

Personalized medicine is a new mantra evolving in health care. Harnessing each person’s clinical, genetic, genomic, and environmental information drives the concept. The idea is simple. We can maximize a patient’s chances of a better outcome if we base treatments on what we know. However, isn’t this in many ways the way it’s always been? Isn’t this in part the basis for that old-time house call? To see how a disease or condition is being cared for in the home environment? Clinicians have long used personalized medicine, without overt use of single nucleotide polymorphisms, but certainly not totally void of genetic information. Of course, physicians of the past did not have gene chips, but they did have family histories often informing their decisions. Today as we raise the hopes for targeted therapies to break us free from the algorithmic treatments often followed by hit and miss approaches, there is a renewed fervor for personalized medicine. How do we get there and how does the average clinician or researcher understand the burgeoning array of information, talking heads, and latest hype of subgroup benefits? A key aspect is to trust your training, your experience, and your instincts, coupled with a few repeated doses of biostatistics.
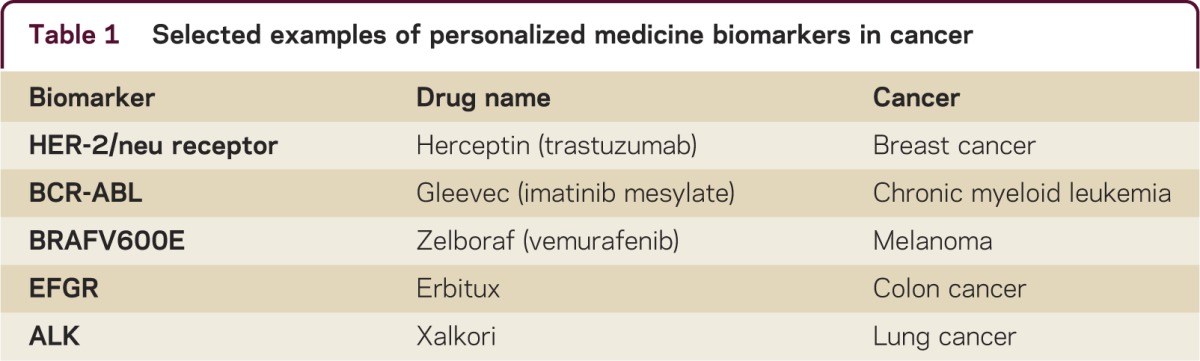
The goal of personalized medicine is to improve treatment outcomes and reduce adverse events that matter to both the clinician and patient. According to the Personalized Medicine Coalition (PMC) in Washington, there were 13 examples of personalized medicine diagnostic biomarkers and medications in 2006, and 72, over 5 times more, in 2011. The focus and limited, but impressive success of personalized medicine has been in cancer and a small number of chronic medical conditions. Specific outcomes in cancer provide promise for similar advances of personalized medicine in neurology. Some examples are outlined in table 1. Herceptin (trastuzumab) used in breast cancer is directed to the 30% of breast cancers with an overexpression of HER-2 protein, which respond to Herceptin. Gleevec (Imatinib mesylate) is used to treat chronic myeloid leukemia, which has increased life expectancy from 5% to 95% at 5 years. Zelboraf (Vemurafenib) is used to treat melanoma, where the late-stage prognosis has been dismal, but 60% of patients have a defect in their DNA, and this drug benefits those with the V600E defect. Other successful personalized medicine examples of “treatment–biomarker” combinations are in colon cancer (Erbitux–EFGR) and lung cancer (Xalkori–ALK).1 There are also successful examples of the prediction of correct personalized dosing. The best known example is the CYP 450 enzyme and its application to Coumadin/warfarin therapy. The correct personalized dosing of warfarin could prevent 17,000 strokes in the United States and avoid 43,000 emergency room visits. The Mayo Clinic and Medco2 tested this prediction in 3,600 patients and found hospitalizations were reduced by 30%. However, in contrast to these successes, the personalized medicine approaches thus far in neurology have failed to replicate these successes.
Table 1. Selected examples of personalized medicine biomarkers in cancer.
A study of interleukin 17F (IL-17F) from Stanford3 reported that pretreatment levels of serum IL-17F could predict poor response of patients with multiple sclerosis (MS) to interferon-β (IFN-β) therapy. Bushnell et al.4 studied IL-17F in stored serum samples from subjects to validate the important finding of Stanford using samples from a large clinical trial. Unfortunately, neither pretreatment nor post-treatment serum levels of IL-17F, IL-17F/F, or their ratio were different in patients classified as good or poor responders to IFN-β, regardless of the definition of treatment response. Investigators from the Stanford study provided aliquots to be certain that the IL-17 assays used in the Bushnell et al. and Stanford studies provided comparable results. Retesting aliquots from the Stanford study demonstrated similar assay performance, eliminating one obvious potential explanation for discrepant results.5 The Bushnell et al. study casts doubt on the future utility of IL-17 as a predictive biomarker or at least in the difficulty of using such a marker with variable predictive validity.
Another avenue is the research in MS. In the largest gene study of MS, the researchers compared DNA from nearly 10,000 people with multiple MS with DNA from more than 17,000 unrelated, healthy individuals. They successfully confirmed 23 previously known genetic links and identified 29 new ones, in addition to 5 strongly suspected genes that contribute to MS. Breakthroughs from MS DNA studies could lead to new treatments or targeted treatments. However, associations with disease occurrence may not predict treatment response. Another study in MS suggests that there are 4 distinct patterns of antibody responses that might be helpful in selecting patient-specific treatments. Progress on predictive biomarkers to guide specific disease-modifying drug therapy for patients with MS has been disappointing, despite extensive efforts.6
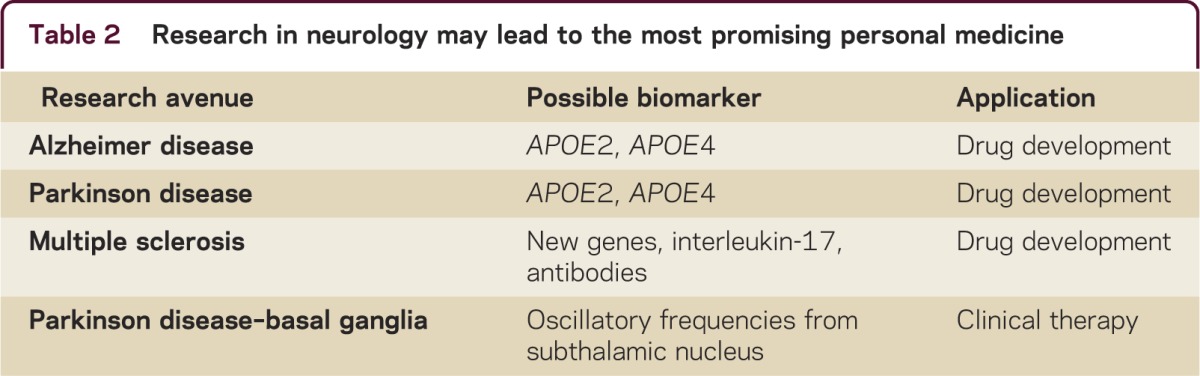
The concept of personalized medicine includes selective genotype-based prescription of drugs to individuals for whom the drug should be safe and effective. For neurologic disorders, the term used is personalized neurology. Several research avenues in neurology may be promising vis-à-vis personal medicine (table 2), but thus far success is at best mixed. Besides the research in MS, another important avenue is the research in Alzheimer disease and Parkinson disease. APOE4 is widely recognized to increase the risk of developing Alzheimer disease, whereas APOE2 is thought to be protective. A new study in Parkinson disease showed that APOE2 may increase the risk of Parkinson disease, indicating that APOE may have varying effects in different neurodegenerative diseases.7
Table 2. Research in neurology may lead to the most promising personal medicine.
A similar situation with few successes appears in personalized medicine research in psychiatry. While disappointing, this may not be surprising, since 80% of 25,000 human genes appear to have some effect on the brain. A third avenue is research in basal ganglia functions and functional connectivities, especially in the specific patterns of oscillatory neuronal discharges which dictate specific motor behaviors. In Parkinson disease, increased endogenous frequencies recorded from the subthalamic nucleus (STN) region are associated with worsening of motor symptoms. These specific oscillatory frequencies could be utilized to tailor a personalized approach to deep brain stimulation in the STN region for effective control while optimizing battery life for a particular patient, better timing battery replacement surgery.
The search for biomarkers is important to neurologic disease. There are few objective methods to predict the effectiveness of specific drugs for individual patients. Patients are treated and when results are not as desired, they are often switched to other drugs, hoping for a satisfactory response. Predictive biomarkers to guide drug selection are certainly needed as the number of available drugs increase. Biomarkers can help us dissect the pathophysiologic processes and promise tremendous value in following aspects: diagnostics and stratification of subcategories of disease stages; prediction of disease course; treatment selection and improved prognosis for treatment success; and the evaluation of novel therapeutics.
Nevertheless, searching for biomarkers is much more difficult that touting their benefits. One key misconception is that many people confuse a statistically significant difference between responders and nonresponders on a biological or clinical variable as sufficient to establish that such is a biomarker and can be used to develop personalized medicine approaches. While such differences are necessary, they are not sufficient. Statistical significance or p values are functions of sample size, not the value of the difference or the clinical importance of the finding or the so-called effect size.
While formulae often make reader’s eyes glaze over, indulge this simple explanation for a moment. Let us suppose among nonresponders to a treatment, the mean IL-17 before treatment initiation was found to be 35 with a standard deviation of 7 (smaller than often seen in the reality for the variability of cytokines, but this is for illustration only). Suppose in responders it was found, on average, to be 42 with a standard deviation of 7 before treatment initiation. The test of significance of this would be calculated from an independent 2-group t test as follows:
t = difference in means/(standard error of the difference)
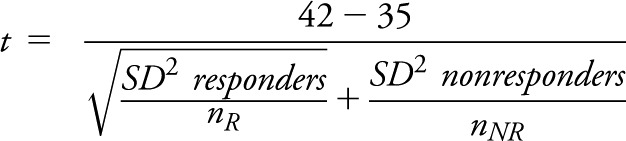 |
t = 7/sqrt(49/nR + 49/nNR)
If nR = 5 and nNR = 5, then our test statistic, t = 7/sqrt (49/5 + 49/5)
t = 7/sqrt (98/5) = 7/4.43 = 1.58 and an associated nonsigificant p value = 0.1528.
However, if we had observed the same values, but with sample sizes of nR, nNR of 50 and 200, then our test statistic would become
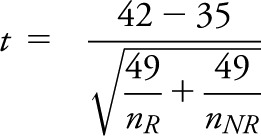 |
= 7/1.107 = 6.32 with an associated highly significant p value of < 0.00001.
So we see that exactly the same differences and variability lead to radically different p values simply because of the size of the samples. Certainly we have more confidence in the estimates of the treatment effects with larger samples, but the effects found are the same in both studies. Thus, basing choices of biomarkers on statistical significance is a risky endeavor. What we really want to know is how comfortably can we predict how a patient will respond to a therapy?
One way to provide estimates of how different 2 groups are, that is independent of the sample size, is to use a quantity called the effect size. The effect size tells us how many standard deviation units apart 2 groups are. It relates the difference between the groups to the inherent variability within a sample. The commonly used definition of effect size is the difference in means/standard deviation within a group. So from the above example, the effect size is 7/7 = 1. An effect size of 1 is a relatively large effect size, indicating the 2 groups are rather different. Most effect sizes of drug treatments are under 0.50.
For a biomarker, we expect to correctly classify to which group an individual is likely to belong, based on the value of the biomarker. There are 4 meaningful values to assess when considering prediction: sensitivity, specificity, positive predictive value (PPV), and negative predictive value (NPV).
Sensitivity is predicting that an individual is a responder or has a condition among those individuals who indeed have the disease. That is, the correct percentage of our predictions of disease. Specificity is predicting that an individual is a nonresponder or does not have a condition among those individuals who indeed do not have the disease. That is, the correct percentage of our predictions of no disease.
PPV is how often our prediction for disease or response is correct among all individuals predicted to have the condition. This value is often more important to patients than is sensitivity. For example, it is comforting to a woman with a positive mammogram that she still has only about 1 in 10 chance of breast cancer (table 3). The PPV is actually low, whereas the sensitivity is about 75%.
Table 3. Mammogram screen test for breast cancer.
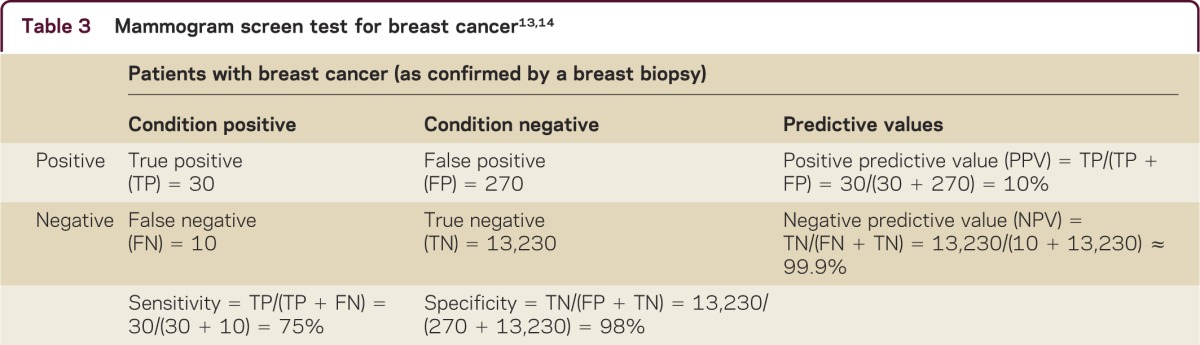
NPV is how often our prediction for not having disease or nonresponse is correct among those predicted not to have the disease. Again, thinking of the patient with a negative mammogram, their NPV is over 99.9% (table 3), whereas the specificity is only about 98%, meaning as a clinician you have 2% chance of missing it, but the patient can assume she is truly disease-free, because the NPV is so high. If we consider the impact of prevalence, we can calculate the NPV using the specificity, sensitivity, and prevalence.8 For example, based on responses to questions about whether a person was ever diagnosed with breast cancer in the Behavioral Risk Factor Surveillance Study in 2009, the overall prevalence of female breast cancer was estimated as 1.52% in Arizona. The recalculated NPV is 99.6%, which provides the same explanation. These measures are critically important in personalized medicine.
Consider our IL-17 example from above. Figure 1 shows graphically the 2 hypothetical distributions where we assume we have 50 responders and 200 nonresponders. This is the highly significant p value and thus, the responder mean is significantly different from the nonresponder mean. However, clinically we have to select a value of the biomarker to decide whether an individual will be a responder or nonresponder. This choice can depend on the situation. In some instances it is important to maximize sensitivity, that is, ensure we do not miss cases when a patient has a condition, such as a rapid HIV test. In other cases, we might focus on specificity. For example, a negative test for troponin may rule out a myocardial infarction. In figure 1, the shaded area might represent a cutpoint to maximize finding responders. In this example, we see that sensitivity is 94%, that is, the area under the curve on the right of the cutpoint defined by the shaded area. However, this cutpoint yields a specificity of nonresponders of only 20%. In terms of personalized medicine, such an approach would indeed identify a high proportion of patients who would respond to a treatment. However, the very low specificity does a poor job of identifying patients who are highly likely not to respond to a therapy.
Figure 1. Interleukin-17 distribution of responders and nonresponders optimized for sensitivity.

The interplay between sensitivity and specificity should be clearly recognized. Except in very rare circumstances (i.e., complete separation of distributions), can specificity be improved at the expense of sensitivity, and vice versa? In figure 2, the area might represent a cutpoint to increase the specificity of nonresponders. In this example, we see that specificity is 90.5%, that is, the area under the curve on the left of the cutpoint. However, the sensitivity of responders decreases to 50%. Obviously, sensitivity or specificity can always be made to be 100%—just say all patients have a disease or are responders (sensitivity will be 100%) or no one has the disease or will respond (specificity is 100%). Thus, it is the mix of these 2 that must be considered in identifying a biomarker. It should be noted that many investigators shy away from cutpoints as individual differing risks themselves are important and it might not be desirable to select a single value.
Figure 2. Interleukin-17 distribution of responders and nonresponders with alternative cutpoint to increase the specificity of nonresponders.

What are the costs of mistakes? If successful therapies can be developed for APOE-positive patients in Alzheimer disease, then specificity (those who do not benefit) may be more important because treating with an ineffective therapy for them may delay other potential brain-sparing therapies. With HIV infection, the public health significance may direct us to focus on screening tests with high sensitivity, but lesser specificity to be verified in more definitive follow-up tests.
Investigators looking to identify or demonstrate a biomarker often use their data to establish a successful outcome. Key to the reader or listener of such results should be how did they define the biomarker as a predictor? Is it the common definition? Do they have an association between 2 variables?
An approach frequently used is to look for significant associations with extreme groups to show that biomarkers differ and that classification (sensitivity and specificity) can be achieved; for example, taking the upper quartile of the biomarker distribution and comparing it to the lowest quartile. This can be used to identify promising candidates, but usually overestimates the predictive qualities of a biomarker. The poor predictive qualities result because it ignores how well the classification works within the 50% of the population that is left out of the analysis. Before looking at an example, consider what measurable characteristics a biomarker may have. A biomarker is a parameter that can be used to measure the progress of disease or the effects of treatment. The parameter can be chemical, physical, or biological. A functional performance measurement of a patient is an indicator of a particular disease state, which is a physical biomarker.
Now, to illustrate, let us consider the following simulated example of a MS study looking at the 25-foot timed walk as a predictive biomarker of the disease. While walking ability is a major way the disease impacts a patient, the question becomes whether you can use a functional test to more easily indicate change over time. You measure 400 patients who have a mean timed walk of 6.9 seconds with a standard deviation of 3.1 seconds. Five years later, 161 (40.25%) of the patients have progressed as measured by the gold standard clinical outcome Expanded Disability Status Scale (EDSS). You want to investigate if the timed walk is a predictive biomarker for who will progress. At baseline, the lowest quartile consists of all those who have a timed walk of 4.8 seconds or less and, of course, there are 100 patients in this lower quartile (25% of the total population). Similarly, the highest (fourth) quartile includes those 100 patients who have a timed walk associated with the value that identifies the highest 100 patients (upper quartile), which are those longer than 9.1 seconds.
To demonstrate you have a found a biomarker, you test whether the progression in the lowest quartile is significantly different from the highest quartile. In the lowest (first) quartile, 17% (17 patients) progress compared to the highest (fourth) quartile, where 61% progress (61 patients). This is highly statistically significant, p < 0.0001, and shows a threefold difference in progression rates. You feel you have found a predictive marker. Stopping here provides strong evidence that you can indeed predict outcome.
However, in the middle 2 quartiles, 42% (81 of the 200 patients) progress, and you attempt to see how good your predictive model works in the total population. This is akin to assessing the timed walk as if you were to use it in the future on all patients who enter your clinic. A common method to evaluate this process is to run a logistic regression analysis with the outcome being progression (yes or no) and the predictor variable (25-foot timed walk) to assess the value of predictor variable on the outcome. The logistic regression analysis shows 25-foot timed walk is significantly associated with the MS progression. Sensitivity is measured as the proportion of patients with progression (yes) who were predicted to progress (yes). Specificity is measured as the proportion of those without progression (no) who were predicted not to progress (no). The predictive power of a logistic regression model can be evaluated by c-statistic (an estimate of the area under a receiver operating characteristic [ROC] curve) along with the classification table, similar to the mammography example in table 3. Using logistic regression you can find an optimal cutoff point for the classification table. This can be determined by finding the probability level that maximizes the sum of sensitivity and specificity in the ROC curve called the Youden Index.9
In our simulated example, we have 161 patients who have progressed as measured by the gold standard clinical outcome EDSS in the 400 patients enrolled 5 years prior. From this analysis you obtain the odds ratio of progressing for each second increase on the timed walk. You also obtain the important estimates of sensitivity and specificity. For these data, the odds ratio is estimated at 1.3 (95% confidence interval = 1.21, 1.42). The estimated sensitivity is 67.4% and the estimated specificity is 67.7%—not the threefold difference in risk of progression seen in the assessment of the highest and lowest extreme quartiles.
Why do the results look so different between the analyses of the extremes vs analyzing the full cohort? There are 2 reasons for this discrepancy. Using quartiles combines all the data below or above a particular cutoff point and ignores the differences in the timed walk among those within the quartile (the variability within the quartile). Secondly, it ignores the individuals in the middle of the distribution for whom the timed walk has more difficulty in sorting out what will happen to them. So, why do researchers use this approach at all? The answer is specifically because of the extreme nature of the results. If there were no relationship between the variable and the outcome at all, then the outcome would be the same irrespective of which quartile one was in. If we performed the same analysis replacing the timed walk with the last 4 digits of someone’s social security number, we would not expect to see differing progression based on the lower last 4 digits compared to the highest 4 digits—the social security number’s last 4 digits are merely sequence numbers. They would be uncorrelated with progression. Thus, using the extremes is a reasonable way to identify potential correlates of outcomes and if one were doing next-generation sequencing one might be able to save enormous resources using this approach, but the results are biased. While this technique can be useful, many researchers stop with the extremes analysis. However, while identifying a correlation using this approach has merit, it is insufficient to demonstrate a general purpose predictive biomarker. It is true that a good biomarker will have the characteristic, but showing good qualities with the extremes is insufficient to declare a biomarker has been found.
Finally, one more important test of a biomarker is what can be called incremental utility or the net reclassification index.10 Incremental utility takes a purported biomarker and asks: how much better are my predictions when I add the putative biomarker to the mix compared to what I can obtain from the simpler model? Paraphrasing from Hlatky et al.,10 it is no longer is it good enough just to report “independently predictive of ….” It is incumbent upon the proponent of the biomarker to demonstrate improved classification is occurring. This can be assessed using a net reclassification index. It is an improvement on simple assessment of classification. Pencina et al.11 extended the idea by examining reclassification of subjects with and without the outcome. Any upward movement in categories for subjects with the outcome implies improved classification, and any downward movement indicates worse reclassification. The interpretation is opposite for subjects without the outcome. The improvement in reclassification is quantified as the sum of differences in proportions of individuals moving up minus the proportion moving down for those with the outcome, and the proportion of individuals moving down minus the proportion moving up for those without the outcome. This sum was labeled the Net Reclassification Improvement. Alternative measures exist that integrate net reclassification over all possible cutoffs for the probability of the outcome (integrated discrimination improvement).11,12
In the coming years, there will be more discussion of personalized medicine. While the days of house calls may have given way to a more convenient way of delivering care, the principles of finding and understanding the environment of the patient may be translated to understanding the environment under which these personalized medicine panaceas have been found. It is especially important to use clinical observation in general populations as a check on overly optimistic enthusiasm. The job of the clinician and researchers alike is to avoid the PEST—premature euphoria in selecting treatments!
STUDY FUNDING
Supported in part by the following NIH grants: 5U01NS042685-06, U01 NS45719-01A1, 3UL1RR025777.
DISCLOSURES
G. Cutter serves as a Contributing Editor to Neurology: Clinical Practice. Y. Liu reports no disclosures. Go to Neurology.org/cp for full disclosures.
REFERENCES
- 1.Gordon E. The state of personalized medicine: the role of biomarkers. Presented at the Personalized Medicine World Congress; January 23, 2012; Stanford University, Stanford, CA.
- 2.Epstein RS, Moyer TP, Aubert RE, et al. Warfarin genotyping reduces hospitalization rates: results from the MM-WES (Medco-Mayo Warfarin Effectiveness study). J Am Coll Cardiol 2010;55:2804–2812. [DOI] [PubMed] [Google Scholar]
- 3.Axtell RC, de Jong BA, Boniface K, et al. T helper type 1 and 17 cells determine efficacy of interferon-beta in multiple sclerosis and experimental encephalomyelitis. Nat Med 2010;16:406–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bushnell SE, Zhao Z, Stebbins CC, et al. Serum IL-17F does not predict poor response to IM IFN-1a in relapsing-remitting MS. Neurology 2012;79:531–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rudick RA. The elusive biomarker for personalized medicine in multiple sclerosis: the search continues. Neurology 2012;79:498–499. [DOI] [PubMed] [Google Scholar]
- 6.Kuznar W. What is the appropriate treatment strategy for MS breakthrough disease? Neurol Rev 2008;16:12–14. [Google Scholar]
- 7.Huang X, Chen PC, Poole C. APOE-[epsilon]2 allele associated with higher prevalence of sporadic Parkinson disease. Neurology 2004;62:2198–2202. [DOI] [PubMed] [Google Scholar]
- 8.Zhou XH, Obuchowski NA, McClish DK. Statistical Methods in Diagnostic Medicine. New York: Wiley-Interscience; 2002. [Google Scholar]
- 9.Youden WJ. Index for rating diagnostic tests. Cancer 1950;3:32–35. [DOI] [PubMed] [Google Scholar]
- 10.Hlatky MA, Greenland P, Arnett DK, et al. ; American Heart Association Expert Panel on Subclinical Atherosclerotic Diseases and Emerging Risk Factors and the Stroke Council. Criteria for evaluation of novel markers of cardiovascular risk: a scientific statement from the American Heart Association. Circulation 2009;119:2408–2416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pencina MJ, D’Agostino RB, Sr, D’Agostino RB, Jr, et al. Evaluating the added predictive ability of a new marker: from area under the ROC curve to reclassification and beyond. Stat Med 2008;27:157–172. [DOI] [PubMed] [Google Scholar]
- 12.Steyerberg EW, Vickers AJ, Cook NR, et al. Assessing the performance of prediction models: a framework for traditional and novel measures. Epidemiology 2010;21:128–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Altman DG, Bland JM. Diagnostic tests 2: predictive values. BMJ 1994;309:102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gunnarsson RK, Lanke J. The predictive value of microbiologic diagnostic tests if asymptomatic carriers are present. Stat Med 2002;21:1773–1785. [DOI] [PubMed] [Google Scholar]