

Abstract
Recently, missense mutations in the gene TARDBP encoding TDP-43 have been linked to familial ALS. The discovery of genes encoding these RNA binding proteins, such as TDP-43 and FUS/TLS, raised the notion that altered RNA metabolism is a major factor underlying the pathogenesis of ALS. To begin to unravel how mutations in TDP-43 cause dysfunction and death of motor neurons, investigators have employed both gain- and loss-of-function studies in rodent model systems. Here, we will summarize major findings from the initial sets of TDP-43 transgenic and knockout rodent models, identify their limitations, and point to future directions toward clarification of disease mechanism(s) and testing of therapeutic strategies that ultimately may lead to novel therapy for this devastating disease.
Keywords: TDP-43, Transgenic, Conditional knockout
1. Introduction
1.1. Amyotrophic lateral sclerosis: no disease-modifying therapy currently available
The motor neuron diseases (MND), including Amyotrophic Lateral Sclerosis (ALS), are chronic, progressive illnesses characterized clinically by severely disabling features involving motor systems (weakness, muscle atrophy, and, in ALS, spasticity); and pathologically by the presence of intracellular protein aggregates (inclusions), alterations in axonal transport, and death of motor neurons. There is a relatively selective involvement of lower motor neurons in MND, and in classical ALS, upper motor neurons are affected. The majority of MND cases appear to be sporadic, but a small percentage of patients have a familial history. Some forms of the disease inherited as autosomal dominants and others as recessives. Studies have shown that in some instances, the presence of specific gene products confers risk for disease. Although symptomatic treatments are available, there are, at present, no effective mechanism-based therapies (Bruijn et al., 2004; Wong et al., 2002). Recent research, particularly studies utilizing animal models, has provided insights into mechanisms of these disorders and identified new potential targets for therapy, thereby facilitating the design and testing of novel treatment strategies.
ALS, often called Lou Gehrig’s disease in the United States, is the most common adult onset form of MND with a prevalence of approximately 2–3 per 100,000 people (Cleveland and Rothstein, 2001; Julien, 2001). Each year in the United States, in excess of 5000 people are diagnosed with ALS. In parts of the United Kingdom, 1 in ~500 deaths is attributed to some form of MND. The principal clinical signs of ALS include: progressive limb weakness, which may be symmetrical or asymmetrical; atrophy of appendicular, bulbar, and respiratory muscles; and spasticity. The paralysis/muscle atrophy and spasticity are the result of degeneration of motor neurons in the spinal cord/brain stem and motor cortex, respectively. The onset of this illness is typically in the 5th or 6th decade of life, and affected individuals die usually within 2–5 years of appearance of symptoms. Both sporadic (sALS) and familial (fALS) forms of illnesses exist; familial cases make up approximately 5–10% of the total. While the causes of the majority of cases of ALS have yet to be identified, shared features of the clinical presentations and pathologies occurring in both sporadic and familial cases suggest the existence of common disease mechanisms.
The identification of causative mutations in specific genes in cases of human MND, including familial ALS (fALS) and spinal muscular atrophy (SMA) (Kabashi et al., 2008; Kwiatkowski et al., 2009; Lefebvre et al., 1995; Puls et al., 2003; Rosen et al., 1993; Sreedharan et al., 2008; Vance et al., 2009), has provided new opportunities to investigate the molecular participants in disease processes using transgenic and gene targeting approaches (Bruijn et al., 2004; Wong et al., 2002). In autosomal dominant fALS, the mutant proteins often acquire toxic properties that directly or indirectly impact on the functions and viability of neurons (Julien, 2001), and introduction of mutant genes into mice reproduces some features of these diseases (Bruijn et al., 1997; Dal Canto and Gurney, 1994; Laird et al., 2008; Wong et al., 1995). In contrast, autosomal recessive diseases, like SMA, which usually lack the functional protein encoded by the mutant gene (Survival Motor Neuron (SMN) in SMA), can often be modeled by gene targeting strategies (Wong et al., 2002).
In models of MND, therapeutic manipulations, manipulation of expression of selected genes in specific cell populations (Lambrechts et al., 2003; Subramaniam et al., 2002), creation of chimeric animals to test whether abnormalities are cell autonomous (Clement et al., 2003), administration of trophic factors to prevent trophic cell death (Henderson et al., 1994; Koliatsos et al., 1993), and testing of a variety of drug therapies (Kriz et al., 2003; Rothstein, 2003; Rothstein et al., 2005; Zhu et al., 2002) have been used to try to ameliorate phenotypes and thus provide insights into disease mechanisms and potential treatment strategies.
2. Recent advances in genetics of ALS provide opportunities for understanding disease mechanisms
Approximately 5–10% of cases of ALS are familial, and, in the majority of these cases, the disease is inherited as an autosomal dominant (Bruijn et al., 2004; Cleveland and Rothstein, 2001; Wong et al., 2002). Since the discovery of mutations in the Cu/Zn superoxide dismutase (SOD1) gene (Rosen et al., 1993), occurring in ~5–10% of autosomal dominant cases of fALS, several other genes or risk factors have been identified: dynactin p150glued linked to autosomal dominant fALS (Puls et al., 2003) and may, as an allelic variant, serve as a risk factor (Munch et al., 2004); autosomal recessive deletion mutations have been identified in ALS2 which encodes Alsin, a protein that regulates GTPases (Hadano et al., 2001; Yang et al., 2001); a rare autosomal dominant form of juvenile ALS, mutations have been identified in the gene (SETX) that encodes senataxin (Chen et al., 2004), which contains a DNA/RNA helicase domain with homology to other proteins known to have roles in the processing of RNA (Moreira et al., 2004); following an observation that deletion of the hypoxia response element in the promoter of the vascular endothelial growth factor (VEGF) gene causes degeneration of motor neurons in mice (Oosthuyse et al., 2001), it has been reported that individuals who are homozygous for certain haplotypes in the VEGF promoter have an increased risk for ALS (Cleveland, 2003; Lambrechts et al., 2003). Although not the subject of this review, recent discoveries of additional genes linked to ALS, including OPTN (Maruyama et al., 2010), VCP (Johnson et al., 2010), UBQLN2 (Deng et al., 2011) and C9ORF72 (DeJesus-Hernandez et al., 2011; Renton et al., 2011) as well as risk factors such as Ataxin-2 polyglutamine expansion (Elden et al., 2010), provide further opportunities to unravel this complex motor neuron disease in the future.
The identification of mutations in TARDBP, a gene encoding a DNA/RNA binding protein TDP-43, that are linked to both sporadic and familial ALS (Kabashi et al., 2008; Lagier-Tourenne and Cleveland, 2009; Neumann et al., 2006; Sreedharan et al., 2008) – the focus of this current review – as well as mutations in FUS/TLS, another member of the RNA-binding protein family, linked to familial ALS (Kwiatkowski et al., 2009; Vance et al., 2009) stimulated the ALS research community to focus efforts in unraveling how mutations in these genes cause motor neuron degeneration. These genetic discoveries raised the possibility as to whether ALS can be a disease of altered RNA metabolism/processing. Indeed, our data showing that TDP-43 regulates the formation and distribution of Survival Motor Neuron (SMN)-containing Gemini of coiled bodies (GEMs), a machinery that is required for the assembly of snRNPs involved in RNA splicing in motor neurons, provide strong support for such a hypothesis (see below). Thus, both gain- and loss-of-function studies will provide the framework to establish the physiological role of TDP-43, particularly its role in motor neurons, information that will be fundamental and critical to assess how ALS-linked mutants cause motor neuron degeneration. Moreover, deep sequencing studies to identify RNA targets of TDP-43 will also begin to clarify pathways that are impacted by TDP-43 and will provide the basis for assessment of pathways that are influenced by ALS-linked mutant TDP-43. Therefore, mouse model systems will provide not only important mechanistic information regarding the neurobiology of TDP-43, but also critical implications for the pathobiology of this RNA binding protein in ALS and potentially other neurodegenerative diseases.
The notion that altered RNA metabolism plays a crucial role in the disease is emphasized by the discovery of a hexanucleotide repeat expansion in an intron of a previously uncharacterized gene, termed C9ORF72, as the cause of a major proportion of cases of ALS and FTD (DeJesus-Hernandez et al., 2011; Renton et al., 2011). How hexanucleotide repeat expansion causes neurodegeneration in ALS or FTD, however, remains unknown at present. The observation that alternative transcripts of C9ORF72 (variants 1 and 3) are greatly reduced in patients (DeJesus-Hernandez et al., 2011) would support the view that loss of C9ORF72 is one major determinant leading to neurodegeneration in ALS and FTD. That RNA foci, a feature common to other noncoding repeat expansion disorders, such as Myotonic Dystrophy (DM-1 and DM-2; refs) and Fragile-X associated Tremor/Ataxia Syndrome (FXTAS; Brook et al., 1992; Mahadevan et al., 1992; Liquori et al., 2001), containing the expanded hexanucleotide repeats was observed in neurons of cases of ALS and FTD (Galloway and Nelson, 2009; Tassone et al., 2004) would offer the possibility of a toxic RNA mechanism via sequestration of RNA binding proteins that underlies neuronal loss. Alternatively, it is also plausible that both mechanisms, i.e., loss of C9ORF72 and gain of RNA toxicity contribute to the pathogenesis of these disorders. Whether and how expansion GGGGCC repeat in C9ORF72 mechanistically linked to TDP-43 proteinopathy will be an important research question for the future.
3. Generation and characterization of initial sets of transgenic rodent models of TDP-43
To understand the pathogenic mechanisms of mutant TDP-43 underlying ALS, it is important to clarify first the physiological roles of TDP-43 in the nervous system. To begin to address this issue, investigators have taken a gain-of-function approach to generate transgenic mice overexpressing wild type or ALS-linked mutant TDP-43 (Table 1) using a neuronal-specific promoter (e.g., Thy1.2), an inducible promoter (e.g., tTA-tetOff) or ubiquitous promoter (e.g. mouse PrP or endogenous human TDP-43).
Table 1.
Rodent models of wild type or ALS-linked mutant TDP-43.
| Investigators | Wegorzewska et al., PNAS 106, 2009 | Stallings et al., Neurobiol Dis 40, 2010 | Xu et al., J Neurosci 30, 2010 Xu et al., Mol Neurodegener 6, 2011 | |||
|---|---|---|---|---|---|---|
|
| ||||||
| Promoter | Mouse prion protein (Prp) | Mouse prion protein (Prp) | Mouse prion protein (Prp) | |||
|
| ||||||
| Transgene | Human flag-TDP-43A315T | Human TDP-43WT | Human TDP-43A315T | Human TDP-43WT | Human TDP-43M337V | |
| Expression levels | ~3× the endogenous murine protein level in spinal cord by non-species-specific TDP-43 antibody | Line 21: significantly higher than the endogenous murine protein level, but lower than the A315T line 23 level | Line 23: 4× the endogenous murine TDP level in spinal cord by non-species specific TDP-43 antibody | Homozygous: 2.5× the endogenous murine protein level in brain by non-species-specific TDP-43 antibody | Line 4, homozygous: 2.7× the endogenous murine protein level in brain by non-species-specific TDP-43 antibody | |
| Phenotype | 0–3 months: normal weight and appearance 4.5 months: weight loss, “swimming” gait Survival: 154±19 days |
10 founders died between 12 and 55 days. Line 21: no motor phenotype up to 11 months | 5 founders died between 14 and 73 days, others can reproduce but die at 52–75 days or have no phenotype. Line 23: declining grip strength and stride length leading to death within 150 days | Hemizygous: similar to nontransgenic. Homozygous: 14 days: lower body weight compared to nontransgenic mice 21 days: hindlimb clasp, body tremors, and “swimming” gait Survival: between 1 and 2 months |
Hemizygous: similar to nontransgenic up through 1 year. Homozygous: 21 days: hindlimb clasp, body tremors, and dragging gait. 1 month: lower body weight compared to nontransgenic mice, early lethality |
|
| TDP-43 and ubiquitin localization | Diffuse nuclear TDP-43 in most neurons and glia. In cytoplasm of neurons in cortical layer 5 and ventral horn, increased diffuse ubiquitin and punctate ubiquitin aggregates. Loss of nuclear TDP-43 staining in some ubiquitin-aggregate-positive neurons | Diffuse nuclear TDP-43 in neurons and glia. Some ventral horn or brainstem neurons with increased diffuse cytoplasmic ubiquitin | Diffuse nuclear TDP-43 in neurons and glia. Colocalization of punctate ubiquitin and phosphorylated TDP (409/410) aggregates in cytoplasm and nuclei of ventral horn and brainstem neurons | Diffuse TDP-43 staining in nuclei, and in cytoplasm of some neurons. Increased diffuse nuclear and cytoplasmic ubiquitin staining. ~15% of spinal motor neurons with phospho-403/404 TDP-43 nuclear aggregates. Rarer cytoplasmic pTDP-43 aggregates | Diffuse TDP-43 staining in nuclei, and in cytoplasm of some neurons. Increased diffuse nuclear and cytoplasmic ubiquitin staining. Many spinal motor neurons with phospho-403/404 TDP-43 nuclear aggregates. Rarer cytoplasmic pTDP-43 aggregates | |
| TDP-43 fragments [antibody used] | 25 and 35 kDa fragments (detergent-soluble) in 1–2 months and older mice [Proteintech 10782-2-AP, anti-body to TDP-43N-260aa] | Some low molecular weight fragments in spinal cord homogenate and cytosolic fractions [Proteintech 10782-2-AP, antibody to TDP-43N-260aa] | Low molecular weight fragments in spinal cord homogenate and cytosolic fractions [Proteintech 10782-2-AP, antibody to TDP-43N-260aa] | 25 and 35 kDa fragments in brain and spinal cord extract from hemizygous and homozygous mice [Proteintech 12892-1-AP, antibody to TDP-43 260aa-C] | 25 and 35 kDa fragments in brain lysates from nontransgenic, hemizygous, and homozygous mice [Proteintech 12892-1-AP, antibody to TDP-43 260aa-C] | |
| Other pathology | Astrogliosis (by GFAP staining) | In cortical layer 5 | Mild, in spinal cord | In ventral horn and brainstem | In spinal cord | In spinal cord and brainstem |
| Motor neuron loss | Decreased number of neurons in cortical layer V. 20% loss of motor neurons in ventral horn | Not quantified | Not quantified | Not quantified | Not quantified | |
| Other aggregates | Unidentified cytoplasmic aggregates displacing nuclei in some neurons | – | – | ~10% of spinal motor neurons contain cytoplasmic mitochondrial clusters. | Mitochondrial clusters in many spinal motor neurons | |
| Investigators | Wils et al., PNAS 107, 2010 | Shan et al., PNAS 107, 2010 | ||||
|---|---|---|---|---|---|---|
|
| ||||||
| Promoter | Murine Thy1.2 | Murine Thy1.2 | ||||
|
| ||||||
| Transgene | Human TDP-43WT | Human TDP-43WT | ||||
| Expression levels | Line TAR4 (hemizygous): human TDP transcript level 1× that of endogenous mouse TDP transcript level in brain | Line TAR6/6 (homozygous): human TDP transcript level 1.2× that of endogenous mouse TDP transcript level in brain | Line TAR4/4 (homozygous): human TDP transcript level 2× that of endogenous mouse TDP transcript level in brain | W3 Line, males: 4.6× endogenous murine TDP level in spinal cord at 4 weeks by non-species-specific TDP-43 antibody | W3 Line, females: 2.3× endogenous murine TDP level in spinal cord by non-species-specific TDP-43 antibody | |
| Phenotype | 14 months: hindlimb clasp 15 months: impaired rotarod performance |
2 months: abnormal hindlimb clasp. 4 months: impaired rotarod performance Survival: avg. 6.7 months |
14–26 days: abnormal hindlimb clasp, decreased stride, impaired rotarod performance, rapid onset spastic paralysis, and death. Survival: <30 days |
14–18 days: hindlimb clasp, tremor, gait abnormalities 4 weeks: ~20% lower body weight compared to nontransgenic mice |
0–3 months: normal weight and appearance 3 months: fine tremor |
|
| Animals characterized | Line TAR 4/4 | W3 males | |
|---|---|---|---|
| TDP-43 and ubiquitin localization | Intranuclear and cytoplasmic ubiquitin aggregates in neurons in cortical layer V and spinal cord. Diffuse nuclear TDP-43 in neurons, with some clearing in neurons with ubiquitin aggregates. Intranuclear ubiquitin aggregates costain for human TDP-43 and phospho-409/410 TDP-43. Some cytoplasmic ubiquitin aggregates costain for phospho-409/410 TDP-43. | Diffuse nuclear TDP-43 staining in spinal cord neurons. Intranuclear aggregates of human TDP-43 colocalized with FUS/TLS and SC35. Diffuse cytoplasmic ubiquitin staining. | |
| TDP-43 fragments [antibody used] | 35 kDa C-terminal fragments in cytoplasmic and nuclear fractions of brain lysates. 25 kDa fragment in nuclear fraction. [Rabbit anti-C-terminal TDP-43 and Proteintech rabbit anti-TDP-43] | No 25 or 35 kDa fragments observed | |
| Other pathology | Astrogliosis (by GFAP staining) | In cortical layer V and spinal cord | In lumbar spinal cord |
| Motor neuron loss | ~30% cortical layer V neuron loss, ~25% neuron loss in lumbosacral region of spinal cord | Not quantified | |
| Other aggregates | – | Mitochondrial cytoplasmic aggregates displacing nuclei in some motor neurons |
| Investigators | Tsai et al., J Exp Med 207, 2010 | Igaz et al., J Clin Invest 121, 2011 | Swarup et al., Brain 134, 2011 | ||
|---|---|---|---|---|---|
| Promoter | Ca2+/calmodulin-dependent kinase II (CaMKII) | CaMKIIa-tTA×tetOff | CaMKIIa-tTA×tetOff | 18-kb full length gene fragment from human bacterial artificial chromosome | |
| Transgene | Mouse TDP-43 cDNA | TetOff human TDP-43WT induced at 28 days | TetOff human TDP-43ΔNLS induced at 28 days | Human TDP-43WT Human TDP-43A315T Human TDP-43G348C |
|
| Expression levels | Homozygous: 2× endogenous murine TDP level in hippocampus and cortex by non-species-specific TDP-43 antibody | Line 12: 0.8× endogenous murine TDP level in cortex by non-species-specific TDP-43 antibody | Line 4: 7.9× endogenous murine TDP level in cortex by non-species-specific TDP-43 antibody | All lines: human TDP transcript level ~3× that of mouse TDP in spinal cord at 3 months | |
| Phenotype | 2 months: impaired performance in Morris water maze and fear conditioning test 6 months: abnormal hind limb clasp, impaired rotarod test Mean survival: 495 days |
1–3 month post-induction: limb clasping | 1 week post-induction: limb clasping | All lines: deficits in passive avoidance test, Barnes maze test, and rotarod at 7–10 months | |
| TDP-43 and ubiquitin localization | Cytoplasmic accumulation, ubiquitin colocalization, and nuclear clearing of TDP-43 in 15–20% of cortical neurons. Diffuse nuclear TDP-43 in other neurons | Mainly nuclear staining in cortical neurons in a mosaic pattern. <0.1% of cortical neurons have phospho-409/410 TDP aggregates. Neurons expressing human TDP have decreased mouse TDP-43 expression. | Mainly cytoplasmic with some nuclear staining in cortical neurons. <1% cortical neurons have cytoplasmic phospho-409/410 TDP-43 aggregates colocalizing with ubiquitin. Neurons expressing human TDP have decreased mouse TDP-43 expression. | Mutant lines, 10 months: diffuse and some punctate cytoplasmic staining of human TDP-43 with ubiquitin in mutant spinal cord, cortex, and hippocampus. Not found in WT mice or in mutant mice at 3 months. | |
| TDP-43 fragments [antibody used] | In urea-soluble brain extract: increased high molecular weight, 25 kDa, and 35 kDa fragments in 6 month vs. 2 month mice [Protein-tech 10782-2-AP, antibody to TDP-43N-260aa] | No C-terminal fragments detected | No C-terminal fragments detected | Mutant lines: 35 kDa and 25 kDa fragments in spinal cord increased at 10 months vs. 3 months. [not stated] | |
| Other pathology | Astrogliosis (by GFAP staining) | In hippocampus and cortex | In hippocampus and cortex | In hippocampus and cortex | Using a luciferase reporter, GFAP promoter activity elevated by 20 weeks in brain and ~30 weeks in spinal cord |
| Motor neuron loss | 24% neuron loss in cortex | ~20% neuron loss in dentate gyrus 1 month post-induction, and ~75% loss 3 month post-induction | ~50% neuron loss in dentate gyrus 1 month post-induction | No difference in number of axons in L5 ventral root | |
| Investigators | Rat model: Zhou et al., PLoS Genet 6, 2010 | ||
|---|---|---|---|
| Promoter | 22 kb minimal human TDP gene from bacterial artificial chromosome | CAG-tTA×tetOff | |
| Transgene | Human TDP-43WT Human TDP-43M337V |
TetOff Human TDP-43M337V | |
| Expression levels | Not quantified, but WT and M337V mice have comparable levels in spinal cord by human-specific TDP-43 antibody. | Not quantified, but has levels comparable to WT mice in spinal cord by human-specific TDP-43 antibody. | |
| Phenotype | WT: no paralysis within 200 days. M337V: founders became paralyzed before 30 days. | Line 16, no Dox treatment: paralysis and death by P20. Line 16, induced at 4 days before delivery: paralysis by P35. Line 7, induced at 4 days before delivery: weakness by P40, paralysis and death by P55, males more severe than females | |
| Animals characterized | WT line 4 | Line 7 | |
| TDP-43 and ubiquitin localization | Diffuse nuclear and cytoplasmic staining of phospho-409/410-TDP. Diffuse ubiquitin staining | Diffuse nuclear and cytoplasmic staining of phospho-409/410-TDP, rare TDP aggregates in the cortex but not spinal cord. Diffuse ubiquitin staining | |
| TDP-43 fragments | Low molecular weight fragments (35 and 15 kDa) [in-house human-specific TDP-43 antibody] | Low molecular weight fragments (35 and 15 kDa) [in-house human-specific TDP-43 antibody] | |
| Other pathology | Astrogliosis (by GFAP staining) | Not observed | Around spinal motor neurons |
| Motor neuron loss | No loss of motor neurons in L3 ventral horn | ~10–15% loss of motor neurons in L3 ventral horn | |
Several groups (Stallings et al., 2010; Wegorzewska et al., 2009; Xu et al., 2010 and Xu et al., 2011) have generated lines of mice expressing human TDP-43 (hTDP-43) under the control of the murine prion protein promoter (moPrP). This promoter drives protein expression in the brain, spinal cord, and heart, and to a lesser extent, in skeletal muscle, lung, liver, and kidney (Borchelt et al., 1996). Each of the moPrP mouse lines expresses either wild-type or the ALS-linked mutants A315T or M337V encoded by the human TARDBP cDNA. In almost every moPrP driven line, an elevated level of hTDP-43 in the spinal cord or brain led to motor deficits and relatively early lethality. High-expressing mouse lines display a more severe and lethal phenotype, and founders with a very high level of protein accumulation perish before reaching sexual maturity. While intranuclear aggregates of TDP-43 and diffuse cytoplasmic ubiquitin staining are common to many lines, cytoplasmic aggregates of TDP-43 colocalized with ubiquitin are rare and generally only detectable using an antibody to phosphorylated TDP-43. Although mice expressing low level of the transgene (hTDP-43WT, line 21) show no motor deficits (Stallings et al., 2010), mice expressing high level of wild type hTDP-43 (hTDP-43WT homozygous line, Xu et al., 2010) show a very similar motor phenotype to the comparably-expressing homozygous mutant M337V line (Xu et al., 2011). Indeed, there appears to be little difference between lines expressing wild-type human TDP-43 and mutant hTDP-43 driven by the moPrP promoter to comparable levels.
In addition to our group (Shan et al., 2010), Wils et al. (2010) have generated TDP-43 mouse lines under the control of a modified murine Thy1 promoter. This Thy1.2 cassette drives the expression almost exclusively in neurons (Caroni, 1997; Vidal et al., 1990). As has been noted previously, mice generated using the Thy1.2 cassette exhibit line-to-line variability in the expression pattern of different populations of neurons (Feng et al., 2000; Wegorzewska and Baloh, 2010). The comparison between these two sets of transgenic lines is difficult because of this line-to-line variability. Indeed, the high-expressing homozygous TAR4/4 mice (Wils et al., 2010) display a spastic paralysis more reminiscent of upper motor neuron degeneration, whereas our lines ultimately develop a flaccid paralysis typical of predominant lower motor neuron disease (Tsao and Wong, pers. comm.). Importantly, we have developed lines of mice expressing hTDP-43 levels slightly above the endogenous mouse TDP-43 level, that develop a robust motor neuron disease phenotype, including flaccid paralysis at around two years of age (Tsao and Wong; pers. comm.). In such lines of mice, the phenotype observed of mice expressing wild type hTDP-43 (hTDP-43WT) is similar to that of ALS linked mutant (G298S) hTDP-43 (hTDP-43G298S; Tsao and Wong; pers. comm.).
The observation that the moPrP and Thy1.2 mice expressing wild type hTDP-43 showed motor deficits and pathology similar to mutant hTDP-43 mice stands as a notable difference between these mouse lines and the SOD1 rodent models. In the SOD1 rodent models, overexpression of ALS-linked mutant human SOD1 caused a paralytic phenotype, but expression of the human wild-type SOD1 to similar levels caused no such phenotype. The toxicity of wild-type hTDP-43 in mice can be explained in two ways. First, as hTDP-43 differs from mouse TDP-43 in several residues, five of which are located in the glycine-rich C-terminal domain that appears to be a hotspot for ALS-linked mutations (Fig. 1), hTDP-43 may act like a mutant, nonfunctional, or dysfunctional protein in a mouse background. However, the strong homology of mouse and human TDP-43 (96% identity, Fig. 1) compared to mouse and human SOD1 (84% identity, Fig. 1), favors the alternative explanation that simply overexpressing any form of TDP-43 to high levels may be toxic in the mice. To assess the relative contribution of overexpression or mutation to the toxicity of hTDP-43 in mice, it is important to analyze a model of overexpression without mutation (overexpressing the mouse form of TDP-43 in mice), and also a model of mutation without overexpression (a knock-in model “humanizing” the endogenous mouse locus, or knocking-in an ALS-associated mutation).
Fig. 1.

Multiple sequence alignments for TDP-43 and SOD1. Amino acid sequences are compared for TDP-43 (RefSeq ID for human: NP_031401.1; mouse: RefSeq ID: NP_663531.1; GenBank no. for rat: EDL81132.1) and SOD1 (RefSeq ID for human: NP_000445.1; mouse: NP_035564.1; rat: NP_058746.1). Nonidentical residues are shown in red text. The yellow highlighted area marks the TDP-43C-terminal domain, and the green highlighted residues mark the positions of ALS-associated mutations in human TDP-43.
A mouse model overexpressing mouse TDP-43 exists, in which the protein is expressed in the hippocampus and cortex using a constitutive Ca2+/calmodulin-dependent kinase II (CaMKII) promoter (Tsai et al., 2010). The investigators using this model demonstrated that the overexpression of the mouse TDP-43 in the forebrain led to behavioral and motor deficits and cortical neuron loss, which indicated that mouse TDP-43 is toxic when overexpressed in mice. This finding supports the view that elevated human TDP-43 is toxic in mice because of an elevated expression level; however, it does not rule out the possibility that hTDP-43 is nonfunctional or dysfunctional in the mouse background.
Indeed, another group advanced the theory that a suppression of normal mouse TDP-43 function was actually the root cause of hTDP-43 toxicity in mice. These investigators used an inducible CaMKII promoter to drive postnatal expression of hTDP-43 (Igaz et al., 2009). Igaz et al. (2009) documented that overexpression of hTDP-43WT led to neuron loss in the dentate gyrus. They also observed that expressing hTDP-43WT in cortical neurons downregulated the level of mouse TDP-43 in those neurons, presumably due to TDP-43 autoregulation (Ayala et al., 2010; Polymenidou et al., 2011). As very few TDP-43 aggregates were observed in these mice, the investigators suggested that the mechanism by which expression of hTDP-43 is toxic is that it suppresses the level of mouse TDP-43 without having the ability to replace the endogenous protein. However, in these mice the total level of TDP-43 is still elevated above the endogenous level, so the assertion that the toxicity arises from loss of normal mouse TDP-43 function, as opposed to an overexpression effect, is difficult to substantiate.
Recognizing the toxicity of highly elevated TDP-43 levels and the importance of expression pattern of the transgene, Swarup et al. (2011) used the TDP-43-containing gene fragment from a human bacterial artificial chromosome (BAC) to drive transgene expression in a low-level pattern mimicking the endogenous expression pattern. The resulting hTDP-43WT, hTDP-43A315T, and hTDP-43G348C lines all have hTDP-43 transcript levels close to 3>× that of the mouse TDP-43 transcript levels in the spinal cord, though it appears that the total TDP-43 level in spinal cord protein extracts are much higher in the mutant lines as compared to that of the wild-type line. All lines exhibit behavioral and motor deficits at 7–12 months of age. Spinal cord sections of 10 month old hTDP-43G348C mice show some punctate staining of hTDP-43 colocalized with ubiquitin, a finding which is not observed in hTDP-43WT mice. Additionally, 25- and 35-kDa fragments are detected in the spinal cord lysate of 10 month old hTDP-43G348C mice, but not of younger hTDP-43G348C mice or of hTDP-43WT mice. There does appear to be significant additional pathological findings associated with the G348C mutation, but it is unknown whether these same findings would occur in hTDP-43WT mice with a more comparable protein level.
Using a similar BAC transgenic approach, a rat model of TDP-43 overexpression has also been generated (Zhou et al., 2010). This model is unique in that the 2 rat founders expressing human TDP-43WT do not show a paralytic phenotype up to 200 days, but the 3 rat founders expressing hTDP-43M337V at comparable levels developed paralysis by 29 days. The investigators also used a tetracycline-inducible system coupled with a CAG promoter to drive ubiquitous expression of hTDP-43M337V postnatally, and found that rats in the lower-expressing line 7 were initially asymptomatic but progressed to paralysis and death by 55 days. While neuronal loss was observed only in the hTDP-43M337V lines, diffuse nuclear and cytoplasmic phosphorylated TDP-43 staining was detected in both mutant and hTDP-43WT rats.
Taken together, these initial sets of TDP-43 transgenic studies revealed several important observations:
Overexpression of human wild type or ALS-linked mutant TDP-43, or mouse TDP-43 is toxic in a dose-dependent manner in mice. In the only TDP-43 rat model study, overexpression of the ALS-linked mutant TDP-43, but not the human wild type TDP-43, is toxic.
The phenotype of each mouse line is sensitive to the pattern of exogenous TDP-43 expression.
Cytoplasmic aggregation of TDP-43 does not appear to be critical to develop a disease phenotype.
4. Abnormal distribution of mitochondria in motor neurons of TDP-43 transgenic mice
To further clarify the mechanism whereby TDP-43 impact on cellular function, it will be important to access the consequence of increased expression of TDP-43 in both the cytoplasmic and nuclear compartments of motor neurons. Consistent with motor deficits observed in our lines of TDP-43 mice, morphological analyses revealed eccentric nuclei with abnormal aggregates/inclusions in cell bodies of motor neurons in the spinal cord (Shan et al., 2010) and brain stem. Although human TDP-43 can be localized to the nuclei of motor neurons, no TDP-43 immunoreactivity is associated with these cytoplasmic inclusions (Shan et al., 2010). In addition, increased ubiquitin immunoreactivity was present in motor neurons of the spinal cord (Shan et al., 2010) and brain stem, particularly associated with these cytoplasmic inclusions. To ascertain the composition of these cytoplasmic aggregates, morphological analysis of motor neurons revealed that they comprised, in part, massive accumulations of mitochondria. These observations suggest that elevated levels of TDP-43 impact on the intracellular trafficking of mitochondria and consequently lead to abnormal distributions of these organelles in motor neurons. Similar findings were also observed in a different line of TDP-43 mice (Xu et al., 2010).
To observe directly the distributions of mitochondria in different compartments of motor neurons, TDP-43 mice were crossbred to Thy1-mitoCFP mice, in which mitochondria are fluorescently labeled with CFP in neurons (Misgeld et al., 2007). MtCFP;TDP-43 compound mice show mitochondria clustered within inclusions of motor neurons (Shan et al., 2010). The observation that mitochondria mainly accumulate within inclusions of cell bodies and are sparsely distributed in neuronal processes in the mtCFP;hTDP-43 compound mice suggests the possibility that trafficking of organelle, particularly mitochondria, is impaired in these nerve cells. If this is the case, nerve terminals of these mice may be deficient in mitochondria. Consistent with this hypothesis, we observed a marked reduction of mitochondria (as indicated by the CFP signal intensity) at nerve terminals of neuromuscular junctions (NMJ) in mtCFP;TDP-43 compound mice (Shan et al., 2010). Moreover, in the NMJ of mtCFP;TDP-43 mice, AChRs form a plaque-like pattern (Shan et al., 2010) similar to that described in a mouse model of SMA (Kong et al., 2009). Such abnormalities are associated with abnormal synaptic transmission in these SMA mice (Kong et al., 2009). These observations suggest the possibility that synaptic transmission is altered at the NMJ of TDP-43 mice. This interpretation is consistent with the weakness and muscle atrophy observed in the TDP-43 mice (Shan et al., 2010).
5. TDP-43 regulates SMN associated GEMs in motor neurons
To determine how elevated levels of TDP-43 in the nucleus lead to motor neuron dysfunction, we examined aspects of TDP-43 related nuclear functions that might be altered in motor neurons of TDP-43 mice. Immunocytochemical analysis of TDP-43 in motor neurons with cytoplasmic inclusions revealed a striking abnormal localization of TDP-43 in the nuclear compartment that is usually associated with two conspicuous intranuclear aggregates (Shan et al., 2010). While there was no co-localization of TDP-43 with ubiquitin in these nuclear inclusions (Shan et al., 2010), we discovered that TDP-43 immunoreactive nuclear aggregates contained both fused in sarcoma (FUS), an RNA-binding protein (Shan et al., 2010) recently linked to cases of ALS (Kwiatkowski et al., 2009; Vance et al., 2009), and SC35, a marker of non-snRNP splicing speckles (Spector, 2001). These results suggest that increased levels of TDP-43 induce its association with FUS and SC35, proteins critical for RNA metabolism.
To examine whether elevated levels of TDP-43 impact on pathways that are involved in RNA splicing, the distributions of the Survival Motor Neuron (SMN) complex was assessed in relation to GEMs and to Cajal bodies, two nuclear structures containing high concentrations of the SMN protein (Battle et al., 2006; Gall, 2000), which is linked to spinal muscular atrophy (SMA), a motor neuron disease of infancy and childhood (Burghes and beattie, 2009; Lefebvre et al., 1995; Zhang et al., 2008). The SMN complex is part of a large multimeric protein assembly essential for biogenesis of small nuclear ribonucleoproteins (snRNPs) required for pre-messenger RNA splicing (Maniatis and Tasic, 2002). Although previous studies of TDP-43 and SMN showed co-localization of these two proteins in the nucleus of transient transfected cells (Wang et al., 2002), we failed to detect co-localization of TDP-43 with SMN-associated GEMs in motor neurons of TDP-43 mice. Rather, our immunocytochemical analysis using SMN antibody showed a significant increase in, the number of GEM bodies in motor neurons of TDP-43 mice (Shan et al., 2010). Since GEM bodies dynamically shuttle between the nucleolus and the nucleoplasm (Dundr et al., 2004; Sleeman et al., 2003), we examined the distributions of GEMs in motor neurons of TDP-43 mice and that while GEMs are normally distributed with one or two discretely associated with the nucleolus in neurons of non-transgenic mice, SMN is present diffusely within the entire nucleolus and SMN-containing GEMs are confined to the perinucleolar region of motor neurons in TDP-43 mice (Shan et al., 2010). The integrity of GEMs is confirmed by the identification of these same nuclear structures with other essential components of GEMs, including Gemin 2 and Gemin 8.
To further examine the role of TDP-43 in motor neurons, we employed a complementary loss-of-function approach to delete the gene encoding TDP-43 (see below). Immunofluorescence analysis revealed in addition to localization of SMN in the cytoplasm, that two prominent SMN-containing GEMs are observed as expected in each spinal motor neuron of control mice (Shan et al., 2010). However, we failed to find such SMN-containing GEMs in motor neurons of homozygous TDP-43 knockout mice, although SMN can be localized in the cytoplasm (Shan et al., 2010). These findings indicate that TDP-43 is required for the formation of GEMs in the nucleus of motor neurons. Taken together, results from both loss- and gain-of-function studies of TDP-43 converge to support the idea that TDP-43 is critical for the generation of SMN-containing GEMs and alteration of TDP-43 could impact on pathways that control RNA splicing.
In future studies, it will be important to determine whether nuclear abnormalities precede the cytoplasmic inclusions containing mitochondria in TDP-43 transgenic mice and characterize the time course of development of nuclear inclusions vs. cytoplasmic inclusions in motor neurons of TDP-43 transgenic mice. We hypothesize that the primary role of TPD-43 is in the nuclear compartment of motor neurons, including their impact on formation and distribution of SMN-GEMs, and we would predict that formation of nuclear TDP-43 positive inclusions precedes accumulation of mitochondria in the cytoplasm of motor neurons. Such outcome would be consistent with the view that perturbation of RNA metabolism/processing would impact on pathways that impact on proper distribution of mitochondria in motor neurons.
6. Development of constitutive and conditional Tardbp null mice
To examine the physiological role of TDP-43, investigators have used a loss-of-function approach to delete the Tardbp gene in mice (Table 2). Several groups (Kraemer et al., 2010; Sephton et al., 2010; Wu et al., 2009) have generated constitutive Tardbp knockout mice and showed that these animals died during early embryogenesis, establishing the essential role this RNA binding protein play during development. In order to bypass embryonic lethality of constitutive Tardbp null mice, conditional Tardbp knockout mouse will be necessary. Toward this end, we have developed a conditional knockout line by engineering a targeting vector in which the 3rd exon of Tardbp was flanked by loxp together with a neomycin resistance gene inserted in the 2nd intron (Chiang et al., 2010). The floxed Tardbp mice were crossbred with a CAG-Cre transgenic mouse line that express the Cre recombinase ubiquitously (Sakai and Miyazaki, 1997) to generate the heterozygous Tardbp knockout (Tardbp+/−) mice (Chiang et al., 2010). The Tardbp+/− mice were fertile and expressed a similar level of TDP-43 in a variety of tissues as compared to those of Tardbp+/+ mice (Chiang et al., 2010), suggesting that the level of TDP-43 is tightly controlled and compensated in the Tardbp+/− mice.
Table 2.
Constitutive or conditional Tardbp knockout mouse models.
| Investigators | Constitutive or conditional Tardbp deletion | Phenotype | Level of Tdp-43 in Tardbp+/− mice |
|---|---|---|---|
| Wu et al., Genesis 48:56, 2009 | Constitutive; deleted exons 2 and 3 | Peri-implantation lethality; blastocysts of homozygotes showed defective outgrowth of inner cell mass in vitro | Tardbp mRNA level similar to wild type level |
| Sephton et al., J. Biol. Chem. 285: 6826, 2010 | Constitutive; gene trap insertion in intron 2 and inframe fusion of exon 2 with beta-galactosidase/neomycin marker | Embryonic lethality between 3.5 and 8.5 days; abnormal expansion of inner cell mass in homozygous blastocysts | mRNA and protein levels similar to those of wild types |
| Kraemer et al., Acta Neuropathol. 119: 409, 2010 | Constitutive; gene trap insertion in intron 2 and inframe fusion of exon 2 with beta-galactosidase/neomycin marker | Embryonic lethality around day 7.5 | Tdp-43 protein level similar to wild type level |
| Chiang et al., PNAS107: 16320, 2010 | Conditional; exon 2 floxed allele | Ubiquitous deletion of Tardbp in adult mice leads to metabolic phenotype and premature death | mRNA and protein levels similar to those of wild types |
7. Loss of body fat in conditional Tardbp knockout mice using ubiquitous ErCre driver lines
To examine the physiological role of TDP-43 in post-natal mice, floxed Tardbp mice were bred with Rosa26-ErCre mice (Badea et al., 2003) to generate inducible Tardbp knockout (ErCre; TardbpF/F) mice in which the Rosa26 enhancer/promoter will direct the expression of ErCre recombinase ubiquitously in the presence of tamoxifen.
In contrast to control, Rosa26-ErCre;TardbpF/F mice unexpectedly die by Day 9 after switching to a tamoxifen-containing diet (Chiang et al., 2010). Because initial necropsy analysis of conditional Tardbp knockout mice indicated a loss of body fat, metabolic analyses of these mice were performed. Upon deletion of Tardbp by diet containing tamoxifen citrate (400 mg/kg diet), body weights of all mice dropped during the first three days (Chiang et al., 2010) as a consequence of reduced food intake (Chiang et al., 2010). Whereas control mice regained some of their weights during the next 4 days correlating with an increase in food intake, the ErCre;TardbpF/F or ErCre;Tardbp+/F mice did not regain their body weights despite the increase in food consumption over this same period (Chiang et al., 2010). Despite significant differences in cumulative weight loss between the control and the Tardbp knockout groups, energy intakes during tamoxifen-dependent deletion of Tardbp were similar among groups (Chiang et al., 2010), suggesting that decreased calorie intake was not the major cause of differences in weight loss. Indirect calorimetry revealed that the altered metabolism contributed to the relatively greater weight loss in the conditional Tardbp knockout mice. Both ErCre;Tardbp+/F and ErCre;TardbpF/F mice showed respiratory exchange ratios (RER=VCO2/VO2) indicative of pure fat oxidation (Chiang et al., 2010), vs. the RER of control mice indicating the high level of carbohydrate oxidation expected based on composition of the specialized tamoxifen-containing diet (Chiang et al., 2010). These findings support the notion that increased fat oxidation rather than reduced energy intake is responsible for the markedly greater weight loss in the conditional Tardbp knockout mice. Since the Rosa26-ErCre;TDPF/F mice usually die by Day 9 after switching to a tamoxifen-containing diet, a weaker driver line of CAG-ErCre mice (Hayashi and McMahon, 2002) was used to confirm the observed lean phenotype and to extend the survival time of tamoxifen treated ErCre;TardbpF/F mice. Most of the CAG-ErCre;TardbpF/F or CAG-ErCre;Tardbp+/F mice survived at least 18 days after switching to the tamoxifen diet. Moreover, the reduction in levels of TDP-43 in these conditional Tardbp knockout mice correlated with the decrease in body weights (Chiang et al., 2010). Importantly, gross examination of mesenteric fat confirmed this dramatic fat loss in CAG-ErCre; TardbpF/F mice (Chiang et al., 2010). Indeed, histological analysis of fatty tissues revealed the absence or reduction, respectively, of fatty vacuoles in adipocytes within subcutaneous tissue (Chiang et al., 2010) and interscapular brown fat (Chiang et al., 2010) in the CAG-ErCre;TardbpF/F or CAG-ErCre; Tardbp+/F mice. Moreover, the positive immunoreactivities of two independent adipocyte markers, ATGL and PPARγ, showed the presence of adipocytes in both white (Chiang et al., 2010) and brown adipose tissues (Chiang et al., 2010), indicating that the loss of fat content is not due to the absence of adipocytes in these CAG-ErCre;TardbpF/F mice, but rather to lack of stored fat within the adipocytes. Taken together, these findings indicate that the decreased level of TDP-43 is responsible for accelerated fat loss in adipocytes of conditional Tardbp knockout mice through increased fat oxidation.
8. Establishment of conditional TDP-43 knockout cells
In order to clarify the mechanism whereby loss of TDP-43 leads to deficits in our conditional knockout mice, it will be important to identify relevant downstream targets of TDP-43. Although CFTR, SMN, NF-L and HDAC6 transcripts have been found to be bound and regulated by TDP-43, misregulation of any of these putative targets would not be predicted to provide a straightforward explanation of the lean phenotype observed in our conditional Tardbp null mice. To identify additional targets of TDP-43, we characterized the Tardbp dependent transcriptome. We engineered a tamoxifen inducible Tardbp knockout ES cell line (termed iTDPKO) by replacing the wild type Tardbp allele of Tardbp+/F ES cells with a CAGErCreEr cassette (Chiang et al., 2010). While both iTDPKO and cTDP ES cells could grow as colonies and amplify after targeting (Chiang et al., 2010), the 4-HT treated iTDPKO ES cells exhibited reduction in a colony size coupled with increased apoptosis (Chiang et al., 2010). These data suggest that TDP-43 is essential for ES cell survival and proliferation and offers an explanation for the embryonic lethality of Tardbp−/− embryos.
9. High throughput DNA sequencing revealed relevant downstream targets of Tardbp
To identify the downstream targets of TDP-43, two independent pairs of the iTDPKO and cTDP ES cells were induced by 100 ng/ml of 4HT for 3 days and total mRNAs were isolated for RNA-seq analysis by Illumina genome analyzer. The raw reads were mapped onto the mm9 mouse genome using a public domain database, and these data were analyzed by the Partek software to identify a set of differentially expressed genes (Chiang et al., 2010). The dramatic reduction in the level of Tardbp mRNA (Chiang et al., 2010) validated the deletion of Tardbp in the ES cells and the methodology of RNA-seq analysis. Significantly, protein blot analysis showed that levels of Tbc1d1 and Rfc2 were nearly abolished in iTDPKO cells (Chiang et al., 2010). Interestingly, among the top 30 hits (genes with the lowest p values and more than 3 fold change), most (25 out of 30) of the genes are down-regulated, suggesting that TDP-43 plays an important role in elevating RNA transcription or maintaining RNA stability. Since Rfc2 is a constitutively expressed protein essential for both DNA repair and replication (Reynolds et al., 1999), the dramatic reduction of this protein in the highly replicating ES cells would provide an explanation for the observed lethality occurring in ES cells lacking Tardbp (Chiang et al., 2010). Since it has been reported that a non-functional Tbc1d1 mutant in the skeletal muscle is responsible for the lean phenotype in mice and that Tbc1d1 is essential for Glut4 translocation to the plasma membrane of skeletal muscle cells for glucose uptake (Chadt et al., 2008), a decrease in Tbc1d1 in skeletal muscle might offer an explanation for the lean phenotype shown in our conditional Tardbp knockout mouse model (Chiang et al., 2010). To test this notion, we assessed the levels of Tbc1d1 in the skeletal muscles of the control, CAG-ErCre;Tardbp+/F, and CAG-ErCre; TardbpF/F mice fed with tamoxifen. Protein blot analysis of muscle extracts showed depletion of Tbc1d1 in CAGErCre; TardbpF/F mice (Chiang et al., 2010) that exhibited marked reduction of fat (Chiang et al., 2010). These findings are consistent with the view that the post-natal deletion of Tardbp led to the lean phenotype through reduction of level of Tbc1d1 protein in the muscle of conditional Tardbp null mice. To test directly whether loss of TDP-43 in skeletal muscle is responsible for the lean phenotype observed in Tardbp null mice, we generated mice lacking TDP-43 selectively in skeletal muscle using a muscle specific Cre driver line, MLC-Cre. Interestingly, preliminary studies indicate that these MLC-Cre;TardbpF/F mice exhibit marked reduction in level of Tbc1d1 muscle degeneration and died between 4 and 5 months of age (Lin and Wong, pers. comm.). Future studies will be required to establish the mechanism whereby TDP-43 dependent regulation of Tbc1d1 impact on metabolism in skeletal muscle.
10. Conditional deletion of Tardbp in the CNS
Because the death that occurred in either Rosa26-ErCre;TardbpF/F or CAG-ErCre;TardbpF/F mice shortly after tamoxifen-induced deletion of Tardbp precluded the evaluation of lack of TDP-43 during aging in the CNS, CNS-specific Cre or ErCre driver lines (eg., CamKII-Cre, CamKII-ErCre, Hb9-ErCre and Isl1-Cre) will be necessary to delete Tardbp in the nervous system, including cortical neurons in the forebrain and motor neurons in the spinal cord. Preliminary studies indicate that Hb9-ErCre;TardbpF/F mice have embryonic lethality, precluding the analysis of adult phenotype in these mice. Likewise, we have begun to examine whether lack of TDP-43 in the forebrain of CamKII-Cre;TardbpF/F or CamKII-ErCre;TardbpF/F mice will lead to a neurodegenerative phenotype associated with memory impairment. Preliminary studies indicate that lack of TDP-43 in the forebrain leads to an age-dependent neurodegeneration in CamKII-Cre;TardbpF/F mice associated with behavioral deficits (Jeong and Wong, pers. comm.). Outcomes from these studies will have important implications regarding disease mechanisms and specifically address whether loss of TDP-43 function contributes to ALS or FTLD-TDP.
11. Current working models for TDP-43 loss- and gain-of function mechanisms
Based on insights gained from the TDP-43 transgenic and knockout studies over the past several years, we propose two working models. For loss-of-function mechanism, we envision that both cell autonomous and non-cell autonomous mechanisms could contribute to the disease by increasing the vulnerability of neuronal systems (Fig. 2). For example, our preliminary data showing that the lack of TDP-43 in forebrains of mice leads to an age-dependent brain atrophy support the view that loss of TDP-43 in CNS neurons could alter RNA metabolism to trigger neurodegeneration in a cell autonomous manner. In parallel, loss of TDP-43 in other cell types or organ systems could likewise contribute to disease in a non-cell autonomous fashion. Our preliminary finding that loss of TDP-43 in skeletal muscle down-regulates Tbc1d1, an outcome that would be predicted to induce hypermetabolism, could compromise neuronal function. Thus, we hypothesize that both cell and non-cell autonomous mechanisms involving loss of TDP-43 converge to increase the vulnerability of neurons in ALS and FTLD-TDP (Fig. 2).
Fig. 2.
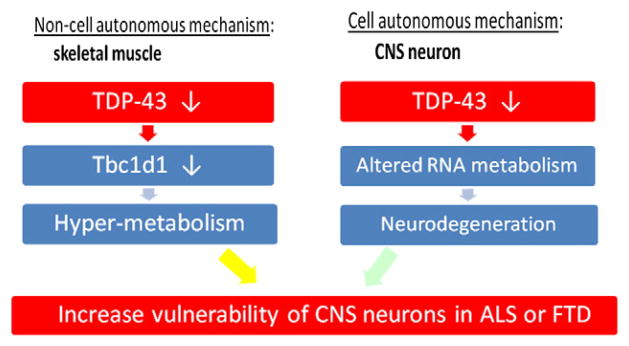
Working model for “loss-of-function” mechanism for ALS-FTD. It is assumed that nuclear inclusions of TDP-43 and its clearance are indicative of loss of TDP-43 function. Both non-cell (skeletal muscle) and cell autonomous (CNS neurons) mechanisms play roles to increase vulnerability of CNS neurons in ALS or FTD.
In contrast, for gain-of-function mechanism, we propose that upregulation of TDP-43 in motor neurons alters RNA metabolism via alternative splicing and RNA stability to increase risk in ALS (Fig. 3). Based on findings from TDP-43 transgenic mice, we suggest that the increase in the level of TDP-43 increases the vulnerability of motor neurons through the formation in the nucleus of TDP-43 inclusions and mislocalization of SMN–GEMs to alter RNA metabolism. Likewise, such upregulation of TDP-43 could also disrupt RNA splicing and stability to alter transport of mitochondria in the cytoplasm and limit their delivery to the terminals. Thus, we envision that factors that disrupt the autoregulation of TDP-43 could lead to its upregulation to increase the vulnerability of motor neurons through altered RNA metabolism in ALS (Fig. 3).
Fig. 3.
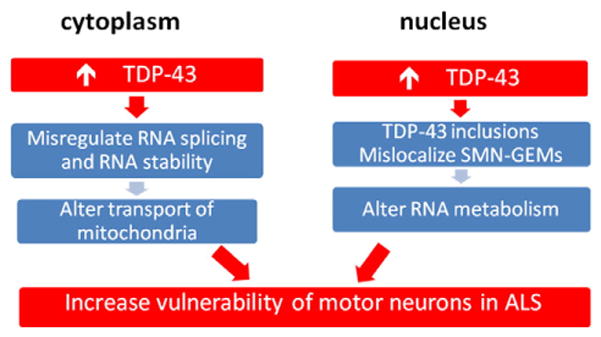
Working model for “gain-of-function” mechanism for ALS. We propose that upregulation of TDP-43 in motor neurons alters RNA metabolism via alternative splicing and RNA stability. This could occur in both the nuclear and cytoplasmic compartment of motor neurons to increase vulnerability of these cells in ALS.
12. Paradigm shift: can ALS be a disease of altered RNA metabolism/processing?
Over the past decade, many pathogenic mechanisms have been proposed for ALS, including altered axonal transport (Griffin et al., 1978; Puls et al., 2005; Williamson and Cleveland, 1999), mitochondrial abnormalities (Liu et al., 2004; Mattiazzi et al., 2002), excitoxicity (Howland et al., 2002; Rothstein, 2009), distal axonopathy (Fischer et al., 2004), disruption of the blood–brain barrier (Zhong et al., 2008), induction of ER stress (Kikuchi et al., 2006), inhibition of the proteasome (Cheroni et al., 2009; Kabashi et al., 2004; Urushitani et al., 2002), toxicity from secreted extracellular SOD1 (Urushitani et al., 2006; Zhao et al., 2010), and excessive production of superoxide from astrocytes and microglia (Harraz et al., 2008). However, the recent discoveries of mutations in TARDBP and FUS/TLS, genes that encode RNA binding proteins, linked to ALS offer the intriguing possibility that altered RNA metabolism or RNA processing may underlie and contribute to motor neuron degeneration (Lagier-Tourenne and Cleveland, 2009). Evidence to date support TDP-43 in regulating the physiology of motor neurons, including those that impact on the proper distributions of SMN in GEMs, nuclear structures that are involved in biosynthesis of snRNPs required for RNA splicing, in motor neurons. Together with results showing the requirement of TDP-43 for the formation of SMN-containing GEMs in motor neurons in our TDP-43 conditional knockout mouse model, our findings implicate a critical role of TDP-43 in regulating SMN/GEMs that may impact on RNA metabolism in motor neurons. Future studies will be necessary to establish the role of TDP-43 in regulating RNA metabolism/processing in motor neurons, and to clarify how ALS-linked TDP-43 mutants impact on these processes and lead to motor neuron degeneration. These efforts will have profound implications for our understanding of pathogenic mechanism of ALS as well as for identification of new therapeutic targets and strategies for the treatment of this devastating illness.
Acknowledgments
The authors would like to thank Xiu Shan, Susan Aja, Venette Nehus and Frances Davenport to their contributions to some of the original TDP-43 work cited in this review. Aspects of this work were supported by grants from the National Institute of Neurological Disorders and Stroke Grant R01 NS41438, the Muscular Dystrophy Association, The Robert Packard Center for ALS Research and The Johns Hopkins Neuropathology Gift Fund.
References
- Ayala YM, De Conti L, Avendano-Vazquez SE, et al. TDP-43 regulates its mRNA levels through a negative feedback loop. EMBO J. 2010;30:277–288. doi: 10.1038/emboj.2010.310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badea TC, Wang Y, Nathans J. A noninvasive genetic/ pharmacologic strategy for visualizing cell morphology and clonal relationships in the mouse. J Neurosci. 2003;23:2314–2322. doi: 10.1523/JNEUROSCI.23-06-02314.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battle DJ, et al. The SMN complex: an assembly machine for RNPs. Cold Spring Harb Symp Quant Biol. 2006;71:313–320. doi: 10.1101/sqb.2006.71.001. [DOI] [PubMed] [Google Scholar]
- Borchelt DR, et al. A vector for expressing foreign genes in the brains and hearts of transgenic mice. Genet Anal. 1996;13:159–163. doi: 10.1016/s1050-3862(96)00167-2. [DOI] [PubMed] [Google Scholar]
- Brook JD, McCurrach ME, Harley HG, et al. Molecular basis of myotonic dystrophy: expansion of a trinucleotide (CTG) repeat at the 3′ end of a transcript encoding a protein kinase family member. Cell. 1992;68:799–808. doi: 10.1016/0092-8674(92)90154-5. [DOI] [PubMed] [Google Scholar]
- Bruijn LI, Becher MW, Lee MK, et al. ALS-linked SOD1 mutant G85R mediates damage to astrocytes and promotes rapidly progressive disease with SOD1-containing inclusions. Neuron. 1997;18:327–338. doi: 10.1016/s0896-6273(00)80272-x. [DOI] [PubMed] [Google Scholar]
- Bruijn LI, Miller TM, Cleveland DW. Unraveling the mechanisms involved in motor neuron degeneration in ALS. Annu Rev Neurosci. 2004;27:723–749. doi: 10.1146/annurev.neuro.27.070203.144244. [DOI] [PubMed] [Google Scholar]
- Burghes AH, Beattie CE. Spinal muscular atrophy: why do low levels of survival motor neuron protein make motor neurons sick? Nat Rev Neurosci. 2009;10:597–609. doi: 10.1038/nrn2670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caroni P. Overexpression of growth-associated proteins in the neurons of adult transgenic mice. J Neurosci Methods. 1997;71:3–9. doi: 10.1016/s0165-0270(96)00121-5. [DOI] [PubMed] [Google Scholar]
- Chadt A, et al. Tbc1d1 mutation in lean mouse strain confers leanness and protects from diet-induced obesity. Nat Genet. 2008;40:1354–1359. doi: 10.1038/ng.244. [DOI] [PubMed] [Google Scholar]
- Chen YZ, Bennett CL, Huynh HM. DNA/RNA helicase gene mutations in a form of juvenile amyotrophic lateral sclerosis (ALS4) Am J Hum Genet. 2004;74:1128–1135. doi: 10.1086/421054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheroni C, et al. Functional alterations of the ubiquitin-proteasome system in motor neurons of a mouse model of familial amyotrophic lateral sclerosis. Hum Mol Genet. 2009;18:82–96. doi: 10.1093/hmg/ddn319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang PM, Ling J, Jeong YH, Price DL, Aja S, Wong PC. Deletion of Tardbp down-regulates Tbc1d1, a gene linked to obesity, and alters body fat metabolism. Proc Natl Acad Sci U S A. 2010;107:16320–16324. doi: 10.1073/pnas.1002176107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clement AM, Nguyen MD, Roberts EA, et al. Wild-type nonneuronal cells extend survival of SOD1 mutant motor neurons in ALS mice. Science. 2003;302:113–117. doi: 10.1126/science.1086071. [DOI] [PubMed] [Google Scholar]
- Cleveland JL. A new piece of the ALS puzzle. Nat Genet. 2003;34:357–358. doi: 10.1038/ng0803-357. [DOI] [PubMed] [Google Scholar]
- Cleveland DW, Rothstein JD. From Charcot to Lou Gehrig: deciphering selective motor neuron death in ALS. Nat Rev Neurosci. 2001;2:806–819. doi: 10.1038/35097565. [DOI] [PubMed] [Google Scholar]
- Dal Canto MC, Gurney ME. Development of central nervous system pathology in a murine transgenic model of human amyotrophic lateral sclerosis. Am J Pathol. 1994;145:1271–1280. [PMC free article] [PubMed] [Google Scholar]
- DeJesus-Hernandez M, Mackenzie IR, Boeve BF, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011;72:245–256. doi: 10.1016/j.neuron.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng HX, Chen W, Hong ST, et al. Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature. 2011;477:211–215. doi: 10.1038/nature10353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dundr M, et al. In vivo kinetics of Cajal body components. J Cell Biol. 2004;164:831–842. doi: 10.1083/jcb.200311121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elden AC, Kim HJ, Hart MP, et al. Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk of ALS. Nature. 2010;466:1069–1075. doi: 10.1038/nature09320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng G, et al. Imaging neuronal subsets in transgenic mice expressing multiple spectral variants of GFP. Neuron. 2000;28:41–51. doi: 10.1016/s0896-6273(00)00084-2. [DOI] [PubMed] [Google Scholar]
- Fischer LR, et al. Amyotrophic lateral sclerosis is a distal axonopathy: evidence in mice and man. Exp Neurol. 2004 Feb;185:232–240. doi: 10.1016/j.expneurol.2003.10.004. [DOI] [PubMed] [Google Scholar]
- Gall JG. Cajal bodies: the first 100 years. Annu Rev Cell Dev Biol. 2000;16:273–300. doi: 10.1146/annurev.cellbio.16.1.273. [DOI] [PubMed] [Google Scholar]
- Galloway JN, Nelson DL. Evidence for RNA-mediated toxicity in the fragile X-associated tremor/ataxia syndrome. Future Neurol. 2009;4:785. doi: 10.2217/fnl.09.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin JW, Hoffman PN, Clark AW, Carroll PT, Price DL. Slow axonal transport of neurofilament proteins: impairment by b, b′-iminodipropionitrile administration. Science. 1978;202:633–635. doi: 10.1126/science.81524. [DOI] [PubMed] [Google Scholar]
- Hadano S, Hand CK, Osuga H, et al. A gene encoding a putative GTPase regulator is mutated in familial amyotrophic lateral sclerosis 2. Nat Genet. 2001;29:166–173. doi: 10.1038/ng1001-166. [DOI] [PubMed] [Google Scholar]
- Harraz MM, et al. SOD1 mutations disrupt redox-sensitive Rac regulation of NADPH oxidase in a familial ALS model. J Clin Invest. 2008;118:659–670. doi: 10.1172/JCI34060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi S, McMahon AP. Efficient recombination in diverse tissues by a tamoxifen-inducible form of Cre: a tool for temporally regulated gene activation/inactivation in the mouse. Dev Biol. 2002;244:305–318. doi: 10.1006/dbio.2002.0597. [DOI] [PubMed] [Google Scholar]
- Henderson CE, Phillips HS, Pollock RA, et al. GDNF: a potent survival factor for motoneurons present in peripheral nerve and muscle. Science. 1994;266:1062–1064. doi: 10.1126/science.7973664. [DOI] [PubMed] [Google Scholar]
- Howland DS, et al. Focal loss of the glutamate transporter EAAT2 in a transgenic rat model of SOD1 mutant-mediated amyotrophic lateral sclerosis (ALS) Proc Natl Acad Sci U S A. 2002;99:1604–1609. doi: 10.1073/pnas.032539299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igaz LM, et al. Expression of TDP-43 C-terminal fragments in vitro recapitulates pathological features of TDP-43 proteinopathies. J Biol Chem. 2009;284:8516–8524. doi: 10.1074/jbc.M809462200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson JO, Mandrioli J, Benatar M, et al. Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron. 2010;68:857–864. doi: 10.1016/j.neuron.2010.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Julien JP. Amyotrophic lateral sclerosis: unfolding the toxicity of the misfolded. Cell. 2001;104:581–591. doi: 10.1016/s0092-8674(01)00244-6. [DOI] [PubMed] [Google Scholar]
- Kabashi E, Agar JN, Taylor DM, Minotti S, Durham HD. Focal dysfunction of the proteasome: a pathogenic factor in a mouse model of amyotrophic lateral sclerosis. J Neurochem. 2004;89:1325–1335. doi: 10.1111/j.1471-4159.2004.02453.x. [DOI] [PubMed] [Google Scholar]
- Kabashi E, et al. TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat Genet. 2008;40 (5):572–574. doi: 10.1038/ng.132. [DOI] [PubMed] [Google Scholar]
- Kikuchi H, et al. Spinal cord endoplasmic reticulum stress associated with a microsomal accumulation of mutant superoxide dismutase-1 in an ALS model. Proc Natl Acad Sci U S A. 2006;103:6025–6030. doi: 10.1073/pnas.0509227103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koliatsos VE, Clatterbuck RE, Winslow JW, Cayouette MH, Price DL. Evidence that brain-derived neurotrophic factor is a trophic factor for motor neurons in vivo. Neuron. 1993;10:359–367. doi: 10.1016/0896-6273(93)90326-m. [DOI] [PubMed] [Google Scholar]
- Kong L, et al. Impaired synaptic vesicle release and immaturity of neuromuscular junctions in spinal muscular atrophy mice. J Neurosci. 2009;29:842–851. doi: 10.1523/JNEUROSCI.4434-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraemer BC, et al. Loss of murine TDP-43 disrupts motor function and plays an essential role in embryogenesis. Acta Neuropathol. 2010;119:409–419. doi: 10.1007/s00401-010-0659-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriz J, Gowing G, Julien JP. Efficient three-drug cocktail for disease induced by mutant superoxide dismutase. Ann Neurol. 2003;53:429–436. doi: 10.1002/ana.10500. [DOI] [PubMed] [Google Scholar]
- Kwiatkowski TJ, Jr, et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science. 2009;323 (5918):1205–1208. doi: 10.1126/science.1166066. [DOI] [PubMed] [Google Scholar]
- Lagier-Tourenne C, Cleveland DW. Rethinking ALS: the FUS about TDP-43. Cell. 2009;136 (6):1001–1004. doi: 10.1016/j.cell.2009.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laird FM, et al. Motor neuron disease occurring in a mutant dynactin mouse model is characterized by defects in vesicular trafficking. J Neurosci. 2008;28:1997–2005. doi: 10.1523/JNEUROSCI.4231-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambrechts D, Storkebaum E, Morimoto M, et al. VEGF is a modifier of amyotrophic lateral sclerosis in mice and humans and protects motoneurons against ischemic death. Nat Genet. 2003;34:383–394. doi: 10.1038/ng1211. [DOI] [PubMed] [Google Scholar]
- Lefebvre S, et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell. 1995;80 (1):155–165. doi: 10.1016/0092-8674(95)90460-3. [DOI] [PubMed] [Google Scholar]
- Liquori CL, Ricker K, Moseley ML, et al. Myotonic dystrophy type 2 caused by a CCTG expansion in intron 1 of ZNF9. Science. 2001;293:864–867. doi: 10.1126/science.1062125. [DOI] [PubMed] [Google Scholar]
- Liu J, et al. Toxicity of familial ALS-linked SOD1 mutants from selective recruitment to spinal mitochondria. Neuron. 2004;43:5–17. doi: 10.1016/j.neuron.2004.06.016. [DOI] [PubMed] [Google Scholar]
- Mahadevan M, Tsilfidis C, Sabourin L, et al. Myotonic dystrophy mutation: an unstable CTG repeat in the 3′ untranslated region of the gene. Science. 1992;255:1253–1255. doi: 10.1126/science.1546325. [DOI] [PubMed] [Google Scholar]
- Maniatis T, Tasic B. Alternative pre-mRNA splicing and proteome expansion in metazoans. Nature. 2002;418:236–243. doi: 10.1038/418236a. [DOI] [PubMed] [Google Scholar]
- Maruyama H, Morino H, Ito H. Mutations of optineurin in amyotrophic lateral sclerosis. Nature. 2010;465:223–236. doi: 10.1038/nature08971. [DOI] [PubMed] [Google Scholar]
- Mattiazzi M, et al. Mutated human SOD1 causes dysfunction of oxidative phosphorylation in mitochondria of transgenic mice. J Biol Chem. 2002;277:29626–29633. doi: 10.1074/jbc.M203065200. [DOI] [PubMed] [Google Scholar]
- Misgeld T, Kerschensteiner M, Bareyre FM, et al. Imaging axonal transport of mitochondria in vivo. Nat Methods. 2007;4:559–561. doi: 10.1038/nmeth1055. [DOI] [PubMed] [Google Scholar]
- Moreira MC, Klur S, Watanabe M, et al. Senataxin, the ortholog of a yeast RNA helicase, is mutant in ataxia-ocular apraxia 2. Nat Genet. 2004;36:225–227. doi: 10.1038/ng1303. [DOI] [PubMed] [Google Scholar]
- Munch C, Sedlmeier R, Meyer T, et al. Point mutations of the p150 subunit of dynactin (DCTN1) gene in ALS. Neurology. 2004;63:724–726. doi: 10.1212/01.wnl.0000134608.83927.b1. [DOI] [PubMed] [Google Scholar]
- Neumann M, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314 (5796):130–133. doi: 10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- Oosthuyse B, Moons L, Storkebaum E, et al. Deletion of the hypoxia-response element in the vascular endothelial growth factor promotor causes motor neuron degeneration. Nat Genet. 2001;28:131–138. doi: 10.1038/88842. [DOI] [PubMed] [Google Scholar]
- Polymenidou M, Lagier-Tourenne C, Hutt KR, et al. Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nat Neurosci. 2011;14:459–468. doi: 10.1038/nn.2779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puls I, Jonnakuty C, LaMonte BH, et al. Mutant dynactin in motor neuron disease. Nat Genet. 2003;33:455–456. doi: 10.1038/ng1123. [DOI] [PubMed] [Google Scholar]
- Puls I, Oh SJ, Sumner CJ, et al. Distal spinal and bulbar muscular atrophy caused by dynactin mutation. Ann Neurol. 2005;57:687–694. doi: 10.1002/ana.20468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renton AE, Majounie E, Waite A, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. 2011;72:257–268. doi: 10.1016/j.neuron.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds N, Fantes PA, MacNeill SA. A key role for replication factor C in DNA replication checkpoint function in fission yeast. Nucleic Acids Res. 1999;27:462–469. doi: 10.1093/nar/27.2.462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen DR, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- Rothstein JD. Of mice and men: reconciling preclinical ALS mouse studies and human clinical trials. Ann Neurol. 2003;53:423–426. doi: 10.1002/ana.10561. [DOI] [PubMed] [Google Scholar]
- Rothstein JD. Current hypotheses for the underlying biology of amyotrophic lateral sclerosis. Ann Neurol. 2009;65 (Suppl 1):S3–9. doi: 10.1002/ana.21543. [DOI] [PubMed] [Google Scholar]
- Rothstein JD, Patel S, Regan MR, et al. Beta-lactam antibiotics offer neuroprotection by increasing glutamate transporter expression. Nature. 2005;433:73–77. doi: 10.1038/nature03180. [DOI] [PubMed] [Google Scholar]
- Sakai K, Miyazaki J. A transgenic mouse line that retains Cre recombinase activity in mature oocytes irrespective of the cre transgene transmission. Biochem Biophys Res Commun. 1997;237:318–324. doi: 10.1006/bbrc.1997.7111. [DOI] [PubMed] [Google Scholar]
- Sephton CF, et al. TDP-43 is a developmentally-regulated protein essential for early embryonic development. J Biol Chem. 2010;285:6826–6834. doi: 10.1074/jbc.M109.061846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan X, Chiang PM, Price DL, Wong PC. Altered distributions of Gemini of coiled bodies and mitochondria in motor neurons of TDP-43 transgenic mice. Proc Natl Acad Sci U S A. 2010;107:16325–16330. doi: 10.1073/pnas.1003459107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sleeman JE, Trinkle-Mulcahy L, Prescott AR, Ogg SC, Lamond AI. Cajal body proteins SMN and Coilin show differential dynamic behaviour in vivo. J Cell Sci. 2003;116 (Pt 10):2039–2050. doi: 10.1242/jcs.00400. [DOI] [PubMed] [Google Scholar]
- Spector DL. Nuclear domains. J Cell Sci. 2001;114 (Pt 16):2891–2893. doi: 10.1242/jcs.114.16.2891. [DOI] [PubMed] [Google Scholar]
- Sreedharan J, et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science. 2008;319 (5870):1668–1672. doi: 10.1126/science.1154584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stallings NR, Puttaparthi K, Luther CM, et al. Progressive motor weakness in transgenic mice expressing human TDP-43. Neurobiol Dis. 2010;40:404–414. doi: 10.1016/j.nbd.2010.06.017. [DOI] [PubMed] [Google Scholar]
- Subramaniam JR, Lyons WE, Liu J, et al. Mutant SOD1 causes motor neuron disease independent of copper chaperone-mediated copper loading. Nat Neurosci. 2002;5:301–307. doi: 10.1038/nn823. [DOI] [PubMed] [Google Scholar]
- Swarup V, et al. Pathological hallmarks of amyotrophic lateral sclerosis/frontotemporal lobar degeneration in transgenic mice produced with TDP-43 genomic fragments. Brain. 2011;134:2610–2626. doi: 10.1093/brain/awr159. [DOI] [PubMed] [Google Scholar]
- Tassone F, Iwahashi C, Hagerman PJ. FMR1 RNA within the intranuclear inclusions of fragile X-associated tremor/ ataxia syndrome (FXTAS) RNA Biol. 2004;1:103–105. doi: 10.4161/rna.1.2.1035. [DOI] [PubMed] [Google Scholar]
- Tsai KJ, et al. Elevated expression of TDP-43 in the forebrain of mice is sufficient to cause neurological and pathological phenotypes mimicking FTLD-U. J Exp Med. 2010;207:1661–1673. doi: 10.1084/jem.20092164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urushitani M, Kurisu J, Tsukita K, Takahashi R. Proteasomal inhibition by misfolded mutant superoxide dismutase 1 induces selective motor neuron death in familial amyotrophic lateral sclerosis. J Neurochem. 2002;83:1030–1042. doi: 10.1046/j.1471-4159.2002.01211.x. [DOI] [PubMed] [Google Scholar]
- Urushitani M, et al. Chromogranin-mediated secretion of mutant superoxide dismutase proteins linked to amyotrophic lateral sclerosis. Nat Neurosci. 2006;9:108–118. doi: 10.1038/nn1603. [DOI] [PubMed] [Google Scholar]
- Vance C, et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science. 2009;323 (5918):1208–1211. doi: 10.1126/science.1165942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidal M, Morris R, Grosveld F, Spanopoulou E. Tissue-specific control elements of the Thy-1 gene. EMBO J. 1990;9:833–840. doi: 10.1002/j.1460-2075.1990.tb08180.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang IF, Reddy NM, Shen CK. Higher order arrangement of the eukaryotic nuclear bodies. Proc Natl Acad Sci U S A. 2002;99:13583–13588. doi: 10.1073/pnas.212483099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wegorzewska I, Baloh RH. TDP-43-based animal models of neurodegeneration: new insights into ALS pathology and pathophysiology. Neurodegener Dis. 2010;8:262–274. doi: 10.1159/000321547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wegorzewska I, Bell S, Cairns NJ, et al. TDP-43 mutant transgenic mice develop features of ALS and frontotemporal lobar degeneration. Proc Natl Acad Sci U S A. 2009;106:18809–18814. doi: 10.1073/pnas.0908767106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson TL, Cleveland DW. Slowing of axonal transport is a very early event in the toxicity of ALS-linked SOD1 mutants to motor neurons. Nat Neurosci. 1999;2:50–56. doi: 10.1038/4553. [DOI] [PubMed] [Google Scholar]
- Wils H, Kleinberger G, Janssens J, et al. TDP-43 transgenic mice develop spastic paralysis and neuronal inclusions characteristic of ALS and frontotemporal lobar degeneration. Proc Natl Acad Sci U S A. 2010;107:3858–3863. doi: 10.1073/pnas.0912417107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong PC, et al. An adverse property of a familial ALS-linked SOD1 mutation causes motor neuron disease characterized by vacuolar degeneration of mitochondria. Neuron. 1995;14:1105–1116. doi: 10.1016/0896-6273(95)90259-7. [DOI] [PubMed] [Google Scholar]
- Wong PC, Cai H, Borchelt DR, Price DL. Genetically engineered mouse models of neurodegenerative diseases. Nat Neurosci. 2002;5:633–639. doi: 10.1038/nn0702-633. [DOI] [PubMed] [Google Scholar]
- Wu LS, et al. TDP-43, a neuro-pathosignature factor, is essential for early mouse embryogenesis. Genesis. 2009;48:56–62. doi: 10.1002/dvg.20584. [DOI] [PubMed] [Google Scholar]
- Xu YF, Gendron TF, Zhang YJ, et al. Wild-type human TDP-43 expression causes TDP-43 phosphorylation, mitochondrial aggregation, motor deficits, and early mortality in transgenic mice. J Neurosci. 2010;30:10851–10859. doi: 10.1523/JNEUROSCI.1630-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu YF, Zhang YJ, Lin WL, et al. Expression of mutant TDP-43 induces neuronal dysfunction in transgenic mice. Mol Neurodegener. 2011;6:73. doi: 10.1186/1750-1326-6-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Hentati A, Deng HX, et al. The gene encoding alsin, a protein with three guanine-nucleotide exchange factor domains, is mutated in a form of recessive amyotrophic lateral sclerosis. Nat Genet. 2001;29:160–165. doi: 10.1038/ng1001-160. [DOI] [PubMed] [Google Scholar]
- Zhang Z, et al. SMN deficiency causes tissue-specific perturbations in the repertoire of snRNAs and widespread defects in splicing. Cell. 2008;133 (4):585–600. doi: 10.1016/j.cell.2008.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao W, et al. Extracellular mutant SOD1 induces microglial-mediated motoneuron injury. Glia. 2010;58:231–243. doi: 10.1002/glia.20919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong Z, et al. ALS-causing SOD1 mutants generate vascular changes prior to motor neuron degeneration. Nat Neurosci. 2008;11:420–422. doi: 10.1038/nn2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H, et al. Transgenic rat model of neurodegeneration caused by mutation in the TDP gene. PLoS Genet. 2010;6 (3):e1000887. doi: 10.1371/journal.pgen.1000887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu S, Stavrovskaya IG, Drozda M. Minocycline inhibits cytochrome c release and delays progression of amyotrophic lateral sclerosis in mice. Nature. 2002;417:74–78. doi: 10.1038/417074a. [DOI] [PubMed] [Google Scholar]