Nakayama H, Uejima M, Shimono K, Kuwahara T, Akimoto S, Hiraoka I, Ohnishi Y. Culture supernatants of Lactobacillus acidophilus and Bifidobacterium adolescentis repress ileal ulcer formation in rats treated with a nonsteroidal antiinflammatory drug by suppressing unbalanced growth of aerobic bacteria and lipid peroxidation. Microbiol Immunol. 1998;42:347–355. doi: 10.1111/j.1348-0421.1998.tb02294.x. [DOI] [PubMed] [Google Scholar]
- 96.Syer SD, McKnight W, Aucouturier A, Martin R, Langella P, Wallace JL. Bifidobacteria exert a protective effect against NSAID-induced enteropathy that is dependent on lactate production. Gastroenterology. 2012;142:S–489. [Google Scholar]
- 97.Elliott SN, Buret A, McKnight W, Miller MJ, Wallace JL. Bacteria rapidly colonize and modulate healing of gastric ulcers in rats. Am J Physiol. 1998;275:G425–G432. doi: 10.1152/ajpgi.1998.275.3.G425. [DOI] [PubMed] [Google Scholar]
- 98.Elliott SN, Wallace JL, McKnight W, Gall DG, Hardin JA, Olson M, Buret A. Bacterial colonization and healing of gastric ulcers: the effects of epidermal growth factor. Am J Physiol Gastrointest Liver Physiol. 2000;278:G105–G112. doi: 10.1152/ajpgi.2000.278.1.G105. [DOI] [PubMed] [Google Scholar]
- 99.Yamada T, Deitch E, Specian RD, Perry MA, Sartor RB, Grisham MB. Mechanisms of acute and chronic intestinal inflammation induced by indomethacin. Inflammation. 1993;17:641–662. doi: 10.1007/BF00920471. [DOI] [PubMed] [Google Scholar]
- 100.Wallace JL, Keenan CM, Granger DN. Gastric ulceration induced by nonsteroidal anti-inflammatory drugs is a neutrophil-dependent process. Am J Physiol. 1990;259:G462–G467. doi: 10.1152/ajpgi.1990.259.3.G462. [DOI] [PubMed] [Google Scholar]
- 101.McCafferty DM, Granger DN, Wallace JL. Indomethacin-induced gastric injury and leukocyte adherence in arthritic versus healthy rats. Gastroenterology. 1995;109:1173–1180. doi: 10.1016/0016-5085(95)90576-6. [DOI] [PubMed] [Google Scholar]
- 102.Chmaisse HM, Antoon JS, Kvietys PR, Grisham MB, Perry MA. Role of leukocytes in indomethacin-induced small bowel injury in the rat. Am J Physiol. 1994;266:G239–G246. doi: 10.1152/ajpgi.1994.266.2.G239. [DOI] [PubMed] [Google Scholar]
- 103.Konaka A, Kato S, Tanaka A, Kunikata T, Korolkiewicz R, Takeuchi K. Roles of enterobacteria, nitric oxide and neutrophil in pathogenesis of indomethacin-induced small intestinal lesions in rats. Pharmacol Res. 1999;40:517–524. doi: 10.1006/phrs.1999.0550. [DOI] [PubMed] [Google Scholar]
- 104.Miura S, Suematsu M, Tanaka S, Nagata H, Houzawa S, Suzuki M, Kurose I, Serizawa H, Tsuchiya M. Microcirculatory disturbance in indomethacin-induced intestinal ulcer. Am J Physiol. 1991;261:G213–G219. doi: 10.1152/ajpgi.1991.261.2.G213. [DOI] [PubMed] [Google Scholar]
- 105.Santucci L, Fiorucci S, Di Matteo FM, Morelli A. Role of tumor necrosis factor alpha release and leukocyte margination in indomethacin-induced gastric injury in rats. Gastroenterology. 1995;108:393–401. doi: 10.1016/0016-5085(95)90065-9. [DOI] [PubMed] [Google Scholar]
- 106.Reuter BK, Wallace JL. Phosphodiesterase inhibitors prevent NSAID enteropathy independently of effects on TNF-alpha release. Am J Physiol. 1999;277:G847–G854. doi: 10.1152/ajpgi.1999.277.4.G847. [DOI] [PubMed] [Google Scholar]
- 107.Bertrand V, Guimbaud R, Tulliez M, Mauprivez C, Sogni P, Couturier D, Giroud JP, Chaussade S, Chauvelot-Moachon L. Increase in tumor necrosis factor-alpha production linked to the toxicity of indomethacin for the rat small intestine. Br J Pharmacol. 1998;124:1385–1394. doi: 10.1038/sj.bjp.0701968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Gadelle D, Raibaud P, Sacquet E. beta-Glucuronidase activities of intestinal bacteria determined both in vitro and in vivo in gnotobiotic rats. Appl Environ Microbiol. 1985;49:682–685. doi: 10.1128/aem.49.3.682-685.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Hayllar J, Smith T, Macpherson A, Price AB, Gumpel M, Bjarnason I. Nonsteroidal antiinflammatory drug-induced small intestinal inflammation and blood loss. Effects of sulfasalazine and other disease-modifying antirheumatic drugs. Arthritis Rheum. 1994;37:1146–1150. doi: 10.1002/art.1780370806. [DOI] [PubMed] [Google Scholar]
- 110.Morris AJ, Murray L, Sturrock RD, Madhok R, Capell HA, Mackenzie JF. Short report: the effect of misoprostol on the anaemia of NSAID enteropathy. Aliment Pharmacol Ther. 1994;8:343–346. doi: 10.1111/j.1365-2036.1994.tb00298.x. [DOI] [PubMed] [Google Scholar]
- 111.Fujimori S, Seo T, Gudis K, Ehara A, Kobayashi T, Mitsui K, Yonezawa M, Tanaka S, Tatsuguchi A, Sakamoto C. Prevention of nonsteroidal anti-inflammatory drug-induced small-intestinal injury by prostaglandin: a pilot randomized controlled trial evaluated by capsule endoscopy. Gastrointest Endosc. 2009;69:1339–1346. doi: 10.1016/j.gie.2008.08.017. [DOI] [PubMed] [Google Scholar]
- 112.Davies GR, Wilkie ME, Rampton DS. Effects of metronidazole and misoprostol on indomethacin-induced changes in intestinal permeability. Dig Dis Sci. 1993;38:417–425. doi: 10.1007/BF01316493. [DOI] [PubMed] [Google Scholar]
- 113.Jenkins RT, Rooney PJ, Hunt RH. Increased bowel permeability to 51Cr-EDTA in controls caused by naproxen is not prevented by cytoprotection. Arthritis Rheum. 1998;31(Suppl 1):R11. [Google Scholar]
- 114.Bjarnason I, Smethurst P, Fenn CG, Lee CE, Menzies IS, Levi AJ. Misoprostol reduces indomethacin-induced changes in human small intestinal permeability. Dig Dis Sci. 1989;34:407–411. doi: 10.1007/BF01536263. [DOI] [PubMed] [Google Scholar]
- 115.Montalto M, Gallo A, Curigliano V, D’Onofrio F, Santoro L, Covino M, Dalvai S, Gasbarrini A, Gasbarrini G. Clinical trial: the effects of a probiotic mixture on non-steroidal anti-inflammatory drug enteropathy - a randomized, double-blind, cross-over, placebo-controlled study. Aliment Pharmacol Ther. 2010;32:209–214. doi: 10.1111/j.1365-2036.2010.04324.x. [DOI] [PubMed] [Google Scholar]
- 116.Endo H, Higurashi T, Hosono K, Sakai E, Sekino Y, Iida H, Sakamoto Y, Koide T, Takahashi H, Yoneda M, et al. Efficacy of Lactobacillus casei treatment on small bowel injury in chronic low-dose aspirin users: a pilot randomized controlled study. J Gastroenterol. 2011;46:894–905. doi: 10.1007/s00535-011-0410-1. [DOI] [PubMed] [Google Scholar]
- 117.Watanabe T, Nishio H, Tanigawa T, Yamagami H, Okazaki H, Watanabe K, Tominaga K, Fujiwara Y, Oshitani N, Asahara T, et al. Probiotic Lactobacillus casei strain Shirota prevents indomethacin-induced small intestinal injury: involvement of lactic acid. Am J Physiol Gastrointest Liver Physiol. 2009;297:G506–G513. doi: 10.1152/ajpgi.90553.2008. [DOI] [PubMed] [Google Scholar]
- 118.Dial EJ, Dohrman AJ, Romero JJ, Lichtenberger LM. Recombinant human lactoferrin prevents NSAID-induced intestinal bleeding in rodents. J Pharm Pharmacol. 2005;57:93–99. doi: 10.1211/0022357055191. [DOI] [PubMed] [Google Scholar]
- 119.Liepke C, Adermann K, Raida M, Mägert HJ, Forssmann WG, Zucht HD. Human milk provides peptides highly stimulating the growth of bifidobacteria. Eur J Biochem. 2002;269:712–718. doi: 10.1046/j.0014-2956.2001.02712.x. [DOI] [PubMed] [Google Scholar]
- 120.Troost FJ, Saris WH, Brummer RJ. Recombinant human lactoferrin ingestion attenuates indomethacin-induced enteropathy in vivo in healthy volunteers. Eur J Clin Nutr. 2003;57:1579–1585. doi: 10.1038/sj.ejcn.1601727. [DOI] [PubMed] [Google Scholar]
- 121.Arakawa T, Watanabe T, Fukuda T, Yamasaki K, Kobayashi K. Rebamipide, novel prostaglandin-inducer accelerates healing and reduces relapse of acetic acid-induced rat gastric ulcer. Comparison with cimetidine. Dig Dis Sci. 1995;40:2469–2472. doi: 10.1007/BF02063257. [DOI] [PubMed] [Google Scholar]
- 122.Mizoguchi H, Ogawa Y, Kanatsu K, Tanaka A, Kato S, Takeuchi K. Protective effect of rebamipide on indomethacin-induced intestinal damage in rats. J Gastroenterol Hepatol. 2001;16:1112–1119. doi: 10.1046/j.1440-1746.2001.02592.x. [DOI] [PubMed] [Google Scholar]
- 123.Niwa Y, Nakamura M, Ohmiya N, Maeda O, Ando T, Itoh A, Hirooka Y, Goto H. Efficacy of rebamipide for diclofenac-induced small-intestinal mucosal injuries in healthy subjects: a prospective, randomized, double-blinded, placebo-controlled, cross-over study. J Gastroenterol. 2008;43:270–276. doi: 10.1007/s00535-007-2155-4. [DOI] [PubMed] [Google Scholar]
- 124.Fujimori S, Takahashi Y, Gudis K, Seo T, Ehara A, Kobayashi T, Mitsui K, Yonezawa M, Tanaka S, Tatsuguchi A, et al. Rebamipide has the potential to reduce the intensity of NSAID-induced small intestinal injury: a double-blind, randomized, controlled trial evaluated by capsule endoscopy. J Gastroenterol. 2011;46:57–64. doi: 10.1007/s00535-010-0332-3. [DOI] [PubMed] [Google Scholar]
- 125.Yasuda M, Kawahara R, Hashimura H, Yamanaka N, Iimori M, Amagase K, Kato S, Takeuchi K. Dopamine D-receptor antagonists ameliorate indomethacin-induced small intestinal ulceration in mice by activating α7 nicotinic acetylcholine receptors. J Pharmacol Sci. 2011;116:274–282. doi: 10.1254/jphs.11037fp. [DOI] [PubMed] [Google Scholar]
- 126.Kawahara R, Yasuda M, Hashimura H, Amagase K, Kato S, Takeuchi K. Activation of α7 nicotinic acetylcholine receptors ameliorates indomethacin-induced small intestinal ulceration in mice. Eur J Pharmacol. 2011;650:411–417. doi: 10.1016/j.ejphar.2010.10.031. [DOI] [PubMed] [Google Scholar]
- 127.Kato S, Matsuda N, Matsumoto K, Wada M, Onimaru N, Yasuda M, Amagase K, Horie S, Takeuchi K. Dual role of serotonin in the pathogenesis of indomethacin-induced small intestinal ulceration: pro-ulcerogenic action via 5-HT3 receptors and anti-ulcerogenic action via 5-HT4 receptors. Pharmacol Res. 2012;66:226–234. doi: 10.1016/j.phrs.2012.06.002. [DOI] [PubMed] [Google Scholar]
- 128.Rautio J, Kumpulainen H, Heimbach T, Oliyai R, Oh D, Järvinen T, Savolainen J. Prodrugs: design and clinical applications. Nat Rev Drug Discov. 2008;7:255–270. doi: 10.1038/nrd2468. [DOI] [PubMed] [Google Scholar]
- 129.Qandil AM. Prodrugs of nonsteroidal anti-inflammatory drugs (NSAIDs), more than meets the eye: a critical review. Int J Mol Sci. 2012;13:17244–17274. doi: 10.3390/ijms131217244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Whittle BJ, Hansen D, Salmon JA. Gastric ulcer formation and cyclo-oxygenase inhibition in cat antrum follows parenteral administration of aspirin but not salicylate. Eur J Pharmacol. 1985;116:153–157. doi: 10.1016/0014-2999(85)90196-7. [DOI] [PubMed] [Google Scholar]
- 131.Wallace JL, McKnight GW. Characterization of a simple animal model for nonsteroidal anti-inflammatory drug induced antral ulcer. Can J Physiol Pharmacol. 1993;71:447–452. doi: 10.1139/y93-066. [DOI] [PubMed] [Google Scholar]
- 132.Estes LL, Fuhs DW, Heaton AH, Butwinick CS. Gastric ulcer perforation associated with the use of injectable ketorolac. Ann Pharmacother. 1993;27:42–43. doi: 10.1177/106002809302700111. [DOI] [PubMed] [Google Scholar]
- 133. Available from: http: //www.logicaltx.com/product-pipeline/nsaid-prodrug.php.
- 134.Goldstein JL, Jungwirthová A, David J, Spindel E, Bouchner L, Pešek F, Searle S, Skopek J, Grim J, Ulč I, et al. Clinical trial: endoscopic evaluation of naproxen etemesil, a naproxen prodrug, vs. naproxen - a proof-of-concept, randomized, double-blind, active-comparator study. Aliment Pharmacol Ther. 2010;32:1091–1101. doi: 10.1111/j.1365-2036.2010.04442.x. [DOI] [PubMed] [Google Scholar]
- 135.Graham DY. Endoscopic ulcers are neither meaningful nor validated as a surrogate for clinically significant upper gastrointestinal harm. Clin Gastroenterol Hepatol. 2009;7:1147–1150. doi: 10.1016/j.cgh.2009.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Lanza FL. Endoscopic studies of gastric and duodenal injury after the use of ibuprofen, aspirin, and other nonsteroidal anti-inflammatory agents. Am J Med. 1984;77:19–24. doi: 10.1016/s0002-9343(84)80014-5. [DOI] [PubMed] [Google Scholar]
- 137.Graham DY, Smith JL, Holmes GI, Davies RO. Nonsteroidal anti-inflammatory effect of sulindac sulfoxide and sulfide on gastric mucosa. Clin Pharmacol Ther. 1985;38:65–70. doi: 10.1038/clpt.1985.136. [DOI] [PubMed] [Google Scholar]
- 138.Carson JL, Strom BL, Morse ML, West SL, Soper KA, Stolley PD, Jones JK. The relative gastrointestinal toxicity of the nonsteroidal anti-inflammatory drugs. Arch Intern Med. 1987;147:1054–1059. [PubMed] [Google Scholar]
- 139.Roth SH, Tindall EA, Jain AK, McMahon FG, April PA, Bockow BI, Cohen SB, Fleischmann RM. A controlled study comparing the effects of nabumetone, ibuprofen, and ibuprofen plus misoprostol on the upper gastrointestinal tract mucosa. Arch Intern Med. 1993;153:2565–2571. [PubMed] [Google Scholar]
- 140.Chan FK, Sung JJ, Ching JY, Wu JC, Lee YT, Leung WK, Hui Y, Chan LY, Lai AC, Chung SC. Randomized trial of low-dose misoprostol and naproxen vs. nabumetone to prevent recurrent upper gastrointestinal haemorrhage in users of non-steroidal anti-inflammatory drugs. Aliment Pharmacol Ther. 2001;15:19–24. doi: 10.1046/j.1365-2036.2001.00890.x. [DOI] [PubMed] [Google Scholar]
- 141.Henry D, Dobson A, Turner C. Variability in the risk of major gastrointestinal complications from nonaspirin nonsteroidal anti-inflammatory drugs. Gastroenterology. 1993;105:1078–1088. doi: 10.1016/0016-5085(93)90952-9. [DOI] [PubMed] [Google Scholar]
- 142.Wallace JL, Ferraz JG. New pharmacologic therapies in gastrointestinal disease. Gastroenterol Clin North Am. 2010;39:709–720. doi: 10.1016/j.gtc.2010.08.020. [DOI] [PubMed] [Google Scholar]
- 143.Wallace JL, Del Soldato P. The therapeutic potential of NO-NSAIDs. Fundam Clin Pharmacol. 2003;17:11–20. doi: 10.1046/j.1472-8206.2003.00125.x. [DOI] [PubMed] [Google Scholar]
- 144.Caliendo G, Cirino G, Santagada V, Wallace JL. Synthesis and biological effects of hydrogen sulfide (H2S): development of H2S-releasing drugs as pharmaceuticals. J Med Chem. 2010;53:6275–6286. doi: 10.1021/jm901638j. [DOI] [PubMed] [Google Scholar]
- 145.Wallace JL. Hydrogen sulfide-releasing anti-inflammatory drugs. Trends Pharmacol Sci. 2007;28:501–505. doi: 10.1016/j.tips.2007.09.003. [DOI] [PubMed] [Google Scholar]
- 146.Fiorucci S, Distrutti E, Cirino G, Wallace JL. The emerging roles of hydrogen sulfide in the gastrointestinal tract and liver. Gastroenterology. 2006;131:259–271. doi: 10.1053/j.gastro.2006.02.033. [DOI] [PubMed] [Google Scholar]
- 147.Wallace JL, Miller MJ. Nitric oxide in mucosal defense: a little goes a long way. Gastroenterology. 2000;119:512–520. doi: 10.1053/gast.2000.9304. [DOI] [PubMed] [Google Scholar]
- 148.Fiorucci S, Antonelli E, Distrutti E, Rizzo G, Mencarelli A, Orlandi S, Zanardo R, Renga B, Di Sante M, Morelli A, et al. Inhibition of hydrogen sulfide generation contributes to gastric injury caused by anti-inflammatory nonsteroidal drugs. Gastroenterology. 2005;129:1210–1224. doi: 10.1053/j.gastro.2005.07.060. [DOI] [PubMed] [Google Scholar]
- 149.Zanardo RC, Brancaleone V, Distrutti E, Fiorucci S, Cirino G, Wallace JL. Hydrogen sulfide is an endogenous modulator of leukocyte-mediated inflammation. FASEB J. 2006;20:2118–2120. doi: 10.1096/fj.06-6270fje. [DOI] [PubMed] [Google Scholar]
- 150.Elliott SN, McKnight W, Cirino G, Wallace JL. A nitric oxide-releasing nonsteroidal anti-inflammatory drug accelerates gastric ulcer healing in rats. Gastroenterology. 1995;109:524–530. doi: 10.1016/0016-5085(95)90341-0. [DOI] [PubMed] [Google Scholar]
- 151.Wallace JL. Physiological and pathophysiological roles of hydrogen sulfide in the gastrointestinal tract. Antioxid Redox Signal. 2010;12:1125–1133. doi: 10.1089/ars.2009.2900. [DOI] [PubMed] [Google Scholar]
- 152.Wallace JL, Ignarro LJ, Fiorucci S. Potential cardioprotective actions of no-releasing aspirin. Nat Rev Drug Discov. 2002;1:375–382. doi: 10.1038/nrd794. [DOI] [PubMed] [Google Scholar]
- 153.Konturek SJ, Brzozowski T, Majka J, Pytko-Polonczyk J, Stachura J. Inhibition of nitric oxide synthase delays healing of chronic gastric ulcers. Eur J Pharmacol. 1993;239:215–217. doi: 10.1016/0014-2999(93)90997-v. [DOI] [PubMed] [Google Scholar]
- 154.Ma L, Wallace JL. Endothelial nitric oxide synthase modulates gastric ulcer healing in rats. Am J Physiol Gastrointest Liver Physiol. 2000;279:G341–G346. doi: 10.1152/ajpgi.2000.279.2.G341. [DOI] [PubMed] [Google Scholar]
- 155.Wallace JL, Caliendo G, Santagada V, Cirino G, Fiorucci S. Gastrointestinal safety and anti-inflammatory effects of a hydrogen sulfide-releasing diclofenac derivative in the rat. Gastroenterology. 2007;132:261–271. doi: 10.1053/j.gastro.2006.11.042. [DOI] [PubMed] [Google Scholar]
- 156.Whittle BJ, Lopez-Belmonte J, Moncada S. Regulation of gastric mucosal integrity by endogenous nitric oxide: interactions with prostanoids and sensory neuropeptides in the rat. Br J Pharmacol. 1990;99:607–611. doi: 10.1111/j.1476-5381.1990.tb12977.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.MacNaughton WK, Cirino G, Wallace JL. Endothelium-derived relaxing factor (nitric oxide) has protective actions in the stomach. Life Sci. 1989;45:1869–1876. doi: 10.1016/0024-3205(89)90540-7. [DOI] [PubMed] [Google Scholar]
- 158.Wallace JL, Dicay M, McKnight W, Martin GR. Hydrogen sulfide enhances ulcer healing in rats. FASEB J. 2007;21:4070–4076. doi: 10.1096/fj.07-8669com. [DOI] [PubMed] [Google Scholar]
- 159.Reuter BK, Cirino G, Wallace JL. Markedly reduced intestinal toxicity of a diclofenac derivative. Life Sci. 1994;55:PL1–PL8. doi: 10.1016/0024-3205(94)90083-3. [DOI] [PubMed] [Google Scholar]
- 160.Davies NM, Røseth AG, Appleyard CB, McKnight W, Del Soldato P, Calignano A, Cirino G, Wallace JL. NO-naproxen vs. naproxen: ulcerogenic, analgesic and anti-inflammatory effects. Aliment Pharmacol Ther. 1997;11:69–79. doi: 10.1046/j.1365-2036.1997.115286000.x. [DOI] [PubMed] [Google Scholar]
- 161.Hawkey CJ, Jones JI, Atherton CT, Skelly MM, Bebb JR, Fagerholm U, Jonzon B, Karlsson P, Bjarnason IT. Gastrointestinal safety of AZD3582, a cyclooxygenase inhibiting nitric oxide donator: proof of concept study in humans. Gut. 2003;52:1537–1542. doi: 10.1136/gut.52.11.1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Schnitzer TJ, Hochberg MC, Marrero CE, Duquesroix B, Frayssinet H, Beekman M. Efficacy and safety of naproxcinod in patients with osteoarthritis of the knee: a 53-week prospective randomized multicenter study. Semin Arthritis Rheum. 2011;40:285–297. doi: 10.1016/j.semarthrit.2010.06.002. [DOI] [PubMed] [Google Scholar]
- 163.Karlsson J, Pivodic A, Aguirre D, Schnitzer TJ. Efficacy, safety, and tolerability of the cyclooxygenase-inhibiting nitric oxide donator naproxcinod in treating osteoarthritis of the hip or knee. J Rheumatol. 2009;36:1290–1297. doi: 10.3899/jrheum.081011. [DOI] [PubMed] [Google Scholar]
- 164.Baerwald C, Verdecchia P, Duquesroix B, Frayssinet H, Ferreira T. Efficacy, safety, and effects on blood pressure of naproxcinod 750 mg twice daily compared with placebo and naproxen 500 mg twice daily in patients with osteoarthritis of the hip: a randomized, double-blind, parallel-group, multicenter study. Arthritis Rheum. 2010;62:3635–3644. doi: 10.1002/art.27694. [DOI] [PubMed] [Google Scholar]
- 165.Schnitzer TJ, Kivitz A, Frayssinet H, Duquesroix B. Efficacy and safety of naproxcinod in the treatment of patients with osteoarthritis of the knee: a 13-week prospective, randomized, multicenter study. Osteoarthritis Cartilage. 2010;18:629–639. doi: 10.1016/j.joca.2009.12.013. [DOI] [PubMed] [Google Scholar]
- 166.Wallace JL, Viappiani S, Bolla M. Cyclooxygenase-inhibiting nitric oxide donators for osteoarthritis. Trends Pharmacol Sci. 2009;30:112–117. doi: 10.1016/j.tips.2009.01.001. [DOI] [PubMed] [Google Scholar]
- 167.Lohmander LS, McKeith D, Svensson O, Malmenäs M, Bolin L, Kalla A, Genti G, Szechinski J, Ramos-Remus C. A randomised, placebo controlled, comparative trial of the gastrointestinal safety and efficacy of AZD3582 versus naproxen in osteoarthritis. Ann Rheum Dis. 2005;64:449–456. doi: 10.1136/ard.2004.023572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Li L, Rossoni G, Sparatore A, Lee LC, Del Soldato P, Moore PK. Anti-inflammatory and gastrointestinal effects of a novel diclofenac derivative. Free Radic Biol Med. 2007;42:706–719. doi: 10.1016/j.freeradbiomed.2006.12.011. [DOI] [PubMed] [Google Scholar]
- 169.Sparatore A, Perrino E, Tazzari V, Giustarini D, Rossi R, Rossoni G, Erdmann K, Schröder H, Del Soldato P. Pharmacological profile of a novel H(2)S-releasing aspirin. Free Radic Biol Med. 2009;46:586–592. doi: 10.1016/j.freeradbiomed.2008.11.013. [DOI] [PubMed] [Google Scholar]
- 170.Li L, Bhatia M, Moore PK. Hydrogen sulphide--a novel mediator of inflammation? Curr Opin Pharmacol. 2006;6:125–129. doi: 10.1016/j.coph.2005.10.007. [DOI] [PubMed] [Google Scholar]
- 171.Whiteman M, Le Trionnaire S, Chopra M, Fox B, Whatmore J. Emerging role of hydrogen sulfide in health and disease: critical appraisal of biomarkers and pharmacological tools. Clin Sci (Lond) 2011;121:459–488. doi: 10.1042/CS20110267. [DOI] [PubMed] [Google Scholar]
- 172.Dufton N, Natividad J, Verdu EF, Wallace JL. Hydrogen sulfide and resolution of acute inflammation: A comparative study utilizing a novel fluorescent probe. Sci Rep. 2012;2:499. doi: 10.1038/srep00499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Wallace JL, Vong L, McKnight W, Dicay M, Martin GR. Endogenous and exogenous hydrogen sulfide promotes resolution of colitis in rats. Gastroenterology. 2009;137:569–578. doi: 10.1053/j.gastro.2009.04.012. [DOI] [PubMed] [Google Scholar]
- 174.Wallace JL, Ferraz JG, Muscara MN. Hydrogen sulfide: an endogenous mediator of resolution of inflammation and injury. Antioxid Redox Signal. 2012;17:58–67. doi: 10.1089/ars.2011.4351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Blackler R, Syer S, Bolla M, Ongini E, Wallace JL. Gastrointestinal-sparing effects of novel NSAIDs in rats with compromised mucosal defence. PLoS One. 2012;7:e35196. doi: 10.1371/journal.pone.0035196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Waldram A, Holmes E, Wang Y, Rantalainen M, Wilson ID, Tuohy KM, McCartney AL, Gibson GR, Nicholson JK. Top-down systems biology modeling of host metabotype-microbiome associations in obese rodents. J Proteome Res. 2009;8:2361–2375. doi: 10.1021/pr8009885. [DOI] [PubMed] [Google Scholar]
- 177.Kodela R, Chattopadhyay M, Kashfi K. NOSH-Aspirin: A Novel Nitric Oxide-Hydrogen Sulfide-Releasing Hybrid: A New Class of Anti-inflammatory Pharmaceuticals. ACS Med Chem Lett. 2012;3:257–262. doi: 10.1021/ml300002m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Lichtenberger LM, Zhou Y, Dial EJ, Raphael RM. NSAID injury to the gastrointestinal tract: evidence that NSAIDs interact with phospholipids to weaken the hydrophobic surface barrier and induce the formation of unstable pores in membranes. J Pharm Pharmacol. 2006;58:1421–1428. doi: 10.1211/jpp.58.10.0001. [DOI] [PubMed] [Google Scholar]
- 179.Lichtenberger LM, Wang ZM, Romero JJ, Ulloa C, Perez JC, Giraud MN, Barreto JC. Non-steroidal anti-inflammatory drugs (NSAIDs) associate with zwitterionic phospholipids: insight into the mechanism and reversal of NSAID-induced gastrointestinal injury. Nat Med. 1995;1:154–158. doi: 10.1038/nm0295-154. [DOI] [PubMed] [Google Scholar]
- 180.Lichtenberger LM, Barron M, Marathi U. Association of phosphatidylcholine and NSAIDs as a novel strategy to reduce gastrointestinal toxicity. Drugs Today (Barc) 2009;45:877–890. doi: 10.1358/dot.2009.45.12.1441075. [DOI] [PubMed] [Google Scholar]
- 181.Cryer B, Bhatt DL, Lanza FL, Dong JF, Lichtenberger LM, Marathi UK. Low-dose aspirin-induced ulceration is attenuated by aspirin-phosphatidylcholine: a randomized clinical trial. Am J Gastroenterol. 2011;106:272–277. doi: 10.1038/ajg.2010.436. [DOI] [PubMed] [Google Scholar]
- 182.Lanza FL, Marathi UK, Anand BS, Lichtenberger LM. Clinical trial: comparison of ibuprofen-phosphatidylcholine and ibuprofen on the gastrointestinal safety and analgesic efficacy in osteoarthritic patients. Aliment Pharmacol Ther. 2008;28:431–442. doi: 10.1111/j.1365-2036.2008.03765.x. [DOI] [PMC free article] [PubMed] [Google Scholar]