

Summary
The metabolic pathway of protein N-glycosylation influences intercellular adhesion by affecting the composition and cytoskeletal association of E-cadherin protein complexes, or adherens junctions (AJs). In sparse cells, E-cadherin is modified extensively with complex N-glycans and forms nascent AJs, while in dense cultures, hypoglycosylated E-cadherin drives the assembly of mature AJs with increased levels of γ- and α-catenins. N-glycosylation of E-cadherin is controlled by the DPAGT1 gene, a key regulator of the N-glycosylation pathway. DPAGT1 is a target of the canonical Wnt signaling pathway, with both β- and γ-catenins binding to Tcf at its promoter. We now report that DPAGT1 senses cell density through canonical Wnt signaling. In dense cells, depletion of β-catenin from the DPAGT1 promoter correlated with downregulation of its cellular abundance, while loss of nuclear γ-catenin reflected its greater recruitment to AJs. DPAGT1 itself affected canonical Wnt signaling, with forced changes in its expression resulting in corresponding changes in transcriptionally active β-catenin and canonical Wnt activity. Remarkably, a 2.4-fold increase in the DPAGT1 mRNA level resulted in increased N-glycosylation and reduced membrane localization of E-cadherin, coincident with dramatic changes in cell morphology. Lastly, we present evidence that N-glycosylation status of E-cadherin controls its antagonism of canonical Wnt signaling. Transfection of hypoglycosylated E-cadherin mutant, V13, but not fully N-glycosylated E-cadherin, into sparse cells inhibited canonical Wnt activity by depleting nuclear β- and γ-catenins. Collectively, our studies show that cells coordinate DPAGT1 expression and protein N-glycosylation with canonical Wnt signaling and E-cadherin adhesion via positive and negative feedback mechanisms.
Key words: DPAGT1, E-cadherin adhesion, N-glycosylation, Wnt signaling, β-catenin, γ-catenin
Introduction
Cell behavior is controlled by highly conserved networks of core signaling pathways that interact with cellular metabolism and cell adhesion. Among them, the canonical Wnt pathway plays a prominent role in regulating proliferation and cell fate (MacDonald et al., 2009; van Amerongen and Nusse, 2009). Canonical Wnt signaling has been shown to regulate, and to be antagonized by, E-cadherin adhesion, a key regulator of cell polarity, proliferation and survival in epithelial tissues (Brembeck et al., 2006; Heuberger and Birchmeier, 2010). Recently, the metabolic pathway of protein N-glycosylation has been shown to affect E-cadherin adhesion and to be a target of canonical Wnt signaling (Liwosz et al., 2006; Nita-Lazar et al., 2010; Sengupta et al., 2010). This suggested reciprocal interactions among canonical Wnt, protein N-glycosylation and E-cadherin adhesion.
Protein N-glycosylation is essential for viability and highly conserved in evolution, but its significance in regulating various cell functions has been relatively unappreciated. Modification of proteins with N-glycans has acknowledged roles in protein folding, targeting, clearance, secretion, intracellular signaling and cell adhesion (Helenius and Aebi, 2001; Zhao et al., 2008). A key regulator of protein N-glycosylation is the evolutionarily conserved DPAGT1 gene. DPAGT1 encodes the dolichol-P-dependent N-acetylglucosamine-1-phosphate-transferase (GPT) that initiates the synthesis of the lipid-linked oligosaccharide (LLO) precursor for protein N-glycosylation in the endoplasmic reticulum (ER) (Helenius and Aebi, 2001; Kukuruzinska and Lennon, 1998). DPAGT1 functions at the rate limiting step in the N-glycosylation pathway, and modest changes in its expression lead to robust changes in the N-glycosylation capacity of cells and N-glycosylation status of proteins (Clark et al., 1983; Hayes and Lucas, 1983; Meissner et al., 1999; Mendelsohn et al., 2005; Sengupta et al., 2010; Welply et al., 1985).
Among numerous proteins affected by DPAGT1 is E-cadherin, the major epithelial cell-cell adhesion receptor and tumor suppressor (Gumbiner, 2005; Jamora and Fuchs, 2002; Takeichi, 1995; Wheelock and Johnson, 2003). The N-glycosylation status of E-cadherin has been shown to regulate the molecular organization and cytoskeletal association of E-cadherin protein complexes, known as adherens junctions (AJs) (Jamal et al., 2009; Liwosz et al., 2006; Nita-Lazar et al., 2009; Nita-Lazar et al., 2010). Increased DPAGT1 expression is associated with extensive N-glycosylation of E-cadherin, which precludes its interaction with stabilizing proteins and with the cytoskeleton and promotes the formation of nascent AJs in proliferating and cancer cells (Liwosz et al., 2006; Nita-Lazar et al., 2009; Nita-Lazar et al., 2010). In contrast, downregulation of DPAGT1 expression leads to reduced N-glycosylation of E-cadherin, which is required for the formation of mature AJs in dense cultures and differentiated cells (Nita-Lazar et al., 2010; Vagin et al., 2008). Accordingly, partial inhibition of DPAGT1 with siRNA results in the production of hypoglycosylated E-cadherin, which organizes mature AJs. In cancer cells, downregulation of DPAGT1 has been shown to reverse their mesenchymal phenotype to an epithelial morphology (Jamal et al., 2012; Nita-Lazar et al., 2009). Likewise, the hypoglycosylated E-cadherin mutant, V13, generated by the deletion of the major complex and high mannose/hybrid N-glycan addition sites, has been shown to form mature AJs in Chinese Hamster Ovary (CHO) cells lacking endogenous E-cadherin (Liwosz et al., 2006). When expressed in Madin-Darby canine kidney (MDCK) cells, V13 also organized mature AJs and drove intercellular adhesion (Liwosz et al., 2006; Nita-Lazar et al., 2010).
Our recent studies demonstrated that DPAGT1 was a target of the canonical Wnt signaling pathway. Activation of Wnt signaling in human, canine and hamster cell lines led to an upregulation of DPAGT1 transcript levels, which was associated with increased abundance of β- and γ-catenins at the DPAGT1 promoter (Sengupta et al., 2010). The canonical Wnt-dependent activation of DPAGT1 expression was recently shown to be a feature of oral tumors in vivo and to be associated with the loss of E-cadherin adhesion (Jamal et al., 2012). N-glycosylation has been reported to be required for the activity of a Wnt ligand, Wnt3a, and for the maturation of the canonical Wnt co-receptor, LRP5/6 (Jung et al., 2011; Khan et al., 2007; Komekado et al., 2007). This suggested that through its effects on the N-glycosylation status of Wnt components, DPAGT1 also affected the canonical Wnt activity.
In contrast to DPAGT1, the genes that function downstream in the N-glycosylation pathway in the ER, ALG1, and in the Golgi, MGAT5, were not direct targets of canonical Wnt signaling (Sengupta et al., 2010). Based on the criteria of sensitivity to different de-glycosylation enzymes, however, we reported that increased DPAGT1 expression correlated with a greater modification of E-cadherin with complex N-glycans, while downregulation of DPAGT1 was associated with diminished complex N-glycans and increased high-mannose/hybrid structures (Nita-Lazar et al., 2009; Nita-Lazar et al., 2010; Sengupta et al., 2010). Since modification with complex N-glycans occurs in the Golgi, this suggested that DPAGT1 was co-regulated with the ER and Golgi N-glycosylation genes.
DPAGT1 and protein N-glycosylation have been shown to be regulated with development and differentiation, and in response to cell density information, but the underlying mechanisms have remained obscure (Clark et al., 1983; Fernandes et al., 1999; Hayes and Lucas, 1983; Meissner et al., 1999; Mota et al., 1994; Welply et al., 1985). Here, using MDCK cells, we report that under normal physiological conditions, DPAGT1 senses cell density via canonical Wnt signaling and AJ maturity. We provide evidence that upregulation in DPAGT1 mRNA was associated with increases in ALG1 and MGAT5 transcript levels. Importantly, both attenuation and amplification of DPAGT1 expression directly influenced cellular levels of transcriptionally active β-catenin and canonical Wnt activity. Remarkably, a modest 2.4-fold increase in DPAGT1 mRNA led to a substantial increase in the N-glycosylation status of E-cadherin and to its reduced membrane localization, coincident with dramatic changes in cell morphology. Moreover, we show that the N-glycosylation status of E-cadherin affects the ability of E-cadherin to antagonize canonical Wnt signaling and DPAGT1 expression. Hypoglycosylated E-cadherin mutant, V13, effectively depleted nuclear β- and γ-catenins, albeit through distinct mechanisms. Our studies identify the first N-glycosylation gene, DPAGT1, as a master control switch that integrates protein N-glycosylation with canonical Wnt signaling and E-cadherin adhesion via positive and negative feedback loops.
Results
DPAGT1 senses cell density information through canonical Wnt signaling
Dense cultures of MDCK cells exhibit decreased endogenous canonical Wnt signaling compared to proliferating cells (Stockinger et al., 2001). Since DPAGT1 has also been shown to be downregulated in growth arrested cells (Fernandes et al., 1999), we examined whether this was a direct consequence of reduced canonical Wnt activity. Analyses of DPAGT1 transcript levels by quantitative PCR revealed a 50% reduction in dense cells compared to sparse cultures (Fig. 1A, DPAGT1). The abundance of the DPAGT1 protein, GPT, was also reduced in dense cells (Fig. 1B, GPT). This decrease in DPAGT1 expression correlated with the reduction of cellular β-catenin levels when normalized to the actin control (Fig. 1B, β-catenin). In contrast, levels of γ-catenin were unchanged between sparse and dense cells (Fig. 1B, γ-catenin). Furthermore, chromatin immunoprecipitation (ChIP) assays revealed that relative to the IgG control, dense cultures displayed a 4.3-fold reduction in the amount of β-catenin and a 4-fold decrease in γ-catenin levels at the DPAGT1 promoter (Fig. 1C). Since cellular levels of γ-catenin were not altered with cell density, this suggested that the depletion of γ-catenin occurred through a mechanism distinct from that of β-catenin.
Fig. 1.

DPAGT1 senses cell density via Wnt/β-catenin signaling. (A) Quantitative PCR of DPAGT1 transcript levels in sparse and dense MDCK cells (***P<0.001). (B) Immunoblot of GPT, β-catenin and γ-catenin expression in sparse and dense cultures. Bar graphs: fold change in GPT, β- and γ-catenin levels after normalization to GAPDH. (C) ChIP analyses of β-catenin and γ-catenin at the DPAGT1 promoter in sparse and dense cells after normalization to the IgG control (**P<0.005). (D) Luciferase reporter activity from the FOP-DPAGT1 vector in sparse and dense cells (*P<0.05). (E) Luciferase reporter activity from the TOP-Flash vector in sparse and dense cells (***P<0.001). (F) Immunoblot of ABC expression in sparse and dense cells. (G) Immunoblot of ICAT in sparse cells after transfection with control vector, pCMV, and vector with ICAT under the CMV promoter. (H) Immunoblot of ABC in sparse cells transfected with either the pCMV control vector or vector with ICAT. Bar graph: fold change in ABC levels after normalization to GAPDH. (I) Luciferase reporter activity from the FOP-Flash vector in sparse cells transfected with either the pCMV control vector or ICAT (*P<0.05). (J) Luciferase reporter activity from the TOP-DPAGT1 vector in sparse cells transfected with either the pCMV control vector or ICAT (*P<0.05). (K) Immunoblot of pGSK-3β and GSK-3β expression in sparse and dense cultures. Bar graph: fold change in pGSK3β/GSK3β levels after normalization to GAPDH. (L) E-cadherins were immunoprecipitated from sparse and dense cultures and their association with β-catenin and α-catenin was assessed by immunoblot. (M) E-cadherins were immunoprecipitated from sparse and dense cultures and their association with γ-catenin was assessed by immunoblot.
Decreased abundance of β- and γ-catenins at the DPAGT1 promoter in dense cultures correlated with 60% lower promoter activity, as reflected by the luciferase reporter activity from the FOP-DPAGT1 vector, containing three tandem repeats of the DPAGT1 Tcf binding region (Fig. 1D) (Sengupta et al., 2010). This was associated with a 93% inhibition of canonical Wnt activity using the TOP-Flash luciferase reporter construct (Fig. 1E). Under conditions of high canonical Wnt activity in sparse cultures, a substantial pool of β-catenin would be expected to be transcriptionally active due to its reduced N-terminal phosphorylation at Ser37 and Thr41, brought about by inhibition of GSK-3β (Kimelman and Xu, 2006; MacDonald et al., 2009). Indeed, immunoblot with an antibody that recognizes active β-catenin, ABC, which is dephosphorylated on Ser37 or Thr41, revealed substantially higher ABC levels in sparse cells compared to dense cultures (Fig. 1F).
To document that density-dependent changes in β-catenin's occupancy of the DPAGT1 promoter were mediated by canonical Wnt activity, we examined the effects of ICAT, an inhibitor of β-catenin and Tcf-4, on FOP-DPAGT1 activity in sparse cells. ICAT is a 9-kDa polypeptide that inhibits β-catenin's nuclear signaling by binding β-catenin and interfering with its interaction with Tcf without substantially affecting E-cadherin junctions (Gottardi and Gumbiner, 2004). Recently, ICAT has been shown to be a downstream target of the E2F1 transcription factor and to reduce the cellular pool of ABC (Wu et al., 2011). Transfection of sparse cells with ICAT driven by the CMV promoter showed a significant increase in its abundance compared to the control, vector expressing cells (Fig. 1G). Such overexpression of ICAT reduced the pool of ABC in sparse cells by 40% (Fig. 1H). Accordingly, both FOP-DPAGT1 and TOP-Flash activities were inhibited by ICAT (Fig. 1I,J).
To determine if downregulation of ABC in dense cells was due to increased activity of GSK-3β, we measured the amount of the inactive form of the enzyme using an antibody to phospho-GSK-3β. Results showed greater amounts of inactive GSK-3β in sparse cells compared to dense cultures, while the total amount of GSK-3β remained unchanged, suggesting an increase in the active enzyme level in dense cells (Fig. 1K). Cell density-dependent downregulation of DPAGT1 expression and canonical Wnt activity was also associated with an increased interaction between E-cadherin and α-catenin, consistent with greater maturity of hypoglycosylated E-cadherin-containing AJs (Fig. 1L) (Liwosz et al., 2006; Nita-Lazar et al., 2010). No significant differences were detected in the association of β-catenin with AJs between sparse and dense cells (Fig. 1L). In contrast to β-catenin, the levels of γ-catenin were not significantly affected by changes in cell density (Fig. 1B). Instead, co-immunoprecipitation studies showed greater abundance of γ-catenin in E-cadherin protein complexes in dense culture compared to sparse cells, as reported by us previously (Fig. 1M) (Liwosz et al., 2006). While the inhibitory effect of cell density on canonical Wnt activity has been previously reported (Steel et al., 2005), this is the first demonstration that cell density-dependent changes in DPAGT1 expression are mediated by the canonical Wnt pathway.
DPAGT1 regulates canonical Wnt signaling
Previously, we showed that partial inhibition of DPAGT1 in MDCK cells and in salivary gland adenocarcinoma A253 cells reduced N-glycosylation of the surface pool of E-cadherin and stabilized AJs (Nita-Lazar et al., 2009; Nita-Lazar et al., 2010). To determine the effects of DPAGT1 on canonical Wnt signaling, we examined whether its partial inhibition affected the levels of β-catenin and its transcriptionally active pool, ABC. Treatment of MDCK cells with 125 nM siRNA (S) resulted in the downregulation of DPAGT1 mRNA by 50% that correlated with lower GPT protein levels (Fig. 2A,B). This modest downregulation of DPAGT1 expression was reflected in a smaller molecular size of E-cadherin on an immunoblot (Fig. 2C). In addition, diminished DPAGT1 expression was associated with a reduction in cellular β-catenin and in ABC, in particular (Fig. 2D). In contrast, no changes in cellular levels of γ-catenin were detected between NS and S cells (Fig. 2E). Likewise, no significant changes in the abundance of GSK-3β or its phosphorylated, inactive form were detected (Fig. 2F). Since only a small pool of GSK-3β participates in the canonical Wnt pathway (Lee et al., 2003), it may not be possible to detect changes in GSK-3β under these conditions. Nonetheless, diminished abundance of both total cellular β-catenin and ABC levels in DPAGT1 siRNA-treated cells suggested that partial inhibition of DPAGT1 led to decreased canonical Wnt signaling. Measurement of the luciferase reporter activity driven from the DPAGT1 promoter in S cells using the FOP-DPAGT1 vector showed more than a 60% reduction (Fig. 2G). Similar attenuation was found for the overall canonical Wnt activity from the TOP-Flash vector in S cells (Fig. 2H). ChIP assays further confirmed that the lower activity of the DPAGT1 promoter reflected its diminished occupancy by β- and γ-catenins in S cells (Fig. 2I,J).
Fig. 2.

Partial silencing of DPAGT1 inhibits canonical Wnt activity. (A) Quantitative PCR of DPAGT1 transcript levels in non-silenced (NS) or DPAGT1 silenced (S) cells (***P<0.001). (B) Immunoblot of GPT in NS and S cells. Bar graph: fold change in GPT levels after normalization to actin. (C) Immunoblot of E-cadherin from NS and S cells. (D) Immunoblot of β-catenin and ABC levels in NS and S cells. Bar graphs: fold change in β-catenin and ABC levels after normalization to GAPDH. (E) Immunoblot of γ-catenin expression in NS and S cells. (F) Immunoblot of pGSK-3β and GSK-3β protein levels in NS and S cells. Bar graph: fold change in pGSK3β/GSK3β levels after normalization to GAPDH. (G) Luciferase reporter activity from the FOP-DPAGT1 vector in NS and S cells (***P<0.001). (H) Luciferase reporter activity from the TOP-Flash vector in NS and S cells (***P<0.001). (I) ChIP analyses of β-catenin and Tcf at the DPAGT1 promoter in NS and S cells after normalization to the IgG control (**P<0.005). (J) ChIP analyses of γ-catenin and Tcf at the DPAGT1 promoter in NS and S cells after normalization to the IgG control (**P<0.005). (K) Immunofluorescence localization of β-catenin, ABC, γ-catenin and LRP5/6, in NS and S cells, counterstained for F-actin and nuclei. Scale bars, 10 µm.
Despite diminished cellular levels of ABC and total β-catenin in S cells, no significant changes were detected in their junctional localization (Fig. 2K, ABC, β-catenin). Both total β-catenin and its active form, ABC, were found at cell-cell borders with some diffuse, cytoplasmic localization. On the other hand, we have shown before that diminished N-glycosylation of E-cadherin enhanced intercellular adhesion by driving the formation of mature AJs that preferentially interacted with γ-catenin. Indeed, diminished nuclear presence of γ-catenin in S cells correlated with its more focused localization to cell-cell contacts and colocalization with F-actin (Fig. 2K, γ-catenin). Importantly, LRP5/6, shown to require N-glycosylation for secretion and membrane localization (Jung et al., 2011; Khan et al., 2007), appeared more abundant and diffusely localized in the cytoplasm in S cells, suggesting that downregulation of N-glycosylation affected its efficient transport to the cell surface (Fig. 2K, LRP5/6).
Since downregulation of DPAGT1 attenuated canonical Wnt signaling, this suggested that amplification of DPAGT1 expression would have the opposite effect and drive canonical Wnt activity. Previously, we and others reported that DPAGT1 had a complex transcription pattern, with two of its mRNAs produced by alternative transcription initiation (Huang et al., 1998; Meissner et al., 1999). We predicted that these transcripts gave rise to two GPT isoforms with different lengths of the N-terminal region (Huang et al., 1998). To test the effects of increased DPAGT1 expression on canonical Wnt signaling, we generated stable transfectants of MDCK cells with recombinant DPAGT1 cDNAs, producing the short variant 2 (TV2) and the full length (TFL) transcripts fused to Myc and DDK tags and under the control of the CMV promoter.
Quantitative PCR analyses showed that compared to the control cells, transfectants with TV2 exhibited a 2.8-fold increase while those with TFL had a 2.4-fold increase in DPAGT1 mRNA levels (Fig. 3A). In addition, both transfectants exhibited significant increases in the steady-state levels of ALG1 and MGAT5, indicating that a modest upregulation in DPAGT1 expression was associated with the amplification of downstream N-glycosylation genes that functioned in the ER and the Golgi (Fig. 3A).
Fig. 3.

Expression of recombinant DPAGT1 drives canonical Wnt signaling and N-glycosylation of E-cadherin. (A) Quantitative PCR of DPAGT1, ALG1 and MGAT5 transcripts from cells transfected with the full length (TFL) and variant 2 (TV2) DPAGT1 cDNA clones. Results represent one of two independent determinations. (B) N-glycosylation of E-cadherin in control cells (C) and TV2 and TFL transfectants before and after PNGaseF treatment. Results are representative of three independent experiments. (C) Immunoblot of recombinant GPT isoforms (Myc tag) and ABC from control cells (C) and TV2 and TFL transfectants. Bar graph: fold change in ABC levels after normalization to GAPDH. Results are representative of one of two independent experiments. (D) Immunoblot of β- and γ-catenins from control cells (C) and TV2 and TFL transfectants. Bar graph: fold change in β- and γ-catenin levels after normalization to GAPDH. Results are representative of one of two experiments. (E) Luciferase reporter activity from the TOP-Flash vector in control (C) cells and TV2 and TFL transfectants. Results are representative of one of three independent determinations. (F) Immunofluorescence localization of GPT (Myc tag) and E-cadherin in control cells (C) and TV2 and TFL transfectants counterstained for F-actin. Scale bars, 10 µm.
Immunoblot analyses using an antibody to Myc tag revealed that TV2 and TFL produced similar levels of recombinant GPT, although under our experimental conditions, no mobility differences between its two isoforms were detected (Fig. 3C, Myc tag). Comparison of endogenous and recombinant GPT levels using an antibody to the C-terminal region of GPT revealed a modest increase, 2- to 3-fold, in TV2 and TFL cells (data not shown). However, immunoblot analyses revealed that E-cadherin from TV2 cells migrated with a higher molecular size compared to the endogenous E-cadherin, while E-cadherin from TFL cells exhibited an even greater increase in size (Fig. 3B). These increases in size were due greater N-glycosylation, since treatment with PNGaseF, an amidase that removes most N-glycans from the asparagine residues on N-glycoproteins, reversed E-cadherin's mobility shifts (Fig. 3B).
Importantly, transfectants with increased DPAGT1 expression displayed greater abundance of active β-catenin, ABC (Fig. 3C). Also, the cellular levels of β-catenin were slightly elevated, while those of γ-catenin exhibited a more pronounced increase (Fig. 3D). Greater ABC levels in TV2 and TFL cells were reflected in a significantly higher TOP-Flash activity, with TFL having a more robust effect (Fig. 3E). This indicated that DPAGT1 directly affected canonical Wnt signaling. To determine if increases in canonical Wnt activity and in the N-glycosylation status of E-cadherin affected E-cadherin junctional organization, we examined immunolocalization of E-cadherin in TV2 and TFL cells. Analysis of recombinant GPT using antibodies to Myc tag revealed diffuse distribution with some perinuclear localization, with a few TFL cells exhibiting a more intense GPT immunostaining (Fig. 3F, Myc tag, TV2, TFL). Whereas E-cadherin displayed well-organized membrane localization in the control cells, it was primarily cytoplasmic in TV2 and TFL cells, suggesting diminished adhesion (Fig. 3F, E-cad). Strikingly, counterstaining for F-actin revealed a greatly altered morphology of TV2 and TFL cells, which included enlarged cell size and a significant increase in stress fibers. Collectively, these results show that DPAGT1 regulates canonical Wnt signaling, and that even modest increases in its expression have dramatic consequences on canonical Wnt activity, E-cadherin localization and cell morphology.
Hypoglycosylated E-cadherin mutant, V13, inhibits canonical Wnt signaling and DPAGT1 expression
We and others have shown that the N-glycosylation status of E-cadherin was attenuated in dense cultures (Liwosz et al., 2006; Vagin et al., 2008). Since dense cultures produce mature AJs containing hypoglycosylated E-cadherin, it seemed likely that the N-glycosylation status of E-cadherin played a role in antagonizing canonical Wnt signaling. Previous studies have shown that the expression of exogenous E-cadherin interfered with cell proliferation and transformation by inhibiting β-catenin transcriptional activity (Gottardi et al., 2001; Maher et al., 2009; Stockinger et al., 2001). Moreover, this suppressive effect of E-cadherin was shown to reflect its ability to increase the turnover of β-catenin in a junction-localized phosphodestruction complex (Maher et al., 2009).
We have reported that complex oligosaccharides modify site 1 in ectodomain 4 (EC4) of E-cadherin, while high mannose/hybrid structures are at site 3 in EC5, with a minor N-glycan at site 2 in EC4 (Liwosz et al., 2006). Our earlier studies also demonstrated that hypoglycosylated E-cadherin mutant, V13, lacking N-glycan addition sites 1 and 3, behaved like E-cadherin in dense cultures, driving the formation of mature AJs by remodeling E-cadherin scaffolds, with increased levels of stabilizing and actin interacting proteins that included γ- and α-catenins (Jamal et al., 2009; Nita-Lazar et al., 2010). Indeed, as shown in Fig. 4A, V13 migrated with a similar molecular size to E-cadherin from dense cultures. Therefore, we reasoned that cells transfected with V13 would organize E-cadherin junctions resembling those formed by endogenous hypoglycosylated E-cadherin in dense cultures. In this way, V13 would serve as a useful tool for examining the effects of hypoglycosylated, mature E-cadherin junctions on canonical Wnt activity and DPAGT1 expression.
Fig. 4.

The hypoglycosylated E-cadherin mutant, V13, inhibits DPAGT1 transcription. (A) Immunoblot comparison of mobilities of E-cadherin and V13 with E-cadherin from sparse and dense cells. (B) Immunoblot comparing exogenous FLAG-tagged E-cadherins, in cells transfected with either wild-type E-cadherin (E-cad) or its hypoglycosylated mutant (V13) (***P<0.001). (C) Quantitative PCR of DPAGT1 transcript levels in untransfected control (C), E-cad and V13 cells. (D) Immunoblot of GPT protein levels in control (C), E-cad and V13 cells. (E) Comparison of DPAGT1 transcript decay rates in untransfected cells and cells transfected with either E-cad or V13. Total RNAs were isolated from cells grown in the presence of 5 µg/ml actinomycin D for 0, 4, 6, 8, 12 and 24 hours. Results are from three independent experiments. (F) ChIP analyses of β-catenin, γ-catenin and Tcf at the DPAGT1 promoter in E-cad and V13 cells after normalization to the IgG control (***P<0.001). (G) Luciferase reporter activity from the FOP-DPAGT1 vector in E-cad and V13 cells (***P<0.001). (H) Luciferase reporter activity from the TOP-Flash vector in E-cad and V13 cells (**P<0.005). (I) Luciferase reporter activity from the TOP-Flash vector in E-cad cells stimulated with either conditioned medium alone (CM) or from cells overexpressing either Wnt3a or Wnt5a (***P<0.001). (J) Luciferase reporter activity from the TOP-Flash vector in V13 cells stimulated with either conditioned medium alone (CM) or from cells overexpressing either Wnt3a or Wnt5a (***P<0.001).
Comparison of E-cadherin abundance in sparse stable transfectants, expressing either wild-type E-cadherin (E-cad) or V13 mutant (V13), showed that V13 was present at reduced levels compared to the fully glycosylated E-cadherin (Fig. 4B). V13 cells, however, exhibited reduced DPAGT1 transcript levels by 50% compared to E-cad cells and to control cells expressing endogenous E-cadherin (Fig. 4C). Accordingly, the abundance of the DPAGT1 protein product, GPT, was decreased in V13 cells (Fig. 4D).
To eliminate the possibility that V13 affected DPAGT1 transcript stability, we examined the DPAGT1 mRNA decay rates following inhibition of transcription with actinomycin D. In untransfected cells, the DPAGT1 mRNA decayed with a half-life (t1/2) of 22 hours (Fig. 4E, C), indicating that it belonged to a class of stable transcripts, typical of metabolic and structural genes (Sharova et al., 2009; Stefanovic et al., 2000). Transfection with E-cadherin led to a small (14%) decrease in the DPAGT1 transcript half-life from 22 hours to 19 hours (Fig. 4E, E-cad). In contrast, V13 did not have a detectable effect on the DPAGT1 mRNA half-life, which remained at 22 hours (Fig. 4E, V13).
We next examined whether V13 inhibited DPAGT1 transcription by affecting the interaction of β- and γ-catenins with the DPAGT1 promoter. Analyses of the DPAGT1 promoter occupancy by β- and γ-catenins using ChIP assays revealed that, relative to the IgG control, V13 caused a 20-fold decrease in the amount of β-catenin and a 50-fold reduction in γ-catenin (Fig. 4F). This reduction in β- and γ-catenin levels at the DPAGT1 promoter by V13 correlated with a 40% inhibition of its promoter activity, measured using the FOP-DPAGT1 vector. In addition, V13 cells exhibited a 50% downregulation of canonical Wnt activity, as assessed with the TOP-Flash reporter construct (Fig. 4G,H). Furthermore, the endogenous canonical Wnt activity in E-cad and V13 cells was stimulated by conditioned medium containing the canonical Wnt ligand, Wnt3a, but not a non-canonical ligand, Wnt5a, indicating that the effect of V13 was specific to canonical Wnt signaling (Fig. 4I,J). Interestingly, while V13 cells exhibited lower basal level of TOP-Flash activity than E-cad cells, the fold increase in their TOP-Flash activity following stimulation with Wnt3a was greater than in E-cad cells. This suggested that V13 suppressed the endogenous Wnt signaling activity to a greater extent than E-cad. Taken together, these results showed that the N-glycosylation status of E-cadherin affected endogenous canonical Wnt signaling.
V13 reduces nuclear pools of β- and γ-catenins through distinct mechanisms
Since V13 cells had diminished levels of transcriptionally active β- and γ-catenins, we next investigated the mechanism(s) underlying the antagonism of canonical Wnt signaling by V13. We reported earlier that V13 preferentially interacted with γ-catenin, but not with β-catenin (Jamal et al., 2009; Liwosz et al., 2006; Nita-Lazar et al., 2010). Likewise, maturation of E-cadherin junctions during salivary gland cytodifferentiation is accompanied by an increased recruitment of γ-catenin to AJs (Walker et al., 2008). Therefore, we compared interactions of E-cadherin and V13 with components of E-cadherin junctions by co-immunoprecipitation (IP) using E-cad and V13 fused to different tags. Routinely, for each IP, controls included input samples, and for specificity, IgG instead of a primary antibody to a protein of interest, as shown in a representative FLAG pull-down of E-cad and V13 (Fig. 5A). Indeed, immunoprecipitation of transfected Myc-tagged E-cadherins with an antibody to Myc tag revealed more γ-catenin in V13 complexes than in those containing fully N-glycosylated E-cadherin (Fig. 5B). On the other hand, a pull-down of FLAG-tagged E-cad and V13 showed no significant difference in their association with β-catenin (Fig. 5C). In contrast, V13 preferentially interacted with α-catenin, confirming our previous reports that V13 organized more mature AJs compared to E-cadherin (Fig. 5D). The maturity of V13-containing AJs was further emphasized by their diminished binding of IQGAP1, a junction destabilizer (Fig. 5E) (Kuroda et al., 1998).
Fig. 5.

The hypoglycosylated E-cadherin, V13, inhibits canonical Wnt signaling through different mechanisms. (A) Representative control for immunoprecipitation experiments. E-cadherins were immunoprecipitated with an antibody to FLAG from E-cad and V13 cells and their association with either control IgG isotypes or FLAG was analyzed by immunoblot. To assure specificity, all immunoprecipitation studies routinely included IgG controls. (B) E-cadherins were immunoprecipitated with an antibody to Myc tag from E-cad and V13 cells and their association with γ-catenin was assessed by immunoblot. (C) FLAG pull-down samples from E-cad and V13 cells and their association with β-catenin was examined by immunoblot. (D) FLAG pull-down samples from E-cad and V13 cells and their association with α-catenin was analyzed by immunoblot. (E) E-cadherins were immunoprecipitated with an antibody to GST from E-cad and V13 cells and their association with IQGAP1 was assessed by immunoblot. (F) Immunoblot of γ-catenin expression in E-cad and V13 cells. (G) Immunoblot of β-catenin expression in E-cad and V13 cells. Bar graph: fold change in β-catenin levels after normalization to GAPDH. (H) Immunoblot of ABC protein levels in E-cad and V13 cells. (I) Immunoblot of ABC and β-catenin levels in nuclear extracts from E-cad and V13 cells. (J) Immunoblot of ABC in E-cad cells transfected with either the pCMV control vector or an active β-catenin inhibitor, ICAT. Bar graph: fold change in ABC levels after normalization to GAPDH. (K) Luciferase reporter activity from the TOP-Flash vector in E-cad cells transfected with either the pCMV control vector or ICAT (**P<0.005). (L) Luciferase reporter activity from the FOP-DPAGT1 vector in E-cad cells transfected with either the pCMV control vector or ICAT (***P<0.001). (M) Immunofluorescence localization of β-catenin, ABC, γ-catenin, α-catenin, IQGAP1, p120 and Myc tag in E-cad and V13 cells counterstained for F-actin. Scale bars, 10 µm.
Immunoblot analyses revealed that E-cad and V13 cells had similar amounts of total cellular γ-catenin (Fig. 5F). Since V13 displayed increased association with γ-catenin compared to E-cad (Fig. 5B), this suggested that V13 depleted γ-catenin from the promoters of Wnt responsive genes by its increased recruitment to AJs. On the other hand, we detected a reduction in cellular β-catenin in V13 cells (Fig. 5G), which corresponded to a pronounced attenuation in the pool of its transcriptionally active form, ABC (Fig. 5H). This attenuation in ABC and β-catenin abundance in V13 cells correlated with a decrease in its nuclear pool when normalized to lamin B receptor (Fig. 5I). We note that the differences in total cellular β-catenin levels between E-cad and V13 cells were most prominent at sparse cell densities, or about 30–40% confluence, after which they became less pronounced. To validate that high ABC levels in E-cad cells reflected transcriptionally active β-catenin, we inhibited its nuclear signaling by transfecting E-cad cells with ICAT. As shown in Fig. 5J, ICAT reduced cellular levels of ABC in E-cad cells. As expected, ICAT inhibited canonical Wnt and DPAGT1 promoter activities, as measured by the luciferase reporter gene expression from the TOP-Flash and FOP-DPAGT1 vectors, respectively (Fig. 5K,L). These results confirmed that high levels of ABC in E-cad cells reflected its signaling function.
Examination of total β-catenin and ABC localization by indirect immunofluorescence and confocal microscopy did not reveal any significant differences between E-cad and V13 cells (Fig. 5M, ABC and β-catenin). In contrast, both γ- and α-catenins were more focused at cell-cell contact sites in V13 cells (Fig. 5M, γ- and α-catenins). As expected, IQGAP1 was less prominent at cell-cell borders in V13 cells (Fig. 5M, IQGAP1). No major differences were detected in the distribution of p120 in E-cad and V13 cells, suggesting that the recycling of E-cadherin did not depend on its N-glycosylation status (Fig. 5M, p120). On the other hand, V13 cells displayed more co-localization with F-actin than E-cad cells, as predicted for cells forming mature E-cadherin junctions (Fig. 5M, Myc tag).
Our results so far have shown that V13 downregulates the activity of the DPAGT1 promoter by reducing the pool of transcriptionally active β-catenin. To examine if V13 influenced the activity of GSK-3β, we compared its cellular abundance with its inactive, phosphorylated form. After normalization to GAPDH levels, we found a decreased ratio of inactive to total GSK-3β in V13 cells (Fig. 6A), which reflected a statistically significant change based on four experiments (P<0.05). Thus, the depletion of β-catenin from the DPAGT1 promoter and diminished canonical Wnt activity could be, at least in part, due to an increased pool of active GSK-3β. Indeed, immunofluorescence localization of GSK-3β showed that it was preferentially distributed to cell-cell contacts in V13 cells (Fig. 6I, GSK-3β), supporting the recent report that increased β-catenin turnover by E-cadherin was associated with enhanced activity of the phosphodestruction complex at the sites of cell-cell contacts (Maher et al., 2009).
Fig. 6.

Potential mechanism of V13 action. (A) Immunoblot of pGSK-3β and GSK-3β expression in E-cad and V13 cells. Bar graph: fold change in the pGSK-3β/GSK-3β ratio after normalization to GAPDH. (B) Immunoblot of co-immunoprecipitation of CD82 tetraspanins with E-cadherin, β-catenin and γ-catenin from E-cad and V13 cells. (C) Immunoblot of β-catenin expression after inhibition of sphingomyelin synthesis by GW4869 in E-cad and V13 cells. (D) Immunoblot of ABC protein levels after inhibition of sphingomyelin synthesis by GW4869 in E-cad and V13 cells. (E) Immunoblot of Dkk1 protein levels in E-cad and V13 cells. (F) Immunoblot of Dkk3 expression in E-cad and V13 cells. (G) Immunoblot of LRP6 before and after treatment with PNGaseF from E-cad and V13 cells. (H) Immunoblot of FLAG pull-down of E-cadherins from E-cad and V13 cells and their association with LRP5/6. Bar graph: fold change in LRP levels after normalization to FLAG. (I) Immunofluorescence localization of GSK-3β, LRP5/6 and Wnt3a in E-cad and V13 cells counterstained for F-actin. Scale bars, 10 µm.
Another potential mechanism for the depletion of β-catenin in V13 cells could involve exosomal extrusion, a recently described mechanism for reducing cellular pools of transcriptionally active β-catenin in a GSK-3β-independent manner (Chairoungdua et al., 2010). Therefore, we examined the interaction of E-cad/β-catenin and V13/β-catenin complexes with CD82 and CD9 tetraspanins. Results showed that in V13 cells, E-cadherin, β- and γ-catenins interacted less with CD82 than in E-cad cells (Fig. 6B). Similar interaction was found for E-cad and V13 with CD9 tetraspanin (data not shown). To determine if diminished association between V13 and CD82 was due to the extrusion of CD82/V13/β-catenin from V13 cells, we inhibited cells with a sphingomyelin synthesis inhibitor, GW4869. No significant accumulation of either ABC or total β-catenin was detected in sphingomyelin-inhibited E-cad or V13 cells, indicating that exosome extrusion was not a likely mechanism of β-catenin depletion by V13 (Fig. 6C,D).
The observed diminished pools of ABC in V13 cells could also arise due to the attenuation of canonical Wnt signaling at the cell membrane. Immunoblot analyses showed no changes in the expression of canonical Wnt inhibitors, DKK1 and DKK3 (Fig. 6E,F). However, LRP6 migrated with a higher molecular size in E-cad than in V13 cells, and this slower migration was reversed by treatment with PNGaseF, indicating that it was due to its increased N-glycosylation status in E-cad cells (Fig. 6G). No differences were detected in the phosphorylation status of LRP6 in E-cad and V13 cells (data not shown). Since another classical cadherin, neuronal N-cadherin has been shown to negatively regulate canonical Wnt signaling by interacting with LRP5 and axin and promoting degradation of β-catenin (Haÿ et al., 2009), we examined interactions of E-cad and V13 with LRP5/6 by co-immunoprecipitation. Although both E-cadherins interacted with LRP5/6, the V13 complexes had increased levels of the co-receptor (Fig. 6H). Moreover, immunofluorescence localization showed that in addition to its membrane association, LRP5/6 exhibited a more pronounced diffuse cytoplasmic staining in V13 cells (Fig. 6I, LRP5/6). This suggested that diminished N-glycosylation of V13 and/or LRP5/6 promoted their interaction, while also interfering with LRP5/6 membrane localization. Although levels of endogenous Wnt3a were too low to evaluate its N-glycosylation status, V13 cells had increased cytoplasmic staining of Wnt3a, suggesting that like LRP5/6, it was more retained in the cytoplasm (Fig. 6I, Wnt3a).
Discussion
Our work demonstrates that cells utilize positive and negative feedback mechanisms to integrate the activities of the metabolic pathway of protein N-glycosylation, canonical Wnt signaling and E-cadherin-mediated adhesion. We show that the first N-glycosylation gene and a key regulator of protein N-glycosylation, DPAGT1, senses cell density information via the canonical Wnt signaling pathway, which, in turn, is associated with the maturity of AJs. We then illustrate that DPAGT1 itself directly affects canonical Wnt signaling, in part, by controlling the cellular retention of its co-receptor, LRP5/6. Moreover, we provide evidence that the ability of E-cadherin to interfere with canonical Wnt signaling by depleting nuclear β- and γ-catenins depends on its N-glycosylation status. A schematic of these interactions is shown in Fig. 7.
Fig. 7.
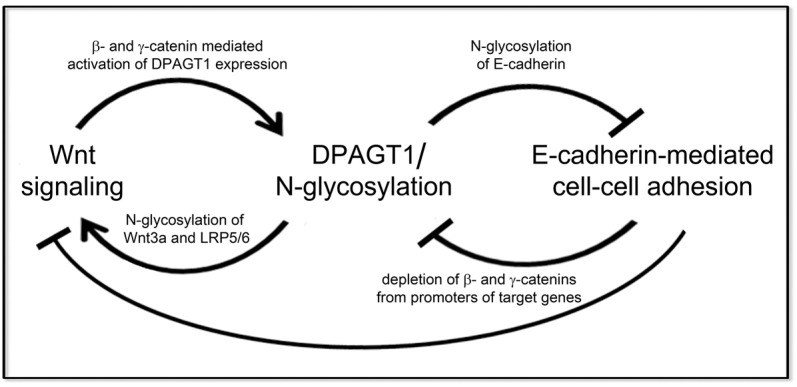
Interactions among canonical Wnt signaling, DPAGT1/N-glycosylation and E-cadherin. Schematic illustrating the positive and negative feedback loops among E-cadherin, canonical Wnt signaling and DPAGT1/N-glycosylation. Canonical Wnt signaling activates DPAGT1 transcription, and DPAGT1 promotes N-glycosylation of Wnt components to further enhance Wnt signaling. High DPAGT1 expression causes extensive N-glycosylation of E-cadherin, which, in turn, inhibits E-cadherin adhesion. Under conditions of diminished canonical Wnt signaling, DPAGT1 expression is reduced, leading to hypoglycosylation of E-cadherin. Hypoglycosylated E-cadherin inhibits canonical Wnt signaling by depleting β- and γ-catenins from promoters of target genes, including the DPAGT1 promoter, thus maintaining low canonical Wnt activity, DPAGT1 expression and the hypoglycosylated status of E-cadherin in mature AJs.
While the inhibitory effect of cell density on canonical Wnt activity has been previously reported (Clevers, 2006; Maher et al., 2009; Steel et al., 2005), our studies provide the first demonstration that cell density-dependent changes in Wnt activity regulate DPAGT1 expression by the depletion of β- and γ-catenins from the TCF/LEF site in its promoter. Downregulation of β-catenin was coincident with the reduction of total cellular β-catenin and ABC, consistent with a recent report showing that attenuation of ABC with cell density was associated with the localization of the phosphodestruction complex to cell-cell contact sites and with increased abundance of E-cadherin (Maher et al., 2009). Our results suggest that increased maturity of E-cadherin junctions in dense cultures promoted the reduction of β-catenin levels. Indeed, E-cadherin protein complexes from dense cells exhibited greater interaction with α-catenin compared to AJs from sparse cells (Fig. 1L) (Liwosz et al., 2006; Nita-Lazar et al., 2010). Furthermore, the depletion of γ-catenin from the DPAGT1 promoter in dense cultures was associated with its increased abundance at AJs that also contained more α-catenin. Since increased interaction of γ- and α-catenins with AJs has been linked to their stabilization (Hartsock and Nelson, 2008; Kobielak and Fuchs, 2004; Leonard et al., 2008), our studies suggest that in dense cells, AJ maturity is linked to the reduced levels of transcriptionally active β-catenin.
The notion that E-cadherin junctional maturity influences levels of active β-catenin is further supported by our results showing that the N-glycosylation status controls E-cadherin's ability to antagonize canonical Wnt signaling and DPAGT1 expression. Transfection of sparse MDCK cells with V13 inhibited DPAGT1 expression by interfering with the transcriptional activities of β- and γ-catenins. In both cases, the consequences of V13 transfection resembled the scenario in dense cultures: ABC was depleted via downregulation of β-catenin's abundance, while γ-catenin was preferentially recruited to AJs. This selective depletion of β-catenin by V13 could be due to GSK-3β-driven increased degradation of β-catenin. Although GSK-3β activity affects numerous cellular functions, with a relatively small pool involved in regulating β-catenin turnover in the destruction complex (Lee et al., 2003), we detected increased localization of GSK-3β to cell-cell contact sites in V13 cells. Thus, mature, hypoglycosylated E-cadherin-containing AJs are likely to promote the turnover of active β-catenin via membrane-localized GSK-3β. This is consistent with the recently reported localization of the phosphodestruction complex to cell-cell contacts being responsible for N-terminal phosphorylation and turnover of β-catenin (Maher et al., 2009). At the same time, since V13 appeared to associate more with LRP5/6, we cannot exclude the possibility that, similar to N-cadherin-dependent regulation of osteoblast function, interaction of V13 with LRP5/6 and axin could also account for its negative regulation of canonical Wnt signaling (Haÿ et al., 2009).
In contrast to β-catenin, the levels of cellular γ-catenin were similar in E-cad and V13 cells. Thus, the depletion of γ-catenin from the DPAGT1 promoter can be explained by its increased sequestration to V13-containing AJs. Previous studies with neuronal PC12 cells showed that Wnt1-dependent increased interactions between γ-catenin and E-cadherin were able to drive cell-cell adhesion and epithelial phenotype in these cells, highlighting the importance of this catenin in E-cadherin adhesion (Bradley et al., 1993). In addition to γ-catenin, V13 preferentially interacted with α-catenin, an important regulator of actin organization and an enhancer of E-cadherin junctional maturity (Kobielak and Fuchs, 2004). Accordingly, V13 exhibited less interaction with IQGAP1, shown to destabilize E-cadherin junctions by competing with α-catenin for binding to β-catenin (Kuroda et al., 1998). Therefore, our results further highlight the importance of N-glycosylation in determining the maturity of E-cadherin junctions and their ability to inhibit canonical Wnt signaling.
In addition to controlling E-cadherin's adhesive function, N-glycosylation has been shown to affect the activity, secretion and cell surface localization of canonical Wnt components, including Wnt3a and LRP5/6 (Khan et al., 2007; Komekado et al., 2007). Indeed, our results demonstrate that partial inhibition of DPAGT1 downregulated endogenous canonical Wnt signaling that correlated with diminished membrane localization of the Wnt co-receptor, LRP5/6. While we could not consistently detect Wnt3a in DPAGT1 siRNA-treated cells, cells transfected with V13 also displayed increased cytoplasmic distribution of both LRP5/6 and Wnt3a. These studies confirm our recent findings that downregulation of DPAGT1 in head and neck squamous cell carcinoma, A253 cells, attenuated canonical Wnt signaling, indicating that positive regulation between DPAGT1 and canonical Wnt signaling represents a feature shared by different cell types, including normal and cancer cells (Jamal et al., 2012).
We also show that increased expression of DPAGT1 had dramatic effects on canonical Wnt activity, E-cadherin's N-glycosylation and distribution, as well as on cell morphology. Stable MDCK cell transfectants with either the full length (TFL) or truncated variant 2 (TV2) cDNAs displayed 2.4- and 2.8-fold increases in the levels of corresponding transcripts. Although complex transcription patterns have been reported for DPAGT1, virtually nothing is known about their functional significance (Huang et al., 1998; Meissner et al., 1999). Our initial studies suggested that in differentiated cells, the shorter TV2 variant predominated, while the full length TFL was more abundant in embryonic and tumor tissues (M. K., unpublished observations). In our present studies, expression of either transcript produced similar levels of recombinant GPT, which led to significant increases in canonical Wnt activity. Interestingly, although TV2 cells had higher mRNA levels compared to TFL cells, the latter exhibited an almost 2-fold greater TOP-Flash activity, suggesting that the longer GPT isoform was more effective in inducing canonical Wnt signaling. Although further studies are needed to elucidate the molecular details underlying functional differences of the GPT isoforms, these results further confirmed that DPAGT1 and canonical Wnt signaling functioned in a positive feedback loop. Furthermore, the N-glycosylation status of E-cadherin was significantly increased in TFL and TV2 transfectants, and this correlated with its reduced membrane localization. The most striking feature of cells with increased DPAGT1 expression was their altered phenotype that included enlarged size and increased abundance of stress fibers, suggesting that they have acquired a mesenchymal phenotype. Studies are in progress to characterize the effects of increased DPAGT1 expression on downstream signaling events leading to these changes in cellular phenotype.
Upregulation of DPAGT1 expression was also associated with increased mRNA levels of ALG1 and MGAT5 genes, suggesting coordinate regulation of N-glycosylation genes in the ER and Golgi. This supports our previous observations showing changes in the sensitivities of N-glycoproteins to different de-glycosylation enzymes associated with changes in DPAGT1 expression. Since modification with complex N-glycans occurs in the Golgi, it is likely that in addition to MGAT5, other Golgi N-glycosylation genes are co-regulated with DPAGT1.
The mechanism via which N-glycosylation affects E-cadherin adhesive strength is not well understood. Our published and current co-immunoprecipitation studies suggest that N-glycosylation of E-cadherin ectodomains promotes conformational changes of its cytoplasmic tail in cis that lead to increased interaction with actin binding and crosslinking proteins and with the actin cytoskeleton (Jamal et al., 2009; Liwosz et al., 2006; Nita-Lazar et al., 2010). It is possible that such conformational changes result in increased compaction of E-cadherin, although, at least in MDCK cells, this compaction would have to occur in the absence of major changes in E-cadherin abundance.
While the role of β-catenin in Wnt signaling is well defined, the function of γ-catenin has been controversial (Shimizu et al., 2008; Zhurinsky et al., 2000b). Depending on cell context, γ-catenin can either facilitate transcriptional activation by β-catenin or function itself as a Wnt-dependent transcription factor (Maeda et al., 2004; Zhurinsky et al., 2000a). The precise function of γ-catenin at the DPAGT1 promoter requires further investigation. Since cytoplasmic accumulation of β- and γ-catenins has been shown to involve different mechanisms, and their interaction with Tcf occurs at different sites (Miravet et al., 2002), it is not surprising that V13 utilized distinct mechanisms to interfere with their transcriptional activities. Taken together, our studies identify β- and γ-catenins as the E-cadherin N-glycosylation-sensitive molecular links that align AJ maturity with protein N-glycosylation and canonical Wnt signaling.
In summary, the work presented here shows that DPAGT1 and the metabolic pathway of protein N-glycosylation interact with canonical Wnt signaling and E-cadherin adhesion via positive and negative feedback loops. DPAGT1 is regulated with cell density through the canonical Wnt signaling pathway. Likewise, DPAGT1 itself affects canonical Wnt signaling by affecting, at least in part, the N-glycosylation status and membrane localization of its components. Moreover, the N-glycosylation status of E-cadherin determines its ability to antagonize canonical Wnt signaling and DPAGT1 expression by depleting β- and γ-catenins from the canonical Wnt target genes. Thus, DPAGT1 and canonical Wnt function in a positive feedback loop, while DPAGT1 and E-cadherin adhesion are inversely related. Collectively, our studies reveal novel molecular mechanisms via which cells coordinate the activities of protein N-glycosylation, Wnt signaling and E-cadherin-mediated adhesion. These interactions are likely to belong to a core network of cellular processes that hardwire cells, and whose dysregulation is associated with disease states.
Materials and Methods
Reagents
Monoclonal antibodies to E-cadherin, p120, IQGAP1, β-catenin, γ-catenin, α-catenin, CD82, CD9 and IgG isotype controls were obtained from BD Transduction Laboratories. Polyclonal antibodies to Lamin B receptor and Wnt3a were from Abcam and LRP5/6 was from Thermo Fisher Scientific. Monoclonal antibodies to Myc tag, GSK-3β and LRP6 and polyclonal antibody to pGSK-3β were from Cell Signaling Technology. Monoclonal antibodies to Tcf3/4 and ABC were from Exalpha Biologicals and Millipore, respectively. Monoclonal antibody to pan-actin Ab-5 was from NeoMarkers while polyclonal antibody to GAPDH was from Sigma. Polyclonal antibody to ICAT was from Santa Cruz Biotechnology and an antibody to GST was purchased from Invitrogen. Polyclonal Dkk1 and Dkk3 antibodies were purchased from Abcam. Rabbit polyclonal antibody to hamster GPT was prepared commercially (Covance Research Products, Inc.).
Cell culture, transfections and cell lysates
MDCK cells (NBL-2, American Type Culture Collection) were maintained in DMEM (Gibco), supplemented with 10% FBS, penicillin and streptomycin. For sparse cultures, cells were plated at 5×103/cm2 and grown for 24 hours, while dense cultures cells were plated at 5×104/cm2 and grown for 72 h.
MDCK transfectants expressing exogenous E-cad and V13 were generated by transfection with pCMV5B vectors containing cDNAs encoding either wild type E-cadherin or its mutated version, V13, at 80–90% confluence using Lipofectamine 2000. Transfectants were enriched for two weeks in the presence of G418 (0.8 µg/ml) and processed for analyses, as described (Nita-Lazar et al., 2010). Transfectants expressing recombinant DPAGT1, either full length (TFL) or variant 2 (TV2), were obtained by transfection of passage 2 MDCK cells with DPAGT1 full length (Refseq NM_001382) and transcript variant (Refseq NM_203316) cDNA clones (Origene) at 80–90% confluence using Lipofectamine 2000. Controls included untransfected cells and cells transfected with a control pCMV6-Entry vector. After 14 h, the media were changed, and cells were divided into several plates and grown in the presence of G418. Media were changed every 2–3 days and supplemented with G418. After two weeks, cells were processed for RNA isolation and preparation of total cell lysates (TCLs). For immunofluorescence analyses, stable transfectant cells were plated in chamber slides at a density of 5–6×103/cm2 and processed as described before (Nita-Lazar et al., 2010).
To compare the effects of Wnt3a and Wnt5a, MDCK cells were grown to 80–90% confluence, cells were serum starved for 24 h and then grown in the presence of 50% conditioned medium isolated from either L-mouse fibroblasts or L-mouse fibroblasts stably expressing either Wnt3a or Wnt5a cDNA (ATCC) for 24 h. In order to examine the effect of ICAT, sparse MDCK cells and E-cadherin cells were transfected with pFLAG-CMV-2 control vector or pFLAG-CMV-2-ICAT (gift from Cara Gottardi, Northwestern University) and the TOP-Flash or FOP-DPAGT1 constructs. In order to investigate the exosomal discharge of β-catenin, E-cad- and V13-transfected MDCK cells were treated with a sphingomyelinase inhibitor, GW4869 (5 µM) (EMD Chemicals), for 16 h. Protein concentrations were determined using BCA assay (Pierce).
Immunoblots
TCLs were fractionated on 7.5% SDS-PAGE, transferred onto PVDF membranes and processed as described (Nita-Lazar et al., 2010). Nuclear extract were prepared from E-cadherin- and V13-transfected MDCK cells according to a published procedure (Dignam et al., 1983).
Immunoprecipitation and FLAG pull-down
Equal amounts of TCL (200 µg) were precleared with IgG isotype control antibodies and 30 µl of protein G-coupled Sepharose beads (GE Healthcare) or protein A-coupled Agarose beads, incubated for 2 h at 4°C with 2.5 µg of antibodies against either E-cadherin, Myc, FLAG, GST or CD82, followed by incubation with protein G or A beads. To assure specificity, all immunoprecipitation studies routinely included IgG controls. The beads were washed three times with lysis buffer (10 mM Tris–HCl, pH 7.5, 1 mM EDTA, 1 mM EGTA and 0.5% Triton X-100), samples were resuspended in 100 µl of 2×SDS sample buffer, boiled for 5 min at 95°C and analyzed by immunoblot. TCLs from MDCK cells transfected with either E-cad- or V13-FLAG- tagged constructs were prepared in RIPA buffer. Exogenous E-cadherins were pulled down by treating TCLs (300 µg) with FLAG-M2 agarose (Sigma) at 4°C for 2 h. Proteins were eluted using 2×SDS sample buffer and analyzed by immunoblot.
RNA interference
SMART-poolTM of siRNAs targeting DPAGT1 (S) was obtained from Dharmacon. Non-silencing (NS) negative control siRNA was from Qiagen. MDCK cells were transfected at 80–90% confluence with 125 nM of either NS or S using Lipofectamine 2000 (Invitrogen) and cultured for 72 h.
Quantitative RT-PCR
Total RNAs, isolated from sparse and dense MDCK cells, or cells transfected with either E-cadherin or V13, and cells transfected with TV2 and TFL construct, were used for cDNA synthesis to assess DPAGT1, MGAT5 and ALG1 expression by real-time PCR, as described before (Sengupta et al., 2010).
mRNA stability
Total RNAs were extracted from untransfected, E-cadherin- or V13-transfected MDCK cells grown in the presence of 5 µg/ml actinomycin D (Sigma) for 0, 4, 6, 8, 12 and 24 hours using an RNeasy RNA isolation kit (Qiagen). Steady state mRNA levels were measured by quantitative RT-PCR, as described above. 18S was used as an endogenous control.
Chromatin immunoprecipitation (ChIP)
Chromatin in sparse and dense, NS and S or E-cadherin- and V13-transfected cells was cross-linked with 1% formaldehyde and prepared for ChIP analyses as described before (Sengupta et al., 2010). The primers surrounding the Wnt responsive element were: forward primer 5′ TTTTCCGCTTTGGGCTATACA3′, reverse primer 5′AATTAAAGACAGACCAACCAAAACG3′, and the Taqman probe was: 6FAMACACGGTGTTAGCAA AGMGBFNQ.
Luciferase assays
Plasmid DNAs (2 µg), TOP-Flash, FOP-Flash and FOP-DPAGT1 (FOP-Flash containing 3×human DPAGT1 sequence), were transfected into untreated sparse and dense cultures, DPAGT1 silenced and non-silenced MDCK cells or cells bearing either wild type E-cadherin or V13 and cells harboring DPAGT1 cDNA clones (TV2 and TFL) and processed for luciferase activity as described. An empty pGL3-Basic vector was used as a control and a reference plasmid, while PSV-beta-gal (0.1 µg, Promega) was used to normalize transfection efficiency. Values were normalized to β-galactosidase activity. In all TOP-Flash and FOP-DPAGT1 assays, the FOP-Flash vector was separately transfected and its luciferase reporter values were determined. These values were then subtracted from experimental reporter readouts to obtain normalized luciferase activities.
Immunofluorescence imaging
Immunofluorescence analyses were carried out with a Zeiss Axiovert 200 M confocal microscope; settings were fixed to the most highly stained sample and all other images were acquired at those settings (Nita-Lazar et al., 2009). Negative controls lacked primary antibodies. Rhodamine-phalloidin was obtained from Molecular Probes. Secondary antibodies included goat anti-mouse or anti-rabbit IgG derivatized with fluorescein isothiocyanate (FITC) (Molecular Probes).
Footnotes
Funding
This work was supported by the National Institute of Dental and Craniofacial Research, National Institutes of Health [grant number RO1 DE015304 to M.A.K.]. Deposited in PMC for release after 12 months.
References
- Bradley R. S., Cowin P., Brown A. M. (1993). Expression of Wnt-1 in PC12 cells results in modulation of plakoglobin and E-cadherin and increased cellular adhesion. J. Cell Biol. 123, 1857–1865 10.1083/jcb.123.6.1857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brembeck F. H., Rosário M., Birchmeier W. (2006). Balancing cell adhesion and Wnt signaling, the key role of beta-catenin. Curr. Opin. Genet. Dev. 16, 51–59 10.1016/j.gde.2005.12.007 [DOI] [PubMed] [Google Scholar]
- Chairoungdua A., Smith D. L., Pochard P., Hull M., Caplan M. J. (2010). Exosome release of β-catenin: a novel mechanism that antagonizes Wnt signaling. J. Cell Biol. 190, 1079–1091 10.1083/jcb.201002049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark G. F., Miller K. R., Smith P. B. (1983). Formation of dolichol-linked sugar intermediates during the postnatal development of skeletal muscle. J. Biol. Chem. 258, 14263–14270 [PubMed] [Google Scholar]
- Clevers H. (2006). Wnt/beta-catenin signaling in development and disease. Cell 127, 469–480 10.1016/j.cell.2006.10.018 [DOI] [PubMed] [Google Scholar]
- Dignam J. D., Lebovitz R. M., Roeder R. G. (1983). Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res. 11, 1475–1489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes R. P., Cotanche D. A., Lennon–Hopkins K., Erkan F., Menko A. S., Kukuruzinska M. A. (1999). Differential expression of proliferative, cytoskeletal, and adhesive proteins during postnatal development of the hamster submandibular gland. Histochem. Cell Biol. 111, 153–162 10.1007/s004180050345 [DOI] [PubMed] [Google Scholar]
- Gottardi C. J., Gumbiner B. M. (2004). Role for ICAT in beta-catenin-dependent nuclear signaling and cadherin functions. Am. J. Physiol. Cell Physiol. 286, C747–C756 10.1152/ajpcell.00433.2003 [DOI] [PubMed] [Google Scholar]
- Gottardi C. J., Wong E., Gumbiner B. M.2001). E-cadherin suppresses cellular transformation by inhibiting β-catenin signaling in an adhesion-independent manner. J. Cell Biol. 153, 1049–1060 10.1083/jcb.153.5.1049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gumbiner B. M. (2005). Regulation of cadherin-mediated adhesion in morphogenesis. Nat. Rev. Mol. Cell Biol. 6, 622–634 10.1038/nrm1699 [DOI] [PubMed] [Google Scholar]
- Hartsock A., Nelson W. J. (2008). Adherens and tight junctions: structure, function and connections to the actin cytoskeleton. Biochim. Biophys. Acta 1778, 660–669 10.1016/j.bbamem.2007.07.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haÿ E., Laplantine E., Geoffroy V., Frain M., Kohler T., Müller R., Marie P. J. (2009). N-cadherin interacts with axin and LRP5 to negatively regulate Wnt/beta-catenin signaling, osteoblast function, and bone formation. Mol. Cell. Biol. 29, 953–964 10.1128/MCB.00349-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes G. R., Lucas J. J. (1983). Stimulation of lipid-linked oligosaccharide assembly during oviduct differentiation. J. Biol. Chem. 258, 15095–15100 [PubMed] [Google Scholar]
- Helenius A., Aebi M. (2001). Intracellular functions of N-linked glycans. Science 291, 2364–2369 10.1126/science.291.5512.2364 [DOI] [PubMed] [Google Scholar]
- Heuberger J., Birchmeier W. (2010). Interplay of cadherin-mediated cell adhesion and canonical Wnt signaling. Cold Spring Harb. Perspect. Biol. 2, a002915 10.1101/cshperspect.a002915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang G. T., Lennon K., Kukuruzinska M. A. (1998). Characterization of multiple transcripts of the hamster dolichol-P-dependent N-acetylglucosamine-1-P transferase suggests functionally complex expression. Mol. Cell. Biochem. 181, 97–106 10.1023/A:1006877929614 [DOI] [PubMed] [Google Scholar]
- Jamal B. T., Nita–Lazar M., Gao Z., Amin B., Walker J., Kukuruzinska M. A. (2009). N-glycosylation status of E-cadherin controls cytoskeletal dynamics through the organization of distinct β-catenin- and γ-catenin-containing AJs. Cell Health Cytoskelet. 2009, 67–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamal B., Sengupta P. K., Gao Z. N., Nita–Lazar M., Amin B., Jalisi S., Bouchie M. P., Kukuruzinska M. A. (2012). Aberrant amplification of the crosstalk between canonical Wnt signaling and N-glycosylation gene DPAGT1 promotes oral cancer. Oral Oncol. 48, 523–529 10.1016/j.oraloncology.2012.01.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamora C., Fuchs E. (2002). Intercellular adhesion, signalling and the cytoskeleton. Nat. Cell Biol. 4, E101–E108 10.1038/ncb0402-e101 [DOI] [PubMed] [Google Scholar]
- Jung H., Lee S. K., Jho E. H. (2011). Mest/Peg1 inhibits Wnt signalling through regulation of LRP6 glycosylation. Biochem. J. 436, 263–269 10.1042/BJ20101512 [DOI] [PubMed] [Google Scholar]
- Khan Z., Vijayakumar S., de la Torre T. V., Rotolo S., Bafico A. (2007). Analysis of endogenous LRP6 function reveals a novel feedback mechanism by which Wnt negatively regulates its receptor. Mol. Cell. Biol. 27, 7291–7301 10.1128/MCB.00773-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimelman D., Xu W. (2006). beta-catenin destruction complex: insights and questions from a structural perspective. Oncogene 25, 7482–7491 10.1038/sj.onc.1210055 [DOI] [PubMed] [Google Scholar]
- Kobielak A., Fuchs E. (2004). Alpha-catenin: at the junction of intercellular adhesion and actin dynamics. Nat. Rev. Mol. Cell Biol. 5, 614–625 10.1038/nrm1433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komekado H., Yamamoto H., Chiba T., Kikuchi A. (2007). Glycosylation and palmitoylation of Wnt-3a are coupled to produce an active form of Wnt-3a. Genes Cells 12, 521–534 10.1111/j.1365-2443.2007.01068.x [DOI] [PubMed] [Google Scholar]
- Kukuruzinska M. A., Lennon K. (1998). Protein N-glycosylation: molecular genetics and functional significance. Crit. Rev. Oral Biol. Med. 9, 415–448 10.1177/10454411980090040301 [DOI] [PubMed] [Google Scholar]
- Kuroda S., Fukata M., Nakagawa M., Fujii K., Nakamura T., Ookubo T., Izawa I., Nagase T., Nomura N., Tani H.et al. (1998). Role of IQGAP1, a target of the small GTPases Cdc42 and Rac1, in regulation of E-cadherin-mediated cell-cell adhesion. Science 281, 832–835 10.1126/science.281.5378.832 [DOI] [PubMed] [Google Scholar]
- Lee E., Salic A., Krüger R., Heinrich R., Kirschner M. W. (2003). The roles of APC and Axin derived from experimental and theoretical analysis of the Wnt pathway. PLoS Biol. 1, E10 10.1371/journal.pbio.0000010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard M., Chan Y., Menko A. S. (2008). Identification of a novel intermediate filament-linked N-cadherin/gamma-catenin complex involved in the establishment of the cytoarchitecture of differentiated lens fiber cells. Dev. Biol. 319, 298–308 10.1016/j.ydbio.2008.04.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liwosz A., Lei T., Kukuruzinska M. A. (2006). N-glycosylation affects the molecular organization and stability of E-cadherin junctions. J. Biol. Chem. 281, 23138–23149 10.1074/jbc.M512621200 [DOI] [PubMed] [Google Scholar]
- MacDonald B. T., Tamai K., He X. (2009). Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev. Cell 17, 9–26 10.1016/j.devcel.2009.06.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda O., Usami N., Kondo M., Takahashi M., Goto H., Shimokata K., Kusugami K., Sekido Y. (2004). Plakoglobin (gamma-catenin) has TCF/LEF family-dependent transcriptional activity in beta-catenin-deficient cell line. Oncogene 23, 964–972 10.1038/sj.onc.1207254 [DOI] [PubMed] [Google Scholar]
- Maher M. T., Flozak A. S., Stocker A. M., Chenn A., Gottardi C. J. (2009). Activity of the beta-catenin phosphodestruction complex at cell-cell contacts is enhanced by cadherin-based adhesion. J. Cell Biol. 186, 219–228 10.1083/jcb.200811108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meissner J. D., Naumann A., Mueller W. H., Scheibe R. J. (1999). Regulation of UDP-N-acetylglucosamine:dolichyl-phosphate N-acetylglucosamine-1-phosphate transferase by retinoic acid in P19 cells. Biochem. J. 338, 561–568 10.1042/0264-6021:3380561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendelsohn R. D., Helmerhorst E. J., Cipollo J. F., Kukuruzinska M. A. (2005). A hypomorphic allele of the first N-glycosylation gene, ALG7, causes mitochondrial defects in yeast. Biochim. Biophys. Acta 1723, 33–44 10.1016/j.bbagen.2005.01.017 [DOI] [PubMed] [Google Scholar]
- Miravet S., Piedra J., Miró F., Itarte E., García de Herreros A., Duñach M. (2002). The transcriptional factor Tcf-4 contains different binding sites for beta-catenin and plakoglobin. J. Biol. Chem. 277, 1884–1891 10.1074/jbc.M110248200 [DOI] [PubMed] [Google Scholar]
- Mota O. M., Huang G. T., Kukuruzinska M. A. (1994). Developmental regulation and tissue-specific expression of hamster dolichol-P-dependent N-acetylglucosamine-1-P transferase (GPT). Biochem. Biophys. Res. Commun. 204, 284–291 10.1006/bbrc.1994.2457 [DOI] [PubMed] [Google Scholar]
- Nita–Lazar M., Noonan V., Rebustini I., Walker J., Menko A. S., Kukuruzinska M. A. (2009). Overexpression of DPAGT1 leads to aberrant N-glycosylation of E-cadherin and cellular discohesion in oral cancer. Cancer Res. 69, 5673–5680 10.1158/0008-5472.CAN-08-4512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nita–Lazar M., Rebustini I., Walker J., Kukuruzinska M. A. (2010). Hypoglycosylated E-cadherin promotes the assembly of tight junctions through the recruitment of PP2A to adherens junctions. Exp. Cell Res. 316, 1871–1884 10.1016/j.yexcr.2010.02.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengupta P. K., Bouchie M. P., Kukuruzinska M. A. (2010). N-glycosylation gene DPAGT1 is a target of the Wnt/beta-catenin signaling pathway. J. Biol. Chem. 285, 31164–31173 10.1074/jbc.M110.149195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharova L. V., Sharov A. A., Nedorezov T., Piao Y., Shaik N., Ko M. S. (2009). Database for mRNA half-life of 19 977 genes obtained by DNA microarray analysis of pluripotent and differentiating mouse embryonic stem cells. DNA Res. 16, 45–58 10.1093/dnares/dsn030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu M., Fukunaga Y., Ikenouchi J., Nagafuchi A. (2008). Defining the roles of beta-catenin and plakoglobin in LEF/T-cell factor-dependent transcription using beta-catenin/plakoglobin-null F9 cells. Mol. Cell. Biol. 28, 825–835 10.1128/MCB.02375-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steel M. D., Puddicombe S. M., Hamilton L. M., Powell R. M., Holloway J. W., Holgate S. T., Davies D. E., Collins J. E. (2005). Beta-catenin/T-cell factor-mediated transcription is modulated by cell density in human bronchial epithelial cells. Int. J. Biochem. Cell Biol. 37, 1281–1295 10.1016/j.biocel.2004.12.010 [DOI] [PubMed] [Google Scholar]
- Stefanovic B., Lindquist J., Brenner D. A. (2000). The 5′ stem-loop regulates expression of collagen alpha1(I) mRNA in mouse fibroblasts cultured in a three-dimensional matrix. Nucleic Acids Res. 28, 641–647 10.1093/nar/28.2.641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stockinger A., Eger A., Wolf J., Beug H., Foisner R. (2001). E-cadherin regulates cell growth by modulating proliferation-dependent beta-catenin transcriptional activity. J. Cell Biol. 154, 1185–1196 10.1083/jcb.200104036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeichi M. (1995). Morphogenetic roles of classical cadherins. Curr. Opin. Cell Biol. 7, 619–627 [DOI] [PubMed] [Google Scholar]
- Vagin O., Tokhtaeva E., Yakubov I., Shevchenko E., Sachs G. (2008). Inverse correlation between the extent of N-glycan branching and intercellular adhesion in epithelia. Contribution of the Na,K-ATPase beta1 subunit. J. Biol. Chem. 283, 2192–2202 10.1074/jbc.M704713200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Amerongen R., Nusse R. (2009). Towards an integrated view of Wnt signaling in development. Development 136, 3205–3214 10.1242/dev.033910 [DOI] [PubMed] [Google Scholar]
- Walker J. L., Menko A. S., Khalil S., Rebustini I., Hoffman M. P., Kreidberg J. A., Kukuruzinska M. A. (2008). Diverse roles of E-cadherin in the morphogenesis of the submandibular gland: insights into the formation of acinar and ductal structures. Dev. Dyn. 237, 3128–3141 10.1002/dvdy.21717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welply J. K., Lau J. T., Lennarz W. J. (1985). Developmental regulation of glycosyltransferases involved in synthesis of N-linked glycoproteins in sea urchin embryos. Dev. Biol. 107, 252–258 10.1016/0012-1606(85)90393-8 [DOI] [PubMed] [Google Scholar]
- Wheelock M. J., Johnson K. R. (2003). Cadherins as modulators of cellular phenotype. Annu. Rev. Cell Dev. Biol. 19, 207–235 10.1146/annurev.cellbio.19.011102.111135 [DOI] [PubMed] [Google Scholar]
- Wu Z., Zheng S., Li Z., Tan J., Yu Q. (2011). E2F1 suppresses Wnt/β-catenin activity through transactivation of β-catenin interacting protein ICAT. Oncogene 30, 3979–3984 10.1038/onc.2011.129 [DOI] [PubMed] [Google Scholar]
- Zhao Y. Y., Takahashi M., Gu J. G., Miyoshi E., Matsumoto A., Kitazume S., Taniguchi N. (2008). Functional roles of N-glycans in cell signaling and cell adhesion in cancer. Cancer Sci. 99, 1304–1310 10.1111/j.1349-7006.2008.00839.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhurinsky J., Shtutman M., Ben–Ze’ev A. (2000a). Differential mechanisms of LEF/TCF family-dependent transcriptional activation by beta-catenin and plakoglobin. Mol. Cell. Biol. 20, 4238–4252 10.1128/MCB.20.12.4238-4252.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhurinsky J., Shtutman M., Ben–Ze’ev A. (2000b). Plakoglobin and beta-catenin: protein interactions, regulation and biological roles. J. Cell Sci. 113, 3127–3139 [DOI] [PubMed] [Google Scholar]