

Summary
Rapidly progressive dementias are conditions that typically cause dementia over weeks or months. They are a particular challenge to neurologists as the differential diagnosis often is different from the more typical, slowly progressive dementias. Early and accurate diagnosis is essential, as many of the etiologies are treatable. The information in this review is in part based on experience through our rapidly progressive dementia program at the University of California San Francisco, Memory and Aging Center. As treatment of a rapidly progressive dementia is entirely dependent on the diagnosis, we present a comprehensive, structured, but pragmatic approach to diagnosis, including key clinical, laboratory, and radiologic features. For the 2 most common causes of rapid dementia, treatment algorithms for the autoimmune encephalopathies and symptomatic management for the neurodegenerative causes are discussed.
Although no formal definition exists for what constitutes a rapidly progressive dementia (RPD), generally we use the term when dementia occurs in less than 1–2 years from illness onset, but more commonly over weeks to months.1 Because these conditions are relatively uncommon, the appropriate diagnostic workup and treatments often are unfamiliar to many neurologists. Accurate, thorough, and prompt diagnosis is important as many RPDs are treatable, and even curable. In this article, we present a practical, systematic approach for RPD diagnosis as well as treatment algorithms for the management of immunotherapy-responsive and other dementias.
Etiology of RPDs
The breakdown of etiologies of RPDs varies among dementia centers. At our center (University of California, San Francisco, Memory and Aging Center), the diagnostic breakdown of RPDs has been 62% prion disease (all forms), 15% other neurodegenerative diseases, 8% autoimmune, 4% infectious, and 2% each psychiatric, cancer, toxic-metabolic, and vascular causes; 4% were of undetermined etiology, often leukoencephalopathies.2 Of all RPDs referred to our dementia program, 17% had potentially treatable etiologies (50% autoimmune, 13% each infectious, psychiatric, and cancer, and 10% toxic-metabolic). Our center has a bias toward prion disease as this is a focus of our clinical research program.3 A major dementia referral center in Greece found the most common cause was dementia (27%) due to reversible causes (toxic/metabolic, infectious, autoimmune, vasculitis, and hydrocephalus included in this group) followed by Alzheimer disease (AD, 18%), frontotemporal dementia (16%), Creutzfeldt-Jakob disease (CJD) (13%), and various other neurodegenerative diseases (13%).4 At the US National Prion Disease Pathology Surveillance Center, of the 1,106 patients autopsied, 68% were diagnosed with prion diseases, 17% with neurodegenerative conditions, 3% vascular, 2% each immune-mediated and neoplasms, 1% each infection and toxic-metabolic, and 4% undetermined (insufficient tissue). Potentially treatable causes, including immune-mediated disorders, neoplasms, infectious diseases, and toxic/metabolic encephalopathies, were found in 6.4% of all cases.5
Diagnostic approach
Clinical assessment
Making the correct diagnosis of an RPD is often difficult, but is the key to appropriate treatment. RPD diagnosis usually requires a systematic and thorough approach. A detailed medical history, including emphasis on elucidating first symptoms, documenting all prescribed and nonprescribed medications and any relevant family history, is imperative. Examination should establish if any other neurologic features are present and determine whether other organ systems are involved, so physical and neurologic examination must be thorough. Cognitive assessment can be done with a brief test, such as the Montreal Cognitive assessment (MoCA; www.mocatest.org), but a more detailed assessment might further refine the localization of cognitive deficits (particularly for neurodegenerative conditions). Use of the mnemonic VITAMINS (table 1) is a useful way to review potential etiologies for RPDs: vascular, infectious, toxic-metabolic, autoimmune, metastases/neoplasm, iatrogenic/inborn error of metabolism, neurodegenerative, or systemic/seizures.
Table 1.
Some causes of rapidly progressive dementia, organized by VITAMINS mnemonica
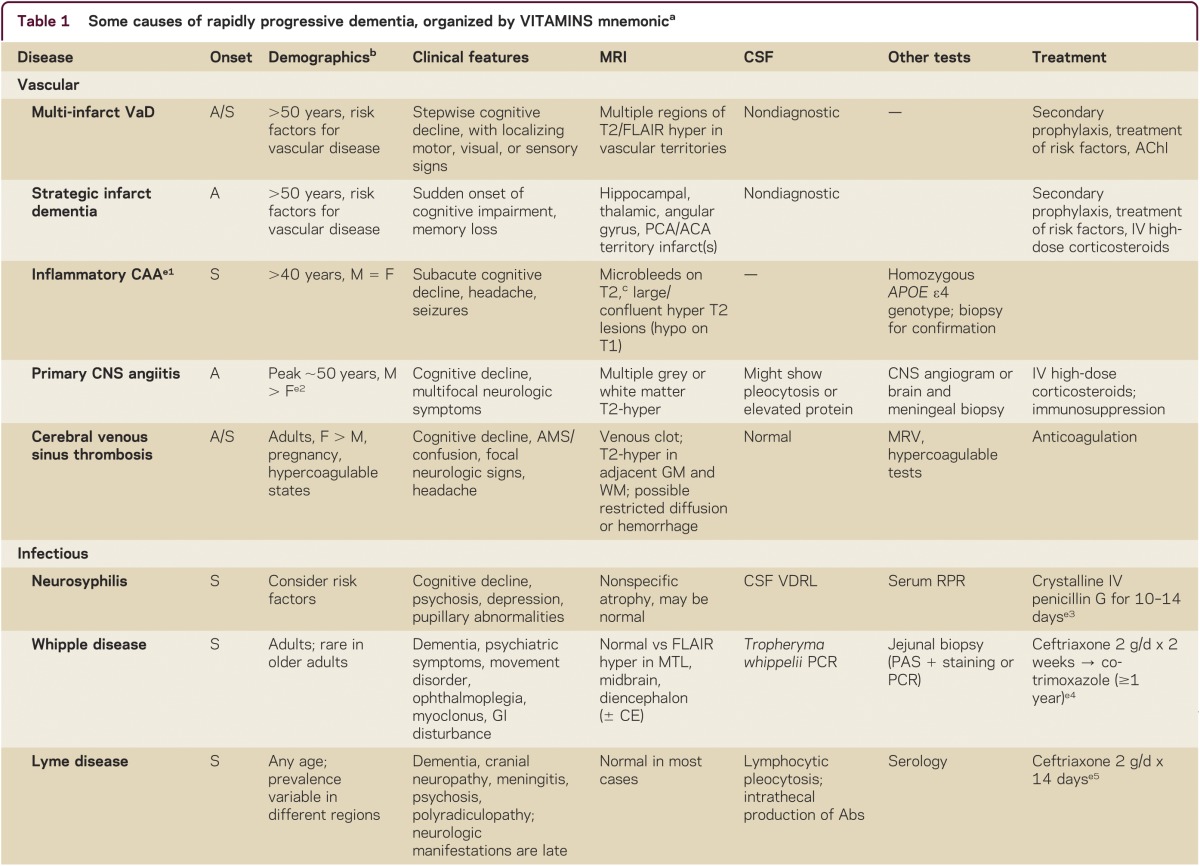
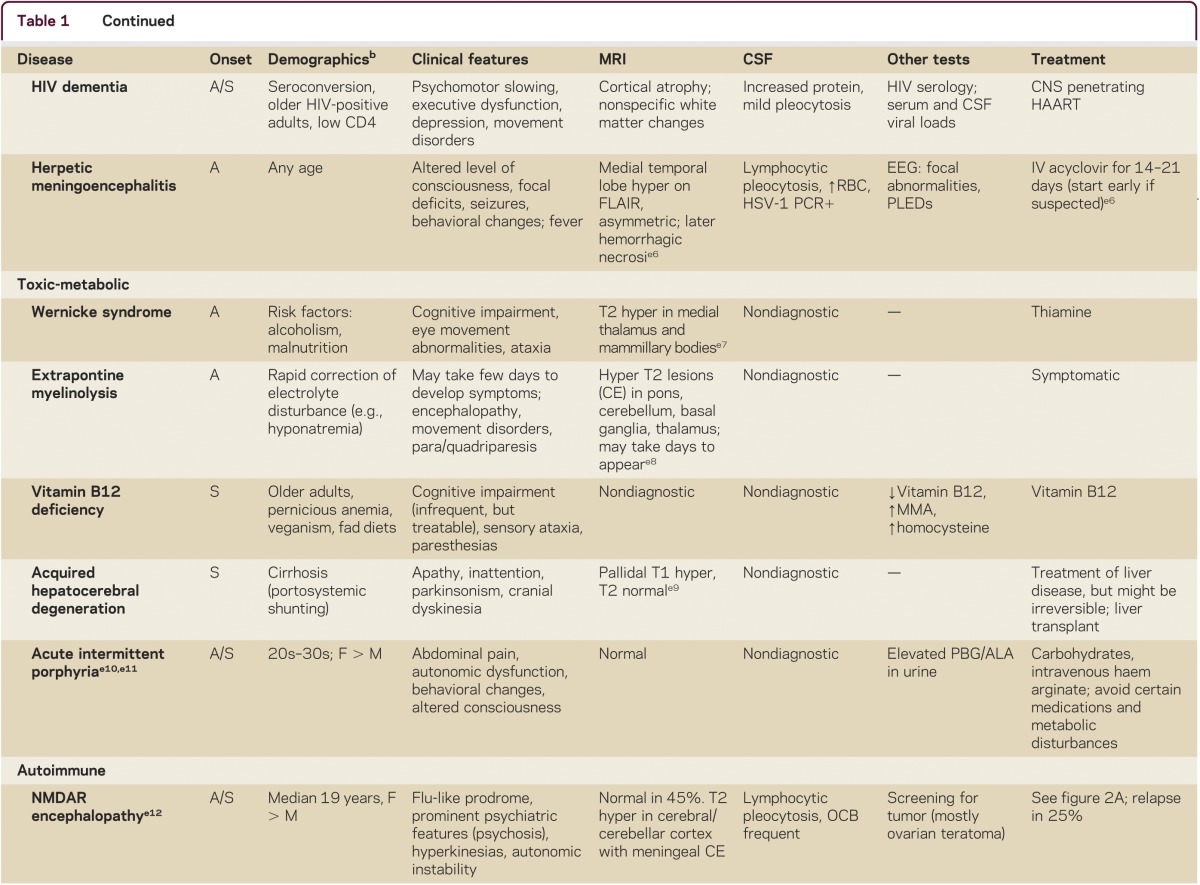
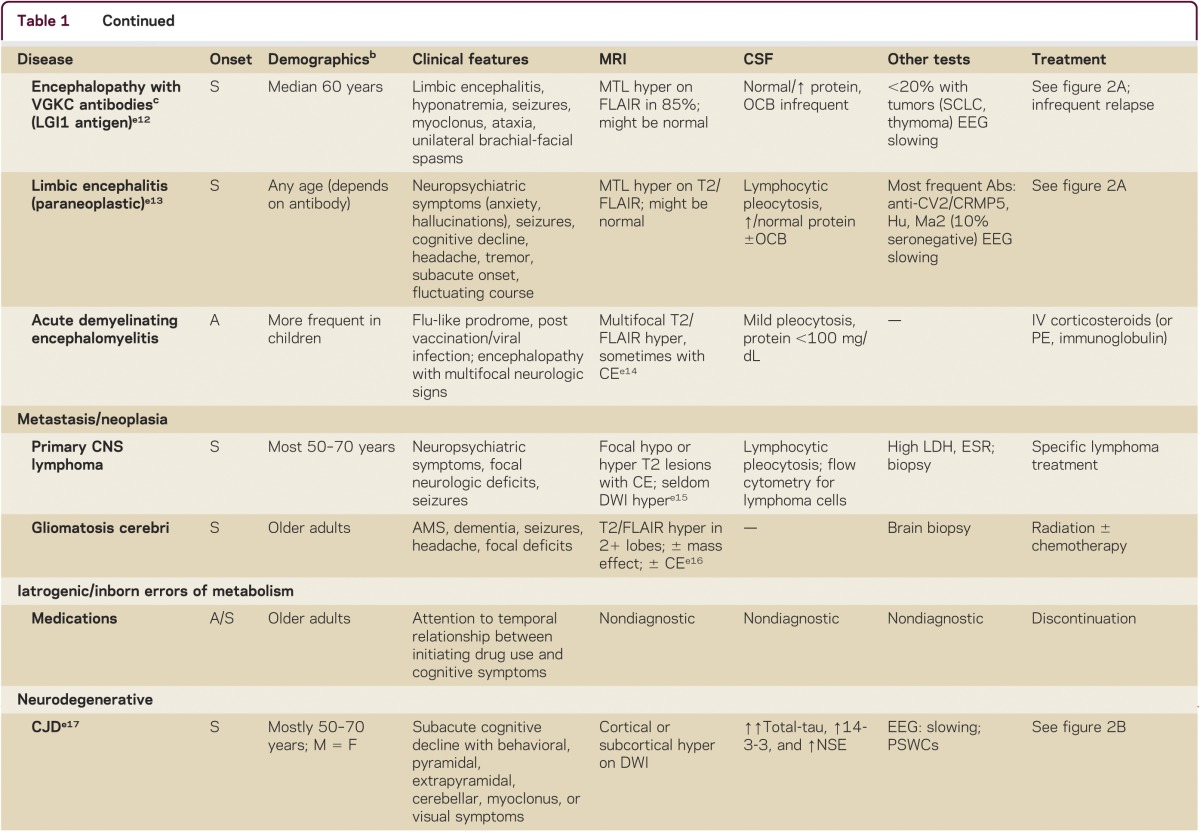
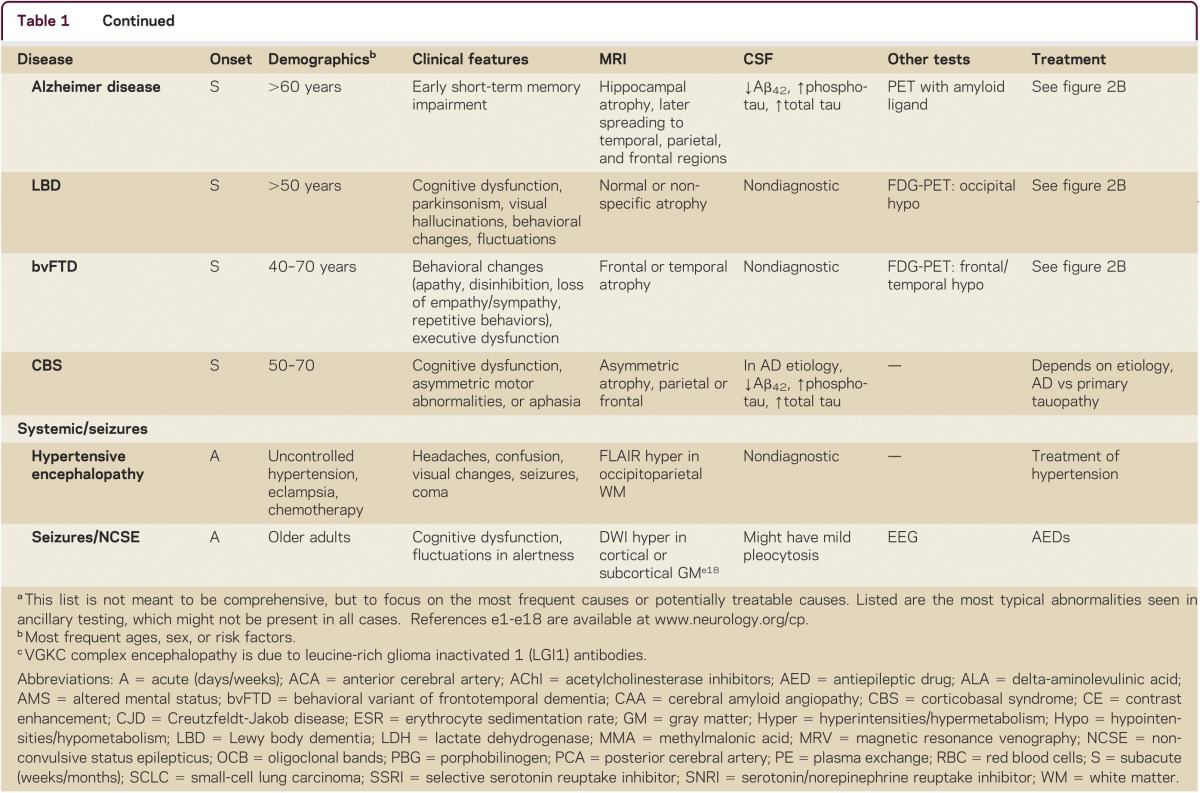
Laboratory tests
As many RPDs have overlapping clinical features, ancillary testing is necessary. Although it is important to be pragmatic and avoid unnecessary costs and testing-associated morbidity, the diagnostic workup should be comprehensive and cover the most frequent causes of RPD, with special attention to potentially treatable conditions.
Figure 1 shows 2 levels of laboratory testing: a first level of tests that should be ordered or at least considered in most RPDs, and a second level, which includes tests to consider in selected cases. For the first level, always remember the mantras that “common things are common” and “don't forget the basics!”—metabolic disturbances are a frequent cause of acute/subacute encephalopathy in older patients (delirium). The second level of tests depends on the results from the first level. In the vast majority of cases, combining clinical findings, the serologic dementia screen, CSF, and neuroimaging will help narrow down the differential diagnoses and determine additional types of tests to be ordered.
Figure 1. Suggested diagnostic tests for initial rapidly progressive dementia evaluation.

ANA = antinuclear antibody; ANCA = anti-neutrophil cytoplasmic antibody; anti-TG = anti-thyroglobulin; anti-TP = anti-thyroperoxidase; CRP = C-reactive protein; ESR = erythrocyte sedimentation rate; NSE = neuron-specific enolase; RF = rheumatoid factor. Modified from Geschwind et al.2
Screening for serum (in some cases, also CSF) antibodies causing autoimmune and paraneoplastic encephalopathies6 is important as many of these conditions are treatable and even reversible. Knowing the specific antibodies might help to better define the syndrome or identify the underlying cancer,7 as well as indicate likelihood of response to treatment.8,9 Probably the most common antibodies associated with autoimmune (paraneoplastic or non-paraneoplastic) encephalopathy are the intraneuronal antibodies anti-Hu (most frequently associated with small-cell lung carcinoma [SCLC]), anti-Ma2 (testicular germ-cell tumors), anti-CV2/CRMP5 (SCLC, thymoma, and others) and the neuronal surface antibodies AMPA receptor (AMPAR) antibody, VGKC-complex (LGI1 and Caspr2) antibodies, and GABAB receptor (GABABR) antibody.6
The clinical presentation of AEs varies greatly and includes cognitive decline and psychiatric changes developing over days, weeks, or months. Seizures, myoclonus, extrapyramidal symptoms, ataxia, or signs of hypothalamic or autonomic dysfunction can also be observed.10,11 The term limbic encephalitis (LE) is often used to describe a classic phenotype of AE characterized by subacute onset of behavioral changes, memory problems, and seizures. But frequently the disease is multifocal and other parts of the CNS are affected (such as hypothalamus or brainstem), characterizing an autoimmune encephalomyelitis.10,12
Clinical features and demographics might give clues as to the antibody causing an AE. Encephalopathy due to VGKC complex antibodies (caused by LGI-1 receptor antibodies)13 can cause RPD that resembles CJD, though hyponatremia due to SIADH might be a clue as it is not typical of CJD, but is found in about 60% of cases with VGKC complex antibody encephalopathy.14 AMPAR antibody encephalitis is more frequent in middle-aged women15; in a male under 50, anti-Ma2 antibodies (usually associated with testicular tumors) and testicular ultrasound should be considered.11,16,17 AMPAR antibody encephalitis is associated with SCLC, breast or thymus tumors, and GABABR antibody encephalitis is associated with LE and frequent seizures.6 Several excellent reviews on these antibody-mediated encephalopathies have recently been published.11,17–19
Encephalopathy associated with NMDA receptor (NMDAR) antibodies has a distinct clinical presentation (table 1).11,18 It occurs more frequently in children or young adults, but has been reported up to the eighth decade of life. Psychiatric symptoms are prominent at onset; hyperkinetic movements such as dystonia, orofacial dyskinesias or chorea, seizures and signs of autonomic instability, and ultimately pulmonary distress necessitating intubation are later clinical features.20
A reasonable basic screen for autoimmune and paraneoplastic encephalopathies in the context of RPDs might include anti-Hu, anti-Ma2, anti-CV2, VGKC complex (LGI1 and anti-CASPR2 if available) antibodies, anti-amphiphysin, GAD65, and NMDAR antibodies. Although less common, in the appropriate clinical context, consider AMPAR and GABABR antibodies. We recommend sending complete panels of antibodies when feasible, as many patients have more than one antibody, and some antibodies identify the syndrome, others the neoplasm. In seronegative cases in which one has a high degree of suspicion for an AE, or NMDAR encephalopathy is possible, CSF testing should complement serum testing, particularly when treatment with IV immunoglobulin (IVIg) or plasma exchange has already been given, as antibody levels in the CSF might remain elevated.20,21
When autoimmune encephalitis is considered, it is critical to screen for tumors (figure 1). Paraneoplastic encephalopathies can precede identification of the associated neoplasia by as many as 3 years (though most often less than 1 year), and so if the initial screening is negative, regular cancer screening is required (figure 2).6,11 The chance of finding a tumor, however, is variable and depends on the antibody: for example, whereas a causative tumor is found in more than 95% of patients with encephalitis associated with anti-Hu or anti-Ma2, it is found in less than 20% of patients with LGI1 receptor antibodies.11,18 AMPAR antibodies are associated with finding a tumor in 70% of cases, but with GAD65 antibodies, which are infrequently associated with encephalopathy, a tumor is found in only 8% of cases.11
Figure 2. Proposed non–evidence-based treatment algorithm for autoimmune paraneoplastic and non-paraneoplastic encephalopathies.

Adapted from Mckeon et al.19
Autoimmune encephalopathies also occur with systemic autoimmune diseases (often associated with vasculitis).22 Serologic screening for rheumatologic disorders is also recommended for RPD evaluation, although often serology will be normal and brain biopsy might be indicated23 (figure 1; table 1).
CSF
A lumbar puncture should be performed in almost all patients with RPD. It may be particularly helpful with infectious, autoimmune, and neoplastic disorders (figure 1). Lymphocytic pleocytosis, elevated protein level, high immunoglobulin G index, or oligoclonal bands are often present in AEs,6 particularly those of paraneoplastic etiology,24 although they are not specific and might occur in patients with CJD, neuroinfections generating a strong immune response (e.g., subacute sclerosing panencephalitis), or neoplasias. In CJD, basic CSF analysis is typically normal, but in our cohort 50% of cases show mildly increased protein levels (usually 50–100 mg/dL).25 Mild pleocytosis or oligoclonal bands sometimes occur in prion disease.26 Markers for evidence of rapid neuronal injury, such as 14-3-3 protein, neuron-specific enolase (NSE), and total tau might be helpful to confirm the rapid course, rapid neuronal injury, and might be helpful in the rare cases of sCJD in which the DWI MRI is nondiagnostic. Testing for Aβ42, phospho-tau, and total-tau might be helpful when AD is in the differential.27
Neuroimaging
A brain MRI in patients with RPD should include T1 with and without contrast, fluid-attenuated inversion recovery (FLAIR), diffusion-weighted imaging (DWI) and apparent diffusion coefficient (ADC) map, and hemosiderin (e.g., gradient echo or susceptibility) sequences (table 1). Space-occupying lesions (such as most tumors or certain infections, such as toxoplasmosis) should prompt a directed investigation (sometimes requiring biopsy). Certain tumors, such as lymphomas and gliomatosis/lymphomatosis cerebri, however, might not present as space-occupying lesions. In sCJD, DWI abnormalities are characterized by cortical (i.e., cortical ribboning) or deep nuclei hyperintensities, usually with concomitant hypointensities on ADC map.28,29 Hyperintensities are typically more prominent on DWI than FLAIR. If abnormalities are brighter on FLAIR than DWI or there is isolated limbic involvement, this suggests against CJD and should prompt investigations into other etiologies (e.g., autoimmune, toxic-metabolic).28
EEG
EEG is necessary to rule out seizures as the cause of RPD. EEG might also reveal a pattern typical of CJD with periodic sharp wave complexes (PSWCs; seen in about 2/3 of sCJD cases30) or the periodic pattern seen in SSPE. PSWCs, however, might also be seen in hepatic encephalopathy, HE, end-stage AD and DLB, and possibly other conditions.25,31
Brain biopsy
Brain biopsy is sometimes necessary when the less invasive workup is nondiagnostic. At our center, we found that brain biopsy was diagnostic in 65% of patients with RPD (excluding nonlymphomatous tumors or patients with HIV), and the results led to initiation of therapy in 44%.23 In our CJD cohort, 14% of brain biopsies were false negative. Brain biopsies in dementia (not necessarily RPD) generally have had diagnostic yields of 20%–57%, finding treatable causes in about 10% of cases.23,32 Schott et al.33 provide a pragmatic guide for physicians when brain biopsy in dementia is being considered.
Treatment
Autoimmune and paraneoplastic encephalopathies
When to give a trial of immunotherapy?
If an autoimmune encephalopathy is suspected, then a trial of immunotherapy is recommended (figure 2). Certain clinical clues might suggest an autoimmune disorder: a fluctuating clinical course; the detection of a neural-specific paraneoplastic or non-paraneoplastic antibody; other serum markers of autoimmunity such as thyroid antibodies; and detection of inflammatory markers in the CSF.34 The diagnosis of an autoimmune dementia, however, essentially hinges on whether an objective clinical improvement can be demonstrated with immunotherapy.19 Usually the improvement in cognition in responders is striking.17 In responsive patients, the shorter the delay to treatment, often the better the clinical outcome.19
Overall, encephalopathies associated with neuronal surface antigens have better response to treatment than those associated with intraneuronal antibodies. Among the intraneuronal antibodies, LE with anti-Ma2, VGKC-associated, anti-CV2, and AMPAR antibodies, for example, often have excellent response to treatment.6 In a recent observational study, the following clinical and laboratory findings predicted a good response to treatment: subacute onset, fluctuating course, headache, tremor, CSF pleocytosis, or protein >100 mg/dL.17
When not to give a trial of immunotherapy?
Immunosuppression should probably be avoided if infections that might be exacerbated by immunosuppression are high on the differential. If lymphoma is in the differential, steroids should be avoided, as they will cause necrosis of the tumor cells, likely rendering future biopsies nondiagnostic and delaying appropriate treatment.1 It is also important to realize that some conditions that are not necessarily primarily immune-mediated sometimes respond well, albeit sometimes only temporarily, to immunosuppression or related treatments. In fact, IVIg has shown some benefit in AD in phase 2 clinical trials and is in phase 3 trials currently (www.clinicaltrials.gov).
How to give steroids
As the steroid trial is sometimes also a diagnostic test as well as a treatment, it is important to give a sufficiently high dose so that a clinical response is clear. Unfortunately, there are no data as to the minimum sufficient steroid dose for treatment of immune-mediated dementias; we usually give 1 g of IV methylprednisolone per day for 5 days or equivalent. It is imperative to document baseline measures of clinical features, EEG, neuropsychology, and imaging so that changes can be objectively measured.17
Alternative first-line immunotherapy
Patients who cannot tolerate steroids or in whom they might be contraindicated should be given a course of IVIg or plasma exchange (PLEX). IVIg or PLEX might also be indicated when corticosteroids initially are effective, but repeat trials fail to show benefit or corticosteroids fail initially, but there is strong suspicion of an autoimmune etiology. PLEX sometimes is reserved for the critical care setting19 and has a higher adverse event rate; it should be used with caution in patients with anti-NMDA encephalitis with autonomic involvement.35
Combined therapy
In cases in which the diagnosis is clear, such as when an explanatory neural-specific autoantibody is identified, then a more aggressive approach to immunosuppression might be appropriate.35 Some experts recommend early treatment using a combination of corticosteroids and IVIg followed by a course of concurrent rituximab and cyclophosphamide if there is no clinical improvement after 1 cycle.35 We tend to use single treatments initially, so that it is clear which agent is effective. It does not make sense to use plasma exchange shortly after IVIg as the potential therapeutic benefit of IVIg would be removed. Clinical response might take days to weeks.
Maintenance therapy
In patients with a positive response to the trial of immunotherapy, relapse rate is high and longer term treatment (figure 2) is usually required to maintain remission.17,19 Appropriate osteoporosis and Pneumocystis jiroveci pneumonia prophylaxis should be considered in all patients,19 as well as screening for latent tuberculosis. For clinicians not comfortable administering immunosuppressive therapy, advice can be sought from a neuroimmunologist or rheumatologist.12
Toxic-metabolic
Treatment of toxic-metabolic etiologies varies depending on the specific disorder. Some disorders, such as thiamine deficiency, are easier and cheaper to treat empirically rather than to test for thiamine levels. When treating a patient with suspected Wernicke encephalopathy, remember never to give glucose alone without thiamine, as this might exacerbate the underlying condition. When considering B12 deficiency, measuring homocysteine and methylmalonic acid, as well as B12, might reveal early signs of a B12 deficiency.2 As some patients do not absorb oral B12, retesting levels after initiating treatment is required. If levels are not sufficiently high, IM treatment is appropriate.
Infectious
Treatment of infectious RPD essentially involves detection of the infection and appropriate treatment in consultation with infectious disease specialists. If viral encephalitis is suspected, antivirals should be commenced immediately before awaiting results of investigation; viral PCR on CSF might remain positive even after treatment has been commenced.36 We routinely screen for HIV, syphilis, and Lyme disease, as they are treatable if diagnosed early.1,25 Opportunistic fungal infections such as aspergillosis and cryptococcus might present as RPD and the opportunity to diagnose and treat should not be missed. Although rare, CNS Whipple disease is probably underdiagnosed as it might not present with gastrointestinal symptoms and classic facial movements; we recently were involved with an RPD case mistaken for CJD and then DLB, but only diagnosed with Whipple at autopsy. As Whipple is treatable with antibiotics, we recommend a low threshold for considering this infection.
Neurodegenerative
CJD is uniformly fatal and more than 85% of patients with CJD die within 1 year of onset.37 Despite active efforts with drug development and clinical trials, currently there is no curative or disease-modifying treatment for CJD or other neurodegenerative causes for RPD. Some symptomatic treatment options for CJD, AD, behavioral variant frontotemporal dementia, and Lewy body dementia are shown in figure 3.
Figure 3. Proposed management algorithm for more common neurodegenerative causes of RPD.

AD = Alzheimer disease; bvFTD = behavioral variant frontotemporal dementia; CJD = Creutzfeldt-Jakob disease; LBD = Lewy body dementia; RPD = rapidly progressive dementia. Quinacrine failed to show benefit in a clinical trial.38 LBD management adapted from: Boeve BF (2005), Clinical, diagnostic, genetic and management issues in dementia with Lewy bodies, Clin Sci 109(4), 343–354.39
CONCLUSIONS
RPDs can be a diagnostic challenge to neurologists because they are infrequent in clinical practice and the spectrum of differential diagnoses is broad. The use of a systematic approach to diagnosis (VITAMINS) is helpful to cover the main diagnostic etiologies. Treatment success depends on accurate and early diagnosis and searching for potentially treatable causes is one of the mainstays of the diagnostic workup. Even when the RPD etiology is not treatable per se, there are still options of symptomatic treatments aiming at a better quality of life for patients. Ten diagnostic pearls for RPD are provided in table 2.
Table 2.
Ten diagnostic pearls for rapidly progressive dementia

Supplementary Material
Footnotes
Supplemental Data: neurology.org/cp
STUDY FUNDING
Information acquired for this review was supported in part by NIH/NIA R01-AG031189, NIH-NINDS contract N01-NS-0-2328, NIH/NCRR UCSF CTSI UL1 RR024131, and the Michal J. Homer Family Fund.
DISCLOSURES
The authors report no disclosures relevant to the manuscript. Go to Neurology.org/cp for full disclosures.

REFERENCES
- 1.Geschwind MD, Haman A, Miller BL. Rapidly progressive dementia. Neurol Clin 2007;25:783–807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Geschwind MD, Shu H, Haman A, Sejvar JJ, Miller BL. Rapidly progressive dementia. Ann Neurol 2008;64:97–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Geschwind MD. Clinical trials for prion disease: difficult challenges, but hope for the future. Lancet Neurol 2009;8:304–306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Papageorgiou SG, Kontaxis T, Bonakis A, Karahalios G, Kalfakis N, Vassilopoulos D. Rapidly progressive dementia: causes found in a Greek tertiary referral center in Athens. Alzheimer Dis Assoc Disord 2009;23:337–346 [DOI] [PubMed] [Google Scholar]
- 5.Chitravas N, Jung RS, Kofskey DM, et al. Treatable neurological disorders misdiagnosed as Creutzfeldt-Jakob disease. Ann Neurol 2011;70:437–444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rosenfeld MR, Dalmau J. Update on paraneoplastic and autoimmune disorders of the central nervous system. Semin Neurol 2010;30:320–331 [DOI] [PubMed] [Google Scholar]
- 7.Pittock SJ, Kryzer TJ, Lennon VA. Paraneoplastic antibodies coexist and predict cancer, not neurological syndrome. Ann Neurol 2004;56:715–719 [DOI] [PubMed] [Google Scholar]
- 8.Graus F, Saiz A, Lai M, et al. Neuronal surface antigen antibodies in limbic encephalitis: clinical-immunologic associations. Neurology 2008;71:930–936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Greenlee JE. Treatment of paraneoplastic neurologic disorders. Curr Treat Options Neurol 2010;12:212–230 [DOI] [PubMed] [Google Scholar]
- 10.Pruitt AA. Immune-mediated encephalopathies with an emphasis on paraneoplastic encephalopathies. Semin Neurol 2011;31:158–168 [DOI] [PubMed] [Google Scholar]
- 11.Titulaer MJ, Soffietti R, Dalmau J, et al. Screening for tumours in paraneoplastic syndromes: report of an EFNS task force. Eur J Neurol 2011;18:19-e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rosenbloom MH, Smith S, Akdal G, Geschwind MD. Immunologically mediated dementias. Curr Neurol Neurosci Rep 2009;9:359–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lai M, Huijbers MG, Lancaster E, et al. Investigation of LGI1 as the antigen in limbic encephalitis previously attributed to potassium channels: a case series. Lancet Neurol 2010;9:776–785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Geschwind MD, Tan KM, Lennon VA, et al. Voltage-gated potassium channel autoimmunity mimicking Creutzfeldt-Jakob disease. Arch Neurol 2008;65:1341–1346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lai M, Hughes EG, Peng X, et al. AMPA receptor antibodies in limbic encephalitis alter synaptic receptor location. Ann Neurol 2009;65:424–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dalmau J, Graus F, Villarejo A, et al. Clinical analysis of anti-Ma2-associated encephalitis. Brain 2004;127:1831–1844 [DOI] [PubMed] [Google Scholar]
- 17.Flanagan EP, Caselli RJ. Autoimmune encephalopathy. Semin Neurol 2011;31:144–157 [DOI] [PubMed] [Google Scholar]
- 18.Lancaster E, Martinez-Hernandez E, Dalmau J. Encephalitis and antibodies to synaptic and neuronal cell surface proteins. Neurology 2011;77:179–189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mckeon A, Lennon VA, Pittock SJ. Dementia: immunotherapy-responsive dementias and encephalopathies. Continuum 2010;16:80–101 [DOI] [PubMed] [Google Scholar]
- 20.Dalmau J, Gleichman AJ, Hughes EG, et al. Anti-NMDA-receptor encephalitis: case series and analysis of the effects of antibodies. Lancet Neurol 2008;7:1091–1098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dalmau J, Lancaster E, Martinez-Hernandez E, Rosenfeld MR, Balice-Gordon R. Clinical experience and laboratory investigations in patients with anti-NMDAR encephalitis. Lancet Neurol 2011;10:63–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lyons MK, Caselli RJ, Parisi JE. Nonvasculitic autoimmune inflammatory meningoencephalitis as a cause of potentially reversible dementia: report of 4 cases. J Neurosurg 2008;108:1024–1027 [DOI] [PubMed] [Google Scholar]
- 23.Josephson SA, Papanastassiou AM, Berger MS, et al. The diagnostic utility of brain biopsy procedures in patients with rapidly deteriorating neurological conditions or dementia. J Neurosurg 2007;106:72–75 [DOI] [PubMed] [Google Scholar]
- 24.Psimaras D, Carpentier AF, Rossi C. Cerebrospinal fluid study in paraneoplastic syndromes. J Neurol Neurosurg Psychiatry 2010;81:42–45 [DOI] [PubMed] [Google Scholar]
- 25.Geschwind MD. Dementia: rapidly progressive dementia: prion diseases and other rapid dementias. Continuum 2010;16:153–175 [DOI] [PubMed] [Google Scholar]
- 26.Green A, Sanchez-Juan P, Ladogana A, et al. CSF analysis in patients with sporadic CJD and other transmissible spongiform encephalopathies. Eur J Neurol 2007;14:121–124 [DOI] [PubMed] [Google Scholar]
- 27.Shaw LM, Vanderstichele H, Knapik-Czajka M, et al. Qualification of the analytical and clinical performance of CSF biomarker analyses in ADNI. Acta Neuropathol 2011;121:597–609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vitali P, Maccagnano E, Caverzasi E, et al. Diffusion-weighted MRI hyperintensity patterns differentiate CJD from other rapid dementias. Neurology 2011;76:1711–1719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Meissner B, Kallenberg K, Sanchez-Juan P, et al. MRI lesion profiles in sporadic Creutzfeldt-Jakob disease. Neurology 2009;72:1994–2001 [DOI] [PubMed] [Google Scholar]
- 30.Steinhoff BJ, Zerr I, Glatting M, Schulz-Schaeffer W, Poser S, Kretzschmar HA. Diagnostic value of periodic complexes in Creutzfeldt-Jakob disease. Ann Neurol 2004;56:702–708 [DOI] [PubMed] [Google Scholar]
- 31.Tschampa HJ, Neumann M, Zerr I, et al. Patients with Alzheimer's disease and dementia with Lewy bodies mistaken for Creutzfeldt-Jakob disease. J Neurol Neurosurg Psychiatry 2001;71:33–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Warren JD, Schott JM, Fox NC, et al. Brain biopsy in dementia. Brain 2005;128:2016–2025 [DOI] [PubMed] [Google Scholar]
- 33.Schott JM, Reiniger L, Thom M, et al. Brain biopsy in dementia: clinical indications and diagnostic approach. Acta Neuropathol 2010;120:327–341 [DOI] [PubMed] [Google Scholar]
- 34.Castillo P, Woodruff B, Caselli R, et al. Steroid-responsive encephalopathy associated with autoimmune thyroiditis. Arch Neurol 2006;63:197–202 [DOI] [PubMed] [Google Scholar]
- 35.Rosenfeld MR, Dalmau J. Anti-NMDA-receptor encephalitis and other synaptic autoimmune disorders. Curr Treat Options Neurol 2011;13:324–332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lakeman FD, Whitley RJ. Diagnosis of herpes simplex encephalitis: application of polymerase chain reaction to cerebrospinal fluid from brain-biopsied patients and correlation with disease: National Institute of Allergy and Infectious Diseases Collaborative Antiviral Study Group. J Infect Dis 1995;171:857–863 [DOI] [PubMed] [Google Scholar]
- 37.Pocchiari M, Puopolo M, Croes EA, et al. Predictors of survival in sporadic Creutzfeldt-Jakob disease and other human transmissible spongiform encephalopathies. Brain 2004;127:2348–2359 [DOI] [PubMed] [Google Scholar]
- 38.Geschwind M, Kuo A, Raudabaugh B, et al. A randomized, double-blind, controlled study of the efficacy of quinacrine in the treatment of sporadic CJD: American Academy of Neurology 62nd annual meeting: abstract S34.002. Neurology 2010;74:A395 [Google Scholar]
- 39.Boeve BF. Clinical, diagnostic, genetic and management issues in dementia with Lewy bodies. Clin Sci 2005;109:343–354 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.