

Summary
Plexin B1, the receptor for Semaphorin 4D (Sema4D), is expressed by melanocytes in the skin. We recently showed that Sema4D suppresses activation of the hepatocyte growth factor receptor, MET, in melanocytes, and that knockdown of Plexin B1 results in activation of MET. MET signaling mediates proliferation, survival and migration in melanocytes, and its activation is associated with transformation of melanocytes to melanoma. In this report we investigated the mechanism by which Plexin B1 inhibits MET activation. Our results show that Plexin B1 and MET exist as an oligomeric receptor-receptor complex in melanocytes, and that receptor association is increased by Sema4D. MET and Plexin B1 receptor complexes were identified along the cell body of melanocytes, and Sema4D increased receptor association on dendrites, suggesting that Sema4D regulates MET-dependent processes at precise locations on the melanocyte. Despite activation of MET, Plexin B1 knockdowns proliferated slowly and showed increased apoptosis compared with controls. Shp2, a non-receptor protein tyrosine phosphatase, translates growth and survival signals from MET and other receptor tyrosine kinases. Plexin B1 knockdowns had markedly lower levels of Shp2 compared with controls, and Sema4D upregulated Shp2 expression at the protein and message level in normal melanocytes. Functional studies showed that blockade of Shp2 activity abrogated MET-dependent activation of Erk1/Erk2 and Akt in melanocytes. These results suggest a complex role for Sema4D and Plexin B1 in orchestrating signaling from the MET receptor in melanocytes. Because Shp2 is a downstream adaptor protein for multiple receptors, Sema4D may control the effects of several growth factors on melanocytes through regulation of Shp2.
Key words: Plexin B1, Melanocyte, MET, HGF, Hepatocyte growth factor, RTK, Receptor tyrosine kinases
Introduction
Melanocytes are highly dendritic cells derived from the neural crest that produce melanin, which is transferred to keratinocytes, providing photoprotection to the skin. Melanocytes are also progenitor cells of melanoma, a tumor that is deadly once it has metastasized. Paracrine and autocrine factors regulate melanocyte function and play a role in transformation of melanocytes to melanoma (Haass et al., 2004; Villanueva and Herlyn, 2008). We have shown that melanocytes express the neural guidance receptor Plexin B1, which regulates melanocyte survival and proliferation, through binding to its ligand, Semaphorin 4D (Sema4D) (Soong et al., 2012). Sema4D is expressed by keratinocytes and melanocytes, thereby functioning as an autocrine and paracrine factor (Soong et al., 2012). Semaphorins are a large family of transmembrane and secreted proteins that are ligands for Plexin receptors and were originally identified in the nervous system (Giger et al., 2010; Pasterkamp and Giger, 2009), and are widely expressed, including in the skin. Plexin B1 has recently been identified as a tumor suppressor protein for melanoma, suggesting a role for Plexin B1 in melanoma initiation or progression (Argast et al., 2009; Stevens et al., 2010). Plexin receptors are type I transmembrane proteins (A–D) that bind to Semaphorins (Tamagnone et al., 1999). While they lack intrinsic kinase activity, the intracellular domain of Plexins contains an intrinsic Ras and Rap1-GTPase activating (GAP) domain, resulting in exchange of GTP for GDP on M-Ras, R-Ras, and Rap1 (Oinuma et al., 2004; Saito et al., 2009; Wang et al., 2012).
MET is a receptor tyrosine kinase (RTK) whose signaling stimulates melanocyte migration, proliferation and cell survival (Damm et al., 2010; Halaban et al., 1992a; Halaban et al., 1992b; Halaban et al., 1993) and its activation is implicated in melanoma progression (Chattopadhyay et al., 2012; Kenessey et al., 2010; Lin et al., 1998; Thomas et al., 2007). MET activation occurs through binding of hepatocyte growth factor (HGF) to the β-chain of MET, inducing oligomerization and transphosphorylation of tyrosine residues 1234 and 1235. Following activation, adaptor proteins, including Gab1, Grb2, Shp2 and SOS are recruited to binding sites on the cytoplasmic domain of MET, resulting in migration, proliferation, survival, and branching morphogenesis. MET downstream effects are mediated, in part, through Shp2, which is required for full activation of the MAP kinase and Akt signaling pathways in response to HGF (Li et al., 2009; Schaeper et al., 2000). Shp2, a non-receptor protein tyrosine phosphatase that is ubiquitously expressed, is recruited to the cytoplasmic domains of RTKs upon receptor activation, and is involved in downstream signaling that ultimately controls many aspects of cellular function, including proliferation, apoptosis, senescence and gene transcription (Chong et al., 2003; Yang et al., 2006). A recent report shows that Shp2 is required for Sema4D-dependent axon repulsion in neurons, although the mechanisms of this interaction have not been defined (Fuchikawa et al., 2009). Sema4D has been reported to stimulate MET receptor activation through Plexin B1 signaling in endothelial and epithelial cells (Conrotto et al., 2005; Giordano et al., 2002; Soong et al., 2012; Stevens et al., 2010). MET activation by Plexin B1 signaling in these cells is accompanied by phosphorylation of Plexin B1 on tyrosine residues in the cytoplasmic domain, which is required for regulation of Rho, a small GTP binding protein involved in actin remodeling (Giordano et al., 2002).
We recently reported that Sema4D abrogates HGF-dependent MET activation in normal human melanocytes and that knockdown of Plexin B1 resulted in robust activation of MET, in the absence of HGF (Soong et al., 2012). Paradoxically, despite enhanced MET activity, knockdowns exhibited slowed proliferation and increased apoptosis compared with controls. In this report we have determined the effect of chronic downregulation of Plexin B1 on MET-dependent migration, and have examined the mechanism by which Plexin B1 signaling regulates MET activation. Through several complementary techniques we show that in normal human melanocytes Plexin B1 and MET exist in a receptor-receptor complex, which is increased by Sema4D. Further, we show that Sema4D positively regulates the expression of Shp2, and that Shp2 is required for HGF-dependent MAP kinase and Akt activation in melanocytes. These studies suggest that Plexin B1 controls MET signaling in two ways, directly through receptor-receptor association that inhibits MET activation, and indirectly through regulation of Shp2 levels that control MET-dependent downstream signaling events.
Results
Plexin B1 suppresses MET-dependent Gab activation and migration
Gab1 is a multi-substrate adaptor protein that docks to the active MET receptor and mediates downstream effects of MET including migration (Hov et al., 2004; Rajadurai et al., 2012). Plexin B1 knockdowns show markedly increased MET activity (Soong et al., 2012). To determine if knockdown of Plexin B1 activates Gab1, melanocytes were placed in serum and growth factor free media for 24 hours, then lysed and blotted for phosphorylated MET and Gab1 (Fig. 1A). As previously observed, knockdown of Plexin B1 induced robust activation of MET. Densitometry analysis showed that an average 2.5-fold knockdown in Plexin B1 expression increased MET phosphorylation 7-fold. Active Gab1 was increased 1.4-fold in Plexin B1 knockdowns compared with non-target controls and activation was MET-dependent as shown by experiments in which Plexin B1 knockdowns were treated with a MET kinase inhibitor followed by western blotting for active Gab1 (Fig. 1B). Densitometry of total MET and GAB1, normalized to actin, did not show any significant effects of Plexin B1 knockdown on levels of total protein. To determine if knockdown of Plexin B1 stimulates MET-dependent migration, scratch assays were performed. Cells were serum and growth factor starved for 24 hours prior to placement of a scratch in a near-confluent lawn of cells. Because melanocytes migrate as individual cells, rather than as sheets, cells that migrated into the scratch were counted. After 24 hours, migration of Plexin B1 knockdowns was significantly higher (P<0.0001) than non-target controls (Fig. 1C). Inclusion of a MET kinase inhibitor reduced migration by more than 50% (P = 0.003) in Plexin B1 knockdowns, but had no effect on cells expressing non-target shRNA. Blockade of MET activation was confirmed by blotting of cell lysates from the scratch assays for phosphorylated MET. Finally, MET-dependent suppression of E-cadherin is associated with increased migration of melanocytes, and other cell types (Han et al., 2005; Koefinger et al., 2011). Levels of E-cadherin were lower in Plexin B1 knockdowns compared with controls, consistent with increased MET activation and MET-dependent migration (Fig. 1D).
Fig. 1.

Knockdown of Plexin B1 leads to MET-dependent phosphorylation of Gab1 and stimulates MET-dependent migration in melanocytes. (A) Total cell lysates of Plexin B1 silenced melanocytes (shPB1, cell line 1) and their non-target controls (shRNA-C) were resolved on 7.5% SDS-PAGE and blotted for phosphorylated Gab1 and MET. Knockdown of Plexin B1 activates MET and Gab1 (arrows). Knockdown of Plexin B1 was confirmed by western blotting. Results are representative of three individual experiments. (B) Plexin B1 knockdowns were treated with MET kinase inhibitor (10 µM) for 15 minutes, and lysates were resolved on 7.5% SDS-PAGE, and blotted for phosphorylated MET and Gab1. Phosphorylated Gab1 is decreased by ∼50% in the presence of MET inhibitor, indicating that activation of Gab1 in Plexin B1 knockdowns is partially MET-dependent. Results are representative of three individual experiments. (C) Representative images from scratch assays of Plexin B1 knockdowns (cell line 1) and controls at time 0 and 24 hours. Arrows highlight cells that have migrated into the scratch. The graph shows averaged number of cells that migrated into the scratch from three experiments ±s.d. Plexin B1 knockdowns (shPB1) migrate significantly faster than controls (shRNA-C), which is blocked by a MET kinase inhibitor. Melanocytes from the scratch assays were lysed and resolved on a 7.5% SDS-PAGE and blotted for Plexin B1 and phosphorylated MET. A representative blot from two experiments confirms Plexin B1 knockdown (1.4-fold), increased MET phosphorylation in Plexin B1 knockdowns (arrows) and loss of MET activation in the presence of MET inhibitor. (D) Cell lysates of Plexin B1 knockdowns (shPB1, cell line 1) or non-target controls (shRNA-C) were resolved on 7.5% SDS-PAGE and blotted for E-cadherin. E-cadherin expression is decreased in knockdowns compared to controls. Results are representative of three individual experiments.
Plexin B1 and MET associate in human melanocytes
We first determined whether chronic loss of Plexin B1 stimulates the synthesis of the MET ligand HGF, resulting in MET activation. western blotting showed that loss of Plexin B1 does not result in de novo synthesis of HGF (Fig. 2A). This was further supported by experiments in which Plexin B1 knockdowns were treated with blocking antibodies to HGF, which had no effect on MET activity (Fig. 2B). We next determined if Plexin B1 and MET receptors are co-localized in melanocytes, which could result in inhibition of MET activation. Co-localization analysis was first carried out in normal human melanocytes in suspension by digital imaging with an ImageStream imaging flow cytometer (George et al., 2004). Melanocytes were dual-labeled with antibodies against MET and Plexin B1. A averaged similarity bright detail score of 34.3% (±2 s.d., n = 3) consistent with association between Plexin B1 and MET, was observed (Fig. 3A). In-situ Proximity Ligation Assay (PLA) detects protein-protein associations within a 40 nM space in intact cells in vitro, and allows for visualization of interactions within discrete areas of the cell. Melanocytes cultured on PureCol-coated glass coverslips were treated with Sema4D for 15 minutes, then labeled with antibodies against MET and Plexin B1, followed by oligo-linked probes, enzymatic ligation, and rolling circle amplification with introduction of a fluorochrome tag on co-localized receptors. Under resting conditions MET and Plexin B1 receptor co-localization was identified on the melanocyte cell body, with some receptor-receptor co-localization detectable along the dendrites (Fig. 3B). One hour following treatment with Sema4D, significantly increased numbers of fluorescent dots (P = 0.017), with numerous receptor-receptor pairs along the length of the dendrites, were identified. Specificity of the assay was confirmed by analysis of Plexin B1 knockdowns, which failed to show PLA-positive dots. Finally, association of receptors was assessed by immunoprecipitation. Melanocytes were treated with Sema4D for 15 minutes, proteins were cross-linked and MET or Plexin B1 immunoprecipitated from cell lysates, followed by western blotting for the complementary receptor. Plexin B1 and MET were identified as a receptor complex in immunoprecipitates under basal conditions (Fig. 3C). Treatment with Sema4D modestly increased receptor-receptor association at 15 minutes (1.5-fold), with sustained association after 1 hour of treatment (Fig. 3D; 3-fold). Taken together, these data are consistent with Plexin B1-dependent suppression of MET activation through direct receptor-receptor interaction.
Fig. 2.
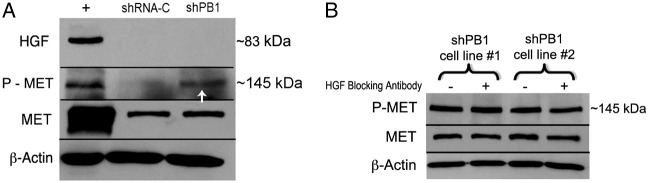
Knockdown of Plexin B1 does not upregulate HGF production in melanocytes. (A) Total cell lysates of Plexin B1 knockdowns (shPB1; cell line 2) and non-target controls (shRNA-C) were resolved on 7.5% SDS-PAGE and blotted for HGF and P-MET. Controls (+) consisted of lysates of 293FT cells. Plexin B1 knockdowns show MET activation (arrow), but do not show de novo production of HGF. Results are representative of two experiments. (B) Plexin B1 knockdowns (shPB1; cell line 1 and cell line 2) were treated with HGF blocking antibody (0.5 µg/ml) or goat IgG for 1 hour. Total cell lysates were resolved on 7.5% SDS-PAGE and blotted for P-MET. Results show no changes in MET phosphorylation with HGF blocking antibody treatment. Results are representative of two experiments.
Fig. 3.

Plexin B1 and MET associate in normal human melanocytes. (A) Representative images from ImageStream analysis of melanocytes dual labeled for MET and Plexin B1 (PB1), and merged images. Bright Detail Similarity histogram shows the percentage of receptors that co-localized in the gated area within the sample (106 cells). Thirty-six percent of the cells show Plexin B1 and MET co-localization within a 0.75 µM radius. Results are representative of three individual experiments. (B) Representative images of melanocytes stained for Plexin B1 and MET, with detection by PLA. Fluorescent dots are identified on the cell body of melanocytes (arrows), which are increased following treatment with Sema4D, particularly along the dendrites (inset). The graph shows the average number of fluorescent dots per cell ±s.d. Sema4D treatment significantly increased association of the MET and Plexin B1 receptors after 1-hour treatment (inset, P = 0.017). Negative controls including PLA performed on Plexin B1-silenced cells stained for Plexin B1 and MET, and melanocytes incubated with non-immune serum instead of primary antibody are shown. Results are representative of three individual experiments. (C) Immunoprecipitation of Plexin B1, or MET from melanocyte lysates show that the receptors are in a complex. Treatment with Sema4D increases the association of MET and Plexin B1 1.2-fold as shown by densitometry analysis of receptor normalized to the complimentary receptor. Shown are immunoprecipitates from 300 µg of protein resolved on 7.5% SDS-PAGE. Results are representative of three individual experiments. (D) Immunoprecipitation of MET from melanocyte lysates show that MET-Plexin B1 remains in a complex following treatment with Sema4D at 1 hour. Shown are immunoprecipitates from 300 µg of protein resolved on 7.5% SDS-PAGE. Results are representative of two individual experiments.
Sema4D regulates Shp2 expression
MET activation stimulates the MAP kinase and PI3-kinase-Akt pathway, resulting in proliferation, migration and survival of melanocytes (Halaban et al., 1993). Although loss of Plexin B1 activated MET, previous work showed that Plexin B1 knockdowns had significantly lower proliferation rates and increased apoptosis even when grown in complete media, compared with non-target controls (Soong et al., 2012). Shp2 transmits signals from multiple RTKs to activate Erk1/Erk2 and Akt. In Plexin B1 knockdowns Shp2 protein was markedly decreased compared with non-target controls (Fig. 4A). Knockdown of Plexin C1, a receptor that shares 30% homology with Plexin B1, had no effect on Shp2 expression in melanocytes (data not shown). To determine if Sema4D upregulates Shp2 expression in normal melanocytes, cells treated with Sema4D were analyzed by western blotting and comparative real-time PCR. Sema4D increased Shp2 protein expression at 48 and 72 hours, and significantly increased Shp2 message after 48 hours treatment (Fig. 4B). Treatment of melanocytes for up to 72 hours with Semaphorin 7A, a Semaphorin expressed by keratinocytes, had no effect on levels of Shp2 protein (Fig. 4C). The time frame of Shp2 regulation by Sema4D suggests activation of downstream intermediate signaling intermediates that stimulate Shp2 message transcription or stability.
Fig. 4.
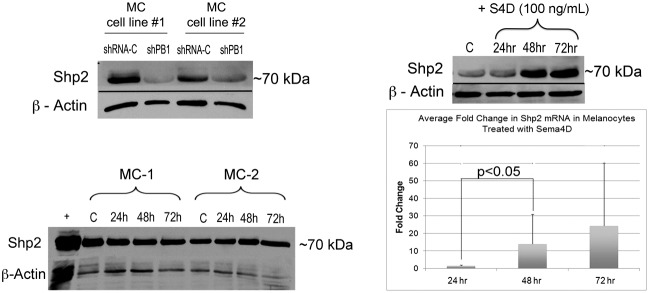
Sema4D stimulates Shp2 expression at the protein and message level. (A) Plexin B1 knockdowns or controls were lysed and resolved on 7.5% SDS-PAGE and blotted for Shp2. Results show that Plexin B1 knockdowns (shPB1) have lower levels of Shp2 compared to controls (shRNA-C). Results shown are from two individual melanocyte cultures silenced for Plexin B1 (cell lines 1 and 2). (B) Treatment of normal human melanocytes (MC) with Sema4D (100 ng/ml) upregulates Shp2 expression at 48 and 72 hours. Shown is a western blot of total cell lysates resolved on 7.5% SDS-PAGE and blotted for Shp2. Results shown are representative of three experiments. The bar graph shows the average fold change in Shp2 message normalized to β-actin, measured by comparative RT-PCR. The bars represent the average of four individual cultures (24 hours) and three individual cultures (48 and 72 hours), ±s.d. (C) Treatment of normal human melanocytes with Sema7A (100 ng/ml) had no effect on Shp2 expression at 48 and 72 hours. Shown is a western blot of total cell lysates resolved on a 7.5% SDS-PAGE and blotted for Shp2.
To determine if inhibition of Shp2 phosphatase activity decreases MET-dependent Erk1/Erk2 and Akt activation in melanocytes, cells were growth factor and serum starved for 24 hours then treated with HGF in the presence of an inhibitor of Shp2 phosphatase activity (50 µM), and Erk1/Erk2 and Akt phosphorylation was assessed by western blotting (Fig. 5). Blockade of Shp2 phosphatase activity rapidly (15 min) attenuated MET activation in response to HGF, shown by decreased levels of phosphorylated MET (Fig. 5A, asterisks). As expected, HGF induced robust activation of Erk1/Erk2 (Fig. 5A) and Akt (Fig. 5B), which was sustained for 60 minutes following HGF treatment. Blockade of Shp2 phosphatase activity resulted in sustained attenuation Erk1/Erk2 and Akt activation in response to HGF, consistent with a role for Shp2 in translation of proliferative and survival signals from MET receptor in melanocytes.
Fig. 5.
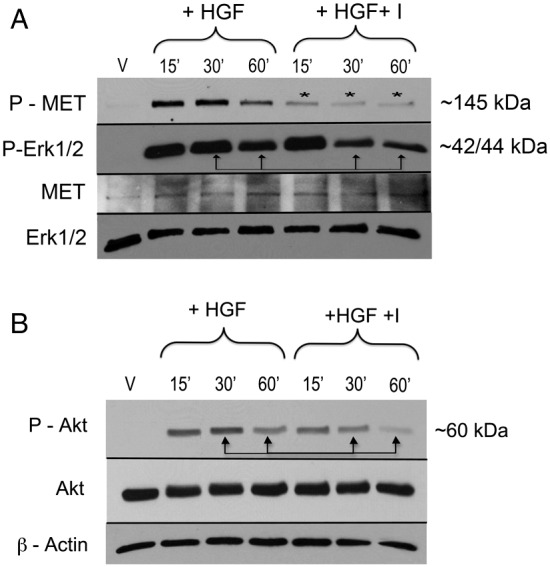
Shp2 regulates MET-dependent Erk1/Erk2 and Akt activation in melanocytes. (A) Melanocytes were treated with HGF (10 ng/ml) in the presence of Shp2 inhibitor (I; 50 µM). Total cell lysates were resolved on 7.5% SDS-PAGE, and blotted for phosphorylated MET and Erk1/2. Inhibition of Shp2 attenuates MET (asterisks) and Erk1/2 phosphorylation in response to HGF at 30 and 60 minutes of treatment (arrows). Results are representative of two individual experiments. (B) Melanocytes were treated as described above and total cell lysates were resolved on 7.5% SDS-PAGE, and blotted for phosphorylated Akt. Inhibition of Shp2 attenuates HGF-dependent Akt phosphorylation at 30 and 60 minutes of treatment (arrows). Results are representative of two individual experiments.
Discussion
We are interested in Plexin B1 because its ligand, Sema4D, blocks HGF-dependent MET activation melanocytes and melanoma (Soong et al., 2012; Stevens et al., 2010). The MET receptor plays a role in melanocyte growth, survival and migration, and dysregulation of MET signaling contributes to melanoma initiation and progression. We show that functional effects of MET activation in response to chronic suppression of Plexin B1 include MET-dependent Gab1 activation, increased migration and suppression of E-cadherin expression. These data suggest that downregulation of Plexin B1, which is seen in melanocytes treated with UVB (Soong et al., 2012), and in melanoma (Argast et al., 2009; Stevens et al., 2010), may stimulate migration in a MET-dependent manner. In this report we examined the mechanisms by which Plexin B1 regulates MET activity in melanocytes. Our data suggest that Plexin B1 inhibits MET activation through direct receptor-receptor interaction. We also identify a second, indirect mechanism for Plexin B1-dependent regulation of MET activity through modulation of Shp2 expression.
Our data indicate that Plexin B1 interacts directly with MET in human melanocytes, inhibiting oligomerization and activation in response to HGF. This was shown through identification of receptor co-localization by ImageStream, PLA analysis, and by co-immunoprecipitation studies. Sema4D treatment increased Plexin B1-MET receptor interactions, particularly on melanocyte dendrites, as shown by PLA studies. This suggests that Sema4D provides localized regulation of migration at the advancing edge of the melanocyte dendrite through inhibition of MET activity. Because melanocytes produce Sema4D, it is possible that MET is auto-inhibited through Sema4D-dependent Plexin B1 association with MET. When Plexin B1 expression is lowered, either secondary to UVB as previously reported (Soong et al., 2012), or during transformation to melanoma, MET activity is predicted to increase at the advancing edge of the dendrite, resulting in Gab1 activation, suppression of E-cadherin expression, and enhanced migration. How Plexin B1 association with MET inhibits its activation is unclear, but may be due to steric interference of oligomerization, or may involve secondary signaling molecules. In epithelial cells (Giordano et al., 2002), MET and Plexin B1 also co-associate, similar to melanocytes, but this interaction results in activation, rather than inhibition of MET. In these cells activation of MET by Sema4D is accompanied by phosphorylation of Plexin B1 on tyrosine residues in the cytoplasmic domain (Giordano et al., 2002), which is required for regulation of Rho dependent signaling. Sema4D does not stimulate phosphorylation of tyrosine residues on Plexin B1 in melanocytes (unpublished observations), highlighting cell type specific differences in Sema4D and Plexin B1 interactions. Whether lack of tyrosine phosphorylation of Plexin B1 in response to Sema4D in melanocytes contributes to observed differences in MET activation in response to Sema4D requires further study.
Shp2 translates growth and survival signals from multiple RTKs and is important in development and oncogenesis (Feng, 2007; Grossmann et al., 2010; Neel et al., 2003). Shp2 expression was markedly lowered in Plexin B1 knockdowns compared with non-target controls, accounting for the slow growth and increased apoptosis in these cells. Sema4D at nanogram concentrations positively regulated Shp2 expression in melanocytes at the protein and mRNA level. We speculate that Sema4D may maintain Shp2 expression in melanocytes through an autocrine signaling loop. The pathways involved in Sema4D-dependent Shp2 regulation in melanocytes require further study. Plexin B1 activates NFκb (Yang et al., 2011), and Shp2 is an NFκb target (Seki et al., 2007), therefore it is possible that loss of Plexin B1 lowers Shp2 through an NFκb-dependent pathway. A recent report shows that bFGF maintains Shp2 levels in endothelial cells through post-translational mechanisms (Hatanaka et al., 2012). Although Sema4D-dependent increases in Shp2 protein were accompanied by increased Shp2 message, we cannot exclude the possibility that enhanced protein stability contributes to regulation of Shp2 levels in Sema4D-treated melanocytes. Further studies are being planned to test this hypothesis. We show that Shp2 is necessary for full activation of Erk1/Erk2 and Akt in melanocytes in response to HGF. We also observed rapid MET dephosphorylation in melanocytes upon Shp2 inhibition, suggesting that Shp2 inhibits the activity of a MET phosphatase. Because Shp2 is an adaptor protein for many RTKs, regulation of Shp2 expression by Sema4D/Plexin B1 may play a regulatory role in melanocyte response to multiple paracrine factors, including stem cell factor, nerve growth factor, and basic fibroblast growth factor which bind to KIT, TrkA and FGFR1, respectively. Interestingly, dominant negative mutations in the phosphatase domain of Shp2 have been identified in LEOPARD syndrome, an autosomal dominant multisystem disorder characterized by development of hundreds of benign lentigines in affected patients (Conti et al., 2003). It is interesting to speculate that Sema4D contributes to the formation or maintenance of lentigines in the general population through its effects on Shp2 expression. Experiments are currently underway to test this notion.
Materials and Methods
Reagents
PureCol was from Inamed Biomaterials (Freemont, CA); the BCA Protein Assay kit was from Pierce Chemical (Rockford, IL). Rabbit polyclonal antibodies to β-actin, rabbit polyclonal antibodies to Plexin B1 for immunoprecipitation (H-300), rabbit polyclonal antibodies to MET (sc-10), mouse monoclonal antibodies to Shp2 (D3), and Protein A agarose beads were purchased from Santa Cruz Biotechnologies (Santa Cruz, CA). Rabbit polyclonal antibodies to Plexin B1 for western blotting were purchased from ECM Biosciences (Versailles, KY). Mouse monoclonal antibodies to MET were purchased from Millipore (Billerica, MA); rabbit monoclonal antibodies to phospho-Met (Y-1234/Y-1235), rabbit polyclonal antibodies for phospho-Gab1 (Y-307) and Gab1 were purchased from Cell Signaling Technology (Danvers, MA). Antibodies against E-cadherin were purchased from R&D Systems (Minneapolis, MN). Rabbit polyclonal antibodies to HGF were purchased from Abcam (Cambridge, MA). Horseradish peroxidase-conjugated goat anti-rabbit and goat anti-mouse antibodies, and hepatocyte growth factor (HGF) were purchased from Sigma Co., (St Louis, MO). MET Kinase Inhibitor III [2-(6-((R)-1-(2,6-dichloro-3-fluorophenyl)ethoxy)quinolin-3-yl)cyclopropanecarboxamide] was purchased from Calbiochem (Darmstadt, Germany). Shp2 inhibitor [8-Hydroxy-7-[(6-sulfo-2-naphthyl)azo]-5-quinolinesulfonic acid] was purchased from Tocris bioscience (Bristol, UK). DSP (Dithiobis[succinimidyl propionate]), a cleavable membrane permeable cross-linker with a spacer arm of 12.0 Angstroms, was purchased from Thermo Scientific (Pittsburgh, PA). Full-range rainbow molecular weight marker was purchased from Amersham Life Sciences (Arlington Heights, IL). Polybrene was purchased from Santa Cruz Biotechnologies. DNA ladder, 20 bp Molecular Ruler, was purchased from Bio-Rad (Hercules, CA).
Cells and cell culture
Neonatal foreskins were obtained according to the University of Rochester Research Subjects Review Board guidelines and were the source of cultured human melanocytes as previously described (Scott et al., 2009). Cells were cultured in melanocyte growth media (MGM) consisting of Opti-MEM (Gibco-BRL) containing: 5% FBS (fetal bovine serum; Atlanta Biologicals; Lawrenceville, GA), 10−4 M iso-butyl-methylxanthine (IBMX), Anti-Anti (Gibco-BRL), 2.5 nM Cholera Toxin, 0.1 mM dbcAMP, 25 ng/ml phorbol ester. All supplements, except FBS and Anti-Anti were purchased from Sigma Co. With the exception of scratch assays and experiments in which real time PCR were performed, pooled cultures of melanocytes initiated from 2–3 foreskins were used at passage 2–3.
Knockdown of Plexin B1 in human melanocytes
Melanocytes from two individual foreskin cultures, ‘cell line 1’ and ‘cell line 2’, were plated in MGM at 105 cells in a 6-well plate. Cells were transduced at 2.5 Multiplicity of Infection with MISSION Sigma Lentivirus particles expressing shRNA targeting human Plexin B1 (Clone TRCN0000061536) using polybrene. Cells infected with non-target shRNA (shRNA-NT) in Lentivirus were used as controls. Stable transductants were selected with puromycin dihydrochloride (Sigma Co., St Louis, MO) at 2.5 µg/ml.
Western blotting
Cells were lysed in RIPA buffer (150 mM NaCl, 1% NP40, 0.5% DOC, 0.1% SDS, 50 mM Tris-HCl) with protease inhibitors (Boehringer Mannheim, Gmbht, Germany) and protein was quantitated using bovine serum albumen (BSA) as a standard (Bio- Rad Laboratories, Hercules, CA). Protein was resolved on SDS-PAGE gels and blotted using standard procedures. Visualization of the immuno-reactive proteins was accomplished with an enhanced chemiluminescence reaction (Pierce Chemical, Rockford, IL).
Comparative real-time polymerase chain reaction (RT-PCR)
Total RNA was isolated using the RNeasy Mini Kit (QIAgen, Valencia, CA). Reverse transcription was performed using 0.75 µg of total RNA with SuperScript II reverse transcriptase (BioRad Laboratories, Hercules, CA). PCR was performed using iQ SYBR Green Supermix (BioRad Laboratories, Hercules, CA) on the Applied Biosystems ABI prism 7700 sequence-detection system (BioRad iCycler). Primers used for amplification of Shp2 were: fwd: 5-GACTTTTGGCGGATGGTGTTCC-3; rvs: 5-CGGCGCTTTCTTTGACGTTCCT-3. Primers used for amplification of β-actin were: fwd: 5-CACGCACGATTTCCCGCTCGG-3; rvs: 5-CAGGCTGTGCTATCCTGTAC-3. The conditions were 95°C 4 minutes, 95°C 30 seconds, 54°C 20 seconds, 72°C 90 seconds, 72°C 5 minutes (all 40 cycles). The PCR products were resolved on 1.5% agarose gels to verify product size. Fold change in mRNA between samples was determined using the ΔΔCT with β-actin as a reference gene, assuming an efficiency of 94.3%. Efficiency of Shp2 amplification was assumed to be 93.6%.
ImageStream analysis
Melanocytes in MGM (106 cells) were fixed with 3.7% methanol-free formaldehyde. The cells were stained in staining media (0.1% BSA and 100 mM EDTA in PBS) with Alexa 488-conjugated mouse monoclonal antibodies against MET (R&D Systems, Minneapolis, MN) and rabbit polyclonal antibodies against Plexin B1 (H300; Santa Cruz Biotechnology) followed by fluorescent Alexa-594 conjugated donkey anti-rabbit antibodies (Invitrogen, Grand Island, NY). Nuclei were stained by incubation of the cells with Vybrant Dye Cycle Violet (Invitrogen). Negative controls consist of cells stained with non-immune serum instead of primary antibody. Cells were analyzed with ImageStream X Mark II (Amnis, Seattle, WA); at least 10,000 images were obtained from each sample. To create data files for use in spectral compensation, images of unstained cells and of cells dual labeled with antibody/fluorophore were acquired without brightfield illumination. Post-acquisition spectral compensation and data analysis was performed using IDEAS® image analysis software package (Amnis Corp.) to determine a similarity bright detail score. Using this technique, co-localization within 0.75 µM or less can be identified (Beum et al., 2006).
Proximity ligation assay
Cells (105) were plated on PureCol coated coverslips and allowed to attach for 24 hours. Cells were placed in growth factor and serum free media for 24 hours and treated with Sema4D (100 ng/µL) for 15 minutes or 1 hour, then fixed in 3.7% formaldehyde (15 minutes), and incubated with rabbit polyclonal antibodies to MET (sc-10; Santa Cruz Biotechnology) and mouse monoclonal primary antibodies to Plexin B1 (A-8; Santa Cruz Biotechnology). Identification of receptor-receptor proximity was accomplished using DuoLink In Situ Proximity Ligation Assay (Olink Bioscience, Uppsala, Sweden). Photographs were taken from a fluorescence microscope with a Spot Digital camera using a filter with an absorbance of 594 nM. For quantitation, fluorescent dots were counted in a minimum of 100 cells for each condition.
Cross-linking and immunoprecipitation
Cells were placed in growth factor and serum free media for 24 hours and were then treated with Sema4D (100 ng/µL), and proteins were cross-linked with DSP (1.5 µM, pH 8) for 30 minutes, and the reaction was stopped with Tris (pH 8.0) for 5 minutes. Cells were lysed and incubated with antibodies against MET or Plexin B1 overnight at 4°C. Protein A beads were used to capture immune complexes. Immunoprecipitates were resolved on 7.5% SDS-PAGE and blotted for either Plexin B1 or MET. Controls consisted of cells incubated with non-immune IgG instead of primary antibodies.
Scratch assay
Plexin B1 knockdowns or non-target controls were plated at 3×105 in a 6-well dish and allowed to grow to near confluence (∼80%). Twenty-four hours after being placed in serum and growth factor free media, three scratches were made on the bottom of each well using a sterile 200 µl pipette tip. Digital images were taken at time 0 and 24 hours later. Quantitation of migrated cells was performed by counting the number of cells (defined as cells with nuclei) that migrated from the edges of the scratches from digital photographs using ImageJ software (version 1.46, NIH). A minimum of three fields from each scratch was analyzed, and the numbers were averaged.
Recombinant Sema4D and Sema7A
Recombinant Sema4D and Sema7A were expressed as Fc-tagged proteins as previously described (Scott et al., 2008; Scott et al., 2009). Protein purity, quantity and identity were assessed by silver staining of gels and western blotting. Controls consisted of culture supernatants of non-transfected 293FT cells (ATCC, Manassas, VA) that were treated identically as culture supernatants of transfected cells (hereafter called ‘Control’).
Statistical analysis
Differences between means were analyzed by two-tailed Student's t-test. A P value <0.05 was considered significant.
Footnotes
Funding
This work was supported by the National Institutes of Health (NIH) [grant number R01CA136499 to G.S.]; and an NIH training grant [grant number 5T32AR007472 to J.S.]. Deposited in PMC for release after 12 months.
References
- Argast G. M., Croy C. H., Couts K. L., Zhang Z., Litman E., Chan D. C., Ahn N. G. (2009). Plexin B1 is repressed by oncogenic B-Raf signaling and functions as a tumor suppressor in melanoma cells. Oncogene 28, 2697–2709 10.1038/onc.2009.133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beum P. V., Lindorfer M. A., Hall B. E., George T. C., Frost K., Morrissey P. J., Taylor R. P. (2006). Quantitative analysis of protein co-localization on B cells opsonized with rituximab and complement using the ImageStream multispectral imaging flow cytometer. J. Immunol. Methods 317, 90–99 10.1016/j.jim.2006.09.012 [DOI] [PubMed] [Google Scholar]
- Chattopadhyay C., Ellerhorst J. A., Ekmekcioglu S., Greene V. R., Davies M. A., Grimm E. A. (2012). Association of activated c-Met with NRAS-mutated human melanomas. Int. J. Cancer 131, E56–E65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong Z. Z., Lin S. H., Kang J. Q., Maiese K. (2003). The tyrosine phosphatase SHP2 modulates MAP kinase p38 and caspase 1 and 3 to foster neuronal survival. Cell. Mol. Neurobiol. 23, 561–578 10.1023/A:1025158314016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrotto P., Valdembri D., Corso S., Serini G., Tamagnone L., Comoglio P. M., Bussolino F., Giordano S. (2005). Sema4D induces angiogenesis through Met recruitment by Plexin B1. Blood 105, 4321–4329 10.1182/blood-2004-07-2885 [DOI] [PubMed] [Google Scholar]
- Conti E., Dottorini T., Sarkozy A., Tiller G. E., Esposito G., Pizzuti A., Dallapiccola B. (2003). A novel PTPN11 mutation in LEOPARD syndrome. Hum. Mutat. 21, 654 10.1002/humu.9149 [DOI] [PubMed] [Google Scholar]
- Damm S., Koefinger P., Stefan M., Wels C., Mehes G., Richtig E., Kerl H., Otte M., Schaider H. (2010). HGF-promoted motility in primary human melanocytes depends on CD44v6 regulated via NF-kappa B, Egr-1, and C/EBP-beta. J. Invest. Dermatol. 130, 1893–1903 10.1038/jid.2010.45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng G. S. (2007). Shp2-mediated molecular signaling in control of embryonic stem cell self-renewal and differentiation. Cell Res. 17, 37–41 10.1038/sj.cr.7310140 [DOI] [PubMed] [Google Scholar]
- Fuchikawa T., Nakamura F., Fukuda N., Takei K., Goshima Y. (2009). Protein tyrosine phosphatase SHP2 is involved in Semaphorin 4D-induced axon repulsion. Biochem. Biophys. Res. Commun. 385, 6–10 10.1016/j.bbrc.2009.05.024 [DOI] [PubMed] [Google Scholar]
- George T. C., Basiji D. A., Hall B. E., Lynch D. H., Ortyn W. E., Perry D. J., Seo M. J., Zimmerman C. A., Morrissey P. J. (2004). Distinguishing modes of cell death using the ImageStream multispectral imaging flow cytometer. Cytometry A 59, 237–245 [DOI] [PubMed] [Google Scholar]
- Giger R. J., Hollis E. R., 2nd, Tuszynski M. H. (2010). Guidance molecules in axon regeneration. Cold Spring Harb. Perspect. Biol. 2, a001867 10.1101/cshperspect.a001867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giordano S., Corso S., Conrotto P., Artigiani S., Gilestro G., Barberis D., Tamagnone L., Comoglio P. M. (2002). The semaphorin 4D receptor controls invasive growth by coupling with Met. Nat. Cell Biol. 4, 720–724 10.1038/ncb843 [DOI] [PubMed] [Google Scholar]
- Grossmann K. S., Rosário M., Birchmeier C., Birchmeier W. (2010). The tyrosine phosphatase Shp2 in development and cancer. Adv. Cancer Res. 106, 53–89 10.1016/S0065-230X(10)06002-1 [DOI] [PubMed] [Google Scholar]
- Haass N. K., Smalley K. S., Herlyn M. (2004). The role of altered cell-cell communication in melanoma progression. J. Mol. Histol. 35, 309–318 10.1023/B:HIJO.0000032362.35354.bb [DOI] [PubMed] [Google Scholar]
- Halaban R., Fan B., Ahn J., Funasaka Y., Gitay–Goren H., Neufeld G. (1992a). Growth factors, receptor kinases, and protein tyrosine phosphatases in normal and malignant melanocytes. J. Immunother. 12, 154–161 [DOI] [PubMed] [Google Scholar]
- Halaban R., Rubin J. S., Funasaka Y., Cobb M., Boulton T., Faletto D., Rosen E., Chan A., Yoko K., White W.et al. (1992b). Met and hepatocyte growth factor/scatter factor signal transduction in normal melanocytes and melanoma cells. Oncogene 7, 2195–2206 [PubMed] [Google Scholar]
- Halaban R., Rubin J. S., White W. (1993). met and HGF-SF in normal melanocytes and melanoma cells. EXS 65, 329–339 [PubMed] [Google Scholar]
- Han S. U., Lee H. Y., Lee J. H., Kim W. H., Nam H., Kim H., Cho Y. K., Kim M. W., Lee K. U. (2005). Modulation of E-cadherin by hepatocyte growth factor induces aggressiveness of gastric carcinoma. Ann. Surg. 242, 676–683 10.1097/01.sla.0000186171.85804.fe [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatanaka K., Lanahan A A., Murakami M., Simons M. (2012). Fibroblast growth factor signaling potentiates VE-cadherin stability and adherens junctions by regulating Shp2. PLoS ONE. 7, e37600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hov H., Holt R. U., Ro T. B., Fagerli U. M., Hjorth–Hansen H., Baykov V., Christensen J. G., Waage A., Sundan A., Borset M. (2004). A selective c-met inhibitor blocks an autocrine hepatocyte growth factor growth loop in ANBL-6 cells and prevents migration and adhesion of myeloma cells. Clin. Cancer Res. 10, 6686–6694 [DOI] [PubMed] [Google Scholar]
- Kenessey I., Keszthelyi M., Krámer Z., Berta J., Adám A., Dobos J., Mildner M., Flachner B., Cseh S., Barna G.et al. (2010). Inhibition of c-Met with the specific small molecule tyrosine kinase inhibitor SU11274 decreases growth and metastasis formation of experimental human melanoma. Curr. Cancer Drug Targets 10, 332–342 10.2174/156800910791190184 [DOI] [PubMed] [Google Scholar]
- Koefinger P., Wels C., Joshi S., Damm S., Steinbauer E., Beham–Schmid C., Frank S., Bergler H., Schaider H. (2011). The cadherin switch in melanoma instigated by HGF is mediated through epithelial-mesenchymal transition regulators. Pigment Cell Melanoma Res. 24, 382–385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J., Reed S. A., Johnson S. E. (2009). Hepatocyte growth factor (HGF) signals through SHP2 to regulate primary mouse myoblast proliferation. Exp. Cell Res. 315, 2284–2292 10.1016/j.yexcr.2009.04.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin S., Rusciano D., Lorenzoni P., Hartmann G., Birchmeier W., Giordano S., Comoglio P., Burger M. M. (1998). C-met activation is necessary but not sufficient for liver colonization by B16 murine melanoma cells. Clin. Exp. Metastasis 16, 253–265 10.1023/A:1006596909948 [DOI] [PubMed] [Google Scholar]
- Neel B. G., Gu H., Pao L. (2003). The ‘Shp’ing news: SH2 domain-containing tyrosine phosphatases in cell signaling. Trends Biochem. Sci. 28, 284–293 10.1016/S0968-0004(03)00091-4 [DOI] [PubMed] [Google Scholar]
- Oinuma I., Ishikawa Y., Katoh H., Negishi M. (2004). The Semaphorin 4D receptor Plexin-B1 is a GTPase activating protein for R-Ras. Science 305, 862–865 10.1126/science.1097545 [DOI] [PubMed] [Google Scholar]
- Pasterkamp R. J., Giger R. J. (2009). Semaphorin function in neural plasticity and disease. Curr. Opin. Neurobiol. 19, 263–274 10.1016/j.conb.2009.06.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajadurai C. V., Havrylov S., Zaoui K., Vaillancourt R., Stuible M., Naujokas M., Zuo D., Tremblay M. L., Park M. (2012). Met receptor tyrosine kinase signals through a cortactin-Gab1 scaffold complex, to mediate invadopodia. J. Cell Sci. 125, 2940–2953 10.1242/jcs.100834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito Y., Oinuma I., Fujimoto S., Negishi M. (2009). Plexin-B1 is a GTPase activating protein for M-Ras, remodelling dendrite morphology. EMBO Rep. 10, 614–621 10.1038/embor.2009.63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaeper U., Gehring N. H., Fuchs K. P., Sachs M., Kempkes B., Birchmeier W. (2000). Coupling of Gab1 to c-Met, Grb2, and Shp2 mediates biological responses. J. Cell Biol. 149, 1419–1432 10.1083/jcb.149.7.1419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott G. A., McClelland L. A., Fricke A. F., Fender A. (2009). Plexin C1, a receptor for semaphorin 7a, inactivates cofilin and is a potential tumor suppressor for melanoma progression. J. Invest. Dermatol. 129, 954–963 10.1038/jid.2008.329 [DOI] [PubMed] [Google Scholar]
- Seki N., Hashimoto N., Taira M., Yagi S., Yoshida Y., Ishikawa K., Suzuki Y., Sano H., Horiuchi S., Yoshida S.et al. (2007). Regulation of Src homology 2-containing protein tyrosine phosphatase by advanced glycation end products: the role on atherosclerosis in diabetes. Metabolism 56, 1591–1598 10.1016/j.metabol.2007.06.029 [DOI] [PubMed] [Google Scholar]
- Soong J., Chen Y., Shustef E. M., Scott G. A. (2012). Sema4D, the ligand for Plexin B1, suppresses c-Met activation and migration and promotes melanocyte survival and growth. J. Invest. Dermatol. 132, 1230–1238 10.1038/jid.2011.414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens L., McClelland L., Fricke A., Williamson M., Kuo I., Scott G. (2010). Plexin B1 suppresses c-Met in melanoma: a role for plexin B1 as a tumor-suppressor protein through regulation of c-Met. J. Invest. Dermatol. 130, 1636–1645 10.1038/jid.2010.13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamagnone L., Artigiani S., Chen H., He Z., Ming G. I., Song H., Chedotal A., Winberg M. L., Goodman C. S., Poo M.et al. (1999). Plexins are a large family of receptors for transmembrane, secreted, and GPI-anchored semaphorins in vertebrates. Cell 99, 71–80 10.1016/S0092-8674(00)80063-X [DOI] [PubMed] [Google Scholar]
- Thomas J., Liu T., Cotter M. A., Florell S. R., Robinette K., Hanks A. N., Grossman D. (2007). Melanocyte expression of survivin promotes development and metastasis of UV-induced melanoma in HGF-transgenic mice. Cancer Res. 67, 5172–5178 10.1158/0008-5472.CAN-06-3669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villanueva J., Herlyn M. (2008). Melanoma and the tumor microenvironment. Curr. Oncol. Rep. 10, 439–446 10.1007/s11912-008-0067-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., He H., Srivastava N., Vikarunnessa S., Chen Y. B., Jiang J., Cowan C. W., Zhang X. (2012). Plexins are GTPase-activating proteins for Rap and are activated by induced dimerization. Sci. Signal. 5, ra6 10.1126/scisignal.2002636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W., Klaman L. D., Chen B., Araki T., Harada H., Thomas S. M., George E. L., Neel B. G. (2006). An Shp2/SFK/Ras/Erk signaling pathway controls trophoblast stem cell survival. Dev. Cell 10, 317–327 10.1016/j.devcel.2006.01.002 [DOI] [PubMed] [Google Scholar]
- Yang Y. H., Zhou H., Binmadi N. O., Proia P., Basile J. R. (2011). Plexin-B1 activates NF-κB and IL-8 to promote a pro-angiogenic response in endothelial cells. PLoS ONE 6, e25826 10.1371/journal.pone.0025826 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]