

Summary
There is increasing need for both consensus definitions and continued research into the causes, clinical spectrum, and treatment of pediatric movement disorders. Treatment has been largely based on experience rather than evidence because clinical trials are limited. With development of consensus definitions, identification of causative genes, understanding of the clinical spectrum of disease, and clinical trials, we can provide overall better care for children with movement disorders. This review highlights 5 areas where progress is being made to achieve these goals in pediatric movement disorders.
Pediatric movement disorders is a relatively new and growing field of child neurology. Whereas hypokinetic disorders such as Parkinson disease predominate in adults, children more commonly demonstrate hyperkinetic disorders such as tics, tremor, chorea, and dystonia. There are a large number of genetic and heredodegenerative diseases which cause secondary movement disorders in childhood. Advances in pediatric movement disorders have been made by solidifying movement disorder definitions, expanding the spectrum of clinical phenotypes, understanding genetic causes of movement disorders, and rigorously evaluating treatment efficacy for common movement disorders. Five areas were chosen to highlight these advances: consensus definitions, newly discovered PRRT2 mutation, clinical and genetic spectrum of GLUT-1 deficiency and neurodegeneration with brain iron accumulation (NBIA) disorders, and comprehensive behavioral intervention for tics (CBIT).
Creation of consensus definitions for childhood movement disorders
For years, lack of a common language surrounding childhood motor disorders hampered diagnosis, medication management, and ability to create clinical rating scales, and made selection of a homogenous population for research challenging. To address this concern, the NIH-funded Taskforce on Childhood Motor Disorders was created in 2001. The Taskforce includes specialists from developmental pediatrics, neurology, neurosurgery, orthopedic surgery, physical therapy, occupational therapy, physical medicine and rehabilitation, neurophysiology, muscle physiology, and biomechanics. One of the initial goals of the taskforce was to define consistent terminology across multiple clinical and research disciplines. Such terminology would facilitate communication between clinicians and researchers and allow for clear entry and outcome criteria for research in childhood motor disorders.
In 2003, the group published classifications and definitions of disorders causing hypertonia in childhood and defined the terms spasticity, dystonia, and rigidity.1 A 2006 publication focused on definition and classification of negative motor signs in childhood and provided definitions for weakness, reduced selective motor control, ataxia, and deficits of praxis.2 The third publication in 2010 focused on hyperkinetic movements in childhood and provided definitions for dystonia, chorea, athetosis, myoclonus, tremor, tics, and stereotypies.3 These definitions will need continuing review as we gain knowledge about the pathophysiologic mechanisms of these movements and develop biometric assessments to further distinguish among them.
Since the initial 2003 publication, these definitions have been cited in numerous articles and are in use across multiple disciplines. The definitions have become the standard for research in childhood motor disorders. As a measure of the impact that these definitions have had on the field, the 2003 consensus definitions alone have been cited in at least 25 articles including clinical trials for medication efficacy, development of animal models, biomechanical studies, physical therapy interventions, and rating scale development.
PRRT2 mutation: Causative gene of paroxysmal kinesigenic dyskinesia, infantile convulsion with choreoathetosis syndrome, and benign familial infantile epilepsy
Paroxysmal kinesigenic dyskinesia (PKD) is an involuntary, paroxysmal movement disorder characterized by brief, frequent episodes of dyskinesias precipitated by sudden movement.4 PKD can be classified as primary or secondary with primary PKD most often inherited as an autosomal dominant trait.4
In some families, PKD is associated with infantile convulsions alone or infantile convulsion and choreoathetosis (ICCA) syndrome. ICCA is characterized by benign infantile convulsions inherited as an autosomal dominant trait with variable expression of PKD.4–6 Individuals develop nonfebrile convulsions between the ages of 3 and 12 months which typically resolve by 2 years of age or PKD which typically presents in adolescence with normal development and a normal neurologic examination between attacks.6 In 1997, the first genetic association between familial infantile convulsions and paroxysmal choreoathetosis was found to be localized to the pericentromeric region of chromosome 16.6 The genetic link was proposed to support the hypothesis that PKD had an epileptic origin.6 Another study confirmed this linkage and proposed that a channelopathy could explain the variable expression and cosegregation of these paroxysmal neurologic disorders.5
In 2011, the proline-rich transmembrane protein 2 (PRRT2) gene located in the pericentromeric region of chromosome 16 was identified as the first causative gene of PKD through exome sequencing and linkage analysis.7 While the families studied shared common features of PKD (movement trigger, duration of attack, no loss of consciousness, good response to treatment with anticonvulsants), there was clinical heterogeneity within and between families.7 Variable features included age at onset, presence of ICCA syndrome, predominant character of the hyperkinetic movement, predominant body region involved, and the presence of additional triggers.7 Another study showed PRRT2 mutations in 14 of 17 families with benign familial infantile epilepsy (BFIE) and 5 of 6 families with ICCA syndrome.8
The function of PRRT2 is an area of continued research. PRRT2 is mainly expressed in the basal ganglia and may interact with synaptosomal-associated protein 25 (SNAP 25), which is involved in neurotransmitter release from synaptic vesicles potentially leading to neuronal hyperexcitability.7,8
The identification of PRRT2 mutation as a causative gene for PKD, ICCA, and BFIE supports a genetic overlap between paroxysmal movement disorders and epilepsy, which has been recognized in other genetic disorders such as GLUT-1 deficiency.8 It highlights the role of non-ion channel mutations in the pathogenesis of both epilepsy and paroxysmal movement disorders.8 The basis for the clinical heterogeneity in the presentation of the paroxysmal disorders due to PRRT2 mutations remains unknown. Future research will likely expand the clinical phenotype of PRRT2 mutations as well as offer the potential for new therapeutic targets.
The expanding spectrum of GLUT-1 deficiency syndrome
GLUT-1 deficiency syndrome, caused by a mutation in the SCL2A1 gene which encodes for the glucose 1 transporter, is classically characterized by infantile epilepsy, developmental delay, decelerated head growth, acquired microcephaly, cognitive impairment, ataxia, and spasticity.9 Laboratory evaluation shows low CSF glucose levels with normoglycemia and can be confirmed by erythrocyte glucose uptake studies and by GLUT-1 gene mutation analysis.
An expanded spectrum of non-classic phenotypes has been described, some with prominent movement disorders and some which present exclusively with movement disorders. These include a phenotype characterized by mental retardation, dysarthric speech, and intermittent ataxia without seizures, a phenotype characterized by choreoathetosis and dystonia, and a phenotype characterized by paroxysmal exercise-induced dyskinesias (PED) with or without epilepsy.9,10
The most common movement disorders described in GLUT-1 deficiency include ataxia, spasticity, and dystonia.9–13 More recently, mutations in GLUT-1 have been described as a cause of paroxysmal dyskinesias including autosomal dominant and sporadic PED (DYT18) and in slowly progressive spastic paraparesis combined with PED (DYT9).12,13
The spectrum and frequency of movement disorders in GLUT-1 deficiency has been described10 (table 1). Video recordings and charts of 57 patients with GLUT-1 deficiency were reviewed and found gait disturbance (89%), dystonia (86%), chorea (75%), cerebellar action tremor (70%), myoclonus (16%), and dyspraxia (21%).10 Additionally, 28% of patients had nonepileptic paroxysmal events which included episodic ataxia, weakness, parkinsonism, exercise-induced dyskinesias, and nonkinesigenic dyskinesia.10 There is marked phenotypic heterogeneity in the presentation of GLUT-1 deficiency. Although the severity of the phenotype may be related to the residual function of GLUT-1, there is no clear correlation with specific movement disorders.11
Table 1.
Movement disorders in GLUT1 deficiency10
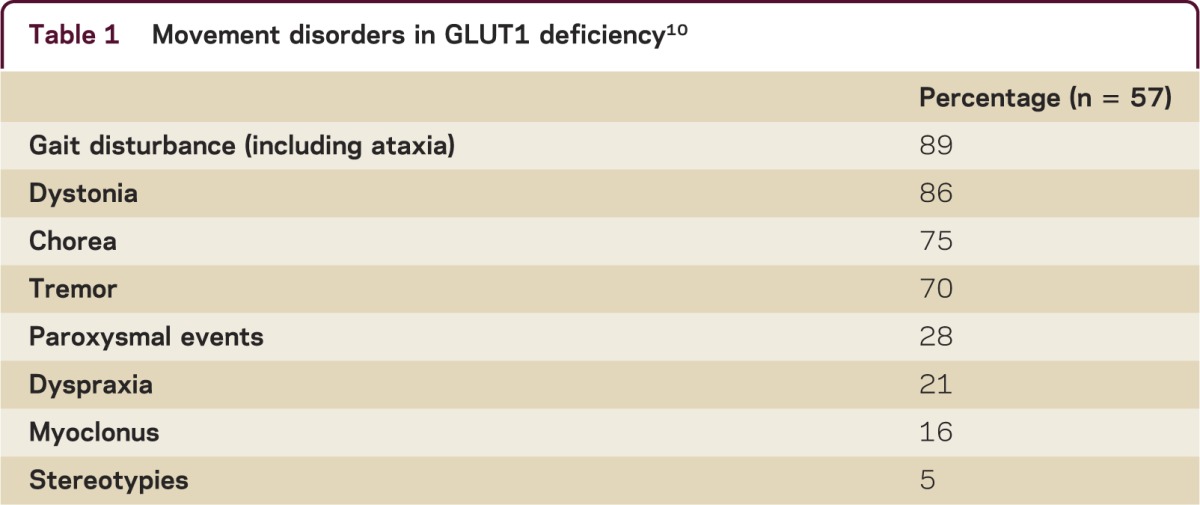
The recognition of the frequency and spectrum of movement disorders in GLUT-1 has expanded the patient population for which GLUT-1 testing will be beneficial. It is especially important because treatment with the ketogenic diet may lead to symptomatic improvement.11 While the classic presentation of GLUT-1 deficiency syndrome is more common, GLUT-1 deficiency should be considered in the differential diagnosis of childhood-onset movement disorders including paroxysmal dyskinesias.
Evolving spectrum and classification of neurodegeneration with brain iron accumulation
NBIA is a group of distinct disorders, each of which results in the excessive accumulation of iron, particularly in the basal ganglia, and presents with a progressive extrapyramidal syndrome. There have been recent advances in understanding the clinical spectrum of these disorders as well as in the discovery of new genes that are now known to cause NBIA. Age-dependent phenotypes have also been recognized. Historically, the terms Hallervorden-Spatz disease, NBIA, pantothenate kinase-associated neurodegeneration (PKAN), and others were used interchangeably, complicating review of older literature. NBIA has become the umbrella term for a group of distinct disorders unified by brain iron accumulation.
There are now 7 established genes known to cause NBIA: PANK2 causing PKAN, PLA2G6 causing phospholipase-associated neurodegeneration (PLAN), FTL causing neuroferritinopathy, ACP causing aceruloplasminemia, FA2H causing fatty acid hydroxylase–associated neurodegeneration (FAHN), ATP13A2 causing Kufor-Rakeb disease, and MMIN causing mitochondrial membrane protein–associated neurodegeneration.14 Clinically, there is overlap between the NBIA syndromes as well as overlap with other diseases, which adds further complexity to their diagnosis.
PKAN (NBIA1) is the most common form of NBIA and has been subdivided into an early-onset classic presentation and an atypical presentation with later onset. Approximately 90% of cases present with the classic phenotype of early-onset gait abnormalities, dystonia, and progressive corticospinal tract dysfunction.14,15 In the atypical form, onset can be in the 20s and 30s and can present with speech abnormalities, unilateral tremor, or focal arm dystonia.14,16
PLAN (NBIA2) is the second most common form of NBIA, which also has an age-dependent phenotype with a classic and atypical form.15 The classic presentation occurs in early childhood with infantile neuroaxonal dystrophy characterized by progressive motor and mental retardation, cerebellar ataxia, marked truncal hypotonia, pyramidal signs, and optic atrophy, while later onset, atypical presentations may be more mild and present with a dystonia-parkinsonism phenotype.14,16
Kufor-Rakeb disease (PARK9) is a rare autosomal recessive disease characterized by parkinsonism with or without pyramidal signs and eye movement abnormalities with onset in adolescence.15 FAHN (SPG25) is characterized by childhood-onset gait impairment, spastic quadriparesis, severe ataxia, and dystonia.15 Mutations in this gene are also known to cause leukodystrophy and a form of hereditary spastic paraplegia.15 Aceruloplasminemia is an autosomal recessive disease caused by a mutation in the ceruloplasmin gene on chromosome 3 and is characterized by adult-onset movement disorders and dementia.15 Neuroferritinopathy is an autosomal dominant disorder caused by a mutation in the FTL gene and is characterized by onset around age 40, chorea, and dystonia.15
While there is considerable clinical overlap in the disorders of NBIA, there are distinctive MRI features which help distinguish among these disorders. The hallmark of all disorders of NBIA is the deposition of iron in the globus pallidus which causes symmetric T2-weighted hypointensity. The distinctive neuroimaging features include the area of greatest iron deposition, white matter involvement, cerebellar atrophy, and brainstem atrophy (table 2).17 Diagnostic algorithms have been proposed which combine clinical features and MRI findings to distinguish among the disorders of NBIA.17,18 Key decision points include evaluation for extrapyramidal involvement on MRI, ophthalmologic findings including pigmentary retinopathy and optic atrophy, and screening blood tests including complete blood count, copper, ceruloplasmin, and serum iron idexes.18
Table 2.
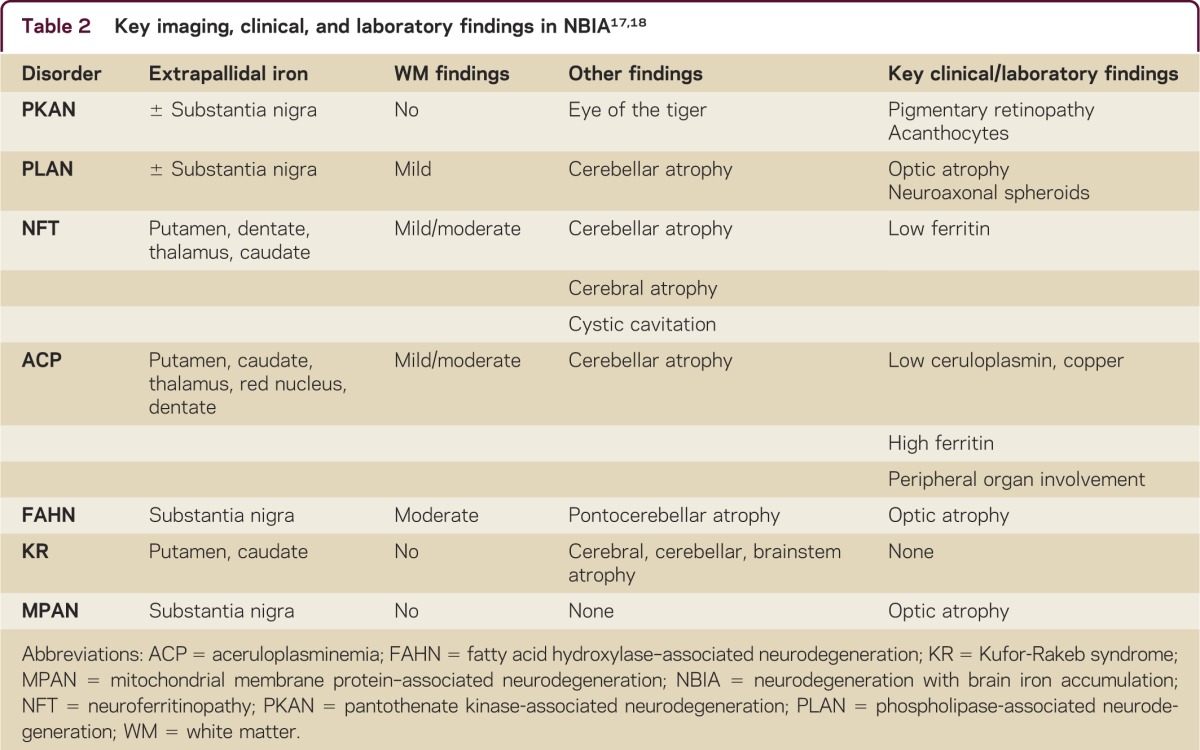
The disorders of NBIA are clinically varied and genetically diverse. There is significant overlap in presentation among these disorders as well as distinctive clinical features and imaging findings that may help differentiate these disorders and guide genetic testing. Often considered to be a pediatric neurologic condition, it is now known that NBIA can present at any age and may be clinically indistinguishable from other neurodegenerative disorders which more commonly present in adulthood such as idiopathic Parkinson disease and Huntington disease. Future research into the metabolic pathways affected in these disorders may ultimately lead to targeted treatment.
Behavioral intervention for tics
Tics are repeated, individually recognizable, intermittent movements or movement fragments that are brief, suppressible, and often associated with a premonitory urge.3 Tourette syndrome (TS) is a neuropsychiatric disorder characterized by childhood onset of chronic motor and phonic tics and is commonly associated with attention-deficit/hyperactivity disorder, obsessive compulsive symptoms, anxiety, and behavioral problems.19,20 Tic disorders are common, with approximately 1 in 5 children experiencing transient tics and 1 in 100 developing TS.19
Pediatric movement disorders: Five new things.
New consensus definitions have become the standard for research in pediatric movement disorders across multiple disciplines. They are now used in clinical trials, biomechanical studies, and development of rating scales.
PRRT2 mutation is a cause of paroxysmal kinesigenic dyskinesia (PKD), infantile convulsion with choreoathetosis syndrome (ICCA), and benign familial infantile epilepsy (BFIE), linking pediatric paroxysmal movement disorders to epilepsy.
Recognition of high incidence and increased spectrum of movement disorders in GLUT-1 deficiency.
Identification of new genes and expanded clinical spectrum in the disorders of neurodegeneration with brain iron accumulation.
Comprehensive behavioral intervention for tics, in a randomized controlled trial, demonstrated efficacy with an effect size similar to that in medication trials.
In many children, tics are mild, do not interfere with daily activities, and therefore do not require treatment. However, tics can be disabling by causing social embarrassment, pain, or inability to perform activities. When treatment of tics is indicated, there are both pharmacologic and behavioral therapies.
A variety of behavioral interventions for tics have been tried for many years including massed practice, relaxation training, self-monitoring, habit reversal therapy, exposure and response prevention, and cognitive behavioral therapy. There have been an increasing number of clinical trials in pediatric movement disorders in recent years but randomized controlled trials for behavioral interventions are uncommon and methodologically challenging. There are few rigorous trials that evaluate the efficacy of the individual components of behavioral interventions or that address combinations of these techniques.
In 2012, the largest randomized controlled trial in TS evaluated a combination of behavioral interventions as a single strategy called CBIT, which is largely based on habit reversal therapy. The first component of habit reversal therapy involves tic awareness, where the patient learns to self monitor tics and to focus on an urge or other early sign a tic is about to occur; the second component is developing a competing response, which involves engaging in a voluntary behavior that is physically incompatible with the tic.21 A second component of CBIT includes relaxation training and function intervention to address situations that exacerbate tics.21 CBIT is most effective when the participant can identify a premonitory urge and is motivated to practice the techniques outside of therapy sessions.
A randomized, controlled trial of 126 children with TS evaluated the efficacy of CBIT to reduce tics and tic-related impairment in children and adolescents as compared to supportive psychotherapy and education. Administration of CBIT resulted in greater improvement in symptom severity among children with TS with a similar efficacy to that found in placebo-controlled medication trials.21 While this study included children on medications, CBIT provides an effective, nonpharmacologic treatment option for children with TS.
Although this study provides evidence of the efficacy of CBIT, the expanding use of CBIT will be limited by the lack of health care providers trained in these interventions. Typically, CBIT is implemented by psychologists or therapists with specific training in the technique. In this study, CBIT was implemented by therapists with master’s level or higher education who underwent systematic training and certification.21 There are currently investigations into the efficacy of abbreviated forms of CBIT provided by nurse practitioners or physicians. In the future, it is possible that a broader range of providers can be trained and certified in CBIT, allowing for more widespread and consistent use.
DISCUSSION
As the field of pediatric movement disorders grows, consensus definitions will allow for streamlined medical management and studies of homogenous populations with the hope of identifying novel treatments. Continued research to identify genetic causes of secondary pediatric movement disorders and to understand the full spectrum of these disorders may to lead to better understanding of the metabolic pathways involved. Clinical trials will help refine treatment by establishing evidence-based medication management. With this information, we can begin to move from symptomatic treatments to potentially disease-modifying treatments.
DISCLOSURES
J. Blackburn has no financial disclosures. J. Mink is funded by NIH grants R01NS060022, R01NS039821, and CDC grant U01DD000510. He serves on the Tourette Syndrome Association Scientific Advisory Board, on the Dystonia Medical Research Foundation Medical and Scientific Advisory Committee, and on the Batten Disease Support and Research Association Medical Advisory Board. He receives honoraria from the American Academy of Neurology for work as Associate Editor of Neurology® and from Edison Pharmaceuticals for serving on a Data and Safety Monitoring Board. E. Augustine is funded by NIH grant K12NS066098, and receives research support from the Food and Drug Administration, Tourette Syndrome Association, and Batten Disease Support and Research Association. She received an honorarium for speaking at the 2012 AAN annual meeting. Go to Neurology.org/cp for full disclosures.

REFERENCES
- 1.Sanger TD, Delgado MR, Gaebler-Spira D, Hallett M, Mink JW. Classification and definition of disorders causing hypertonia in childhood. Pediatrics 2003;111:e89–e97. [DOI] [PubMed] [Google Scholar]
- 2.Sanger TD, Chen D, Delgado MR, Gaebler-Spira D, Hallett M, Mink JW. Definition and classification of negative motor signs in childhood. Pediatrics 2006;118:2159–2167. [DOI] [PubMed] [Google Scholar]
- 3.Sanger TD, Chen D, Fehlings DL, et al. Definition and classification of hyperkinetic movements in childhood. Mov Disord 2010;25:1538–1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lotze T, Jankovic J. Paroxysmal kinesigenic dyskinesias. Semin Pediatr Neurol 2003;10:68–79. [DOI] [PubMed] [Google Scholar]
- 5.Lee WL, Tay A, Ong HT, Goh LM, Monaco AP, Szepetowski P. Association of infantile convulsions with paroxysmal dyskinesias (ICCA syndrome): confirmation of linkage to human chromosome 16p12-q12 in a Chinese family. Hum Genet 1998;103:608–612. [DOI] [PubMed] [Google Scholar]
- 6.Szepetowski P, Rochette J, Berquin P, Piussan C, Lathrop GM, Monaco AP. Familial infantile convulsions and paroxysmal choreoathetosis: a new neurological syndrome linked to the pericentromeric region of human chromosome 16. Am J Hum Genet 1997;61:889–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang JL, Cao L, Li XH, et al. Identification of PRRT2 as the causative gene of paroxysmal kinesigenic dyskinesias. Brain 2011;134:3493–3501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Heron SE, Grinton BE, Kivity S, et al. PRRT2 mutations cause benign familial infantile epilepsy and infantile convulsions with choreoathetosis syndrome. Am J Hum Genet 2012;90:152–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang D, Pascual JM, Yang H, et al. GLUT-1 deficiency syndrome: clinical, genetic, and therapeutic aspects. Ann Neurol 2005;57:111–118. [DOI] [PubMed] [Google Scholar]
- 10.Pons R, Collins A, Rotstein M, Engelstad K, De Vivo DC. The spectrum of movement disorders in GLUT-1 deficiency. Mov Disord 2010;25:275–281. [DOI] [PubMed] [Google Scholar]
- 11.Brockmann K. The expanding phenotype of GLUT1-deficiency syndrome. Brain Dev 2009;31:545–552. [DOI] [PubMed] [Google Scholar]
- 12.Weber YG, Kamm C, Suls A, et al. Paroxysmal choreoathetosis/spasticity (DYT9) is caused by a GLUT1 defect. Neurology 2011;77:959–964. [DOI] [PubMed] [Google Scholar]
- 13.Schneider SA, Paisan-Ruiz C, Garcia-Gorostiaga I, et al. GLUT1 gene mutations cause sporadic paroxysmal exercise-induced dyskinesias. Mov Disord 2009;24:1684–1688. [DOI] [PubMed] [Google Scholar]
- 14.Keogh MJ, Chinnery PF. Current concepts and controversies in neurodegeneration with brain iron accumulation. Semin Pediatr Neurol 2012;19:51–56. [DOI] [PubMed] [Google Scholar]
- 15.Schneider SA, Hardy J, Bhatia KP. Syndromes of neurodegeneration with brain iron accumulation (NBIA): an update on clinical presentations, histological and genetic underpinnings, and treatment considerations. Mov Disord 2012;27:42–53. [DOI] [PubMed] [Google Scholar]
- 16.Schneider SA, Bhatia KP. Syndromes of neurodegeneration with brain iron accumulation. Semin Pediatr Neurol 2012;19:57–66. [DOI] [PubMed] [Google Scholar]
- 17.Kruer MC, Boddaert N, Schneider SA, et al. Neuroimaging features of neurodegeneration with brain iron accumulation. AJNR Am J Neuroradiol 2012;33:407–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kruer MC, Boddaert N. Neurodegeneration with brain iron accumulation: a diagnostic algorithm. Semin Pediatr Neurol 2012;19:67–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jankovic J, Kurlan R. Tourette syndrome: evolving concepts. Mov Disord 2011;26:1149–1156. [DOI] [PubMed] [Google Scholar]
- 20.Kurlan R. Clinical practice: Tourette's syndrome. N Engl J Med 2010;363:2332–2338. [DOI] [PubMed] [Google Scholar]
- 21.Piacentini J, Woods DW, Scahill L, et al. Behavior therapy for children with Tourette disorder: a randomized controlled trial. JAMA 2010;303:1929–1937. [DOI] [PMC free article] [PubMed] [Google Scholar]