

Abstract
Recent genome-wide association studies have described many loci implicated in type 2 diabetes (T2D) pathophysiology and beta-cell dysfunction, but contributed little to our understanding of the genetic basis of insulin resistance. We hypothesized that genes implicated in insulin resistance pathways may be uncovered by accounting for differences in body mass index (BMI) and potential interaction between BMI and genetic variants. We applied a novel joint meta-analytical approach to test associations with fasting insulin (FI) and glucose (FG) on a genome-wide scale. We present six previously unknown FI loci at P<5×10−8 in combined discovery and follow-up analyses of 52 studies comprising up to 96,496non-diabetic individuals. Risk variants were associated with higher triglyceride and lower HDL cholesterol levels, suggestive of a role for these FI loci in insulin resistance pathways. The localization of these additional loci will aid further characterization of the role of insulin resistance in T2D pathophysiology.
Introduction
In contrast to the recent progress in the discovery of genetic variants underlying type 2 diabetes (T2D) pathophysiology and beta-cell function, our understanding of the genetic basis of insulin resistance remains limited1. Partly because early case-control studies of T2D were designed to maximize the likelihood of detecting variants increasing T2D risk directly rather than through mediation of adiposity, most of the associated loci discovered in these studies uncovered genes related to beta cell dysfunction2. More recently, we have shown that the genetic architecture of quantitative indices of beta-cell function and of insulin resistance differs markedly: given the same individuals, sample sizes and biochemical measurements, we described a larger number of signals for beta cell function than for insulin resistance3,4. While this observation is consistent with the higher reported heritability of insulin secretion compared to resistance, overall heritability estimates of insulin resistance in individuals of European ancestry of 25%–44% suggest that many loci remain to be discovered and that novel strategies are required for their localization5.
Obesity is an important determinant of insulin resistance6. It was postulated that adiposity might modulate the genetic determinants of insulin resistance and contribute to the heterogeneity of T2D etiology. Wang et al. demonstrated that heritability of insulin resistance increases with higher body mass index (BMI)7, while some candidate gene studies have observed that genetic effect size varies with adiposity level8–10: findings compatible with the presence of underlying interaction between BMI and genetic variants for insulin resistance. Furthermore, hormones, adipokines and proinflammatory cytokines produced by adipose tissue can influence insulin signaling via diverse mechanisms11,12, which may interact with genetic variants influencing insulin resistance pathways. Therefore, to localize variants associated with insulin resistance it may also be important to account for gene variant by BMI interaction, which would allow for the potential of adiposity levels to perturb the physiological milieu in which genetic variants in insulin signaling pathways operate. Adiposity may also hinder the localization of genetic variants influencing insulin resistance by introducing extra variance in the outcome that is not attributable to genetic variation5, suggesting that adjustment for adiposity per se may be necessary.
Kraft et al. proposed a joint test that investigates the association between an outcome and a genetic variant, while allowing for possible effect modification by an environmental variable13. Manning et al. developed a statistical method that extends this joint test to a meta-analysis context14. This enabled us to simultaneously test both the genetic main effect, adjusted for BMI, and potential interaction between each genetic variant and BMI. This joint meta-analysis (JMA) approach can provide increased power for detecting the genetic loci when underlying interaction effects are suspected but unknown13 and importantly, as demonstrated in simulation studies, does not lose power to detect genetic main effects in the absence of interaction14. Within the Meta-Analyses of Glucose- and Insulin-related traits Consortium (MAGIC), we implemented this approach and performed a genome-wide JMA to search for SNPs significantly associated with glycemic traits while simultaneously adjusting for BMI and allowing for interaction with BMI. Using this method, we successfully identify loci that are associated with fasting insulin at genome-wide levels of significance.
Results
As a first phase, we conducted discovery genome-wide JMA of SNP main effects and SNP by BMI (SNP×BMI) interaction for four diabetes-related quantitative traits: fasting insulin, fasting glucose, and surrogate measures of beta-cell function (HOMA-B) and insulin resistance (HOMA-IR)15; fasting insulin, HOMA-B and HOMA-IR were log transformed.
The fasting insulin discovery-stage JMA of approximately 2.4 million SNPs in 51,750 non-diabetic individuals from 29 studies (Supplementary Table 1) showed previously reported associations of variants in IGF1 and GCKR with fasting insulin at genome-wide significance (Figure 1a, Supplementary Table 2), and revealed 31 previously unreported loci tentatively associated (P<10−5) with fasting insulin (Figure 1a, Supplementary Table 3a; Supplementary Figure 1)3. The fasting glucose discovery JMA in 58,074 individuals showed association of 16 loci previously described in MAGIC meta-analyses of fasting glucose (Figure 1b, Supplementary Table 2; Supplementary Figure 1), and revealed 20 previously unreported loci tentatively associated with fasting glucose (P<10−5) (Figure 1b, Supplementary Table 3b). The JMA approach revealed an excess of previously undetected signals relative to the number expected by chance (Supplementary Figure 1). The SNPs that showed association P<10−5 in the discovery JMA for HOMA-IR and HOMA-B largely overlapped with those localized for fasting insulin and fasting glucose, without describing additional loci. We therefore focused all subsequent analyses on fasting insulin and fasting glucose. Results for associations with HOMA-IR and HOMA-B are available in Supplementary Table 4.
Figure 1.

Genome-wide plots of the discovery joint meta-analysis (JMA) for fasting insulin (1a top) and fasting glucose (1b top): 17 loci with known associations were observed (red) and 50 loci were taken to the follow-up analysis (light and dark blue). Of these, 12 reached genome-wide significance in the combined discovery and follow-up JMA (dark blue). The P values of these 12 loci from the models fit in the combined discovery and follow-up analyses are presented in Figure 1a (bottom) for fasting insulin and 1b (bottom) for fasting glucose: JMA (red), main effects adjusting for BMI (orange), interaction with continuous BMI (green) and interaction with dichotomous BMI (blue). * G6PC2 JMA P value: 1.7 × 10−113, ** GCK JMA P value: 8.3 ×10−56, *** MTNR1B JMA P value: 4.38 × 10−105.
At each locus that reached P<10−5 in the discovery JMA, an index SNP was chosen to represent the association signal in the genomic region. A total of 50 SNPs (30 SNPs associated with fasting insulin, 19 SNPs associated with fasting glucose, and 1 SNP associated with both) were taken forward to the second phase follow-up analysis. The follow-up analysis included 23 studies with fasting glucose data, 22 of which also had measurements of fasting insulin, and comprised up to 38,422 and 33,823 individuals for fasting glucose and fasting insulin, respectively (Supplementary Table 1). We combined study-specific results from both the discovery (phase 1) and the follow-up (phase 2) studies for the 50 index SNPs (Supplementary Table 3), for total sample sizes of 96,496 for fasting glucose and 85,573for fasting insulin. In the combined JMA, we describe six loci associated with fasting insulin and seven loci associated with fasting glucose at genome-wide levels of significance (P<5×10−8), including PPP1R3B, which was associated with both fasting insulin and fasting glucose (Table 1; Figure 1; Supplementary Table 3). Descriptions of interesting genes around each association signal appear in Box 1 and Box 2 and to facilitate discussion of the quantitative trait loci, gene names are used as labels in the text, tables and figures.
Table 1.
Genetic loci significantly associated at genome-wide level (P<5×10−8) with fasting insulin or fasting glucose from the JMA in the combined discovery and follow-up analysis
| FASTING INSULIN | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| alleles | Stage 1: Discovery (26 cohorts) N=51,750 |
Stage 2: Follow-up (22 cohorts) N=31,450 |
Combined (48 cohorts) N=83,116 |
|||||||||
| Nearest Gene | Index SNP | R Freq | O Freq | BMI=25 Effect (SE) | BMI=30 Effect (SE) | JMA P (Het P) | BMI=25 Effect (SE) | BMI=30 Effect (SE) | JMA P (Het P) | BMI=25 Effect (SE) | BMI=30 Effect (SE) | JMA P (Het P) |
| COBLL1-GRB14 | rs7607980 | T 0.88 | C 0.12 | 0.021 (0.004) | 0.04 (0.005) | 6.1×10−14 (0.25) | 0.026 (0.006) | 0.039 (0.008) | 4.9×10−7 (0.04) | 0.023 (0.003) | 0.039 (0.004) | 4.3×10−20 (0.06) |
| IRS1 | rs2943634 | C 0.66 | A 0.34 | 0.018 (0.003) | 0.026 (0.004) | 1.7×10−12 (0.62) | 0.018 (0.008) | 0.021 (0.010) | 3.6×10−3 (0.53) | 0.018 (0.003) | 0.025 (0.004) | 2.5×10−14 (0.70) |
| PPP1R3B | rs4841132 | A 0.10 | G 0.90 | 0.02 (0.004) | 0.031 (0.006) | 1.4×10−7 (0.50) | 0.034 (0.013) | 0.032 (0.016) | 7.3×10−4 (0.45) | 0.021 (0.004) | 0.031 (0.006) | 1.7×10−10 (0.52) |
| PDGFC | rs4691380 | C 0.67 | T 0.33 | 0.016 (0.003) | 0.021 (0.004) | 1.5×10−8 (0.36) | 0.017 (0.008) | 0.020 (0.010) | 7.2×10−2 (0.31) | 0.016 (0.003) | 0.021 (0.004) | 5.3×10−9 (0.37) |
| UHRF1BP1 | rs4646949 | T 0.75 | G 0.25 | 0.016 (0.003) | 0.021 (0.004) | 5.9×10−8 (0.08) | 0.0009 (0.008) | 0.009 (0.010) | 1.6×10−1 (0.24) | 0.014 (0.003) | 0.020 (0.004) | 3.7×10−8 (0.05) |
| LYPLAL1 | rs2785980 | T 0.67 | C 0.33 | 0.016 (0.003) | 0.017 (0.004) | 7.7×10−8 (0.03) | 0.011 (0.009) | 0.018 (0.010) | 9.7×10−2 (0.20) | 0.016 (0.003) | 0.017 (0.004) | 2.0×10−8 (0.03) |
| FASTING GLUCOSE | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| alleles | Stage 1: Discovery (29 cohorts) N=58,074 |
Stage 2: Follow-up (23 cohorts) N=38,422 |
Combined (52 cohorts) N=96,496 |
|||||||||
| Nearest Gene | Index SNP | R Freq | O Freq | BMI=25 Effect (SE) | BMI=30 Effect (SE) | JMA P (Het P) | BMI=25 Effect (SE) | BMI=30 Effect (SE) | JMA P (Het P) | BMI=25 Effect (SE) | BMI=30 Effect (SE) | JMA P (Het P) |
| ARAP1 | rs11603334 | G 0.83 | A 0.17 | 0.025 (0.004) | 0.033 (0.005) | 4.4×10−11 (0.25) | 0.016 (0.005) | 0.026 (0.007) | 2.2×10−4 (0.004) | 0.022 (0.003) | 0.030 (0.004) | 2.4×10−14 (0.01) |
| FOXA2 | rs6048205 | A 0.95 | G 0.05 | 0.044 (0.007) | 0.033 (0.009) | 7.1×10−10 (0.14) | 0.033 (0.009) | 0.023 (0.012) | 1.4×10−3 (0.03) | 0.040 (0.005) | 0.029 (0.007) | 1.6×10−12 (0.03) |
| DPYSL5 | rs1371614 | T 0.25 | C 0.75 | 0.020 (0.004) | 0.026 (0.005) | 2.9×10−9 (0.19) | 0.019 (0.005) | 0.015 (0.006) | 2.1×10−4 (0.47) | 0.020 (0.003) | 0.022 (0.004) | 2.3×10−12 (0.23) |
| PCSK1 | rs13179048 | C 0.69 | A 0.31 | 0.021 (0.003) | 0.016 (0.004) | 1.6×10−9 (0.24) | 0.028 (0.010) | 0.027 (0.013) | 2.2×10−2 (0.16) | 0.022 (0.003) | 0.018 (0.004) | 1.6×10−10 (0.18) |
| PDX1 | rs2293941 | A 0.22 | G 0.78 | 0.022 (0.004) | 0.015 (0.005) | 1.3×10−8 (0.47) | 0.013 (0.005) | 0.016 (0.006) | 7.8×10−3 (0.04) | 0.019 (0.003) | 0.016 (0.004) | 5.3×10−10 (0.10) |
| PPP1R3B | rs4841132 | A 0.10 | G 0.90 | 0.026 (0.005) | 0.028 (0.007) | 7.9×10−7 (0.94) | 0.033 (0.015) | 0.054 (0.021) | 3.1×10−3 (0.02) | 0.027 (0.005) | 0.030 (0.007) | 7.6×10−9 (0.55) |
| OR4S1 | rs1483121 | G 0.86 | A 0.14 | 0.027 (0.005) | 0.022 (0.006) | 6.5×10−8 (0.70) | 0.014 (0.006) | 0.006 (0.008) | 3.4 ×10−2 (0.42) | 0.021 (0.004) | 0.015 (0.005) | 1.6×10−8 (0.56) |
Directly genotyped and imputed SNPs were tested for association with fasting glucose or fasting insulin by Joint Meta-Analysis (JMA) of the SNP and SNP×BMI effects. The effect estimates presented are derived from the JMA for the specified BMI in kg/m2. A test of heterogeneity (Het P) was performed for each locus. From the discovery analysis, 50 loci were promoted to the follow-up analysis (31 for fasting insulin and 20 for fasting glucose; PPP1R3B was followed-up in both traits). R/O Freq = Trait raising allele/other allele and frequency of trait raising allele; SNP = single nucleotide polymorphism; JMA = joint meta-analysis; het = heterogeneity. N = Sample size presented is the maximum observed among the SNPs in the table.
BOX 1. Genes of biological interest within 500kb of Fasting Insulin index SNPs.
COBLL1-GRB14
The index SNP (rs7607980) identifies a missense mutation that induces a change in amino acid (N [Asn] ®D [Asp]) in position 939 of cordon-bleu protein-like 1, encoded by COBBL1. Cordon-bleu is a protein involved in neural tube development31. The FI-raising allele at the index SNP was associated with higher expression of COBLL1 (P = 0.02) in skeletal muscle in the Malmo Exercise Study (personal communication, O. Hansson). A SNP at this locus (rs10195252) is associated with waist-hip-ratio17 but is in low LD with the index SNP (r2=0.148). GRB14 encodes growth factor receptor-bound protein 14, which inhibits signaling of the insulin receptor(OMIM number: 601524) 32. In addition to being associated with triglyceride levels33, rs10195252 is reported to have a cis acting association with transcript level of GRB14 in omental fat (p= 1.0×10−13)18.
IRS1
IRS1 encodes insulin receptor substrate 1, a critical docking protein in the insulin signaling cascade, which, when phosphorylated by the insulin receptor, activates downstream signaling pathways34. SNPs in or near IRS1 are associated with T2D, HOMA-IR, FI and CAD3,35,36 but have not previously been shown to be associated with FI at the genome-wide level of significance. The index SNP reported here (rs2943634) is the same intergenic SNP associated with CAD36 and is in LD with the nearby insulin resistance and T2D SNP rs29436413 (r2 = 0.782) and T2D SNP rs75783264 (r2=0.815). The variant identified by the lipid GWAS (rs2972146; associated with both TG and HDL) is also reported to have a cis acting association with transcript level of IRS1 in omental adipose tissue (p =2.0 × 10−8)17 and this SNP is in strong LD with the index SNP (r2=0.751). Furthermore, the index SNP (rs2943634) is in moderate LD (r2=0.438) with three SNPs (rs1849878, rs2673148, rs2713547) that tag a known CNV (CNVR1152.1) (r2= 0.51, 0.52, 0.51, respectively).
PPP1R3B-TNKS
This locus is associated with both FI and FG in the present study. PPP1R3B encodes protein phosphatase 1 regulatory (inhibitor) subunit 3B, which prevents glycogen breakdown by regulating the interaction of phosphorylase protein 1 (PP1) with glycogen metabolism enzymes (OMIM number: 610541)32. Two SNPs in/near PPP1R3B are also associated with lipids (rs998728918 and rs212625937) and C-reactive protein (rs998728921) and these are both in strong LD with the index SNP [r2 = 1.0 (rs9987289) and r2= 0.803 (rs2126259), respectively]. SNP rs9987289 is also reported to have a cis acting association with transcript level of PPP1R3B in human liver (p = 1 × 10−14)18. In the present study, we observed that SNP rs19334, associated with expression of PPP1R3B (p= 2.34 × 10−12) in the liver eQTL dataset38, is in low LD with the index SNP (r2= 0.001) but showed moderate association with fasting insulin in the discovery sample (p = 2.87 × 10−4). TNKS encodes TRF1-interacting, ankyrin-related ADP-ribose polymerase which interacts with TRF1 (telomeric repeat binding factor 1) to regulate telomere length39 and also interacts with insulin-responsive amino peptidase (IRAP) in GLUT4 vesicles (OMIM number: 603303)40. Tankyrases are also thought be involved in the wnt signaling pathway41.
PEPD-CEBPA-KCTD15
PEPD encodes peptidase D, an enzyme responsible for the recycling of proline and likely essential for collagen production (OMIM number: 613230)42. A SNP in PEPD (rs731839) is associated with adiponectin levels (personal communication, B. Richards) and is in strong LD with the index SNP (r2 = 0.77). The index SNP is associated with PEPD expression in the adipocyte eQTL dataset (p =9.96 × 10−10)43, but most likely this association is driven by the association between rs17226118 and PEPD expression (p =2.2 × 10−55) despite the weak LD between the index SNP and rs17226118 (r2=0.145). CEBPA encodes CCAAT/enhancer binding protein (C/EBP), alpha, which may control the expression of leptin, an adipokine implicated in weight regulation (OMIN number: 116897)44. KCTD15 encodes potassium channel tetramerisation domain containing 15. A SNP in/near this gene (rs29941) is associated with BMI16, but is in very low LD with the index SNP (r2=0.015).
UHRF1BP1-PPARD
UHRF1BP1 encodes Ubiquitin-like containing PHD and RING finger domains 1 (UHRF1)-binding protein 1. UHRF1 is a protein necessary for DNA methylation (OMIM number: 607990)45. The index SNP in UHRF1BP1 (rs4646949) is in strong LD (r2=0.724) with a SNP (rs2293242) that causes a nonsense change in ANKS1A. The same index SNP is in moderate LD (r2 = 0.363) with a coding SNP that results in a missense change (M1098T) in UHRF1BP1 that is considered damaging based on analysis of homologous sequences46 (Supplementary Table 6). Index SNP rs4646949 in UHRF1BP1 is in strong LD (r2 = 0.95) with two perfect proxies (rs2477508 and rs2814922) for a known CNV (CNVR2857.1). A liver tissue eQTL dataset revealed that SNP rs12173920 is associated with UHRF1BP1 and STEAP4 (six transmembrane epithelial antigen of prostate 4) expression levels (P=9.56×10−6 and P= 2.91×10−8, respectively) and is in moderate LD (r2= 0.334) with the index SNP38. Another SNP in the region, rs2814944, is associated with HDL levels and is reported to have a cis acting association with transcript level of UHRF1BP1 in both omental (p =3.0 × 10−25) and subcutaneous (p=2.0 × 10−18) adipose tissues18. PPARD encodes peroxisome proliferator-activated receptor-delta, a protein involved in the breakdown of fat(OMIM number: 600409)47.
PDGFC-GLRB
PDGFC encodes platelet derived growth factor C, a ligand that binds to specific PDGF receptors (alphaalpha and alphabeta) and has a growth factor domain homologous to VEGF (OMIM number: 608452)48. Tyrosine phosphorylation is activated by the binding of these growth factors to receptors. GLRB encodes glycine receptor, beta, a subunit of glycine receptors (neurotransmitter-gated ion channels) likely involved in glycine receptor structure (OMIM number: 138492)49.
LYPLAL1- SLC30A10
LYPLAL1 encodes lysophospholipase-like 1, a protein that may be involved in adiposity and fat distribution17,50−52. A SNP near LYPLAL1 (rs4846567) is associated with waist-hip-ratio and is in strong LD with the index SNP (r2= 0.796). Furthermore, a variant in this vicinity (rs3001032) is associated with adiponectin levels (personal communication, B. Richards) and is in strong LD with the index SNP (r2=0.828). SLC30A10 encodes solute carrier family 30, member 10 of the zinc transporter subfamily of cation-diffusion facilitators that allows for the outward flow of zinc from cells (OMIM number: 611146)53.
BOX 2. Genes of biological interest within 500kb of Fasting Glucose index SNPs.
PCSK1
PCSK1 encodes proprotein convertase, subtilisin/kexin-type, 1. This protein initiates proinsulin processing to insulin. The expression of this protein is regulated by glucose (OMIM number: 162150)54. The nonsynonymous variant rs6232, which encodes N221D, and rs6234-rs6235, a pair of variants that encode Q665E-S690T, have been shown to be associated with obesity in children and adults55. The index SNP is in strong LD with rs6234 (r2 =0.814). A report from MAGIC demonstrated that rs6235 in PCSK1 is associated with proinsulin levels at a genome-wide level of significance29.
OR4S1-PTPRJ
OR4S1 encodes olfactory receptor, family 4, subfamily S, member 1. These receptors set off neuronal responses for smell perception. PTPRJ encodes protein tyrosine phosphatase (PTP), receptor type, J. By hindering phosphorylation, this protein is thought to inhibit T cell receptor signaling.
ARAP1-INPPL1-STARD10
A SNP in this region, rs1552224 is associated with T2D4 and is in perfect LD with the index SNP (r2 =1.0). The genetic variant associated with increased risk of T2D is also associated with lower fasting proinsulin (adjusted for insulin levels) suggesting that the variant might cause a defect in the early steps of insulin production29. ARAP1 encodes ankyrin repeat, and pleckstrin homology domains-containing protein 1 that controls ARF-, RHO-, and CDC42-dependent cell functions56. INPPL1 encodes inositol polyphosphate phosphatase like−1. In vivo mouse studies indicate that this protein is a negative regulator of insulin signaling and sensitivity (OMIM number: 600829)57. STARD10 encodes StAR-related lipid transfer (START) domain containing 10, a protein thought to be involved in sperm cell maturation but highly expressed in liver and pancreas tissues58 (NF Expression Atlas 2 Data from U133A and GNF1H Chips). Levels of STARD10 expression are stronger in pancreatic and islet tissue than in any other human tissue type29.
FOXA2
FOXA2 encodes forkhead box A2, a DNA-binding protein which regulates the expression of key genes active in glucose sensing in beta cells (OMIM number: 600288). When FOXA2 was continually activated in mice, this led to increases in neuronal MCH and orexin expression, insulin sensitivity, food consumption and metabolism59. It has previously been suggested that mutations in FOXA2 may affect glucose homeostasis79.
GRB10
GRB10 encodes growth factor receptor-bound protein 10 (OMIM number: 601523). This protein forms a complex with insulin receptors and inhibits their signaling60. This protein also interacts with GIGYFs (Grb10 interacting GYF proteins) to regulate insulin-like growth factor receptor 1 signaling61. The risk allele at rs2237457 showed an association with T2D (P=1.1×10−5) and glucose excursion during OGTT (P=0.001) in the Old Order Amish Study19.
DPYSL5-KHK-PPM1G
DPYSL5 encodes dihydropyrimidinase-like 5 and is thought to be involved in directing neuronal growth cones in development(OMIM number 608383). KHK encodes ketohexokinase and is the first enzyme acting in the breakdown of fructose(OMIM number: 229800). PPM1G encodes protein phosphatase, magnesium-dependent, 1 which carries out a dephosphorylation event essential for forming the spliceosome (OMIM number: 605119)62. There is moderate LD (r2 = 0.404) between the index SNP in DPYSL5 (rs1371614) and a SNP (rs2384572) that causes a missense change (I116I or I20I [depending on transcript]) in CGREF1 (cell growth regulator with EF-hand domain 1) that is considered damaging based on analysis of homologous and orthologous sequences46 (Supplementary Table 6).
PDX1
PDX1 encodes pancreatic and duodenal homeobox 1 and is responsible for the development of the pancreas, determining maturation and differentiation of common pancreatic precursor cells (OMIM number: 600733). As pancreatic morphogenesis proceeds, PDX1 action is eventually restricted to beta and delta cells of the islets, where it appears to regulate expression of the insulin and somatostatin genes, respectively. A deletion (FS123TER) and missense changes (Glu164Asp, Glu178Lys) in PDX1 are associated with pancreas agenesis63. While the deletion FS123TER causes pancreas agenesis in homozygotes, heterozygosity is associated with MODY464. The variant Glu224Lys (E224K) was also shown to cause MODY465. Other variants were suspected in increased risk of T2D in some family studies.
PPP1R3B/TNKS
see summary in Box 1
As the JMA simultaneously tests both genetic main effects and interaction effects, in order to further characterize the association signals observed using the JMA, we performed additional analyses to examine main effects and interaction effects separately. These additional analyses included meta-analyzed regression models of main effects, with and without adjustment for BMI, univariate meta-analysis of interaction effects using continuous BMI, and SNP main effects meta-analyses in strata defined by a BMI cut-off of 28 kg/m2, chosen based on the median BMI of the largest cohorts included in our discovery stage JMA (BMI strata are categorized as “leaner” where BMI < 28 kg/m2 and “heavier” where BMI ≥ 28 kg/m2).
Associations with fasting insulin
The follow-up loci associated with fasting insulin (natural log transformed) at genome-wide levels of significance in the combined JMA are presented in Table 1 and described in Box 1. SNP rs7607980 located near COBLL1-GRB14 provided the strongest signal for fasting insulin, with a JMA P value of 4.3×10−20. Analyses performed to characterize this association further revealed that the significant association of rs7607980 with fasting insulin was driven primarily by a BMI-adjusted genetic main effect (P = 1.7×10−16), and showed some suggestion of interaction with BMI (P for interaction = 1.6×10−4; Figure 2; Supplementary Table 3a). Modeled from the combined JMA, the estimated effect size of the genetic variant was greater at a BMI of 30 kg/m2 (JMA β=0.039) compared a BMI of 25 kg/m2 (JMA β=0.023; Table 1). These estimates were supported by BMI-stratified analyses: the genetic effect was larger in heavier (main genetic effect β=0.041, P=2.98×10−10) than in leaner individuals (main genetic effect β= 0.018, P=1.77×10−5; P value for difference between strata = 0.02; Figure 2).
Figure 2.
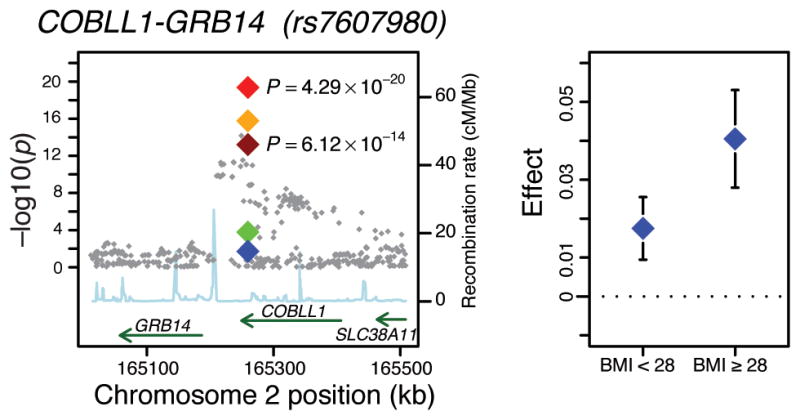
Regional plot of the COBLL1-GRB14 genomic locus. The left plot shows the discovery JMA P values in the background and for the SNP taken forward to the follow-up analyses (rs7607980), the P values of the discovery JMA (dark red) and the combined discovery and follow-up JMA (light red), main effects adjusting for BMI (orange), interaction with continuous BMI (green) and interaction with dichotomous BMI (blue). The right plot shows the beta estimates with 95% confidence interval of the increasing allele in the two BMI strata: BMI<28 and BMI≥28 kg/m2. Effect = Beta estimates from regression models of ln(FI) adjusting for age, sex and other study-specific covariates.
In addition to COBLL1-GRB14, the combined JMA showed five loci to be associated with fasting insulin (IRS1, PPP1R3B, PDGFC, LYPLAL1, UHRF1BP1) at genome-wide significance levels (Table 1; Supplementary Figure 2). For these loci, the additional models to characterize the association demonstrated that the JMA association signals were largely driven by their main genetic effects, adjusted for BMI (P < 5.5×10−9), and showed little evidence of interaction (P > 0.02; Supplementary Table 3a). Additionally, the combined JMA at the PEPD locus showed evidence suggestive of association, albeit just below the conventional significance threshold (P=8.69×10−8). The results of the JMA and the additional meta-analyses for all loci included in the follow-up (phase 2) are presented in Supplementary Table 3a.
As we were interested in identifying loci associated with insulin resistance, we investigated associations between genome-wide significant variants and other traits related to insulin resistance in genome-wide meta-analysis results provided by other consortia: the Diabetes Genetics Replication And Meta-analysis Consortium (DIAGRAM)4, the Genomewide Investigation of ANThropometric measures (GIANT)16,17 consortium, and the Global Lipid Genetic Consortium (GLGC)18 (Table 2). At five out of six loci associated with fasting insulin, the insulin-raising allele was associated with both lower HDL cholesterol and higher triglycerides – a dyslipidemic profile typical of insulin resistance. The insulin-raising alleles near COBLL1-GRB14, LYPLAL1 and PPP1R3B were also associated with a greater waist-hip ratio (WHR adjusted for BMI). Individuals carrying insulin-raising alleles at the index SNP in or near COBLL1-GRB14, LYPLAL1, IRS1 or PDGFC were at increased risk of T2D, albeit with relatively small odds ratios (Table 2).
Table 2.
Associations with type 2 diabetes, lipid profile, and anthropometric measures of loci significantly associated with fasting insulin (FI) or fasting glucose (FG)
| Lipid profile | Anthropometric measures | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Type 2 diabetes | TC | LDL | HDL | Triglycerides | BMI (kg/m2) | WHR (adj BMI) | |||||||||||||
| Trait | Index SNP | Nearest Gene(s) | Alleles R/O | OR | P | Dir | P | Dir | P | Dir | P | Dir | P | β | se | P | β | se | P |
| FI | rs7607980 | COBLL1- GRB14 | T/C | 1.08 | 0.02 | + | 0.09 | + | 0.05 | − | 3.9×10−10 | + | 4.1×10−8 | − 0.016 | 0.007 | 0.03 | 0.031 | 0.007 | 2.7×10−5 |
| FI | rs2943634 | IRS1 | C/A | 1.09 | 2.7×10−5 | + | 0.91 | + | 0.14 | − | 2.3×10−9 | + | 5.2×10−8 | − 0.014 | 0.005 | 0.01 | 0.005 | 0.005 | 0.33 |
| FI/FG | rs4841132 | PPP1R3B | A/G | 1.04 | 0.24 | − | 2.2×10−19 | − | 1.5×10−12 | − | 8.4×10−23 | + | 0.017 | 0.007 | 0.008 | 0.40 | 0.020 | 0.008 | 0.02 |
| FI | rs4691380 | PDGFC | C/T | 1.05 | 0.03 | − | 0.14 | + | 0.70 | − | 1.4×10−4 | + | 0.008 | − 0.005 | 0.005 | 0.34 | 0.007 | 0.005 | 0.17 |
| FI | rs4646949 | UHRF1BP1 | T/G | 1.04 | 0.13 | − | 0.09 | − | 0.04 | + | 0.37 | + | 0.81 | − 0.009 | 0.005 | 0.09 | 0.008 | 0.005 | 0.14 |
| FI | rs2785980 | LYPLAL1 | T/C | 1.04 | 0.05 | + | 0.22 | + | 0.05 | − | 9.0×10−4 | + | 0.0023 | − 0.007 | 0.005 | 0.19 | 0.030 | 0.005 | 2.3×10−9 |
| FG | rs11603334 | ARAP1 | G/A | 1.13 | 7.8×10−6 | − | 0.87 | − | 0.97 | + | 0.32 | − | 0.28 | − 0.012 | 0.006 | 0.07 | 0.001 | 0.006 | 0.83 |
| FG | rs6048205 | FOXA2 | A/G | 1.05 | 0.39 | + | 0.07 | + | 0.15 | + | 0.16 | + | 0.21 | − 0.020 | 0.012 | 0.09 | 0.007 | 0.011 | 0.55 |
| FG | rs1371614 | DPSYL5 | T/C | 0.98 | 0.32 | − | 0.29 | + | 0.51 | − | 0.11 | − | 5.5×10−4 | − 0.008 | 0.006 | 0.13 | 0.001 | 0.005 | 0.84 |
| FG | rs13179048 | PCSK1 | C/A | 1.02 | 0.34 | − | 0.09 | − | 0.09 | + | 0.64 | − | 0.44 | − 0.011 | 0.005 | 0.03 | 0.006 | 0.005 | 0.25 |
| FG | rs2293941 | PDX1 | A/G | 1.01 | 0.76 | + | 0.48 | − | 0.97 | + | 0.02 | − | 0.34 | − 0.007 | 0.006 | 0.23 | 0.006 | 0.006 | 0.28 |
| FG | rs1483121 | OR4S1 | G/A | 1.00 | 0.91 | − | 0.14 | − | 0.05 | − | 0.77 | + | 0.23 | 0.017 | 0.007 | 0.02 | 0.004 | 0.007 | 0.63 |
Associations with type 2 diabetes were assessed in the DIAGRAM meta-analysis of up to 8130 cases and 38987 controls. Associations with lipid profile were performed using publicly available data and included up to 99,900 individuals. Associations with anthropometric measures were performed in the GIANT dataset including up to 123,685 individuals.
SNP = single nucleotide polymorphism; Alleles R/O= Trait raising allele/other allele; TC = total cholesterol; LDL= low density lipoprotein cholesterol; HDL = high density lipoprotein cholesterol; BMI =body mass index; WHR (adj BMI) = waist-to-hip ratio (adjusted for BMI); OR = Odd Ratio; Dir = direction of effect; se = standard error; FI = fasting insulin; FG = fasting glucose
Associations with fasting glucose
Genetic variants associated with fasting glucose at genome-wide significance in the combined JMA are presented in Table 1 and described in Box 2: index SNPs were in or near ARAP1, PCSK1, FOXA2, DPYSL5, OR4S1, PDX1, and PPP1R3B. The combined JMA results for the index SNP near GRB10 demonstrated a suggestive association with fasting glucose, but the P value did not reach genome-wide significance (P=8.3×10−8). Interestingly, a SNP in GRB10 (rs2237457) was previously found to be associated with T2D (P=1.1×10−5) in the Old Order Amish19.
Additional characterization analyses revealed that genetic main effects were the main contributors to these associations (P<3.6×10−8) (Supplementary Table 3b), with little evidence observed for interaction at these loci (P values for interaction>0.06). The combined results of the JMA and the additional meta-analyses for all loci included in the follow-up (phase 2) are presented in Supplementary Table 3b.
In contrast to associations observed at fasting insulin loci, fasting glucose index SNPs did not show compelling associations with insulin resistance related metabolic traits (Table 2), but fasting glucose-raising alleles near PCSK1 and PPP1R3B showed nominal associations with lower 2-hour glucose levels; see Supplementary Table 4.
Functional exploration, the results of expression and eQTL lookups, and conditional analyses based on available databases are presented in the Supplementary Note.
Discussion
Our study demonstrates that insulin resistance loci exist and can be localized when appropriate analysis methods are used to control for the influence of adiposity on insulin resistance. In a discovery dataset that was approximately 35% larger than our previous meta-analysis, we describe six loci not previously known to be associated with fasting insulin, adding substantially to the two loci that were observed using standard association analyses without adjustment for BMI. We also describe seven additional loci associated with fasting glucose. In addition, we detected all previously reported associations for fasting glucose (16 loci) and insulin (2 loci) (Supplementary Table 2)3. With the initial explosion in the number of loci found to be associated with T2D1, we expected to discover similar numbers of loci implicated in insulin resistance and insulin secretion, which has not been the case until now. Our approach was successful at revealing loci implicated in insulin resistance in numbers proportional to those implicated in insulin secretion. Our results underline the importance of taking adiposity into account to understand the heterogeneity of T2D etiology.
The association of fasting insulin with the genetic variant located at the COBLL1-GRB14 locus is of particular interest, based on its concomitant and directionally consistent association with insulin resistance-related traits and its plausible mechanism of action. GRB14 is a tissue-specific negative regulator of insulin receptor signaling (see also Box 1). The risk allele at the index SNP rs7607980 is associated with higher triglycerides and lower HDL cholesterol levels, a dyslipidemic profile characteristic of the insulin resistant state, advocating for a putative role in insulin resistance pathways. Furthermore, this pattern of association was observed for nearly all index SNPs associated with fasting insulin, increasing our confidence that our approach localized genetic loci involved in insulin resistance pathogenesis. As shown in a previous GIANT report17, risk alleles at COBLL1-GRB14 and LYPLAL1 were also associated with increased waist-hip ratio (WHR) adjusted for BMI (Table 2), suggesting that they could be influencing the regulation of adipose tissue distribution to induce insulin resistance. In line with plausible biological actions in adipose tissue, eQTL results in omental adipose tissue demonstrated that expression of GRB14 is associated with SNP rs10195252 (r 2=0.17 with the index SNP rs7607980) in the report from GLGC18.
Our results suggest a SNP×BMI interaction with fasting insulin at COBLL1-GRB14, with a larger genetic effect estimate in heavier individuals, compatible with the notion that some genetic variants implicated in insulin resistance pathways are more likely to be found when studying genetic effects in an ‘obesogenic’ environment. Our observation supports the analyses in the TwinsUK study reporting greater heritability estimates for insulin resistance traits at higher levels of BMI7. Moreover, some candidate gene studies demonstrated that genetic variants implicated in insulin signaling pathways are more readily observable in heavier populations20. Prudente et al. proposed that higher BMI potentiates the effect of genetic variants on insulin resistance pathways and suggested that this effect could be due to tissue specific response to the obesogenic environment5. It is currently unknown how a genetic variant could influence a different response to an obesogenic exposure within specific tissues – liver, muscles, adipose or even central nervous system – but our data add to the current literature by identifying one potential example and arguing for functional studies to pursue the hypothesis proposed by Prudente et al.5.
Among the other loci, PPP1R3B is likely to act via hepatic metabolism to influence fasting insulin and glucose, as well as lipid profile (HDL, LDL, total cholesterol) and CRP levels21. PEPD encodes peptidase D and is likely to have a role in adipokine biology supported by eQTL data in adipose tissue (Supplementary Table 5) and an observed association with adiponectin levels (ADIPOgen consortium, B. Richards, personal communication). Adiponectin is suspected to act as an insulin sensitizer with low levels conferring T2D risk only among individuals who are also insulin resistant22; the independent PEPD associations with this adipokine are supportive of a direct role in insulin resistance. Additionally, other genes located near the loci reported in Table 1 also represent plausible biologic candidates to be involved in various processes related to insulin resistance (see Box 1).
The JMA approach has the potential to reveal genetic variants whose effects differ depending on levels of adiposity, with interactions being tested in either direction. As pointed out previously, a search for T2D genes in leaner populations is more likely to reveal genes implicated in beta-cell function by focusing on pathophysiologic mechanisms independent of adiposity and insulin resistance23. As a corollary of this observation, insofar as adiposity and insulin resistance have a stronger environmental component, main effect sizes for beta-cell function loci may tend to be larger in leaner individuals, although this was equivocal in our findings (Supplementary Table 2).
There have been few previous efforts investigating SNP×BMI interaction and its impact on the risk of T2D or related glycemic traits at a genome-wide level. Timpson et al. investigated genetic variants associated with T2D in the Wellcome Trust Case Control Consortium by dividing the cases into strata of obese and non-obese individuals: in addition to TCF7L2 (of larger effect in non-obese cases), the only other signal they detected and replicated was the well-known obesity-mediated FTO association with T2D in the obese strata24. As expected, we did not detect an association between FTO and fasting insulin or glucose because the JMA approach includes an intrinsic adjustment for BMI.
Strengths and potential limitations
The major strengths of our study are the large sample size, that all cohorts were composed of individuals from European descent, and importantly, that we used a validated statistical approach and successfully applied it at genome-wide level for the first time. Importantly, we identify all previously described associations with fasting insulin and fasting glucose, which highlights the utility of the JMA approach even in the absence of underlying gene-environment interaction.
We used a two-phase analytical approach, using the conventional P<5.0×10−8 significance threshold in combined analyses to determine genome-wide significance for the identified SNPs. This approach has been shown to have greater power when the size of the follow-up sample is smaller than the discovery phase25,26 and has previously been successful applied by our group27–29 and others30. All 12 loci described in Table 1 show consistent directions of effect in the follow-up phase. However, employing a replication strategy that declares statistical significance on the basis of the follow-up phase alone, only 4 of the 12 loci would reach conventional statistical significance accounting for multiple testing (P<0.05/51=0.00098; with significance threshold α/N1, where N1 is the number of SNPs taken forward for replication and one SNP is identified for both fasting glucose and fasting insulin).
Only 12 significant loci were identified after the second stage, despite the fact that 50 loci were taken forward from the discovery stage and only 2 or 3 associations at P<0.00001 would be expected per trait under the hypothesis of no association. Among the reasons for the fewer than expected significant associations is most importantly the size of the follow-up sample. To maximize sample size we took advantage of studies that offered either de novo genotyping, access to genome-wide SNP arrays or had performed genotyping on the Metabochip. While our study represents the largest effort of its kind, it should be acknowledged that the follow-up sample size is still approximately one half of the size of the discovery sample and that false negatives may remain amongst those loci not reaching genome-wide significance.
We used the JMA as the primary model and further characterized only those loci in additional models that reached genome-wide significance. However, consideration of results from the follow-up stage alone showed that the interaction term at the COBBL1-GRB14 locus did not reach statistical significance in additional models, although we observed consistency in direction and effect size in phases 1 and 2.
Our main JMA model assumes a linear interaction, where the per allele effect of a SNP changes across the continuous spectrum of BMI. If the interaction effect is nonlinear, or a threshold effect exists, in which case the association would only be present in one extreme of the BMI distribution, the interaction test with continuous BMI might not agree with that of dichotomous BMI. This could explain the inconsistencies of the interaction results with IRS1, PCSK1 and OR4S1, where we do observe some suggestion of interactions in the stratified models not supported by models including BMI as a continuous variable.
We used the JMA approach to test for SNP main effects adjusted for BMI and to allow for interaction between BMI and SNPs, but the results we observed might be due to one of many factors correlated with BMI, including lifestyle. Additionally, although we identify genes that highlight potential new pathways to insulin resistance and T2D development, we recognize that we have not localized the associations to specific genes.
Conclusion
Previous attempts to identify loci associated with insulin resistance have been hindered partly by methodological limitations. In the present study, by using the newly developed JMA approach, we observed six additional loci associated with fasting insulin and other insulin resistance-associated traits. We used the JMA approach because we hypothesized that the degree of adiposity may mask, positively confound, or modify the association between genetic variants and insulin resistance traits. While the associations we observed resulted mainly from adjustment for BMI, the JMA method allowed the flexibility and power to observe these main effect-driven associations, whilst allowing us to simultaneously test our hypothesis of interaction. The identification of these loci offers the potential to further characterize the etiology and causality of T2D and the role of insulin resistance in that process.
Online Methods
Cohorts
We recruited 29 studies involved in MAGIC (Meta-Analysis of Glucose and Insulin related traits Consortium) for the first phase of discovery analysis, totaling up to 58,070 individuals of European ancestry. Many of these studies were involved in the previous MAGIC effort on fasting glucose and fasting insulin3. In the second phase, follow-up included 23 cohorts with fasting glucose data and 22 cohorts with fasting insulin data, comprising up to 38,422and 33,823 non-diabetic individuals of European ancestry, respectively (detailed discovery and follow-up cohort characteristics are presented in Supplementary Table 1). We excluded individuals with T2D on the basis of known T2D or anti-diabetic treatment, and/or a fasting glucose ≥7 mmol/L so as to remove their influence on misclassification of trait levels66. Local research ethic committees approved all studies and all participants gave informed consent to each original study.
Phenotypes/Quantitative Traits
Fasting glucose and fasting insulin were measured from whole blood, plasma or serum using assays specific for each cohort (Supplementary Table 1). Homeostatic model assessment indices of beta-cell function (HOMA-B) and insulin resistance (HOMA-IR) were derived from fasting glucose and fasting insulin15. Fasting insulin, HOMA-B, and HOMA-IR were log transformed for analyses. Anthropometric measurements such as body mass index (BMI, kg/m2) were obtained in each cohort following standardized procedures. Trait values were not imputed and outliers were not excluded.
Phase 1: Discovery Genotyping
All cohorts included in the discovery phase had genome-wide arrays performed, details of which are specified in Supplementary Table 1. We implemented the following quality control inclusion criteria for the SNPs in all cohorts: 1) Hardy-Weinberg equilibrium P>10−6, 2) minor allele frequency >1%, and 3) SNP call rate >95%. Imputation was performed using MACH67,68 (r2 >0.3) or IMPUTE69,70 (proper_info>0.3) based on the HapMap CEU population (build 36). Both genotyped and imputed SNPs were used in the analysis, with priority given to genotyped SNPs when both were available. Consequently, up to 2.4 million SNPs (genotyped or imputed) were included in the discovery phase meta-analyses.
Phase 2: Follow-up Genotyping
Studies were invited to participate in the follow-up phase of the analysis through either in silico lookups with existing genotype data or de novo genotyping. A large percentage of the follow-up cohorts used the Illumina Cardio-Metabo Chip. Study-specific details for follow-up genotyping are described in Supplementary Table 1.
Statistical Models
Each study submitted regression summary statistics for the meta-analyses, all of which assumed an additive genetic effect and were adjusted for age, sex and study-level covariates at a minimum (Supplementary Table 1). The first regression model included SNP, BMI and SNP×BMI product terms to allow for SNP×BMI interaction effects (Model 1). This regression model was used in the JMA, which is described below, and is considered the primary analysis. Additional regression models were meta-analyzed, solely to characterize the associations observed in the JMA as being driven by either SNP main effects or by SNP by BMI interactions. Main effects models estimating the effect of each SNP on the dependent variables were performed, both with (Model 2) and without (Model 3) adjustment for BMI. Additionally, the continuous BMI measure was dichotomized at 28 kg/m2; this cut-off was chosen based on the median BMI in of the largest cohorts included in our discovery analysis. An interaction model using the dichotomized BMI was fit (Model 4) and stratified models were used to obtain stratum specific estimates in leaner (BMI <28 kg/m2, Model 5) and heavier individuals (BMI ≥28 kg/m2, Model 6). For Models 1 and 4, both the SNP and SNP×BMI regression coefficients and their covariance matrix were reported by each study and robust variance estimates were used to correct the observed inflation of the false positive rate observed for the interaction P-value71.
In phase 1, models 1, 2 and 3 were performed genome-wide, while models 4, 5 and 6 were performed on SNPs with discovery JMA P values less than 10−5. For phase 2, models 1–6 were performed using SNPs taken forward to the follow-up. Regression statistics were obtained with R, SAS, Quicktest (http://toby.freeshell.org/software/quicktest.shtml) or ProbABEL72 (Supplementary Table 1).
The models were fit as:
Model 1. Y = β0 + βC × Cov + βSNP × SNP + βBMI × BMI + βSNP×BMI × SNP×BMI + ε. (BMI as continuous variable)
Model 2. Y = β0 + βC × Cov + βSNP × SNP + ε.
Model 3. Y = β0 + βC × Cov + βSNP × SNP + βBMI × BMI + ε.
Model 4. Y = β0 + βC × Cov + βSNP × SNP + βBMI-di × BMI + βSNP×BMI-di × SNP×BMI + ε. (BMI as a dichotomous variable)
Model 5. Y = β0 + βC × Cov + βSNP × SNP + ε. (in individuals of BMI < 28)
Model 6. Y = β0 + βC × Cov + βSNP × SNP + ε. (in individuals of BMI ≥ 28)
Phase 1 and 2 Meta-analyses
Our objective was to identify genetic loci associated with fasting insulin, as a surrogate for insulin resistance, which may be masked by variation in, or interaction with, BMI. In the primary analysis we performed a joint meta-analysis (JMA) of both the SNP effect and the SNP×BMI interaction effect from Model 1, using a recently developed method14. The JMA is an effective screening tool when the underlying interaction model is unknown and, importantly, retains power when there is no interaction effect. The JMA provides estimates of βSNP and βSNP×BMI and allows for a 2 degree of freedom joint test of the null hypothesis H0: βSNP =βSNP×BMI =0, as well as a test of heterogeneity of regression coefficients. Additionally, the JMA detects association in the presence of either a significant SNP effect, adjusted for BMI, or a SNP×BMI interaction effect. We implemented the JMA method in METAL (v. 2010-02-08 http://www.sph.umich.edu/csg/abecasis/Metal/ with patch provided by Manning et al.14). SNPs that reached a discovery threshold in the JMA of P<10−5 with Pheterogeneity >0.001 and were available in at least one third of the total sample size were taken forward to follow-up analyses. In order to characterize the association signals observed in the JMA, additional meta-analyses were performed that examined main effects and interaction effects separately. The inverse-variance weighted meta-analyses73 approach and heterogeneity test74 were used in the univariate meta-analyses of the SNP main effects in Models 2, 3, 5 and 6 and the SNP×BMI interaction effect from models 1 and 4.
In each genomic region identified from the JMA, the index SNP was chosen as the SNP with the strongest association (lowest P value) unless the region contained a SNP known to be associated with a metabolic condition or trait (and with P<10−5), in which case the latter SNP was selected. In case a follow-up cohort was not able to provide results for an index SNP, several proxy SNPs were chosen using LD information from HapMap (http://hapmap.ncbi.nlm.nih.gov) and the 1000 Genomes project75. SNPs identified by the JMA, but which had previously reported associations with diabetes and/or diabetes-related quantitative traits in MAGIC were not selected for follow-up and are reported in Supplementary Table 2.
Models 1–6 were performed within each follow-up study and the SNPs were meta-analyzed within the follow-up cohorts and in a combined meta-analysis with the discovery cohorts using the methods detailed above. Meta-analyses were performed with proxy SNPs if the index SNP was not available. A SNP was considered significant if the JMA in the combined sample yielded a P value less than 5×10−8. Effect estimates described in the results and displayed in Supplementary Table 3 were obtained from a combined meta-analysis that excluded proxy SNPs.
Two of the index SNPs (near CHL1 and TUBA3C) showed significant associations in the discovery JMA. We excluded these loci from the combined analysis because we observed low heterogeneity P values and because neither the main effects nor interaction models supported the associations observed in the JMA at these loci.
Conditional Analyses
We conducted conditional analyses for index SNPs that were within approximately 1 Mb of established SNPs (known associations with T2D or glycemic traits). We conditioned on the established SNPs for a chromosome-specific analysis in the Framingham Heart Study, one of the largest cohorts in the meta-analyses in which the associations of both the established SNPs and the index SNP were observed. If the SNP retained nominal significance in the conditional analysis, we considered the association signal to be not solely caused by LD with the established SNPs. Because only one study was involved in this conditional analysis, to account for the limited power we used a significance threshold of 0.05.
Associations with related metabolic traits
As we were interested in the effect of the index SNPs on insulin resistance-related traits, associations of each index SNP with related glycemic traits and T2D were sought using results from previous DIAGRAM and MAGIC meta-analyses: T2D4, 2-hour glucose27, and glycated hemoglobin28. We also performed lookups of these SNPs in publicly available data from previous meta-analyses of lipid quantitative traits18. Associations with BMI16 and WHR adjusted for BMI17 were also sought from published GIANT meta-analyses.
Functional Exploration
To determine if any index SNPs were in LD with nearby functional coding variants, we used SNPper76 (http://snpper.chip.org/) to extract all SNPs that fell within 500 kb of the index SNP (1 Mb total). From this list we chose all coding SNPs and used SNAP (http://www.broadinstitute.org/mpg/snap/ldsearch.php) to determine if any of these coding SNPs were in LD (CEPH (Utah residents with ancestry from northern and western Europe) (CEU), 1000 Genomes Project, 500 kb distance) with the index SNP. We used the online tool SIFT dbSNP (http://sift.jcvi.org/www/SIFT_dbSNP.html) to predict potential damage to the protein.
To establish whether index SNPs are in LD with known copy number variants (CNVs), we used a database of 7,411 SNPs that tag 3,188 CNVs from the Wellcome Trust Case Control Consortium (WTCCC)77 in addition to a list of 422 SNPs tagging 261 deletions78. All known SNPs in LD with these tagging SNPs were retrieved using the SNAP tool (http://www.broadinstitute.org/mpg/snap/ldsearch.php) (CEU, 1000 Genomes, within 500 kb). We determined if any index SNPs are in LD with those SNPs tagging CNVs.
Expression Studies and EQTL
We searched for genes potentially implicated in glycemic regulation and insulin resistance in the region flanking 500 kb on each side of index SNPs (1 Mb total). Functions of these genes were investigated using online resources such as OMIM (http://www.ncbi.nlm.nih.gov/omim) or GeneCards V3 (http://www.genecards.org/).
We examined expression levels of candidate genes nearest the index SNP and within 500 kb of the index SNP using available online micro-array expression data (GNF Expression Atlas 2 Data from U133A and GNF1H Chips, http://genome.ucsc.edu). Potential fasting insulin candidates were further pursued with eQTL studies in available datasets.
We queried all SNPs in the 1 Mb region surrounding the fasting insulin index SNPs to see if they were associated with expression levels of any genes in a liver tissue gene expression database38, which used a sample size of 427 subjects of European ancestry and where SNPs with association p-values less than 0.003 were listed. We used the SNAP tool (http://www.broadinstitute.org/mpg/snap/ldsearch.php) to determine if eQTLs were in LD with fasting insulin index SNPs. Fasting insulin index SNPs were also queried in the MuTHER (Multiple Tissue Human Expression Resource) expression database43 to determine if any were eQTLs in subcutaneous adipose tissue (N=776). Further eQTL look-ups were performed searching for top eQTL findings for any SNPs located within 1 Mb of each index SNP (P values <10−3). Data were analyzed in genABEL and probABEL72. A genome-wide FDR of 1% corresponded to a P-value threshold of 5.1×10−5 and data were corrected for multiple testing.
Supplementary Material
Acknowledgments
A full list of acknowledgments is provided in the Supplementary Note.
Footnotes
AUTHOR CONTRIBUTIONS
AKM, JD and JBM conceived of the study, AKM and RAS performed the analysis, AKM, MFH and RAS wrote the manuscript, JBM and CL directed the work, and JLG, NBN, HC, DR, CTL, LFB, IP, RMW, JCF JD, JBM and CL provided analytical advice and revised the manuscript. A full list of author contributions is provided in the Supplementary Note.
References
- 1.Billings LK, Florez JC. The genetics of type 2 diabetes: what have we learned from GWAS? Ann N Y Acad Sci. 2010;1212:59–77. doi: 10.1111/j.1749-6632.2010.05838.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sladek R, et al. A genome-wide association study identifies novel risk loci for type 2 diabetes. Nature. 2007;445:881–885. doi: 10.1038/nature05616. [DOI] [PubMed] [Google Scholar]
- 3.Dupuis J, et al. New genetic loci implicated in fasting glucose homeostasis and their impact on type 2 diabetes risk. Nature genetics. 2010;42:105–116. doi: 10.1038/ng.520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Voight BF, et al. Twelve type 2 diabetes susceptibility loci identified through large-scale association analysis. Nat Genet. 2010;42:579–89. doi: 10.1038/ng.609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Prudente S, Morini E, Trischitta V. Insulin signaling regulating genes: effect on T2DM and cardiovascular risk. Nat Rev Endocrinol. 2009;5:682–93. doi: 10.1038/nrendo.2009.215. [DOI] [PubMed] [Google Scholar]
- 6.Kahn SE, Hull RL, Utzschneider KM. Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature. 2006;444:840–846. doi: 10.1038/nature05482. [DOI] [PubMed] [Google Scholar]
- 7.Wang X, et al. Heritability of insulin sensitivity and lipid profile depend on BMI: evidence for gene-obesity interaction. Diabetologia. 2009;52:2578–84. doi: 10.1007/s00125-009-1524-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Florez JC, et al. Effects of the type 2 diabetes-associated PPARG P12A polymorphism on progression to diabetes and response to troglitazone. The Journal of clinical endocrinology and metabolism. 2007;92:1502–1509. doi: 10.1210/jc.2006-2275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ludovico O, et al. Heterogeneous effect of peroxisome proliferator-activated receptor gamma2 Ala12 variant on type 2 diabetes risk. Obesity (Silver Spring, Md ) 2007;15:1076–1081. doi: 10.1038/oby.2007.617. [DOI] [PubMed] [Google Scholar]
- 10.Cauchi S, et al. The genetic susceptibility to type 2 diabetes may be modulated by obesity status: implications for association studies. BMC medical genetics. 2008;9:45. doi: 10.1186/1471-2350-9-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Trujillo ME, Scherer PE. Adipose tissue-derived factors: impact on health and disease. Endocrine reviews. 2006;27:762–778. doi: 10.1210/er.2006-0033. [DOI] [PubMed] [Google Scholar]
- 12.Shoelson SE, Lee J, Goldfine AB. Inflammation and insulin resistance. The Journal of clinical investigation. 2006;116:1793–1801. doi: 10.1172/JCI29069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kraft P, Yen YC, Stram DO, Morrison J, Gauderman WJ. Exploiting gene-environment interaction to detect genetic associations. Human heredity. 2007;63:111–119. doi: 10.1159/000099183. [DOI] [PubMed] [Google Scholar]
- 14.Manning AK, et al. Meta-analysis of gene-environment interaction: joint estimation of SNP and SNP x environment regression coefficients. Genetic epidemiology. 2011;35:11–18. doi: 10.1002/gepi.20546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Matthews DR, et al. Homeostasis model assessment: insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia. 1985;28:412–419. doi: 10.1007/BF00280883. [DOI] [PubMed] [Google Scholar]
- 16.Speliotes EK, et al. Association analyses of 249,796 individuals reveal 18 new loci associated with body mass index. Nat Genet. 2010;42:937–48. doi: 10.1038/ng.686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Heid IM, et al. Meta-analysis identifies 13 new loci associated with waist-hip ratio and reveals sexual dimorphism in the genetic basis of fat distribution. Nat Genet. 2010;42:949–60. doi: 10.1038/ng.685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Teslovich TM, et al. Biological, clinical and population relevance of 95 loci for blood lipids. Nature. 2010;466:707–13. doi: 10.1038/nature09270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rampersaud E, et al. Identification of novel candidate genes for type 2 diabetes from a genome-wide association scan in the Old Order Amish: evidence for replication from diabetes-related quantitative traits and from independent populations. Diabetes. 2007;56:3053–3062. doi: 10.2337/db07-0457. [DOI] [PubMed] [Google Scholar]
- 20.Stolerman ES, et al. Haplotype structure of the ENPP1 Gene and Nominal Association of the K121Q missense single nucleotide polymorphism with glycemic traits in the Framingham Heart Study. Diabetes. 2008;57:1971–1977. doi: 10.2337/db08-0266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dehghan A, et al. Meta-analysis of genome-wide association studies in >80 000 subjects identifies multiple loci for C-reactive protein levels. Circulation. 2011;123:731–8. doi: 10.1161/CIRCULATIONAHA.110.948570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hivert MF, et al. Insulin resistance influences the association of adiponectin levels with diabetes incidence in two population-based cohorts: the Cooperative Health Research in the Region of Augsburg (KORA) S4/F4 study and the Framingham Offspring Study. Diabetologia. 2011;54:1019–1024. doi: 10.1007/s00125-011-2067-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Florez JC. Newly identified loci highlight beta cell dysfunction as a key cause of type 2 diabetes: where are the insulin resistance genes? Diabetologia. 2008;51:1100–1110. doi: 10.1007/s00125-008-1025-9. [DOI] [PubMed] [Google Scholar]
- 24.Timpson NJ, et al. Adiposity-related heterogeneity in patterns of type 2 diabetes susceptibility observed in genome-wide association data. Diabetes. 2009;58:505–510. doi: 10.2337/db08-0906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Skol AD, Scott LJ, Abecasis GR, Boehnke M. Optimal designs for two-stage genome-wide association studies. Genetic epidemiology. 2007;31:776–788. doi: 10.1002/gepi.20240. [DOI] [PubMed] [Google Scholar]
- 26.Skol AD, Scott LJ, Abecasis GR, Boehnke M. Joint analysis is more efficient than replication-based analysis for two-stage genome-wide association studies. Nature genetics. 2006;38:209–213. doi: 10.1038/ng1706. [DOI] [PubMed] [Google Scholar]
- 27.Saxena R, et al. Genetic variation in GIPR influences the glucose and insulin responses to an oral glucose challenge. Nature genetics. 2010;42:142–148. doi: 10.1038/ng.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Soranzo N, et al. Common variants at 10 genomic loci influence hemoglobin A(C) levels via glycemic and nonglycemic pathways. Diabetes. 2010;59:3229–3239. doi: 10.2337/db10-0502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Strawbridge RJ, et al. Genome-wide association identifies nine common variants associated with fasting proinsulin levels and provides new insights into the pathophysiology of type 2 diabetes. Diabetes. 2011;60:2624–34. doi: 10.2337/db11-0415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lango Allen H, et al. Hundreds of variants clustered in genomic loci and biological pathways affect human height. Nature. 2010;467:832–838. doi: 10.1038/nature09410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Carroll EA, et al. Cordon-bleu is a conserved gene involved in neural tube formation. Dev Biol. 2003;262:16–31. doi: 10.1016/s0012-1606(03)00323-3. [DOI] [PubMed] [Google Scholar]
- 32.Depetris RS, et al. Structural basis for inhibition of the insulin receptor by the adaptor protein Grb14. Mol Cell. 2005;20:325–33. doi: 10.1016/j.molcel.2005.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ridker PM, et al. Polymorphism in the CETP gene region, HDL cholesterol, and risk of future myocardial infarction: Genomewide analysis among 18 245 initially healthy women from the Women’s Genome Health Study. Circulation Cardiovascular genetics. 2009;2:26–33. doi: 10.1161/CIRCGENETICS.108.817304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.White MF. The IRS-signalling system: a network of docking proteins that mediate insulin action. Mol Cell Biochem. 1998;182:3–11. [PubMed] [Google Scholar]
- 35.Rung J, et al. Genetic variant near IRS1 is associated with type 2 diabetes, insulin resistance and hyperinsulinemia. Nat Genet. 2009;41:1110–5. doi: 10.1038/ng.443. [DOI] [PubMed] [Google Scholar]
- 36.Samani NJ, et al. Genomewide association analysis of coronary artery disease. N Engl J Med. 2007;357:443–53. doi: 10.1056/NEJMoa072366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Waterworth DM, et al. Genetic variants influencing circulating lipid levels and risk of coronary artery disease. Arterioscler Thromb Vasc Biol. 2010;30:2264–76. doi: 10.1161/ATVBAHA.109.201020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schadt EE, et al. Mapping the genetic architecture of gene expression in human liver. PLoS Biol. 2008;6:e107. doi: 10.1371/journal.pbio.0060107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Smith S, Giriat I, Schmitt A, de Lange T. Tankyrase, a poly(ADP-ribose) polymerase at human telomeres. Science. 1998;282:1484–7. doi: 10.1126/science.282.5393.1484. [DOI] [PubMed] [Google Scholar]
- 40.Chi NW, Lodish HF. Tankyrase is a golgi-associated mitogen-activated protein kinase substrate that interacts with IRAP in GLUT4 vesicles. J Biol Chem. 2000;275:38437–44. doi: 10.1074/jbc.M007635200. [DOI] [PubMed] [Google Scholar]
- 41.Huang SM, et al. Tankyrase inhibition stabilizes axin and antagonizes Wnt signalling. Nature. 2009;461:614–20. doi: 10.1038/nature08356. [DOI] [PubMed] [Google Scholar]
- 42.Royce PM, Steinmann B. Prolidase deficiency. Wiley-Liss; New York: 2002. pp. 727–738. [Google Scholar]
- 43.Nica AC, et al. The architecture of gene regulatory variation across multiple human tissues: the MuTHER study. PLoS Genet. 2011;7:e1002003. doi: 10.1371/journal.pgen.1002003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.The Atherosclerosis Risk in Communities (ARIC) Study: design and objectives. The ARIC investigators. American Journal of Epidemiology. 1989;129:687–702. [PubMed] [Google Scholar]
- 45.Bostick M, et al. UHRF1 plays a role in maintaining DNA methylation in mammalian cells. Science. 2007;317:1760–4. doi: 10.1126/science.1147939. [DOI] [PubMed] [Google Scholar]
- 46.Ng PC, Henikoff S. Predicting deleterious amino acid substitutions. Genome Res. 2001;11:863–74. doi: 10.1101/gr.176601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Evans RM, Barish GD, Wang YX. PPARs and the complex journey to obesity. Nat Med. 2004;10:355–61. doi: 10.1038/nm1025. [DOI] [PubMed] [Google Scholar]
- 48.Reigstad LJ, et al. Platelet-derived growth factor (PDGF)-C, a PDGF family member with a vascular endothelial growth factor-like structure. J Biol Chem. 2003;278:17114–20. doi: 10.1074/jbc.M301728200. [DOI] [PubMed] [Google Scholar]
- 49.Handford CA, et al. The human glycine receptor beta subunit: primary structure, functional characterisation and chromosomal localisation of the human and murine genes. Brain Res Mol Brain Res. 1996;35:211–9. [PubMed] [Google Scholar]
- 50.den Hoed M, et al. Genetic susceptibility to obesity and related traits in childhood and adolescence: influence of loci identified by genome-wide association studies. Diabetes. 2010;59:2980–8. doi: 10.2337/db10-0370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hotta K, et al. Polymorphisms in NRXN3, TFAP2B, MSRA, LYPLAL1, FTO and MC4R and their effect on visceral fat area in the Japanese population. J Hum Genet. 2010;55:738–42. doi: 10.1038/jhg.2010.99. [DOI] [PubMed] [Google Scholar]
- 52.Lindgren CM, et al. Genome-wide association scan meta-analysis identifies three Loci influencing adiposity and fat distribution. PLoS genetics. 2009;5:e1000508. doi: 10.1371/journal.pgen.1000508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Seve M, Chimienti F, Devergnas S, Favier A. In silico identification and expression of SLC30 family genes: an expressed sequence tag data mining strategy for the characterization of zinc transporters’ tissue expression. BMC Genomics. 2004;5:32. doi: 10.1186/1471-2164-5-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ohagi S, et al. Human prohormone convertase 3 gene: exon-intron organization and molecular scanning for mutations in Japanese subjects with NIDDM. Diabetes. 1996;45:897–901. doi: 10.2337/diab.45.7.897. [DOI] [PubMed] [Google Scholar]
- 55.Benzinou M, et al. Common nonsynonymous variants in PCSK1 confer risk of obesity. Nat Genet. 2008;40:943–5. doi: 10.1038/ng.177. [DOI] [PubMed] [Google Scholar]
- 56.Miura K, et al. ARAP1: a point of convergence for Arf and Rho signaling. Mol Cell. 2002;9:109–19. doi: 10.1016/s1097-2765(02)00428-8. [DOI] [PubMed] [Google Scholar]
- 57.Clement S, et al. The lipid phosphatase SHIP2 controls insulin sensitivity. Nature. 2001;409:92–7. doi: 10.1038/35051094. [DOI] [PubMed] [Google Scholar]
- 58.Kent WJ, et al. The human genome browser at UCSC. Genome Res. 2002;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Silva JP, et al. Regulation of adaptive behaviour during fasting by hypothalamic Foxa2. Nature. 2009;462:646–50. doi: 10.1038/nature08589. [DOI] [PubMed] [Google Scholar]
- 60.Liu F, Roth RA. Grb-IR: a SH2-domain-containing protein that binds to the insulin receptor and inhibits its function. Proc Natl Acad Sci U S A. 1995;92:10287–91. doi: 10.1073/pnas.92.22.10287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Giovannone B, et al. Two novel proteins that are linked to insulin-like growth factor (IGF-I) receptors by the Grb10 adapter and modulate IGF-I signaling. J Biol Chem. 2003;278:31564–73. doi: 10.1074/jbc.M211572200. [DOI] [PubMed] [Google Scholar]
- 62.Murray MV, Kobayashi R, Krainer AR. The type 2C Ser/Thr phosphatase PP2Cgamma is a pre-mRNA splicing factor. Genes Dev. 1999;13:87–97. doi: 10.1101/gad.13.1.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Schwitzgebel VM, et al. Agenesis of human pancreas due to decreased half-life of insulin promoter factor 1. The Journal of clinical endocrinology and metabolism. 2003;88:4398–4406. doi: 10.1210/jc.2003-030046. [DOI] [PubMed] [Google Scholar]
- 64.Stoffers DA, Ferrer J, Clarke WL, Habener JF. Early-onset type-II diabetes mellitus (MODY4) linked to IPF1. Nature genetics. 1997;17:138–139. doi: 10.1038/ng1097-138. [DOI] [PubMed] [Google Scholar]
- 65.Cockburn BN, et al. Insulin promoter factor-1 mutations and diabetes in Trinidad: identification of a novel diabetes-associated mutation (E224K) in an Indo-Trinidadian family. The Journal of clinical endocrinology and metabolism. 2004;89:971–978. doi: 10.1210/jc.2003-031282. [DOI] [PubMed] [Google Scholar]
- 66.Rasmussen-Torvik LJ, et al. Impact of repeated measures and sample selection on genome-wide association studies of fasting glucose. Genet Epidemiol. 2010;34:665–73. doi: 10.1002/gepi.20525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Li Y, Willer C, Sanna S, Abecasis G. Genotype imputation. Annual review of genomics and human genetics. 2009;10:387–406. doi: 10.1146/annurev.genom.9.081307.164242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Li Y, Willer CJ, Ding J, Scheet P, Abecasis GR. MaCH: using sequence and genotype data to estimate haplotypes and unobserved genotypes. Genetic epidemiology. 2010;34:816–834. doi: 10.1002/gepi.20533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Howie BN, Donnelly P, Marchini J. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS genetics. 2009;5:e1000529. doi: 10.1371/journal.pgen.1000529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Marchini J, Howie B, Myers S, McVean G, Donnelly P. A new multipoint method for genome-wide association studies by imputation of genotypes. Nature genetics. 2007;39:906–913. doi: 10.1038/ng2088. [DOI] [PubMed] [Google Scholar]
- 71.Voorman A, Lumley T, McKnight B, Rice K. Behavior of QQ-Plots and Genomic Control in Studies of Gene-Environment Interaction. PloS one. 2011;6:e19416. doi: 10.1371/journal.pone.0019416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Aulchenko YS, Struchalin MV, van Duijn CM. ProbABEL package for genome-wide association analysis of imputed data. BMC bioinformatics. 2010;11:134. doi: 10.1186/1471-2105-11-134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Petitti DB. Meta-analysis, Decision Analysis, and Cost-effectiveness Analysis. Vol. 2. Oxford University Press; New York: 2000. Statistical Methods in meta-analysis; pp. 94–118. [Google Scholar]
- 74.Higgins JP, Thompson SG. Quantifying heterogeneity in a meta-analysis. Statistics in medicine. 2002;21:1539–1558. doi: 10.1002/sim.1186. [DOI] [PubMed] [Google Scholar]
- 75.Genomes Project C etal. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Riva A, Kohane IS. A SNP-centric database for the investigation of the human genome. BMC bioinformatics. 2004;5:33. doi: 10.1186/1471-2105-5-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wellcome Trust Case Control C et al. Genome-wide association study of CNVs in 16,000 cases of eight common diseases and 3,000 shared controls. Nature. 2010;464:713–720. doi: 10.1038/nature08979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.McCarroll SA, et al. Common deletion polymorphisms in the human genome. Nat Genet. 2006;38:86–92. doi: 10.1038/ng1696. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.