

Summary
The integration of transmembrane (TM)-spanning regions of many channels and ion transporters is potentially compromised by the presence of polar and charged residues required for biological function. Although the two TMs of the ATP-gated ion channel subunit P2X2 each contain charged/polar amino acids, we found that each TM is efficiently membrane inserted when it is analysed in isolation, and uncovered no evidence for cooperativity between these two TMs during P2X2 integration. However, using minimal N-glycosylation distance mapping, we find that the positioning of TM2 in newly synthesized P2X2 monomers is distinct from that seen in subunits of the high-resolution structures of assembled homologous trimers. We conclude that P2X2 monomers are initially synthesised at the endoplasmic reticulum in a distinct conformation, where the extent of the TM-spanning regions is primarily defined by the thermodynamic cost of their membrane integration at the Sec61 translocon. In this model, TM2 of P2X2 subsequently undergoes a process of positional editing within the membrane that correlates with trimerisation of the monomer, a process requiring specific polar/charged residues in both TM1 and TM2. We postulate that the assembly process offsets any energetic cost of relocating TM2, and find evidence that positional editing of TM2 in the acid-sensing ion channel (ASIC1a) is even more pronounced than that observed for P2X2. Taken together, these data further underline the potential complexities involved in accurately predicting TM domains. We propose that the orchestrated repositioning of TM segments during subunit oligomerisation plays an important role in generating the functional architecture of active ion channels, and suggest that the regulation of this underappreciated biosynthetic step may provide an elegant mechanism for maintaining ER homeostasis.
Key words: ASIC, Endoplasmic reticulum, Membrane insertion, Oligomerisation, P2X channels
Introduction
The endoplasmic reticulum (ER) is a major site of membrane protein biogenesis producing an array of folding and assembly intermediates with a variety of topologies and complexities. In most cases these distinct classes of precursors are generated via the co-translational pathway that relies on signal recognition particle-dependent delivery of the ribosome-bound polypeptide to the Sec61 translocation complex of the ER membrane (Rapoport, 2007; Cross et al., 2009; Skach, 2009). The Sec61 translocon is capable of integrating a wide variety of membrane protein precursors including those with an N-terminal cleavable signal sequence, signal-anchor proteins of both topologies, and multi-spanning (polytopic) proteins with a range of TM spans (Booth and High, 2004; von Heijne, 2006; Hegde and Kang, 2008; Skach, 2009). In contrast, definitive roles for ER translocon associated components, such as TRAM and the TRAP complex, during the membrane integration process remain ill defined (Hegde and Kang, 2008). Acting in combination with the ribosome, the Sec61 translocon can facilitate complex nascent chain rearrangements and even accommodate the re-orientation of a signal-anchor sequence prior to its committed integration into the bilayer (Devaraneni et al., 2011). The Sec61 translocon has also been implicated in co-ordinating the assembly of pairs, or higher-order bundles, of transmembrane-spanning regions present in polytopic proteins prior to complete lateral exit and membrane integration (Booth and High, 2004; Skach, 2007). It is suggested that such TM assemblies can offset the suboptimal insertion properties of individual TM spans that, in many cases, reflect the presence of functionally important charged or polar residues located within the TM regions of channels and transporters (Sadlish et al., 2005; Sauri et al., 2005; Meindl-Beinker et al., 2006; Ismail et al., 2008; Cross and High, 2009). Indeed, the integration of a TM domain with a strong orientational preference can even modulate the efficiency of insertion of a neighbouring TM span, further illustrating the potential importance of interactions between different TM domains during the biogenesis of multi-spanning membrane proteins (Öjemalm et al., 2012). Many channel proteins are in fact oligomers comprised of several polytopic membrane protein subunits that assemble into a functional complex. In the case of an archaebacterial glutamate transporter homologue, specific TM spans appear to be repositioned relative to the plane of the membrane during the assembly of the subunits, adding a further level of complexity to the biogenesis of such components (Kauko et al., 2010).
P2X receptors are ATP-gated ion channels (Murrell-Lagnado and Qureshi, 2008; Surprenant and North, 2009) for which high-resolution structures of the P2X4 trimer have provided detailed insights into the closed (Kawate et al., 2009) and, more recently, open (Hattori and Gouaux, 2012) states. Each P2X subunit has two TM spans, and in this study we first investigated the thermodynamic cost of their integration into the ER membrane via the Sec61 translocon. Using P2X2 as our model, we find no evidence for the previously suggested cooperativity between TM segments during P2X2 membrane insertion (Cross and High, 2009). However, glycosylation mapping (cf. Kauko et al., 2010) revealed that the membrane disposition of TM2 in newly synthesised P2X monomers is distinct from that observed in assembled trimers (Kawate et al., 2009; Hattori and Gouaux, 2012). Whilst this novel positioning of P2X2-TM2 is not dependent upon two conserved polar residues, Q37 of TM1 and D349 of TM2, both of these residues are critical for the efficient assembly of native P2X2 trimers. This phenotype cannot be rescued by exchanging the two residues to create a Q37D+D349Q double mutant of P2X2, further suggesting an electrostatic interaction between TM1 and TM2 is not essential for P2X2 biogenesis. We conclude that TM2 alters its positioning during P2X2 oligomerisation, and speculate that residues Q37 and D349 each contribute to assembly at some stage during the oligomerisation process. In order to determine if post-integration TM repositioning is relevant to the architecture of other ion channels, we also analysed the related acid-sensing ion channel (ASIC1a) (Jasti et al., 2007; Gonzales et al., 2009). In this case we observe efficient in vitro N-glycosylation of two residues that are actually located within the second TM span of ASIC1a subunits present in an assembled trimer adopting a closed desensitised-like state (Jasti et al., 2007; Gonzales et al., 2009). This suggests that substantial positional editing of TM2 takes place during the assembly and maturation of the ASIC1a trimer, and we propose that these rearrangements are also driven by subunit oligomerisation.
Results
Sec61-mediated insertion of P2X2 transmembrane domains is energetically favourable and non-cooperative
On the basis of a previous cross-linking based study of P2X2 biogenesis at the ER membrane (Cross and High, 2009), it was suggested that a prolonged association between TM1 and the Sec61 translocon may reflect a requirement for its assembly with TM2 in order to offset the thermodynamic cost of integrating charged and polar residues present in these two TM segments (Cross and High, 2009). To test this hypothesis we measured the thermodynamic cost of inserting each TM of P2X2 using a well-established in vitro assay (Hessa et al., 2007; Lundin et al., 2008). Surprisingly, we found that, even when isolated from the rest of the P2X2 sequence by incorporation into the Lep reporter system (Hessa et al., 2007; Lundin et al., 2008), TM1 is a very efficient membrane insertion sequence. Hence, notwithstanding the presence of three charged/polar residues in TM1, its apparent free energy of membrane insertion at the ER (ΔGapp) is −1.4 kcal/mol (Table 1; see also supplementary material Fig. S1, Table S1). We conclude that the previously observed TM1 retention at the Sec61 translocon (Cross and High, 2009) is unlikely to be a direct consequence of the high thermodynamic cost of its membrane insertion.
Table 1. Thermodynamic cost of P2X2-TM integration.
| Region analysed | ΔGpred | ΔGapp | Sequence |
| TM1prestruc | −0.45 | −1.4 | FVHRMVQLLILLYFVWYVFI |
| TM1struc | −0.17 | n.d. | RMVQLLILLYFVWYVFIVQ |
| TM1ΔGpred (std) | −0.82 | n.d. | LGFVHRMVQLLILLYFVWYVFIV |
| TM2prestruc | +0.22 | +1.1 | IINLATALTSIGVGSFLCDWILL |
| TM2prestruc (D349A) | −0.50 | −1.1 | IINLATALTSIGVGSFLCAWILL |
| TM2struc | +2.50 | +1.4 | LIPTIINLATALTSIGVGSFL(CD) |
| TM2ΔGpred (std) | −0.12 | n.d. | LATALTSIGVGSFLCDWILLTFM |
| TM2ΔGpred (opt) | −0.66 | +0.7 | TIINLATALTSIGVGSFLCDWILLTFM |
| TM2ΔGpred (opt) with flanks | −0.66 | −2.0 | GIRIDVIVHGQAGKFSLIPTIINLATALTSIGVGSFLCDWILLTFMNKNKLYSHKKFDKVRTP |
The predicted (ΔGpred) and experimentally determined (ΔGapp) energetic cost in kcal/mol of inserting versions of the first (TM1) and second (TM2) transmembrane-spanning regions of P2X2 when located in a Lep-derived model precursor (cf. supplementary material Fig. S2). In each case, the underlined sequences indicate residues presumed to reside within the plane of the membrane. Residue D349 is shown in bold, and in the case of TM2struc is bracketed and in italics to indicate it is excluded from this sequence together with the preceding cysteine residue. In the case of TM2ΔGpred with flanks, the P2X2-derived sequences adjacent to the core TM region are not underlined. Certain predicted values are provided solely for the basis of comparison and, in these cases, the actual values have not been experimentally determined (n.d.).
We next asked whether TM1 retention at Sec61 was rather a reflection of a potential contribution to TM2 insertion. Shortly after our previous study of P2X2 biogenesis (see Cross and High, 2009), a high-resolution structure for the closely related P2X4 became available (Kawate et al., 2009). This new information suggested that TM2 of P2X2 differs somewhat from the models employed during our previous study, and for this reason we experimentally determined the ΔGapp for both pre- and post-structure TM2 spans (see Table 1). In both cases the membrane insertion of TM2 proved to be far less energetically favourable than that of TM1 (Table 1, TM2prestruc and TM2struc; see also supplementary material Fig. S1, Table S1). The energetic cost of TM2 insertion could be reduced by using a longer sequence [Table 1, see TM2ΔGpred (opt)], derived from the optimal in silico predicted TM span (http://dgpred.cbr.su.se). We also observed that replacing residue D349 of the TM2 prestructure sequence with alanine greatly reduced the empirically determined ΔGapp for membrane insertion [Table 1, see TM2prestruc (D349A)]. Interestingly, all of the predictions for TM2 suggest that residue D349 is located within the membrane-spanning region [Table 1, see TM2prestruc, TM2ΔGpred (std) TM2ΔGpred (opt)], yet in the high-resolution structure of the assembled P2X4 trimer, the equivalent residue, D357, is located just beyond the C-terminal boundary of TM2 (Table 1, TM2struc; see also Kawate et al., 2009). However, although the TM2struc sequence lacks residue D349, the measured ΔGapp for membrane insertion is actually higher than any of the other TM2 variants tested (Table 1). ΔG predictions suggest this is primarily a consequence of the inclusion of additional unfavourable residues at the N-terminal end of the TM span (Table 1, cf. ΔGpred values).
Despite this detailed analysis of the effects of different compositions of TM2 upon its efficiency to act as a TM span, the most striking effect that we observed upon TM2 insertion was obtained by simply incorporating the N- and C-terminal flanking regions present in the wild-type sequence. This produced a substantial stabilizing effect on TM2 resulting in efficient membrane insertion even in the presence of D349 [Table 1, see TM2ΔGpred (opt) with flanks; see also supplementary material Fig. S1, Table S1]. We further explored any potential contribution of an association between P2X2 TMs during membrane insertion by analysing the integration of TM2 within its natural context. In full length P2X2, the integration efficiency for TM2 is 100% (see supplementary material Fig. S2), and we therefore engineered an artificial situation that would better report TM1 mediated effects on TM2 insertion by replacing the normal P2X2 C-terminus with a region from Lep containing an N-glycosylation reporter. Consistent with our previous analysis of P2X2-TM2 spliced into the complete Lep reporter (see Table 1), we find that the efficiency of membrane insertion of TM2 in this P2X2-Lep hybrid protein is reduced to 62% (see supplementary material Fig. S2). This P2X2-Lep hybrid was then used to study the membrane insertion of TM2 when each of the charged/polar residues present in TM1 had been replaced with alanine. Even under conditions where the strongly stabilising effect of the P2X2 C-terminal domain is artificially removed, the capacity of the polar/charged residues to influence TM2 integration was marginal (supplementary material Fig. S2). Thus, both TM1 and TM2 of P2X2 are capable of efficient membrane insertion in isolation and we find no evidence that the charged/polar residues of TM1 contribute to any cooperativity with TM2 during P2X2 integration.
The positioning of TM2 in newly synthesised P2X2 is novel
Strikingly, our in vitro analysis of a range of variants for the second TM span of P2X2 showed that, although in each case their intrinsic hydrophobicity is comparatively low, it is the inclusion of the naturally occurring flanking sequences that has the most pronounced effect on the stable integration of this region. To better understand the precise disposition of TM2 in a newly synthesised P2X2 monomer, we used the minimal glycosylation distance (MGD) approach with full length P2X2 in order to map the location of the TM2 boundary at the ER luminal side of the membrane (Fig. 1A) (see also Nilsson and von Heijne, 1993; Kauko et al., 2010). From the results of these experiments, we estimate that the N-terminal boundary of TM2 in newly synthesised P2X2 is residue Ile331 (Fig. 1A–C). This is somewhat different to the location of the TM2 boundary observed in the crystal structure where the four preceding amino acid residues, LIPT, are located within the plane of the bilayer (Fig. 1A; Table 1). These data suggest that the precise conformation of P2X2-TM2 at biosynthesis is distinct from that seen in assembled P2X trimers (Kawate et al., 2009; Hattori and Gouaux, 2012).
Fig. 1.

Defining the extra-cytoplasmic boundary of P2X2-TM2 using minimal glycosylation distance (MGD) mapping. (A) Wild-type rat P2X2 was engineered to contain one readily accessible N-glycosylation site at residue 182, together with a second potential site introduced at a range of locations between residues 301 and 320 (see branched structures), to enable the location of the extra-cytoplasmic boundary of the second TM of P2X2 to be estimated. A third cryptic N-glycosylation site that was introduced as a reporter at residue 381 in the cytoplasmic tail of P2X2 (see dashed line branched structure), although no evidence for its modification was observed. Arrows labelled 314 and 320 indicate the luminal region of P2X2 that was analysed by N-glycosylation scanning (see also B), while the two red arrows indicate the extra-cytoplasmic boundary of P2X2 TM2 as deduced from the high-resolution crystal structure (labelled TM2struct) and the empirical minimal glycosylation distance approach (MGD). (B) The levels of singly glycosylated (filled circle) and doubly glycosylated (two filled circles) forms of each P2X2 variant were quantified by calculating the peak areas of the corresponding radiolabelled proteins from a 2D intensity profile of each lane. The amount of the doubly glycosylated species was then expressed as a percentage of the total glycosylated protein present (i.e. singly and doubly glycosylated forms). Unglycosylated proteins are indicated by an open circle. (C) The values obtained from quantification of the products shown in B were plotted as a function of the relative locations of the variable N-glycosylation site, and the point at which 50% modification occurred was used to provide an estimate of a location 14 residues form the extra-cytoplasmic boundary TM2 (cf. Nilsson and von Heijne, 1993; Kauko et al., 2010). The outcome of altering key residues in TM1 and TM2 to alanine was investigated by analysing the N-glycosylation efficiency of residue N317 (see colour coding). For wild-type TMs (black diamonds), the values shown are the mean of three independent experiments with the standard deviation indicated.
We explored the possibility that TM2 membrane disposition, rather than membrane insertion per se, can be influenced by cooperative interactions with TM1; carrying out the MGD mapping analysis with various point mutants of key residues and specifically choosing a ‘sensitive’ N-glycosylation reporter site (residue 317) located near the mid-point for modification efficiency in the wild-type P2X2-TMs. We find that substituting H33, R34 and Q37 of TM1 or D349 of TM2 for alanine has no detectable effect on the location of the extra-cytoplasmic boundary of TM2 (see Fig. 1C). Thus, the initial positioning of P2X2-TM2 observed during the in vitro biogenesis of the monomer appears to be determined primarily by its intrinsic amino acid sequence, most likely in combination with the effect of several basic residues in the cytoplasmic flanking region to its C-terminus (cf. Table 1; supplementary material Fig. S2) (see also Discussion).
Specific polar residues of P2X2-TM1 and TM2 are required for authentic trimerisation
A previous study of P2X5 highlighted the importance of TM2 in P2X trimerisation at the ER and defined a specific role for D355 (the equivalent of D349 of P2X2) in initial subunit homotrimerisation, in addition to its role in receptor function (Duckwitz et al., 2006). In the case of a newly synthesised P2X2 monomer, our data suggest that the inclusion of D349 would represent a fairly substantial energetic cost to TM2 insertion that is effectively offset by its C-terminal flanking region (cf. Table 1), and we therefore investigated whether this residue was of any importance to P2X2 assembly using blue native gel electrophoresis (Duckwitz et al., 2006). When wild-type P2X2 is transiently expressed in HeLa cells, and total digitonin soluble material analysed by blue-native gel electrophoresis, we observe a substantial amount of the protein migrating as a discrete ∼230 kDa species (Fig. 2A, lane 1), consistent with the previously described behaviour of authentic P2X5 trimers (Duckwitz et al., 2006). We also observe additional, higher molecular weight species that are most likely misassembled subunits (see below), perhaps resulting from high-level expression (cf. Duckwitz et al., 2006).
Fig. 2.
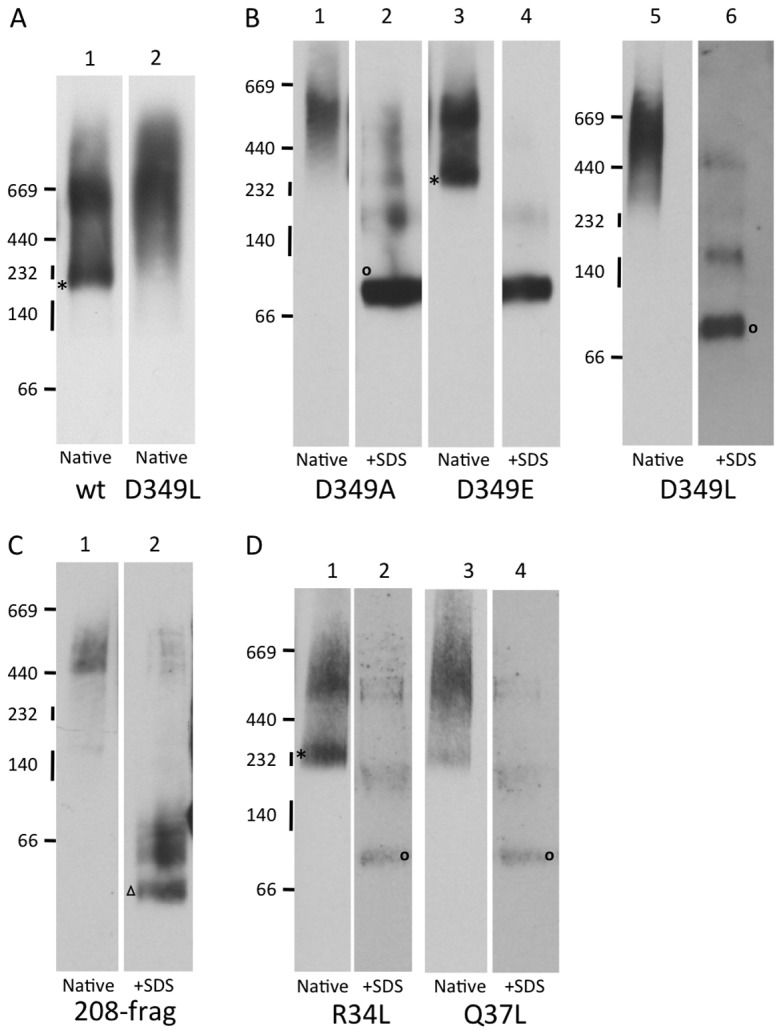
Key residues in TM1 and TM2 are required for authentic P2X2 trimeri sation. (A,B,D) HeLa cells were transiently transfected with plasmids expressing wild type rat P2X2 with a C-terminal GluGlu tag (wt) or a range of P2X2 point mutants. Alternatively, a plasmid encoding an HA-tagged 208 residue fragment of P2X2 lacking the second transmembrane domain was used (C). After harvesting of the cells, digitonin soluble material was analysed by blue native gel analysis in the absence (native) or presence of SDS (+SDS) as indicated. Following electrophoresis, samples were immunoblotted and analysed using antibodies recognising the GluGlu- (A,B,D) or HA-tag (C). The migration of native molecular weight markers (kDa) is indicated to the left of the immunoblots, and lanes sharing a common set of markers are derived from a single native gel. Individual lanes have been rearranged to facilitate direct comparisons. An asterisk (*) indicates the location of presumptive native P2X2 trimers, and an open circle the monomer resulting from SDS treatment. The open triangle indicates the migration of the P2X2-208 fragment after SDS treatment.
Upon altering D349 to Leu only putative misassembled species (>600 kDa) were detected (Fig. 2A, cf. lanes 1 and 2). Likewise replacement of D349 with Ala had the same effect (Fig. 2B, lane 1). In contrast, the conservative substitution D349E resulted in levels of the presumptive P2X2 trimer akin to that observed with the epitope tagged wild-type protein (Fig. 2B, lane 3). It should be noted that much of the 230 kDa trimer, and the larger misassembled oligomers, were converted into monomers upon SDS treatment (Fig. 2B, cf. native and +SDS lanes) consistent with previous studies of P2X5 (Duckwitz et al., 2006). In the case of the >600 kDa P2X2 species this suggests that they represent inappropriate and/or non-productive oligomers rather than terminally misfolded/misassembled aggregates. This proposal is further supported by the behaviour of a 208 amino acid long fragment of P2X2 that completely lacks TM2 and which we previously established to be aggregation prone on the basis of both cross-linking and sucrose gradient centrifugation (Cross and High, 2009). The P2X2-208 fragment generates only a novel high molecular weight species of >500 kDa that is converted to the truncated monomer, and related products, upon SDS treatment (Fig. 2C). This behaviour of the P2X2-208 fragment provides further evidence of its propensity to self assemble, and supports our hypothesis that it could have a dominant negative effect upon the assembly of wild-type P2X2 trimers (Cross and High, 2009).
In contrast to H33 of P2X2-TM1, residues R34 and Q37 display fairly strong conservation, albeit that this does not extend to P2X7 of higher eukaryotes or P2X homologues in lower eukaryotes (Kawate et al., 2009; Surprenant and North, 2009). We therefore focused on the potential roles of R34 and Q37 during P2X2 assembly, again using blue native gel analysis. The substitution of R34 with Leu has no discernible effect on P2X2 assembly and the behaviour of this mutant mirrors that of the wild-type protein (cf. Fig. 2A,D, lane 1). In contrast, the substitution of Q37 for Leu results in almost undetectable levels of the 230 kDa trimer, whilst the larger misassembled species appear unaffected (Fig. 2D, cf. lanes 1 and 3). Taken together, these data suggest that the efficient assembly of P2X2 involves orchestrated interactions between TMs of different monomers.
Misassembled P2X2 oligomers are incorrectly trafficked
Misassembled subunits of oligomers are typically retained at the ER by the cellular quality control machinery (Booth and High, 2004; Roboti et al., 2009), and the analysis of subcellular localisation has the potential to provide a sensitive reporter for the authentic assembly of P2X subunits destined for the plasma membrane. For this reason, we examined the localisation of wild type and mutant P2X2 using immunofluorescence microscopy. Wild-type P2X2 is clearly present at both the plasma membrane and various intracellular locations when transiently expressed in HeLa cells (Fig. 3A, see also inset). Double labelling analysis confirmed that some of this intracellular material was located at the ER, consistent with it biosynthesis here prior to delivery to the cell surface (supplementary material Fig. S3, see wild type) (cf. Ray-Sinha et al., 2009). In contrast, no cell surface labelling was apparent when the P2X2-208 fragment was analysed (Fig. 3B), and the vast majority of the polypeptide was found at the ER (supplementary material Fig. S3, see 208).
Fig. 3.

Misassembled P2X2 subunits are retained at the endoplasmic reticulum. The subcellular localisation of transiently expressed wild-type and mutant P2X2 derivatives was determined by indirect immunofluorescence microscopy using antibodies specific for a C-terminal epitope tag (A, C–G, GluGlu; B, HA). P2X2 derivatives analysed were: A, wild-type; B, P2X2-208 fragment; C, D349L; D, D349A; E, D349E; F, R34L and G, Q37L. Insets show magnified regions illustrating staining of the plasma membrane (A,E,F) and ER (G). All scale bars are 10 µm.
The behaviour of the 208-fragment confirms that aberrant forms of P2X2 are potential substrates for the ER quality control machinery, and we next analysed the fate of the three D349 mutations previously analysed by blue-native gel analysis (cf. Fig. 2). Strikingly, the D349L and D349A mutants both showed ER like staining with no obvious plasma membrane localisation (Fig. 3C,D), whilst the behaviour of the D349E mutant appeared similar to the wild-type protein and displayed clear labelling of the plasma membrane (Fig. 3E). The intracellular staining of the D349L mutant overlapped with that of an ER marker (supplementary material Fig. S3, see D349L), consistent with its retention via ER quality control. Likewise, the subcellular localisation of the two TM1 mutants also correlated with their behaviour upon blue native gel analysis, in that detectable levels of the R34L mutant were observed at the plasma membrane (Fig. 3F), whilst the Q37L mutant appeared almost exclusively in the ER (Fig. 3G, see also inset; supplementary material Fig. S3, Q37L). Hence, residues Q37 of TM1 and D349 of TM2 are important for both the authentic oligomerisation of the P2X2 receptor, and its ability to traffic to the plasma membrane, and alterations to either residue result in retention at the ER, most likely as a consequence of its quality control function (cf. Ray-Sinha et al., 2009).
Although we found no evidence for cooperativity between Q37 of TM1 and D349 of TM2 (cf. Fig. 1; supplementary material Fig. S2), an electrostatic interaction between these two residues might be important during the assembly of P2X2, consistent with the ‘wild-type’ phenotype that we observed with the conservative D349E substitution (cf. Figs 2, 3). To investigate this possibility we generated a Q37D+D349Q double mutant that maintained the possibility of any electrostatic interaction and analysed its behaviour following expression in HeLa cells. In contrast to the wild-type and D349E versions of P2X2, the Q37D/D349E double mutant is unable to assemble into a native trimer and does not traffic to the plasma membrane (supplementary material Fig. S4). These data suggest that the contributions of Q37 and D349 during P2X2 assembly do not reflect a direct electrostatic interaction between the two residues.
TM2 repositioning also occurs in the acid-sensing ion channel
Our observation that TM2 of P2X2 appears to reposition during its trimerisation led us to ask whether this process may also occur during the assembly of other ion-channel subunits, and we identified the structurally related acid-sensing ion channel (ASIC1a) as a candidate for further study (Jasti et al., 2007). In this case, predictions based on empirical analyses of the Sec61 mediated insertion of TM helices (Hessa et al., 2007; Lundin et al., 2008) suggested that the optimal version of the second TM span of ASIC1a might be significantly shorter than that observed in the crystal structure (see supplementary material Table S2, cf. TM2ΔGpred and TM2struc). This predicted difference is primarily a consequence of an increase in the length of TM2 in the final structure resulting from a substantial alteration in the location of the N-terminal extra-cytoplasmic boundary (see supplementary material Table S2), akin to that we had already established for P2X2.
Given the potentially significant difference between the positioning of ASIC1a-TM2 in a newly synthesised monomer and an assembled trimer, we again used the MGD approach to empirically determine the location of its extra-cytoplasmic boundary after incorporation into ER-derived microsomes (see Fig. 4A). Using this approach, and applying the normal criterion of ∼14 residues for the MGD from the N-terminal end of a TM span (cf. Kauko et al., 2010), the experimentally derived location for the extra-cytoplasmic boundary would be residue Ala443, well within TM2 of the structural model for ASIC1a (Fig. 4A), and in fact even within the ΔG based prediction for the optimal TM span (supplementary material Table S2, see TM2ΔGpred). We note that the transition from efficient to inefficient N-glycosylation of the MGD reporter sites in ASIC1a derivatives is rather sharp (Fig. 4B,C), and the MGD-derived location of the extracytoplasmic boundary is of course an approximation. However, it is striking that we observed efficient N-glycosylation of Asn residues replacing Leu429 and Leu430 (Fig. 4B,C) that are actually located within the TM-spanning region of TM2 in the high-resolution crystal structure for ASIC1a (see Fig. 4A; supplementary material Table S2) (Jasti et al., 2007). These data indicate that ASIC1a-TM2 is extremely unlikely to be synthesised in the same conformation as that found in the subunits of an assembled channel, and suggest that the degree of positional editing required for the ASIC1a-TM2 upon subunit assembly is even more striking than that observed for P2X2 (see also Fig. 5).
Fig. 4.

Defining the extra-cytoplasmic boundary of ASIC1a-TM2 using MGD mapping. (A) Chicken ASIC1a was engineered to contain two N-glycosylation sites (cf. Fig. 1), one fixed at residue 402 and a second variable site located between residues 411 and 434 as indicated. The location of a third cryptic N-glycosylation site at residue 476 of the cytoplasmic tail is indicated. Arrows labelled 429 and 434 indicate the luminal region of ASIC1a analysed by N-glycosylation scanning (cf. B), whilst the two arrows labelled TM2struct and MGD indicate the extra-cytoplasmic boundary of TM2 as deduced from the crystal structure and minimal glycosylation mapping, respectively. (B,C) The amounts of the variously modified species were quantified and plotted as for P2X2 analysis (see legend to Fig. 1). All symbols are as previously defined in the legend to Fig. 1.
Fig. 5.
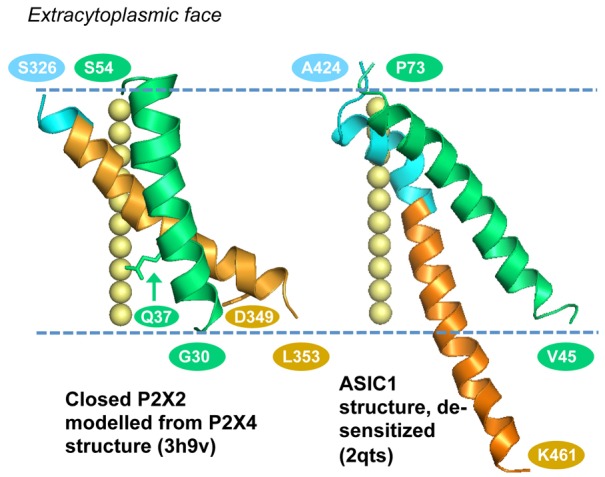
TM helices in P2X2 (modelled from P2X4) and ASIC1a structures. The TM1 (green) and TM2 (orange) segments of a single monomer are shown in each case, with the membrane boundaries (dashed lines) corresponding to membrane placement in the P2X4 and ASIC1a structural reports. Blue represents amino acids that are transmembrane in the known structures, but indicated as aqueous phase accessible by MGD mapping (cf. Figs 1, 4). For P2X2 this region covers residues S326 to P329, and for ASIC1a residues A424 to Q437. Residue numbers are shown for the helix termini in P2X2 and chicken ASIC1a. Relative to modelled P2X2, the equivalent residue numbers in the P2X4 structure are incremented by 2 for TM1 and by 8 for TM2. The locations of Q37 and D349 in the P2X2 model are denoted by the labelled side chains. The yellow spheres show the trimer axis of each system. Closed P2X2 was comparative modelled using the closed P2X4 structure as the template (PDB id 3h9v; Kawate et al., 2009). The structure of a low pH, de-sensitised ASIC1a form (2qts; Jasti et al., 2007) is shown. There is variability in the extent of ordered structure at the C-terminal end of TM2 in this structure, and hence the monomer with the greatest extent (chain B of 2qts) is drawn.
Discussion
Our observation that the TM spans of P2X2 are delayed at the ER translocon during in vitro biogenesis led us to propose that this behaviour might reflect a requirement for their assembly prior to full membrane integration (Cross and High, 2009). In this scenario, the thermodynamic cost of inserting individual TMs would be offset by associations between polar residues in different TMs (Cross and High, 2009). Our analysis of the energetic cost of P2X2-TM insertion using a well-defined Lep-based system (Hessa et al., 2007; Lundin et al., 2008) does not support such a model. Hence, TM1 acts as a very efficient TM span, even when analysed in isolation from the rest of P2X2. Since the system used necessitates the inclusion of at least one additional Lep-derived TM located N-terminal of the TM span being analysed (Hessa et al., 2007; Lundin et al., 2008), we cannot completely exclude some influence of the analytical system on the behaviour of P2X2-TM1 (see also Öjemalm et al., 2012). However, a strength of the Lep system is that it provides a comparative scale for the energetic cost of inserting a diverse range of model and actual TM regions, and by these criteria we find no evidence that TM1 of P2X2 is significantly compromised by the presence of three charged/polar residues.
In the case of TM2, we did obtain evidence that there is a substantial potential thermodynamic cost to incorporating residue D349 into the ER membrane [Table 1, cf. ΔGapp for TM2prestruc and TM2prestruc (D349A)]. However, in practice this cost is efficiently offset by the C-terminal flanking region of P2X2, which contains a number of basic residues characteristic of so called ‘stop-transfer’ signals. Although the contribution of such residues is not taken into account by the ΔG predictor algorithm (Hessa et al., 2007; Lundin et al., 2008), they are known to promote the Sec61 mediated integration of otherwise suboptimal TM-spanning regions (Lerch-Bader et al., 2008). In summary, whilst we cannot completely exclude a role for the cooperative integration of P2X2 TMs, we find no evidence to support a model where their coordinated assembly is required in order to offset the energetic cost of inserting the charged/polar residues they contain into the ER membrane. Interestingly, residue D357 of P2X4, the equivalent of D349 in P2X2, is located in the region of TM2 showing the biggest change in conformation upon transition between the closed and open states of the trimeric channel (Hattori and Gouaux, 2012).
Positional editing of P2X2-TM2
Having analysed the membrane insertion of the P2X2-TMs, we next analysed the positioning of TM2 within the ER membrane, focusing on its extra-cytoplasmic boundary. This analysis showed that the experimentally determined boundary of TM2 in newly synthesised P2X2 is somewhat different to that seen in homologous assembled trimers (Kawate et al., 2009; Browne et al., 2010; Hattori and Gouaux, 2012). Whilst our studies were performed in vitro, it is worth noting that a study using mammalian cells to analyse P2X2 derivatives found that a K324N mutant could be N-glycosylated (Newbolt et al., 1998). This amino acid is located only three residues from the extra-cytoplasmic boundary of TM2 deduced from the crystal structure (Fig. 1A) (cf. Kawate et al., 2009). The N-glycosylation of K324N strongly supports our model in which newly synthesised P2X2 adopts a conformation where this region of polypeptide would be comparatively accessible to the ER luminal oligosaccharyltransferase (cf. Fig. 5, P2X2), notwithstanding suggestions that the minimal glycosylation distance in vivo can be somewhat shorter than that defined using the in vitro system employed here (Hasler et al., 2000).
TM2 conformation and ion channel assembly
How might the conformation of TM2 as synthesised at the ER be altered to achieve the arrangement observed in assembled trimeric channels? We favour the possibility that it is the assembly of P2X2 subunits into a trimer that facilitates this positional editing, via a process akin to that previously suggested for an archaeal glutamate transporter homologue (Kauko et al., 2010). Strikingly, the assembly of P2X2 subunits is dependent upon two key residues, Q37 and D349, that are located near the cytoplasmic boundaries of TM1 and TM2, respectively, as observed in the trimeric P2X4 channel (cf. Fig. 5, P2X2; see also Kawate et al., 2009; Hattori and Gouaux, 2012). Furthermore, we see a close correlation between the efficient assembly of native P2X2 trimers and the ability of the protein to be delivered to the plasma membrane. A requirement for D349 of TM2 during P2X2 assembly is entirely consistent with both a previous study of P2X5 oligomerisation (Duckwitz et al., 2006), and the effect of mutating this residue on P2X2 function (Cao et al., 2009). However, in contrast to previous work based solely on co-immunoprecipitation (Torres et al., 1999), we conclude that TM1, and specifically residue Q37, does play an important role during productive P2X2 oligomerisation. A Q37D+D349Q double mutant of P2X2 is unable to form native trimers, suggesting that the contribution of these two residues to P2X2 assembly is not simply a consequence of an electrostatic interaction between them. Taken together, our data support a model where the efficient assembly of P2X2, and most likely all members of the mammalian P2X family, involves orchestrated interactions between both TM1 and TM2 of different subunits following their initial synthesis at the ER (Murrell-Lagnado and Qureshi, 2008). We speculate that the assembly process would favour the acquisition of a closed P2X conformation (Kawate et al., 2009; Hattori and Gouaux, 2012), thereby providing a mechanism that could protect the characteristic ionic and redox environments of the ER lumen during the obligatory synthesis and assembly of ion channels at the ER membrane (Murrell-Lagnado and Qureshi, 2008).
ASICs are structurally related to P2X receptors (Gonzales et al., 2009) and our preliminary analysis of TM2 from chicken ASIC1a suggests a remarkable extent of positional editing must occur following the initial biosynthesis of the monomeric subunits at the ER (see Fig. 5, ASIC1a). Hence we estimate a difference of up to 14 residues between the experimentally determined extra-cytoplasmic boundary of TM2 (this study) and that observed in the high-resolution structure of the assembled trimer (Jasti et al., 2007). Theoretical calculations indicate that neither the predicted nor the structural version of ASIC1a-TM2 is particularly efficient for Sec61 mediated membrane insertion (cf. supplementary material Table S2). However, like the P2X family, the ASICs all contain a significant number of basic amino acid residues just to the cytosolic C-terminus of TM2 (Jasti et al., 2007), and these residues most likely contribute to its efficient membrane integration as already discussed for P2X2 (cf. Lerch-Bader et al., 2008). In simple thermodynamic terms, this positional editing of TM2 may have some modest cost (cf. supplementary material Table S2), however, we suggest that as for P2X2, it is most likely the assembly of ASIC subunits into functional trimers that drives such re-arrangements (cf. Fig. 5, ASIC1a). This study adds to increasing evidence that the process of TM domain positional editing is comparatively widespread and, as has been previously noted, such phenomena require further levels of complexity to be built into predictions for the structure of multi-spanning membrane proteins that currently rely solely on their amino acid sequence (Kauko et al., 2010).
Materials and Methods
Reagents and enzymes
Cell culture reagents were from GIBCO BRL, Cambrex or Lonza, TNT Quick transcription/translation system from Promega and EasyTag l-[35S]methionine, and l-[35S]methionine from PerkinElmer (UK and Boston, MA). All enzymes were from Fermentas (Burlington, Ontario, Canada) and Finnzymes (Espoo, Finland) and the QuikChange Site-directed Mutagenesis kit was from Stratagene (La Jolla, CA). All other chemicals were from Sigma-Aldrich or BDH/Merck.
Ion channel constructs
Full-length rat P2X2 and its TM segments were sourced as previously described (Cross and High, 2009), whilst the full-length chicken ASIC1a cDNA was a kind gift from E. Gouaux. Individual TMs were inserted in frame with regions of the Lep protein so as to conserve their original transmembrane topology in P2X2, and the estimated cost of membrane insertion determined as previously described (Hessa et al., 2007; Lundin et al., 2008). The analysis of minimal glycosylation distance (MGD) was carried out using full-length ion channel subunits, cloned into pGEM1 (Promega) and engineered to contain specific N-glycosylation sites (AsnAlaThr) using site-directed mutagenesis (cf. Figs 1, 4; supplementary material Table S1). The P2X2-Lep chimera was constructed by replacing the N-terminal region of the Lep protein in the pGEM1 vector (Hessa et al., 2007) with residues 1-353 of P2X2. Individual amino acids within TM1 of P2X2-Lep were altered by site-directed mutagenesis (see supplementary material Fig. S2). In all cases sequence alterations were confirmed by sequencing of the plasmid DNA at Eurofins MWG Operon (Ebersberg, Germany).
For P2X2 expression in mammalian cells all constructs were cloned into pcDNA3.1 (Invitrogen). The wild-type rat P2X2 receptor with a C-terminal GluGlu tag, and a 208 residue fragment lacking the second transmembrane domain and bearing a C-terminal HA tag, were both as previously described (see Cross and High, 2009; and references therein). All other P2X2 derivatives were generated by site-directed mutagenesis of the GluGlu tagged wild-type plasmid and verified by DNA sequencing.
Estimating membrane insertion efficiency and minimal glycosylation distance
Full-length ion channel subunits (P2X2 or ASIC1a), P2X2-Lep chimeras and Lep constructs containing P2X2-TMs, were synthesised using TNT Quick coupled transcription/translation system. Briefly, 150–200 ng DNA template, 5 µCi l-[35S]methionine (1175 mCi/mmol), and 1 µl nuclease treated canine pancreatic rough microsomes (∼40 A280/ml) were added to 10 µl reticulocyte lysate and incubated for 90 minutes at 30°C. All samples were then subjected to SDS-PAGE. The protein bands were visualized using a Fuji FLA-3000 phosphoimager (Fujifilm, Tokyo, Japan) and a 2D intensity profile of each lane was generated using Image Reader V1.8J/Image Gauge V3.45 software. The multi-Gaussian fit program from the Qtiplot software package was used to calculate the peak area of the radiolabelled products from the intensity profiles. To calculate the fraction (fi) of P2X2-TMs that inserted into the microsomal membrane, the peak area of the protein band corresponding to Lep proteins with membrane-spanning P2X2-TMs was divided by the summed peak areas of Lep proteins with membrane-spanning plus membrane-translocated TMs. The apparent free energy of P2X2-TM helix insertion into the membrane (ΔGapp) was obtained using the relation ΔGapp = −RTln(fi/(1−fi)), where R is the gas constant and T the absolute temperature in K (Hessa et al., 2007; Lundin et al., 2008). To obtain the minimal glycosylation distance from the luminal face of the ER membrane for TM2 of both the P2X2 and the ASIC1a, the fraction of each variant where the reporter glycosylation site was modified (i.e. the doubly glycosylated product) was calculated as a percentage of the total glycosylated material (i.e. singly and doubly glycosylated products) using phosphoimaging and quantification as described above (cf. Nilsson and von Heijne, 1993; Kauko et al., 2010). To compare the insertion efficiency of TM2 in different P2X2-Lep variants, the fraction of the monoglycosylated protein was calculated as percentage of the total glycosylated material as above (supplementary material Fig. S2).
Mammalian cell culture and transfection
HeLa cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with l-glutamine and 10% fetal calf serum. For BN-PAGE and immunofluorescence, cells were transfected with P2X2 DNA constructs using TransIT-LT1 transfection reagent (Mirus Bio) according to manufacturer's instructions. Cells for BN-PAGE and immunofluorescence were transfected in 6-well and 24-well plates respectively and both were incubated for 24 hours.
Blue Native-PAGE
HeLa cells expressing P2X2 mutants were trypsinised and pelleted. Cell pellets were washed in PBS and resuspended in BN sample buffer (Invitrogen) supplemented with 1% digitonin. Cells were then solubilised at 4°C for 20 minutes and then centrifuged at 16,000 g for 10 minutes at 4°C to remove any unsolubilised material. Denatured samples were instead resuspended in denaturing SDS PAGE sample buffer and heated at 37°C for 30 minutes. Samples were loaded on Native PAGE 4–16% Bis Tris gels (Invitrogen) and run according to manufacturer's instructions. Samples were then transferred to nitrocellulose membrane for western blotting and membranes were blocked overnight at 4°C in TBS supplemented with 0.05% Tween 20 and 5% low fat dried milk. Western blotting was carried out using either rabbit anti-HA, 1∶500 (Sigma) or rabbit anti-GluGlu, 1∶200 (Abcam) primary antibodies, followed by goat anti-rabbit IgG HRP conjugated secondary antibody, 1∶4000 (Sigma). All antibodies were diluted in TBS supplemented with 0.05% Tween 20 and incubated at room temperature for 4 hours (primary) or 1 hour (secondary). Products were visualized by enhanced chemiluminescence (Pierce).
Immunofluorescence
HeLa cells were transfected with P2X2 constructs on glass coverslips as described above. Cells were either fixed in ice-cold methanol, washed, and blocked with PBS supplemented with 2% low fat dried milk for 30 minutes (Fig. 3; supplementary material Fig. S3), or fixed with 3% formaldehyde and 0.2% glutaraldehyde for 20 minutes, and permeabilised with 0.1% Triton X-100 and 0.05% SDS for 4 minutes at room temperature (supplementary material Fig. S4). All antibodies were diluted in PBS and incubated for 1 hour at room temperature. Primary antibodies and dilutions were as follows: rabbit anti-GluGlu, 1∶50 (Abcam); rabbit anti-HA, 1∶50 (Sigma) and a mouse monoclonal anti-KDEL antibody (1D3, a gift from Prof. Stephen Fuller), 1∶3 to stain the endoplasmic reticulum and early secretory pathway. Coverslips were washed in PBS and treated with fluorophore conjugated secondary antibodies. Coverslips were mounted in Mowiol and viewed with an Olympus BX60 microscope using a 60× oil objective and MetaMorph software (Universal Imaging Corporation).
Acknowledgments
We thank Alan North for his advice and encouragement, Bernhard Dobberstein for canine pancreatic microsomes and Eric Gouaux for the ASIC1a cDNA.
Footnotes
Funding
This work was supported by project grant funding from the Wellcome Trust [grant number 088163/Z/09/Z] to S.H. and J.W., and by grants from the Swedish Cancer Foundation [grant number 120837], the Swedish Research Council [grant number 621-2010-5250], the Swedish Foundation for Strategic Research [grant number A3-05:200], and the European Research Council (ERC-2008-AdG 232648) to G.v.H. Deposited in PMC for immediate release.
Supplementary material available online at http://jcs.biologists.org/lookup/suppl/doi:10.1242/jcs.111773/-/DC1
References
- Booth P. J., High S. (2004). Polytopic membrane protein folding and assembly in vitro and in vivo. Mol. Membr. Biol. 21, 163–170 10.1080/09687680410001697215 [DOI] [PubMed] [Google Scholar]
- Browne L. E., Jiang L. H., North R. A. (2010). New structure enlivens interest in P2X receptors. Trends Pharmacol. Sci. 31, 229–237 10.1016/j.tips.2010.02.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao L., Broomhead H. E., Young M. T., North R. A. (2009). Polar residues in the second transmembrane domain of the rat P2X2 receptor that affect spontaneous gating, unitary conductance, and rectification. J. Neurosci. 29, 14257–14264 10.1523/JNEUROSCI.4403-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross B. C., High S. (2009). Dissecting the physiological role of selective transmembrane-segment retention at the ER translocon. J. Cell Sci. 122, 1768–1777 10.1242/jcs.046094 [DOI] [PubMed] [Google Scholar]
- Cross B. C., Sinning I., Luirink J., High S. (2009). Delivering proteins for export from the cytosol. Nat. Rev. Mol. Cell Biol. 10, 255–264 10.1038/nrm2657 [DOI] [PubMed] [Google Scholar]
- Devaraneni P. K., Conti B., Matsumura Y., Yang Z., Johnson A. E., Skach W. R. (2011). Stepwise insertion and inversion of a type II signal anchor sequence in the ribosome-Sec61 translocon complex. Cell 146, 134–147 10.1016/j.cell.2011.06.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duckwitz W., Hausmann R., Aschrafi A., Schmalzing G. (2006). P2X5 subunit assembly requires scaffolding by the second transmembrane domain and a conserved aspartate. J. Biol. Chem. 281, 39561–39572 10.1074/jbc.M606113200 [DOI] [PubMed] [Google Scholar]
- Gonzales E. B., Kawate T., Gouaux E. (2009). Pore architecture and ion sites in acid-sensing ion channels and P2X receptors. Nature 460, 599–604 10.1038/nature08218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasler U., Greasley P. J., von Heijne G., Geering K. (2000). Determinants of topogenesis and glycosylation of type II membrane proteins. Analysis of Na,K-ATPase beta 1 AND beta 3 subunits by glycosylation mapping. J. Biol. Chem. 275, 29011–29022 10.1074/jbc.M002867200 [DOI] [PubMed] [Google Scholar]
- Hattori M., Gouaux E. (2012). Molecular mechanism of ATP binding and ion channel activation in P2X receptors. Nature, 485, 207–212 10.1038/nature11010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegde R. S., Kang S. W. (2008). The concept of translocational regulation. J. Cell Biol. 182, 225–232 10.1083/jcb.200804157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hessa T., Meindl–Beinker N. M., Bernsel A., Kim H., Sato Y., Lerch–Bader M., Nilsson I., White S. H., von Heijne G. (2007). Molecular code for transmembrane-helix recognition by the Sec61 translocon. Nature 450, 1026–1030 10.1038/nature06387 [DOI] [PubMed] [Google Scholar]
- Ismail N., Crawshaw S. G., Cross B. C., Haagsma A. C., High S. (2008). Specific transmembrane segments are selectively delayed at the ER translocon during opsin biogenesis. Biochem. J. 411, 495–506 10.1042/BJ20071597 [DOI] [PubMed] [Google Scholar]
- Jasti J., Furukawa H., Gonzales E. B., Gouaux E. (2007). Structure of acid-sensing ion channel 1 at 1.9 � resolution and low pH. Nature 449, 316–323 10.1038/nature06163 [DOI] [PubMed] [Google Scholar]
- Kauko A., Hedin L. E., Thebaud E., Cristobal S., Elofsson A., von Heijne G. (2010). Repositioning of transmembrane alpha-helices during membrane protein folding. J. Mol. Biol. 397, 190–201 10.1016/j.jmb.2010.01.042 [DOI] [PubMed] [Google Scholar]
- Kawate T., Michel J. C., Birdsong W. T., Gouaux E. (2009). Crystal structure of the ATP-gated P2X(4) ion channel in the closed state. Nature 460, 592–598 10.1038/nature08198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerch–Bader M., Lundin C., Kim H., Nilsson I., von Heijne G. (2008). Contribution of positively charged flanking residues to the insertion of transmembrane helices into the endoplasmic reticulum. Proc. Natl. Acad. Sci. USA 105, 4127–4132 10.1073/pnas.0711580105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundin C., Kim H., Nilsson I., White S. H., von Heijne G. (2008). Molecular code for protein insertion in the endoplasmic reticulum membrane is similar for N(in)-C(out) and N(out)-C(in) transmembrane helices. Proc. Natl. Acad. Sci. USA 105, 15702–15707 10.1073/pnas.0804842105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meindl–Beinker N. M., Lundin C., Nilsson I., White S. H., von Heijne G. (2006). Asn- and Asp-mediated interactions between transmembrane helices during translocon-mediated membrane protein assembly. EMBO Rep. 7, 1111–1116 10.1038/sj.embor.7400818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murrell–Lagnado R. D., Qureshi O. S. (2008). Assembly and trafficking of P2X purinergic receptors (Review). Mol. Membr. Biol. 25, 321–331 10.1080/09687680802050385 [DOI] [PubMed] [Google Scholar]
- Newbolt A., Stoop R., Virginio C., Surprenant A., North R. A., Buell G., Rassendren F. (1998). Membrane topology of an ATP-gated ion channel (P2X receptor). J. Biol. Chem. 273, 15177–15182 10.1074/jbc.273.24.15177 [DOI] [PubMed] [Google Scholar]
- Nilsson I. M., von Heijne G. (1993). Determination of the distance between the oligosaccharyltransferase active site and the endoplasmic reticulum membrane. J. Biol. Chem. 268, 5798–5801 [PubMed] [Google Scholar]
- Öjemalm K., Halling K. K., Nilsson I., von Heijne G. (2012). Orientational preferences of neighboring helices can drive ER insertion of a marginally hydrophobic transmembrane helix. Mol. Cell 45, 529–540 10.1016/j.molcel.2011.12.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapoport T. A. (2007). Protein translocation across the eukaryotic endoplasmic reticulum and bacterial plasma membranes. Nature 450, 663–669 10.1038/nature06384 [DOI] [PubMed] [Google Scholar]
- Ray–Sinha A., Cross B. C., Mironov A., Wiertz E., High S. (2009). Endoplasmic reticulum-associated degradation of a degron-containing polytopic membrane protein. Mol. Membr. Biol. 26, 448–464 10.3109/09687680903333839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roboti P., Swanton E., High S. (2009). Differences in endoplasmic-reticulum quality control determine the cellular response to disease-associated mutants of proteolipid protein. J. Cell Sci. 122, 3942–3953 10.1242/jcs.055160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadlish H., Pitonzo D., Johnson A. E., Skach W. R. (2005). Sequential triage of transmembrane segments by Sec61alpha during biogenesis of a native multispanning membrane protein. Nat. Struct. Mol. Biol. 12, 870–878 10.1038/nsmb994 [DOI] [PubMed] [Google Scholar]
- Sauri A., Saksena S., Salgado J., Johnson A. E., Mingarro I. (2005). Double-spanning plant viral movement protein integration into the endoplasmic reticulum membrane is signal recognition particle-dependent, translocon-mediated, and concerted. J. Biol. Chem. 280, 25907–25912 10.1074/jbc.M412476200 [DOI] [PubMed] [Google Scholar]
- Skach W. R. (2007). The expanding role of the ER translocon in membrane protein folding. J. Cell Biol. 179, 1333–1335 10.1083/jcb.200711107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skach W. R. (2009). Cellular mechanisms of membrane protein folding. Nat. Struct. Mol. Biol. 16, 606–612 10.1038/nsmb.1600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surprenant A., North R. A. (2009). Signaling at purinergic P2X receptors. Annu. Rev. Physiol. 71, 333–359 10.1146/annurev.physiol.70.113006.100630 [DOI] [PubMed] [Google Scholar]
- Torres G. E., Egan T. M., Voigt M. M. (1999). Identification of a domain involved in ATP-gated ionotropic receptor subunit assembly. J. Biol. Chem. 274, 22359–22365 10.1074/jbc.274.32.22359 [DOI] [PubMed] [Google Scholar]
- von Heijne G. (2006). Membrane-protein topology. Nat. Rev. Mol. Cell Biol. 7, 909–918 10.1038/nrm2063 [DOI] [PubMed] [Google Scholar]