

Abstract
Local delivery systems that provide sustained, high concentrations of antitumor cytokines in the tumor microenvironment while minimizing systemic dissemination are needed to realize the potential of cytokine-based immunotherapies. Recently, co-formulations of cytokines with chitosan solutions have been shown to increase local cytokine retention and bioactivity. In particular, intratumoral (i.t.) injections of chitosan/IL-12 can eliminate established tumors and generate tumor-specific immune responses. In the present study, we explored the mechanisms by which chitosan potentiated IL-12’s antitumor activity. The location of chitosan/IL-12 injection was found to be critical for optimal cytokine delivery. I.t. injections eliminated 9 of 10 MC38 adenocarcinomas while contralateral and peritumoral injections delayed tumor growth but could not eliminate tumors. Microdosing studies demonstrated that IL-12 depots, simulated through daily i.t. injections with IL-12 alone, were not as effective as weekly i.t. chitosan/IL-12. 50–75% of mice receiving daily IL-12 microdoses and 87.5% of mice receiving weekly chitosan/IL-12 were cured of MC38 tumors. Chitosan was found to increase IL-12-mediated leukocytic expansion in tumors and tumor-draining lymph nodes (TDLNs) by 40% and 100%, respectively. Immunophenotyping studies demonstrated that chitosan co-formulation amplified IL-12-induced increases in important effector populations, such as CD8+IFN-γ+ and NKT cells, in tumors and dendritic cell populations in TDLNs. Remarkable increases in Gr-1+CD11b+ tumor infiltrates were also observed in mice receiving chitosan or chitosan/IL-12. This population does not appear be suppressive and may facilitate the local antitumor response. Presented data suggest that chitosan-mediated depot formation and enhanced local cytokine retention is significantly, but not entirely, responsible for increased cytokine bioactivity.
Keywords: chitosan, paracrine delivery, interleukin-12, intratumoral, immunotherapy
1. Introduction
Since the discovery of ‘endogenous pyrogen,’ now known as IL-1, in 1953 [1], the use of exogenous cytokines to treat malignant neoplasms has been well studied and heavily pursued. However, only 2 of 40+ identified cytokines, IFN-α and IL-2, are approved as single agent immunotherapies for a limited number of indications. IFN-α therapy yields an 80% overall response rate with 10% complete responses in hairy cell leukemia [2], 40% objective response rate in AIDS-related Kaposi’s sarcoma [3, 4], and 10–20% complete response rate in chronic myelogenous leukemia [5, 6]. IL-2 therapy yields 10–20% overall response rates with about 5% complete responses in both metastatic renal cell carcinoma [7–9] and metastatic melanoma [8, 10, 11].
Both IFN-α and IL-2 are administered as systemic injections and cause significant adverse events. In particular, systemic IL-2 often requires intensive care due to grade 3 and 4 adverse events including fever, transaminase elevation, hypotension and edema. Nearly all clinical trials, both past and present, evaluating cytokine monotherapies utilize systemic (i.v., s.c. or i.m) injections. However, cytokines function primarily through paracrine and autocrine mechanisms and thus are rarely measurable in the circulation of healthy individuals.
We and others have noted that cytokine-based immunotherapies would be more effective and less toxic if delivered locally and maintained in a tissue of interest, i.e. the tumor. Furthermore, a growing mountain of evidence demonstrates that locally administered cytokines can generate adaptive immunological memory capable of controlling metastasis and preventing recurrence [12–14]. This “local-to-systemic” antitumor immunity encourages re-evaluation of systemic cytokine delivery and justifies the development of localized delivery strategies capable of maximizing cytokine delivery to the tumor microenvironment while minimizing toxicities associated with systemic dissemination of potent, pro-inflammatory cytokines.
Here, we continue our investigation of simple co-formulations of chitosan solution with recombinant cytokines for local administration. Chitosan is a nontoxic (LD50 > 16 g/kg) [15], biodegradable, natural polysaccharide derived from the exoskeletons of crustaceans. Chitosan is a widely used biomaterial with an established safety profile in humans. It is used as a pharmaceutical excipient [16], a weight-loss supplement, an experimental mucosal adjuvant [17] and in an FDA-approved hemostatic dressing [18].
Our previous studies have demonstrated that simple, viscous chitosan solutions are able to maintain high concentrations of co-formulated recombinant cytokines and/or protein antigens following intratumoral (i.t.) or s.c. administration [19–22]. In particular, i.t. injections of co-formulated chitosan and recombinant IL-12 (chitosan/IL-12) were found to eliminate flank MC38 and Panc02 tumors [22]. The ability of chitosan to enhance the retention of IL-12 in the tumor microenvironment was thought to be primarily responsible for the increased antitumor activity. However, the importance of injecting the depot formulation directly into the tumor was not explored. It is possible that sustained systemic release of IL-12 from a distal site, similar to gene-based IL-12 delivery strategies [23–25], may have produced comparable results. In addition, although chitosan alone exhibited no antitumor activity [21, 22], we did not explicitly rule out the possibility that chitosan modulates the immunologic potential of IL-12 through manipulation of local immune cell phenotypes.
The goal of the present study was to understand the mechanisms by which chitosan potentiates cytokine bioactivity. Does chitosan simply amplify cytokine bioactivity through enhanced retention or does chitosan induce an immune response capable of synergizing with cytokine function? To explore the effect of local versus distal IL-12 depots, we characterized the antitumor activities of chitosan/IL-12 injected adjacent to the tumor, contralateral to the tumor or i.t. To isolate the effect of enhanced IL-12 retention in the tumor on antitumor efficacy, we simulated sustained release of IL-12 from a chitosan-based depot through a series of daily injections with fractionated doses of IL-12 alone. Specifically, weekly i.t. injections of chitosan/IL-12 were replaced by five daily injections with equal or decreasing microdoses of IL-12. Finally, to determine if chitosan potentiates the immunologic activity of IL-12, we performed a phenotypic analysis of immune cell subsets in spleens, tumors and tumor draining lymph nodes (TDLNs) during immunotherapy.
2. Materials and Methods
2.1. Laboratory Animals
Female C57BL/6J mice, 8- to 12-weeks old, were obtained from the Jackson Laboratory (Bar Harbor, ME). All mice were housed and maintained under pathogen-free conditions in microisolator cages. All experimental procedures were approved by the Institutional Animal Care and Use Committee at the University of Arkansas. Animal care was in compliance with The Guide for Care and Use of Laboratory Animals (National Research Council).
2.2. Reagents
Dulbecco’s modified Eagle’s Medium (DMEM) was obtained from Hyclone Laboratories (Logan, Utah). Fetal bovine serum (FBS) was purchased from PAA Laboratories (Dartmouth, MA). Trypsin-versene/EDTA, penicillin, streptomycin, nonessential amino acid solution (NEAA), sodium pyruvate, HEPES and ACK lysis buffer were purchased from Lonza (Walkersville, MD). Dulbecco’s phosphate-buffered saline (DPBS) and Hanks’ balanced salt solution (HBSS) were purchased from Mediatech (Manassas, VA). Histopaque 1077 was obtained from MP Biomedicals (Solon, OH, USA). Cell aggregate dissociation buffer Accumax was purchased from Global Cell Solutions (Charlottesville, VA). Glucose, ethylenediaminetetraacetic acid (EDTA), and bovine serum albumin (BSA) were purchased from Amresco (Solon, OH). Sodium azide was purchased from Alfa Aesar (Ward Hill, MA). FACS buffer was comprised of PBS supplemented with 5mM EDTA, 0.2% BSA, and 0.2% sodium azide. Chitosan glutamate, 200–600 kDa, 75% to 90% deacetylated (Protosan G 213) was purchased from NovaMatrix (Sandvika, Norway) and reconstituted in DPBS. Chitosan glutamate was reconstituted in DPBS to a final concentration of 1.5% (w/v) and henceforth referred to as chitosan solution. Recombinant murine IL-12 was purchased from PeproTech (Rocky Hill, NJ).
Fluorescence-labeled antibodies for flow cytometry were purchased from BD Biosciences (San Diego, CA) and include: anti-CD25 (clone: 7D4), anti-CD54 (clone: 3E2), anti-CD19 (clone: 1D3), anti-FoxP3 (clone: MF23), anti-I-Ab (clone: AF6-120.1), anti-CD3 (clone: 500A2), anti-IFN-γ (clone: XMG1.2), anti-CD11b (clone: M1/70), anti-CD25 (clone: PC61), anti-NK1.1 (clone: PK136), anti-CD45 (clone: 30-F11), anti-CD4 (clone: RM4-5), anti- CD11c (clone: HL3), anti-CD8 (clone: 53-6.7), and anti-Gr-1 (clone: RB6-8C5).
2.3. Cells
The murine colon carcinoma cell line, MC38, was generously provided by Dr. Jeffrey Schlom (Laboratory of Tumor Immunology and Biology, National Cancer Institute, Bethesda, MD). Cells were maintained in DMEM supplemented with 10% FBS, 1% NEAA, 1% HEPES, 1% L-glutamine, penicillin (100 IU/ml), and streptomycin (100μg/ml).
2.4. Immunotherapy studies
C57BL/6J mice were inoculated s.c. in the shaved flank with 3 × 105 MC38 cells. For antitumor studies investigating the effect of injection location, tumor-bearing mice (n=10 per group) received injections of 1μg IL-12 in chitosan solution (chitosan/IL-12) i.t., adjacent to the tumor or contralaterally on days 7, 14, 21 and 28. Mice receiving DPBS served as controls. For antitumor studies investigating the depot effect of chitosan, tumor-bearing mice (n=8 per group) were treated i.t. with chitosan/IL-12 on days 7, 14, 21 and 28 or with equal microdoses of IL-12 (0.2μg) alone on days 7–11, 14–18, 21–25 and 28–32. A third cohort of mice received decreasing doses of IL-12 alone to approximate the amount of IL-12 in the tumor following a single injection with chitosan/IL-12 as shown in our previous study [22]. Specifically, mice received 1μg, 0.25μg, 0.12μg, 0.08μg and 0.05μg on days 7, 8, 9, 10 and 11, respectively. The same dose-schedule was repeated starting on days 14, 21 and 28. Mice receiving DPBS served as controls. Tumor volumes were calculated using the modified ellipsoidal formula: tumor volume = ½ × length × width2.
2.4. Immunophenotyping studies
C57BL/6J mice bearing established s.c. tumors were treated with DPBS, chitosan solution, 1μg IL-12 or chitosan/IL-12(1μg) on days 9 and 14 after tumor implantation. On day 17, tumors, tumor draining lymph nodes (TDLNs), i.e. the inguinal lymph node ipsilateral to the tumor, and spleens were harvested. Spleens and lymph nodes were mechanically disrupted with a syringe plunger and passed through a 70μm nylon mesh strainer (BD Biosciences; Bedford, MA). Erythrocytes were lysed with ACK lysis buffer. Tumors were minced into 2 mm cubes and digested in Accumax at room temperature on an orbital shaker for 3 hours. Viable cells were then collected on a histopaque gradient. Viable leukocytes from all tissues were quantified under trypan blue exclusion with an automated cell counter (Cellometer Auto T4; Nexcelom Bioscience; Lawrence, MA).
For flow cytometry studies, cells were washed twice with cold FACS buffer. FcγII and FcγIII receptors on leukocytes were blocked via incubation with 1 μg purified anti-mouse CD16/CD32 (clone: 2.4G2) (BD Biosciences; San Jose, CA) per 1 × 106 cells for 15 min on ice. Cell surface markers were stained with 1μg fluorescence-labeled antibodies per 1 × 106 cells for 30 min on ice. A subset of cells were permeabilized with FACS buffer with 0.1% saponin and stained with fluorescence-labeled antibodies against intracellular markers for another 30 minutes on ice. Cells were then washed twice with cold FACS buffer prior to fixation in 1% paraformaldehyde, 2% glucose, and 5mM sodium azide in PBS. Samples were rinsed twice and analyzed on a BD FACSCanto II (BD Biosciences; San Jose, CA) within 48 hours. Data analyses were performed using FlowJo software v7.6.5 (Tree Star, Ashland, OR).
2.5. Statistical Analysis
Differences in overall survival were analyzed using the logrank test. Differences in mean percentages of immune cell subsets were analyzed using Student’s t-test with unpaired samples. P-values and hazard ratios were computed using GraphPad Prism 5.0 (GraphPad Software, Inc; La Jolla, CA).
3. Results
3.1. Effect of chitosan/IL-12 injection location
To determine if the location of chitosan/IL-12 injection relative to tumor impacts antitumor activity, mice bearing established s.c. MC38 tumors received weekly doses of chitosan/IL-12 injected s.c. adjacent to the tumor, s.c. contralateral to the tumor or i.t. The growth of tumors in mice treated either adjacent or contralateral were delayed compared to DPBS-treated controls (Figure 1A). Adjacent injections were more effective than contralateral injections particularly at later timepoints. A survival analysis indicated that both adjacent (P<0.001 vs. DPBS) and contralateral (P<0.001 vs. DPBS) injections significantly increased overall survival (Figure 1B). Nevertheless, all mice treated with chitosan/IL-12 outside of the tumor developed progressive disease and had to be euthanized within 45 days. In contrast, 9 of 10 mice receiving i.t. injections of chitosan/IL-12 experienced complete tumor regression (Figure 1A). These mice remained tumor-free beyond 90 days (Figure 1B).
Fig. 1.
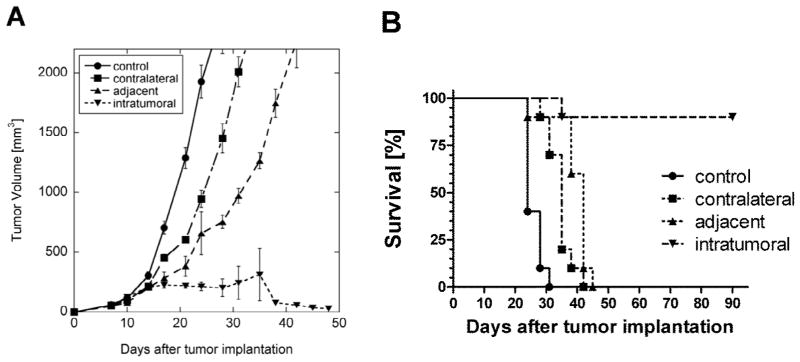
Effect of injection location on antitumor activity. A, tumor growth, and B, survival curves from C57BL/6J mice (n=10 per group) bearing MC38 flank tumors injected on days 7, 14, 21 and 28 with chitosan/IL-12(1μg) s.c. adjacent to the tumor, s.c. contralateral to the tumor or i.t. Control mice were treated i.t. with DPBS. Tumor volume data are presented as mean ± standard deviation.
3.2. Simulating chitosan/IL-12 depot by microdosing
Our previous data demonstrated that chitosan solutions facilitated retention of co-formulated IL-12 for at least 5–6 days [22]. To explore the effect of enhanced retention on antitumor activity, an IL-12 depot was simulated through a series of daily i.t. injections. One cohort of mice received fractionated doses of 0.2μg IL-12 alone while a second cohort received decreasing daily doses of 1μg, 0.25μg, 0.12μg, 0.08μg and 0.05μg IL-12 alone. Both depot simulations were administered for 5 consecutive days beginning on day 7, 14, 21 and 28 after tumor implantation. The fractionated doses simulated an even, sustained delivery of IL-12 to the tumor microenvironment. The intention of the decreasing dose schedule was to approximate the amount IL-12 remaining in the tumor following chitosan/IL-12 i.t. injection as determined previously [22]. Individual tumor growth curves revealed that 4/8 mice receiving the fractionated doses experienced complete tumor regression (Figure 2B) and long-term, tumor-free survival for at least 90 days (Figure 2E). The treatment regimen with decreasing doses led to tumor regression and long-term, tumor-free survival in 6/8 mice (Figures 2C,E). It should be noted that the total amount of IL-12 administered during the decreasing dose treatment was 50% greater than the fractionated dose schedule.
Fig. 2.

Intratumoral IL-12 depot simulated through microdosing. Individual MC38 tumor growth curves from C57BL/6J mice (n=8 per group) treated i.t. with: A, weekly DPBS (control); B, daily fractionated doses of 0.2ug IL-12 alone; C, decreasing daily doses of 1μg, 0.25μg, 0.12μg, 0.08μg and 0.05μg IL-12 alone; or D, weekly chitosan/IL-12 (1μg). E, survival curves for the same mice. Weekly treatments were given on days 7, 14, 21 and 28. Daily treatments were given on days 7–11, 14–18, 21–25 and 28–32.
Similar to the injection location studies (Figure 1), i.t. chitosan/IL-12(1μg) eliminated tumors in 7/8 mice (Figures 2D,E). These mice remained tumor-free for at least 90 days (Figure 2E). There was no statistical difference in survival among the three IL-12 treated groups (P=0.26). However, a hazard ratio assessment indicated that mice treated i.t. with even fractionated doses or decreasing doses were 2.0 times and 4.4 times more likely to die, respectively, than mice treated with chitosan/IL-12.
3.3. Effect of i.t. treatment on leukocytic expansion
Figures 1 and 2 demonstrate that chitosan was effective in augmenting the antitumor activity of IL-12 through sustained delivery to the tumor microenvironment. In an attempt to uncover additional antitumor mechanisms of chitosan beyond enhanced IL-12 retention, we looked for differences in immune cell populations in the spleens, tumor draining lymph nodes and tumors of mice treated with DPBS, chitosan alone, IL-12 alone or chitosan/IL-12. Our current and previous data indicated that differences in tumor growth among these treatment groups start to become apparent 2–3 days after the second weekly injection. Therefore, immunophenotyping studies were focused 3 days after the second injection, i.e. day 17, when differences in leukocyte populations were expected to be greatest.
An assessment of the total number of leukocytes, revealed no differences between treatment groups in the spleen (Figure 3). In the tumor, chitosan alone injections resulted in an increase to 12.6±2.4 × 106 leukocytes per tumor from 9.1±2.0 × 106 leukocytes per tumor in the control (DPBS) group (P=0.04). A similar but not statistically significant increase was observed following IL-12 alone injections which resulted in 12.3±3.1 × 106 leukocytes per tumor (P=0.09 vs. DPBS). When combined, chitosan/IL-12 injections appeared to have an additive effect on leukocytic infiltrates. The resulting 17.2±1.0 × 106 leukocytes per tumor were significantly greater than tumors treated with either chitosan (P=0.004) or IL-12 alone (P=0.01).
Fig. 3.

Effect of treatment on leukocyte expansion in different tissues. C57BL/6J mice (n=6 per group) bearing MC38 flank tumors received i.t. injections of DPBS, chitosan alone, IL-12(1μg) alone or chitosan/IL-12(1μg) on days 7 and 14. On day 17, spleens, tumors and tumor draining lymph nodes (TDLNs) were harvested and viable leukocytes enumerated via automated cell counting. Data for individual mice as well as mean ± standard deviation are presented. *P<0.05 vs. DPBS; ** P<0.05 vs. chitosan; *** P<0.05 vs. IL-12.
In the TDLN, leukocyte increases were more pronounced. The total number of leukocytes in the TDLN of tumor-bearing mice treated with DPBS, chitosan alone, IL-12 alone or chitosan/IL-12 were 2.3±0.7 × 106, 5.4±1.6 × 106, 9.9±2.7 × 106 and 19.8±4.8 × 106, respectively. Unlike tumor infiltrates, the number of leukocytes in the TDLN was significantly different following chitosan alone and IL-12 alone treatments (P=0.02). Similar to tumor infiltrates, chitosan/IL-12 injections elicited significant increases vs. either chitosan (P=0.005) or IL-12 alone (P=0.001).
3.4. Effect of i.t. treatment on immune cell phenotype
In the spleen, phenotypic characterization of leukocyte populations revealed no significant differences in the percentages of CD4+, CD8+, CD19+, CD4+CD25+, NK1.1+, Gr-1+, CD11b+ or CD11c+ cells among the different treatment groups (data not shown).
In the tumor, we observed increases in percentages of four potential effector populations, CD4+, CD8+, CD8+IFN-γ+ and NK1.1+CD3+ cells (Figure 4A), due to i.t. injections with chitosan alone, IL-12 alone and chitosan/IL-12. In particular, chitosan/IL-12 treatment produced statistically significant increases when compared to DPBS-treated controls. The percentage of CD4+ cells in the tumor increased from 1.0±1.0% after DPBS to 6.2±2.6% after chitosan/IL-12 (P=0.003), while CD8+ cells in the tumor increased from 3.2±3.5% after DPBS to 21.5±12.5% after chitosan/IL-12 (P=0.01), CD8+IFN-γ+ cells increased from 0.4±0.2% after DPBS to 3.8±2.5% after chitosan/IL-12 (P=0.02) and NK1.1+CD3+ cells increased from 0.5±0.3% after DPBS to 5.5±4.3% after chitosan/IL-12 (P=0.03). Increases in the same effector populations following chitosan alone and IL-12 alone treatments were documented. However, only IL-12 alone induced statistically significant changes in percentages of CD4+ and CD8+IFN-γ+ cells (P=0.003 and P=0.007 vs. DPBS, respectively).
Fig. 4.

Effect of treatment on leukocyte subset changes. C57BL/6J mice (n=6 per group) bearing MC38 flank tumors received i.t. injections of DPBS, chitosan alone, IL-12(1μg) alone or chitosan/IL-12(1μg) on days 7 and 14. On day 17, A, tumors, and B, TDLNs were harvested and viable leukocytes stained with fluorescence-labeled antibodies for subsequent flow cytometry analysis. Data are presented as mean ± standard deviation. *P<0.05 vs. DPBS; ** P<0.05 vs. chitosan; *** P<0.05 vs. IL-12.
In the TDLN, CD8+IFN-γ+ cells increased from 0.08±0.01% after DPBS to 1.0±0.5% after chitosan/IL-12 (Figure 4B). In contrast to tumor infiltrates, there were no significant differences in CD8+ or NK1.1+CD3+ cells following treatment (data not shown). However, the percentages of total dendritic cells (CD11c+I-Ab+) increased in the TDLN from 1.9±0.4% after DPBS to 5.9±0.5% following chitosan/IL-12 injections (P=0.001) while mature dendritic cells (CD11c+CD54+) increased from 1.2±0.1% after DPBS to 3.4±1.0% following chitosan/IL-12 injections (P=0.001). Treatment with chitosan alone was found to increase the percentage of total dendritic cells (P=0.0003 vs. DPBS), but not the percentage of mature dendritic cells (Figure 4B). IL-12 alone, on the other hand, increased the percentages of both total and mature dendritic cell subsets (P<0.0001 and P=0.01, respectively). The percentage of CD4+ cells decreased from 35.2±5.4% after DPBS to 23.3±5.6%, 18.8±2.4% and 19.6±2.6% following chitosan alone, IL-12 alone and chitosan/IL-12 injections, respectively. All CD4+ decreases were significant vs. DPBS (P<0.014).
We unexpectedly observed a large increase in Gr-1+CD11b+ cells in the tumor but not in spleens or TDLNs. Representative dot plots revealed strong double-positive populations in both chitosan and chitosan/IL-12 treated tumors (Figure 5A). The percentage of Gr-1+CD11b+ cells increased 3.9-fold in chitosan-treated tumors and 5.1-fold in chitosan/IL-12-treated tumors (Figure 5B). IL-12 alone treatment had no effect on this population.
Fig. 5.

Gr-1+CD11b+ cells in tumors of mice receiving i.t. injections. MC38 tumors were processed as in Fig. 4. A, representative dot plots showing increases in Gr-1+CD11b+ cells in tumors treated with chitosan alone or chitosan/IL-12. B, percentages of Gr-1+CD11b+ cells for individual mice as well as mean ± standard deviation. *P<0.05 vs. DPBS; *** P<0.05 vs. IL-12.
4. Discussion
Our results indicate that injection site significantly influences antitumor activity of chitosan/IL-12. An injection of chitosan/IL-12 contralateral to the tumor functions as a systemic controlled release platform. In fact, we have previously shown that a s.c. injection of chitosan/IL-12 results in measurable levels of IL-12, as well as the downstream effector cytokine, IFN-γ, in circulation [22]. Furthermore, our tumor growth delay data with contralateral chitosan/IL-12 are consistent with other delays observed using vector-based IL-12 delivery systems which also provide sustained systemic IL-12 [23–25].
Placing the injection adjacent to the tumor resulted in better tumor control. This may be due to local increases in effector populations as observed in Figures 3 and 4. Therefore, peritumoral injections should be preferred over contralateral or distal injections due to their immunostimulatory influences on local draining lymph nodes and other immune cells in close proximity to the tumor. Nevertheless, for both contralateral and adjacent injections, it is clear that a sufficient amount of IL-12 is not available in the tumor microenvironment for complete tumor control.
In contrast, i.t. injections eliminated tumors in 9 of 10 (Figure 1B) and 7 of 8 mice (Figure 2D). Not only does the i.t. route maximize IL-12 delivery to the tumor, but the i.t. injection itself may help disrupt the highly immunosuppressive tumor microenvironment. The literature contains numerous examples of cancer vaccines and immunotherapies which are more effective when delivered i.t. [25, 26]. I.t. injections are capable of producing a number of beneficial mechanical changes to the tumor including: 1) tumor necrosis through damage to tumor microvessels; 2) enhanced immune infiltrate through both damaged vessels and the subsequent wound healing response; and 3) disruption of the highly pressurized and poorly vascularized tumor architecture. Furthermore, the local availability of IL-12 is important as it has been shown to directly reverse immunosuppressive or tumor-supporting elements such as TAMs [27], dysfunctional tumor-resident T cells [28, 29], and T suppressor cells [30–32].
Overcoming local immune suppression is critical to the success of cancer immunotherapies. It is suggested that the immunosuppressive tumor microenvironment limits the clinical responses of many cancer vaccines despite measurable adaptive responses in peripheral blood [33]. Our data support the notion that the i.t. route of administration is ideal for IL-12 and perhaps other antitumor cytokines.
Microdosing studies demonstrated that an IL-12 depot simulated through daily i.t. injections was nearly as effective in controlling tumors as weekly i.t. chitosan/IL-12. The evenly fractionated dose schedule, i.e. 0.2μg IL-12 per day, appeared to perform slightly worse than the decreasing dose schedule in controlling tumor growth (Figure 2). However, it is noted that the decreasing dose schedule delivers 50% more IL-12 than the evenly fractionated schedule. Nevertheless, either microdosing schedule was more effective than weekly i.t. injections of IL-12 alone which resulted in only 10% long-term survival [22]. These data underscore the importance of sustained, local IL-12 for effective antitumor immunotherapy.
While the survival percentages of mice treated with daily microdoses and chitosan/IL-12 were statistically indistinguishable via logrank analysis, a hazard ratio assessment revealed that mice treated with even fractionated doses and decreasing doses were two to four times more likely to die than mice treated with chitosan/IL-12. Another substantial benefit of chitosan/IL-12 is the weekly administration schedule. Daily i.t. injections are, in general, not clinically practical.
In both the injection location and microdosing studies, mice that experienced complete tumor regression remained tumor-free for the duration of the study. This is consistent with our previous experiences with IL-12-based immunotherapies. We have treated a range of transplantable tumor models, including MC38, CT26, Panc02, B16 and 4T1, with various permutations of chitosan and IL-12-based immunotherapies (unpublished observation). We have yet to observe primary tumor recurrence following complete regression and 40–100% of cured mice are completely protected from tumor-specific rechallenge [22]. This is also consistent with the preclinical IL-12 literature [25, 34, 35].
The finding that chitosan/IL-12 outperformed the simulated IL-12 depots (Figure 2) indicated a potential additional immunostimulatory role for chitosan that was investigated through phenotypic analyses of immune cell populations. In order to maximize the opportunity to discover differences in immune cells with treatment, spleens, tumors and TDLNs were harvested shortly after two weekly i.t. injections. Our previous studies [22], as well as Figure 1, indicated that treatment-related differences in tumor volumes become apparent after the second treatment.
As a rough approximation of immune stimulation, leukocytic expansion in both the tumor and TDLN were most pronounced with chitosan/IL-12 treatment (Figure 3). Chitosan alone and IL-12 alone also increased the number of leukocytes in tumors and TDLNs, albeit to a lesser extent. The finding that chitosan alone induced leukocyte expansion indicated that the polysaccharide had a stimulatory effect. Gross changes in leukocyte numbers were restricted to the local environment as none of the i.t. treatments affected splenocytes counts (Figure 3A).
While the TH1-polarizing and antitumor mechanisms of IL-12 are well documented [35, 36], the importance of immune effector populations during IL-12-mediated tumor regression is model-dependent. For example, IL-12-induced rejection of Sa1 ascites is driven by macrophages [37], NK cells control metastatic MADB106 and 4T1 mammary adenocarcinomas [38, 39], NK and NKT cells contribute to the control of B16 melanomas and RM-1 prostate carcinoma [40, 41] while CD8+ T cells are the primary effectors against Renca tumors [42] and Meth A sarcomas [43].
For the MC38 colon adenocarcinoma model, our previous studies demonstrated that CD8+ and NK cells, but not CD4+ cells, are required for chitosan/IL-12-mediated tumor regression [22]. As a result, we anticipated large percentage increases in CD8+ and NK cells but not CD4+ cells in the tumor following chitosan/IL-12 immunotherapy. Indeed IL-12-mediated increases in the percentages of CD8+ and CD8+IFN-γ+ cells in the tumor were amplified through co-formulation with chitosan. However, we observed no significant changes in the percentages of NK cells, which were identified as NK1.1+CD3− (data not shown). If one factors in the overall leukocyte expansion in the tumor (Figure 3), the total number of NK cells increased substantially with chitosan/IL-12 immunotherapy. The percentage of NK1.1+CD3+ (NKT) cells increased with chitosan/IL-12 treatment, however, it is not know if these cells were critical effectors since only NK cells were depleted with anti-asialo-GM1 in the previous study [22]. Percent CD4+ cells, which were not necessary for chitosan/IL-12-mediated elimination of MC38 tumors [22], were found to increase in tumors but decrease in TDLNs following i.t. chitosan/IL-12 (Figure 4). In the TDLN, the percentages of total and mature dendritic cells, which increased following IL-12 treatment, were further amplified by chitosan/IL-12.
Overall, results of the immunophenotyping studies implied that chitosan does not radically alter the distribution of immune effector cells but rather it amplifies IL-12-induced differences. It is not clear if these changes in distribution were mediated by the sustained presence of IL-12 in the tumor or if they were due to a unique property of chitosan. As shown in Figure 2, daily injections could have simulated sustained IL-12 in the tumor microenvironment, however, the potential of repeated injections to induce significant tumor damage and skew tumor infiltrates toward innate populations outweighed the benefit of such a study.
Perhaps the most intriguing finding of the phenotyping studies was the massive increase in Gr-1+CD11b+ cells in the tumor with chitosan alone or chitosan/IL-12 treatment. Recent literature indicates that significant populations of Gr-1+CD11b+ cells in tumor-bearing mice are most likely myeloid-derived suppressor cells (MDSCs) [44]. However, a number of immunostimulatory functions of Gr-1+CD11b+ have been described recently [45]. Nausch et al., found that Gr-1+CD11b+F4/80+ MDSCs isolated from tumor-bearing mice can activate NK cells [46]. Tomihara et al., recently demonstrated that immunostimulatory Gr-1+CD11b+ cells associated with ovarian carcinoma could cross-prime CTLs and elicit potent antitumor activity following adoptive transfer [47].
In our experiments, Gr-1+CD11b+ cells are not likely to be suppressive as nearly all chitosan/IL-12 treated tumors are eliminated. At the same time, these cells cannot control MC38 tumors by themselves, since all chitosan-treated tumors grew as if untreated [22]. Separately, we (unpublished observation) and others [48] have found that mice injected with chitosan develop neither antibodies against chitosan nor chitosan-specific lymphoproliferative responses. Therefore, the Gr-1+CD11b+ increases are not associated with an adaptive response against chitosan. Consequently, in our context, Gr-1+CD11b+ cells are likely to be associated with an innate immune response that has no direct effector function, but may potentiate the adaptive response elicited by IL-12. Ongoing studies are focused on isolation and functional characterization of these cells.
5. Conclusion
Co-formulations of chitosan were found to enhance the antitumor efficacy of intratumorally administered IL-12. Chitosan did not appear to act via a novel effector pathway, but rather it amplified the traditional antitumor activities of IL-12 by providing sustained, high levels of the cytokine in the tumor microenvironment. In particular, IL-12-mediated increases in effector populations infiltrating the tumor and antigen presenting populations in draining lymph nodes were significantly enhanced through chitosan co-formulation. This localized delivery approach may overcome previous clinical failures with IL-12 that were due in part to the inability of i.v.- or s.c.-administered IL-12 to achieve biologically relevant concentrations of IL-12 in the tumor microenvironment at the maximum tolerated dose in humans. In addition, chitosan may also provide immunostimulatory signals which enhance antigen presentation in the regional environment. Beyond IL-12, the ease of co-formulation of chitosan with soluble proteins, make this an attractive platform for local, sustained delivery of any number of cytokines. Such a local delivery strategy is necessary for cytokine-based immunotherapies to fulfill their potential in the clinical management of cancer.
Acknowledgments
This work was supported by the National Cancer Institute (K22CA131567 to D.A.Z.)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bennett IL, Jr, Beeson PB. Studies on the pathogenesis of fever. II. Characterization of fever-producing substances from polymorphonuclear leukocytes and from the fluid of sterile exudates. J Exp Med. 1953;98:493–508. doi: 10.1084/jem.98.5.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Golomb HM, Ratain MJ, Fefer A, Thompson J, Golde DW, Ozer H, et al. Randomized study of the duration of treatment with interferon alfa-2B in patients with hairy cell leukemia. J Natl Cancer Inst. 1988;80:369–373. doi: 10.1093/jnci/80.5.369. [DOI] [PubMed] [Google Scholar]
- 3.Groopman JE, Gottlieb MS, Goodman J, Mitsuyasu RT, Conant MA, Prince H, et al. Recombinant alpha-2 interferon therapy for Kaposi’s sarcoma associated with the acquired immunodeficiency syndrome. Ann Intern Med. 1984;100:671–676. doi: 10.7326/0003-4819-100-5-671. [DOI] [PubMed] [Google Scholar]
- 4.Real FX, Oettgen HF, Krown SE. Kaposi’s sarcoma and the acquired immunodeficiency syndrome: treatment with high and low doses of recombinant leukocyte A interferon. J Clin Oncol. 1986;4:544–551. doi: 10.1200/JCO.1986.4.4.544. [DOI] [PubMed] [Google Scholar]
- 5.Kantarjian HM, Smith TL, O’Brien S, Beran M, Pierce S, Talpaz M. Prolonged survival in chronic myelogenous leukemia after cytogenetic response to interferon-alpha therapy. The Leukemia Service. Ann Intern Med. 1995;122:254–261. doi: 10.7326/0003-4819-122-4-199502150-00003. [DOI] [PubMed] [Google Scholar]
- 6.Ozer H, George SL, Schiffer CA, Rao K, Rao PN, Wurster-Hill DH, et al. Prolonged subcutaneous administration of recombinant alpha 2b interferon in patients with previously untreated Philadelphia chromosome-positive chronic-phase chronic myelogenous leukemia: effect on remission duration and survival: Cancer and Leukemia Group B study 8583. Blood. 1993;82:2975–2984. [PubMed] [Google Scholar]
- 7.Rosenberg SA, Lotze MT, Muul LM, Chang AE, Avis FP, Leitman S, et al. A progress report on the treatment of 157 patients with advanced cancer using lymphokine-activated killer cells and interleukin-2 or high-dose interleukin-2 alone. N Engl J Med. 1987;316:889–897. doi: 10.1056/NEJM198704093161501. [DOI] [PubMed] [Google Scholar]
- 8.Rosenberg SA, Yang JC, Topalian SL, Schwartzentruber DJ, Weber JS, Parkinson DR, et al. Treatment of 283 consecutive patients with metastatic melanoma or renal cell cancer using high-dose bolus interleukin 2. JAMA. 1994;271:907–913. [PubMed] [Google Scholar]
- 9.Fyfe G, Fisher RI, Rosenberg SA, Sznol M, Parkinson DR, Louie AC. Results of treatment of 255 patients with metastatic renal cell carcinoma who received high-dose recombinant interleukin-2 therapy. J Clin Oncol. 1995;13:688–696. doi: 10.1200/JCO.1995.13.3.688. [DOI] [PubMed] [Google Scholar]
- 10.Atkins MB, Kunkel L, Sznol M, Rosenberg SA. High-dose recombinant interleukin-2 therapy in patients with metastatic melanoma: long-term survival update. Cancer J Sci Am. 2000;6 (Suppl 1):S11–14. [PubMed] [Google Scholar]
- 11.Atkins MB, Lotze MT, Dutcher JP, Fisher RI, Weiss G, Margolin K, et al. High-dose recombinant interleukin 2 therapy for patients with metastatic melanoma: analysis of 270 patients treated between 1985 and 1993. J Clin Oncol. 1999;17:2105–2116. doi: 10.1200/JCO.1999.17.7.2105. [DOI] [PubMed] [Google Scholar]
- 12.Brunda MJ, Luistro L, Rumennik L, Wright RB, Wigginton JM, Wiltrout RH, et al. Interleukin-12: murine models of a potent antitumor agent. Ann N Y Acad Sci. 1996;795:266–274. doi: 10.1111/j.1749-6632.1996.tb52676.x. [DOI] [PubMed] [Google Scholar]
- 13.Hill HC, Conway TF, Jr, Sabel MS, Jong YS, Mathiowitz E, Bankert RB, et al. Cancer immunotherapy with interleukin 12 and granulocyte-macrophage colony-stimulating factor-encapsulated microspheres: coinduction of innate and adaptive antitumor immunity and cure of disseminated disease. Cancer Res. 2002;62:7254–7263. [PubMed] [Google Scholar]
- 14.Egilmez NK, Kilinc MO, Gu T, Conway TF. Controlled-release particulate cytokine adjuvants for cancer therapy. Endocr Metab Immune Disord Drug Targets. 2007;7:266–270. doi: 10.2174/187153007782794335. [DOI] [PubMed] [Google Scholar]
- 15.Arai K, Kinumaki T, Fujita T. Toxicity of chitosan. Bull Tokai Reg Fish Lab. 1968;43:89–94. [Google Scholar]
- 16.Singla AK, Chawla M. Chitosan: some pharmaceutical and biological aspects--an update. J Pharm Pharmacol. 2001;53:1047–1067. doi: 10.1211/0022357011776441. [DOI] [PubMed] [Google Scholar]
- 17.McNeela EA, Jabbal-Gill I, Illum L, Pizza M, Rappuoli R, Podda A, et al. Intranasal immunization with genetically detoxified diphtheria toxin induces T cell responses in humans: enhancement of Th2 responses and toxin-neutralizing antibodies by formulation with chitosan. Vaccine. 2004;22:909–914. doi: 10.1016/j.vaccine.2003.09.012. [DOI] [PubMed] [Google Scholar]
- 18.Wedmore I, McManus JG, Pusateri AE, Holcomb JB. A special report on the chitosan-based hemostatic dressing: experience in current combat operations. J Trauma. 2006;60:655–658. doi: 10.1097/01.ta.0000199392.91772.44. [DOI] [PubMed] [Google Scholar]
- 19.Zaharoff DA, Rogers CJ, Hance KW, Schlom J, Greiner JW. Chitosan solution enhances both humoral and cell-mediated immune responses to subcutaneous vaccination. Vaccine. 2007;25:2085–2094. doi: 10.1016/j.vaccine.2006.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zaharoff DA, Rogers CJ, Hance KW, Schlom J, Greiner JW. Chitosan solution enhances the immunoadjuvant properties of GM-CSF. Vaccine. 2007;25:8673–8686. doi: 10.1016/j.vaccine.2007.10.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zaharoff DA, Hoffman BS, Hooper HB, Benjamin CJ, Jr, Khurana KK, Hance KW, et al. Intravesical immunotherapy of superficial bladder cancer with chitosan/interleukin-12. Cancer Res. 2009;69:6192–6199. doi: 10.1158/0008-5472.CAN-09-1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zaharoff DA, Hance KW, Rogers CJ, Schlom J, Greiner JW. Intratumoral immunotherapy of established solid tumors with chitosan/IL-12. J Immunother. 2010;33:697–705. doi: 10.1097/CJI.0b013e3181eb826d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhu S, Lee DA, Li S. IL-12 and IL-27 sequential gene therapy via intramuscular electroporation delivery for eliminating distal aggressive tumors. J Immunol. 2010;184:2348–2354. doi: 10.4049/jimmunol.0902371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cavallo F, Signorelli P, Giovarelli M, Musiani P, Modesti A, Brunda MJ, et al. Antitumor efficacy of adenocarcinoma cells engineered to produce interleukin 12 (IL-12) or other cytokines compared with exogenous IL-12. J Natl Cancer Inst. 1997;89:1049–1058. doi: 10.1093/jnci/89.14.1049. [DOI] [PubMed] [Google Scholar]
- 25.Zitvogel L, Tahara H, Robbins PD, Storkus WJ, Clarke MR, Nalesnik MA, et al. Cancer immunotherapy of established tumors with IL-12. Effective delivery by genetically engineered fibroblasts. J Immunol. 1995;155:1393–1403. [PubMed] [Google Scholar]
- 26.Egilmez NK, Jong YS, Sabel MS, Jacob JS, Mathiowitz E, Bankert RB. In situ tumor vaccination with interleukin-12-encapsulated biodegradable microspheres: induction of tumor regression and potent antitumor immunity. Cancer Res. 2000;60:3832–3837. [PubMed] [Google Scholar]
- 27.Watkins SK, Egilmez NK, Suttles J, Stout RD. IL-12 rapidly alters the functional profile of tumor-associated and tumor-infiltrating macrophages in vitro and in vivo. J Immunol. 2007;178:1357–1362. doi: 10.4049/jimmunol.178.3.1357. [DOI] [PubMed] [Google Scholar]
- 28.Wesa A, Kalinski P, Kirkwood JM, Tatsumi T, Storkus WJ. Polarized type-1 dendritic cells (DC1) producing high levels of IL-12 family members rescue patient TH1-type antimelanoma CD4+ T cell responses in vitro. J Immunother. 2007;30:75–82. doi: 10.1097/01.cji.0000211316.15278.6e. [DOI] [PubMed] [Google Scholar]
- 29.Egilmez NK, Kilinc MO. Tumor-resident CD8+ T-cell: the critical catalyst in IL-12-mediated reversal of tumor immune suppression. Arch Immunol Ther Exp (Warsz) 2010;58:399–405. doi: 10.1007/s00005-010-0097-7. [DOI] [PubMed] [Google Scholar]
- 30.Kilinc MO, Aulakh KS, Nair RE, Jones SA, Alard P, Kosiewicz MM, et al. Reversing tumor immune suppression with intratumoral IL-12: activation of tumor-associated T effector/memory cells, induction of T suppressor apoptosis, and infiltration of CD8+ T effectors. J Immunol. 2006;177:6962–6973. doi: 10.4049/jimmunol.177.10.6962. [DOI] [PubMed] [Google Scholar]
- 31.Kilinc MO, Rowswell-Turner RB, Gu T, Virtuoso LP, Egilmez NK. Activated CD8+ T-effector/memory cells eliminate CD4+ CD25+ Foxp3+ T-suppressor cells from tumors via FasL mediated apoptosis. J Immunol. 2009;183:7656–7660. doi: 10.4049/jimmunol.0902625. [DOI] [PubMed] [Google Scholar]
- 32.Kilinc MO, Gu T, Harden JL, Virtuoso LP, Egilmez NK. Central role of tumor-associated CD8+ T effector/memory cells in restoring systemic antitumor immunity. J Immunol. 2009;182:4217–4225. doi: 10.4049/jimmunol.0802793. [DOI] [PubMed] [Google Scholar]
- 33.Gajewski TF. Cancer immunotherapy. Mol Oncol. 2012;6:242–250. doi: 10.1016/j.molonc.2012.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nastala CL, Edington HD, McKinney TG, Tahara H, Nalesnik MA, Brunda MJ, et al. Recombinant IL-12 administration induces tumor regression in association with IFN-gamma production. J Immunol. 1994;153:1697–1706. [PubMed] [Google Scholar]
- 35.Colombo MP, Trinchieri G. Interleukin-12 in anti-tumor immunity and immunotherapy. Cytokine Growth Factor Rev. 2002;13:155–168. doi: 10.1016/s1359-6101(01)00032-6. [DOI] [PubMed] [Google Scholar]
- 36.Trinchieri G. Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat Rev Immunol. 2003;3:133–146. doi: 10.1038/nri1001. [DOI] [PubMed] [Google Scholar]
- 37.Tsung K, Dolan JP, Tsung YL, Norton JA. Macrophages as effector cells in interleukin 12-induced T cell-dependent tumor rejection. Cancer Res. 2002;62:5069–5075. [PubMed] [Google Scholar]
- 38.Schwartz Y, Avraham R, Benish M, Rosenne E, Ben-Eliyahu S. Prophylactic IL-12 treatment reduces postoperative metastasis: mediation by increased numbers but not cytotoxicity of NK cells. Breast Cancer Res Treat. 2008;107:211–223. doi: 10.1007/s10549-007-9540-9. [DOI] [PubMed] [Google Scholar]
- 39.Sabel MS, Su G, Griffith KA, Chang AE. Intratumoral delivery of encapsulated IL-12, IL-18 and TNF-alpha in a model of metastatic breast cancer. Breast Cancer Res Treat. 2010;122:325–336. doi: 10.1007/s10549-009-0570-3. [DOI] [PubMed] [Google Scholar]
- 40.Cui J, Shin T, Kawano T, Sato H, Kondo E, Toura I, et al. Requirement for Valpha14 NKT cells in IL-12-mediated rejection of tumors. Science. 1997;278:1623–1626. doi: 10.1126/science.278.5343.1623. [DOI] [PubMed] [Google Scholar]
- 41.Smyth MJ, Taniguchi M, Street SE. The anti-tumor activity of IL-12: mechanisms of innate immunity that are model and dose dependent. J Immunol. 2000;165:2665–2670. doi: 10.4049/jimmunol.165.5.2665. [DOI] [PubMed] [Google Scholar]
- 42.Brunda MJ, Luistro L, Warrier RR, Wright RB, Hubbard BR, Murphy M, et al. Antitumor and antimetastatic activity of interleukin 12 against murine tumors. J Exp Med. 1993;178:1223–1230. doi: 10.1084/jem.178.4.1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Noguchi Y, Richards EC, Chen YT, Old LJ. Influence of interleukin 12 on p53 peptide vaccination against established Meth A sarcoma. Proc Natl Acad Sci U S A. 1995;92:2219–2223. doi: 10.1073/pnas.92.6.2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gabrilovich DI, Bronte V, Chen SH, Colombo MP, Ochoa A, Ostrand-Rosenberg S, et al. The terminology issue for myeloid-derived suppressor cells. Cancer Res. 2007;67:425. doi: 10.1158/0008-5472.CAN-06-3037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pastula A, Marcinkiewicz J. Myeloid-derived suppressor cells: a double-edged sword? Int J Exp Pathol. 2011;92:73–78. doi: 10.1111/j.1365-2613.2010.00754.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nausch N, Galani IE, Schlecker E, Cerwenka A. Mononuclear myeloid-derived “suppressor” cells express RAE-1 and activate natural killer cells. Blood. 2008;112:4080–4089. doi: 10.1182/blood-2008-03-143776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tomihara K, Guo M, Shin T, Sun X, Ludwig SM, Brumlik MJ, et al. Antigen-specific immunity and cross-priming by epithelial ovarian carcinoma-induced CD11b(+)Gr-1(+) cells. J Immunol. 2010;184:6151–6160. doi: 10.4049/jimmunol.0903519. [DOI] [PubMed] [Google Scholar]
- 48.Sorlier P, Hartmann DJ, Denuziere A, Viton C, Domard A. Preparation and development of anti-chitosan antibodies. J Biomed Mater Res A. 2003;67:766–774. doi: 10.1002/jbm.a.10132. [DOI] [PubMed] [Google Scholar]