

Abstract
RNA editing is a powerful way to recode genetic information. Because it potentially affects RNA targets that are predominantly present in neurons, it is widely hypothesized to affect neuronal structure and physiology. Across phyla, loss of the enzyme responsible for RNA editing, Adar, leads to behavioral changes, impaired locomotion, neurodegeneration and death. However, the consequences of a loss of Adar activity on neuronal structure and function have not been studied in detail. In particular, the role of RNA editing on synaptic development and physiology has not been investigated. Here we test the physiological and morphological consequences of the lack of Adar activity on the Drosophila neuromuscular junction (NMJ). Our detailed examination of synaptic transmission showed that loss of Adar increases quantal size, reduces the number of quanta of neurotransmitter released and perturbs the calcium dependence of synaptic release. In addition, we find that staining for several synaptic vesicle proteins is abnormally intense at Adar deficient synapses. Consistent with this finding, Adar mutants showed a major alteration in synaptic ultrastructure. Finally, we present evidence of compensatory changes in muscle membrane properties in response to the changes in presynaptic activity within the Adar mutant NMJs.
Keywords: Neuromuscular Junction, RNA editing, quantal size, quantal scaling
Introduction
RNA editing is a potent form of information recoding that, through the selective deamination of specific adenosines, converts adenosines to inosines within target RNAs (Bass and Weintraub, 1988, Wagner et al., 1989). This process is carried out by an evolutionary conserved family of enzymes: ADARs -Adenosine Deaminase acting on RNAs- (Bass, 2002, Keegan et al., 2004, Valente and Nishikura, 2005). Because inosines are read as guanosines by the translation machinery (Basilio et al., 1962), RNA editing can lead to changes in protein sequences and properties.
In mammals, RNA editing is essential, since mouse mutants for the Adar2 gene show seizures and early mortality (Higuchi et al., 2000). Nevertheless, the function of Adar on synaptic development and function has not been described in Vertebrate systems. In both C. elegans and Drosophila, reduction of Adar function perturbs behaviors ranging from motility to courtship (Palladino et al., 2000b, Tonkin et al., 2002, Jepson and Reenan, 2009, Jepson et al., 2011), while complete lack of Adar leads to early death, severe uncoordination and seizures in Drosophila. In addition, neurodegeneration has been observed in the CNS of Adar mutants (Palladino et al., 2000b) and recent work linking Adar to the Drosophila fragile X mental retardation protein (dFMR1) suggests that Adar plays a role in controlling NMJ growth (Bhogal et al., 2011). In particular, the latter study has shown that dFMR1 modulates Adar function and the efficiency at which it edits a subset of transcripts encoding for synaptic proteins.
Sustained efforts have been made to isolate targets of RNA editing in Drosophila (Hoopengardner et al., 2003, Keegan et al., 2005, Stapleton et al., 2006) and recently, the analysis of the entire Drosophila transcriptome identified a total of 972 distinct editing sites within the transcripts of 597 genes (Graveley et al., 2011). The majority of these editing sites change codons, and a large number occur in ion channels and in molecules that are part of the synaptic vesicle release machinery. However, despite clear evidence of RNA editing within nervous system function, little is known about its role in regulating synaptic function and physiology. Even though earlier reports found that editing is robust in adults and reduced at early stages of development (Palladino et al., 2000a), recent work has shown that a significant subset of RNAs are edited during early stages of Drosophila development (Graveley et al., 2011). This finding validates the choice of the Drosophila larval neuromuscular junction (NMJ) as a model system that is ideal for addressing the question of whether synaptic physiology is affected by the lack of Adar.
Using the larval NMJ system, we show that Adar is essential for optimal neurotransmission, as well as for spontaneous and calcium-dependent transmitter release. These results are consistent with RNA editing being an essential regulator of neuronal communication. In addition, we observe in Adar mutants an increase in presynaptic markers endophilin, synaptotagmin and synapsin. These changes are also accompanied by severe alterations in synaptic ultrastructure, confirming a major role of RNA editing for the development and structure of the NMJ. Finally we observe a compensatory decrease in postsynaptic glutamate receptor subunit IIA (GluRIIA) and provide evidence that quantal scaling might be a common answer to homeostatic challenge at the NMJ.
Materials and Methods
Fly genetics
Drosophila melanogaster were reared on cornmeal media and raised at 22-25°C. The control stock used in this study were y1w1 for Adar5G1 experiments and elav-Gal4C155/+ for the vGlut over-expression experiments. To observe rescue animals, female Adar5G1 were crossed to males 1407-Gal4/SM6-GFP; UAS-Adar/TM6b. The rescued animals were selected as males: Adar5G1/Y; 1407-Gal4/+; UAS-Adar/+. The animals over-expressing vGlut were female reared at 25°C: elav-Gal4C155/+; UAS-vGlut-1/+. The y1w1 stock, elav-Gal4C155 stock as well as the 1407-Gal4 enhancer trap stock (expression in neurons) was obtained from the Bloomington Stock center. The Adar5G1 (Palladino et al., 2000b) null mutant and the UAS-Adar rescue construct (Keegan et al., 2005) were a kind gift from Dr. Rosenthal. The UAS-vGlut1 flies were a kind gift from Dr. DiAntonio.
Immunohistochemistry
Bouton counting
Synaptic bouton numbers were imaged and quantified as previously described (Marie et al., 2010). Wandering third instar larvae reared at 25°C were dissected in HL3 saline, fixed with Bouin’s fixative (Sigma) for 1min and primary antibodies were applied at 4°C overnight. The mouse monoclonal anti-synapsin antibody (Klagges et al., 1996) was used at 1:10 dilution (Developmental Studies Hybridoma Bank, University of Iowa). Alexa-488 labeled anti mouse secondary antibody (Jackson Immunoresearch Laboratories) was used at a dilution of 1:300. The number of boutons per NMJ revealed by the synapsin immunoreactivity was quantified at the synapse on muscle 6/7 in abdominal segment A3.
Quantification of synaptic proteins
Preparations were treated as above. When fluorescence intensities of mutant, wild-type and rescue genotypes were compared, the larval fillets were processed in the same tube, treated identically, mounted on the same slide and imaged using identical settings. Imaging was performed using a Zeiss LSM5 Pascal confocal microscope. Individual entire muscle 4 NMJ synapses were optically sectioned (0.2 μm; series of 12 to 18 sections per synapse). A 2D projection was generated that projected maximal pixel intensity within the z axis of the optical stacks. The entire synaptic area present on muscle 4 was masked using the “Image J” software (http://rsbweb.nih.gov/ij/) and the average fluorescence of the 2D projection was calculated over the entire synaptic area. A muscle area devoid of synaptic boutons was also masked and quantified in order to establish the background intensity level. The fluorescence intensity values represent the difference between the synaptic intensity and muscle intensity (ΔF) over the intensity of the muscle (F) normalized to wild-type values (Marie et al., 2004; 2010). To quantify synaptic protein abundance at the NMJ, the rabbit polyclonal anti-Dap160 antibody was used at 1/200; the mouse monoclonal anti-bruchpilot at 1:50, the mouse monoclonal antibody against cysteine string protein at 1/10, the mouse monoclonal anti-synapsin antibody at 1:10 dilution, the guinea pig polyclonal anti-endophilin antibody, the mouse monoclonal anti-synaptotagmin1 antibody at 1/10; the mouse monoclonal antibody against Dlg at 1:10, the mouse monoclonal anti GluRIIA antibody at 1/10. Secondary antibodies were used at a concentration of 1/300. The antibodies against bruchpilot, cysteine string protein, synapsin, synaptotagmin1, Dlg and GluRIIA were obtained from Developmental Studies Hybridoma Bank, University of Iowa. The anti-Dap160 and anti-endophilin were gifts from Dr. Kelly and Dr. Bellen laboratories.
Electrophysiological recordings
Larval NMJ electrophysiology
Third instar larvae were dissected as previously described (Davis et al., 1998, Marie et al., 2004). Whole-muscle recordings were performed on muscle 6 in abdominal segment A3 using sharp microelectrodes (12–16 MΩ). Only the recordings with resting membrane potentials exceeding -60 mV and with input resistances greater than 5 MΩ were selected for the analysis. The average mEPSP amplitude was quantified by measuring the amplitude of 100–200 individual sequential spontaneous mEPSP events per NMJ, using Mini Analysis software (Synaptosoft). The average per-NMJ mEPSP amplitudes were then calculated for each genotype. The average of 20 supra-threshold evoked EPSP amplitude (stimulation at 0.5 Hz) was calculated for each synapse ensuring that both motor axons innervating muscle 6 in segment A3 were recruited. Quantal content was calculated as the average of EPSP amplitude divided by the average mEPSP amplitude. Quantal content was determined for each synapse and then averaged across synapses to generate the average quantal content for each genotype. Data for steady-state synaptic transmission were acquired in HL3 saline (70 mM NaCl, 5 mM KCl, 10 mM NaHCO3, 115 mM Sucrose, 5 mM Trehalose, 5 mM HEPES, 0.4 mM CaCl2, 10 mM MgCl2).
Electron microscopy
Preparations were fixed in 5% glutaraldehyde, 4% paraformaldehyde in 90 mM cacodylate buffer with 90 mM CaCl2 added for 15 min at room temperature and, after a brief wash in cacodylate buffer, incubated in the fixative at 4°C overnight. After washing in 90 mM cacodylate buffer (two times for 10 min), samples were post-fixed for 30 min in 1% osmium tetroxide in 90 mM cacodylsate buffer with KFeCN added, washed, osmicated for another 30 min, washed in distilled H2O and then incubated in 2% uranylacetate for 1 hour. After washing in H2O, samples were dehydrated through a graded series of ethanol and acetone, and embedded in Epon/Spurr epoxy resin (Electron Microscopy Sciences). Thin sections (50-60 nm) were collected using a Leica Ultracut microtome on Formvar/carbon coated copper slot grids, and contrasted with lead citrate. Samples were examined on a JEOL1020 transmission electron microscope at 60 kV.
Results
Adar affects Quantal size
Despite evidence that Adar is expressed at lower levels at early stages of development (Jepson et al., 2011), it is clear that RNA editing is present both at the embryonic and larval stages (Keegan et al., 2005, Graveley et al., 2011). We therefore turned to the larval NMJ preparation to analyze the role of Adar on synaptic physiology. We first examined whether the average amplitude of spontaneous miniature release events (mEPSP or quantal size) was perturbed in Adar5G1 null mutant synapses. The analysis established that there is indeed a significant 14% increase in the quantal size of Adar5G1 mutants when compared to controls (p < 0.02; Fig. 1A, B). We examined the frequency of miniature release and found that there is no significant difference between control and Adar5G1 mutants. The increase in mEPSP amplitude could be due to an increase in post-synaptic neurotransmitter receptor abundance, increased vesicle size, increased neurotransmitter loading within the vesicle, or simultaneous release of two or more vesicles. To start distinguishing between these possibilities, we analyzed the “neuronal rescue” flies (Adar5G1/y; 1407-Gal4/+; UAS-Adar/+). In an otherwise Adar5G1 mutant background, these flies contain a 1407-Gal4 line used to drive UAS-Adar expression. The 1407-Gal4 driver promotes expression throughout the embryonic nervous system (Sweeney et al., 1995, Fergestad and Broadie, 2001) as well as larval CNS neurons. These flies, that express the Adar cDNA in neurons in an otherwise Adar5G1 mutant background, showed a quantal size (0.76 +/- 0.08 mV; n = 10) that was significantly different from the Adar5G1 mutants (p < 0.02) but undistinguishable from control values (p > 0.25). This showed that it is presynaptic Adar that is essential for proper quantal size and rule out that the increase in quantal size observed in the Adar5G1 mutants is due to the lack of editing of a postsynaptic target.
Figure 1. Adar mutants show an increase in the amplitude of spontaneous miniature release events.
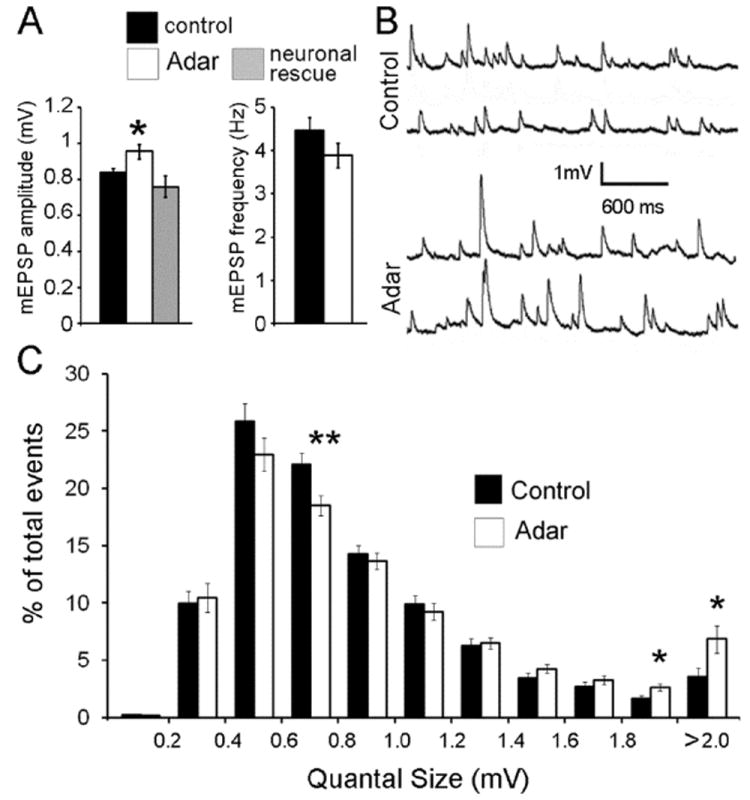
(A) Quantification of average mEPSP amplitude in control and Adar5G1 mutants. The average amplitude spontaneous miniature release events of Adar5G1 mutants (0.96 +/- 0.06 mV; n = 51) is increased by 14% (Student’s t-test; p < 0.02) when compared to controls (0.84 +/- 0.03 mV; n = 55). The frequency of miniature release is unchanged in control (4.5 +/- 0.3 Hz; n = 45) and Adar5G1 mutants (3.9 +/- 0.3 Hz; n = 45; p > 0.18).
(B) Representative traces of miniature spontaneous release events in wild type and Adar5G1 mutants.
(C) Frequency distribution profiles of controls and Adar5G1 mutant mEPSPs. Analysis of 4100 Adar5G1 mutant miniature events and 4200 control miniature events (100 mEPSPs per NMJ; 41 and 42 NMJs) shows that higher amplitude events (between 1.8 and 2 mV and superior to 2mv) are statistically more frequent (Student’s t-test; p < 0.023) in Adar5G1 mutants than in controls. In contrast, events with an amplitude between 0.6 and 0.8 mV are statistically less frequent (Student’s t-test; p < 0.007) in Adar5G1 mutants than in controls.
To understand whether subtler aspects of the mEPSP frequency distribution were altered, we measured the amplitude of 4100 individual mEPSPs (from 41 Adar5G1 mutant NMJs) and 4200 individual mEPSPs (from 42 control NMJs) (Fig. 1C). This analysis showed that the spontaneous miniature release events of high amplitudes (superior to 1.8 mV) are statistically more frequent in Adar5G1 mutants than in controls while the mEPSPs that have amplitudes ranging between 0.6 and 0.8 mV are less abundant in the mutants. Because the mean frequency of mEPSP release is unchanged (Fig. 1A), we conclude that lack of Adar activity leads to more frequent large-amplitude miniature release events.
Adar regulates neurotransmitter release
In order to determine whether Adar is necessary for evoked synaptic release, we recorded from control and Adar5G1 animals in 0.4 mM extracellular calcium saline. A low level of extracellular calcium allows for quantal analysis and the detection of small differences in the number of synaptic vesicles released per action potential (Davis and Goodman, 1998).
We found that the most noticeable change in synaptic transmission in Adar5G1 mutants is a decrease of the number of vesicles released per action potential. This was estimated by calculating the quantal content (average EPSP amplitude/average mEPSP amplitude). The quantal content of the Adar5G1 mutant synapses is reduced by 28% (p < 4.2 10-5; Fig.2A) when compared to controls. The resulting EPSP amplitude in Adar5G1 mutants is 37.9 +/- 2 mV (n = 19) in Adar compared to 45.1 +/- 2.2 mV in controls (n = 17; p < 0.02). These data indicate that the mechanism of neurotransmitter release depends on RNA editing.
Figure 2. Adar mutants show a decrease in transmitter release.
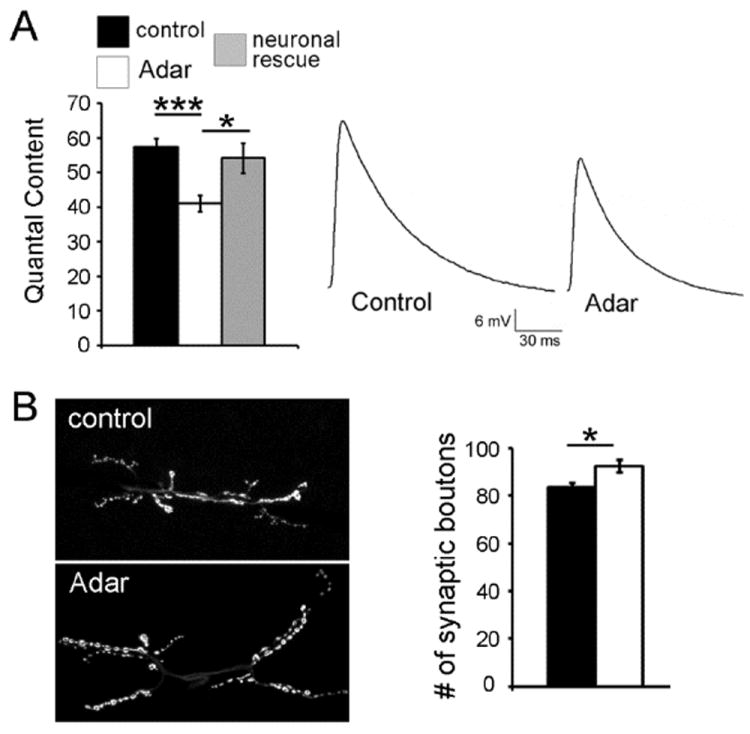
(A) (left) Quantification of average quantal content. Values of Adar5G1 mutants (open bars), control (filled black bars) and neuronal rescue (filled grey bars). We find a significant (p < 4.2 10-5) decrease in the amount of vesicles released in the Adar5G1 mutants: the quantal content of the Adar5G1 mutant synapses is 41.1 +/- 2.4 (n=19) while it is 57.3 +/- 2.6 in controls (n= 17). Homozygous Adar5G1 mutant animals that are rescued by neuronal expression of Adar cDNA show normal vesicle release quantal content of 54.2 +/- 4.3 (n = 10), indistinguishable from control flies (p > 0.55) but significantly different from Adar5G1 mutants (p < 0.02). Data collected in 0.4 mM extracellular Ca2+. Each genotype is identified in the legend at the top of the chart. Error bars represent average ± SEM. (right) Representative traces of control and Adar5G1 mutant EPSPs.
(B) Adar mutant synapses show a slight but significant increase in growth. (Left) Two representative photographs of synapses at muscles 6/7, abdominal segment A3 are revealed by immunofluorescence against Synapsin. (Right) Quantification of the number of synaptic boutons at the synapse 6/7 segment A3 show a slight but significant 10.6% increase in Adar mutants (n = 25) when compared to controls (n = 25). Each genotype is identified in the legend at the top of the chart (A). Error bars represent average ± SEM.
Because Adar is specifically enriched in neurons, we investigated the neuron-specific function of Adar. We examined the “neuronal rescue” animals expressing the UAS-Adar cDNA within neurons in an otherwise Adar null mutant background. Transmitter release was rescued in these animals which showed a quantal content value indistinguishable from control flies (p > 0.55; Fig. 2A) but significantly different from Adar5G1 mutants (p < 0.02; Fig. 2A). This result demonstrates that Adar is affecting presynaptic targets regulating neurotransmitter release and raises the interesting possibility that mRNA editing could be modulating presynaptic function.
This deficit in neurotransmitter release in Adar5G1 mutants could be the consequence of perturbed synaptic development. It is possible that loss of Adar could affect synaptic growth and/or development and indirectly impair synaptic efficacy. Therefore, we assessed NMJ growth by counting the number of synaptic boutons at an identified synapse (muscle 6/7; abdominal segment A3). We find that there is a slight (12.5%) but significant (p < 0.001) increase in synaptic growth in Adar5G1 mutant synapses (86.2 boutons +/- 1.9; n = 25; Fig. 2B) when compared to control synapses (76.6 boutons +/- 1.9; n = 25; Fig. 2B). This agrees qualitatively with a recent study reporting an overgrowth in Adar5G1 mutants (Bhogal et al., 2011), although the latter study reports a more prominent effect than observed in the present study (64 boutons in Adar5G1 versus 40 boutons in WT). It should be noted, however, that previous studies by other laboratories have established control numbers of boutons that are generally on the order of 80 (Ruiz-Canada et al., 2004, Miech et al., 2008), in agreement with what was found in the present study. In summary, our data demonstrate that Adar5G1 mutants have an increased number of synaptic boutons while showing compromised quantal release. Thus, we hypothesize that the defect in vesicular transmitter release in Adar mutants may be due to the lack of editing in one or several of the presynaptic proteins involved in this process.
Lack of Adar leads to changes in abundance of several presynaptic proteins
In order to explain the effect of Adar on transmitter release, and given that we did not observe any reduction in synaptic growth that could explain these differences, we examined the localization and abundance of a subset of synaptic proteins within terminal synaptic boutons using quantitative immunocytochemistry (Marie et al., 2004, Marie et al., 2010). More specifically, we examined the expression of (1) Dap160, a protein homologous to intersectin, a central component of the presynaptic endocytic machinery (Roos and Kelly, 1998, Marie et al., 2004); (2) bruchpilot (Brp), a protein homologous to ELKS/CAST that promotes active zone assembly and vesicle release (Kittel et al., 2006, Wagh et al., 2006); (3) cysteine string protein (Csp), a homolog of the DnaJ/Hsp40 family of co-chaperones that promotes SNARE complex assembly and has a neuroprotective role (Zinsmaier, 2010, Sharma et al., 2011); (4) synapsin, a neuro-specific protein associated with synaptic vesicles (Greengard et al., 1993, Bykhovskaia, 2011); (5) endophilin (Endo), a membrane-binding protein that participates in the endocytosis of clathrin-coated vesicles (Verstreken et al., 2002, Milosevic et al., 2011); and (6) synaptotagmin 1 (Syt) that senses synaptic Ca2+ concentration and regulates synaptic vesicle release (Chapman, 2008, Martens and McMahon, 2008).
We did not notice any obvious differences in the sub-synaptic localization of these presynaptic markers, and quantified the intensities of immunostaining for these proteins at the synapses. While the levels of expression of Dap160, Brp and CSP were not different in controls and Adar5G1 mutants, other proteins showed statistically significant increases in expression in Adar5G1 mutants (Fig. 3). Synapsin showed an increase of 35% in Adar5G1 mutants (staining intensity in control is 100 +/- 4.4 %; n = 34 and 135 +/- 7.8 %; n = 30; p < 3.10-4 in Adar5G1 mutants). Similarly in mutant synapses, endophilin is increased by 37% (n = 36; p < 6.10-4). Interestingly, the amount of synaptotagmin1 doubled in the mutant (staining intensity in control is 100 +/- 6.8 %, n = 26 and 199 +/- 17%, n = 30, p < 3.10-4 in Adar5G1 mutants). The abundance of these synaptic proteins is restored towards control values when we express Adar cDNA in the neurons in an otherwise Adar5G1 mutant background (neuronal rescue; Fig. 3).
Figure 3. The abundance of synapsin, endophilin and synaptotagmin is increased at Adar mutant NMJs.
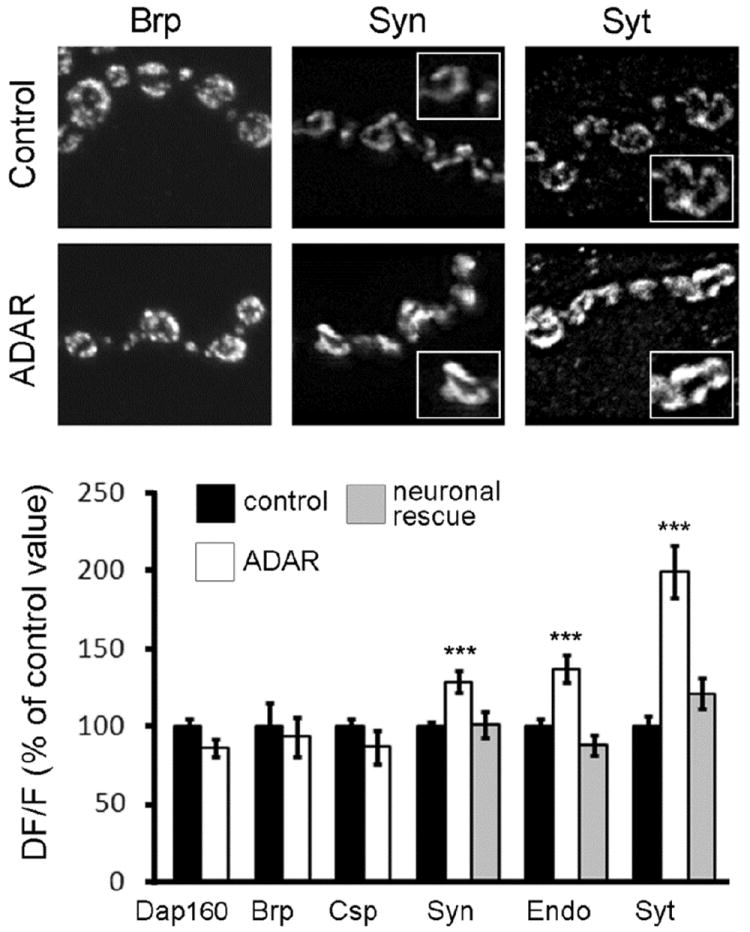
(A) Representative control and Adar5G1 mutant synaptic boutons from muscle 4 show that synapsin, endophilin and synaptotagmin abundance increased in Adar5G1 mutants. Note that in both mutant and controls the proteins seem absent from inter-bouton synaptic segment and that, within boutons, the expression is mainly localized close to the plasma membrane. Insets show synaptic boutons at higher magnification.
(B) Quantification of fluorescence intensity as a measure of Dap160, bruchpilot, cysteine string protein, synapsin, endophilin and synaptotagmin abundance at the NMJ of muscle 4. While levels of Dap160, bruchpilot and cysteine string protein do not vary between control and Adar5G1 mutant, synapsin, endophilin and synaptotagmin levels are increased by 35%, 37% and 99% respectively in Adar5G1 mutant when compared to controls. Levels of expression are restored towards control values when Adar5G1 mutants are rescued by neuronal expression of Adar cDNA (neuronal rescue). Each genotype is identified in the legend at the top of the chart. Error bars represent average ± SEM.
These data suggest that RNA editing is required within motoneurons for the proper expression of a subset of presynaptic proteins. Because Syt1 abundance at the NMJ is strongly affected in the Adar5G1 mutants and because synaptotagmins are essential to the calcium sensing and exocytosis (Pang and Sudhof, 2010), we decided to ask whether Ca2+ dependence of transmitter release was affected in Adar5G1 mutants.
Adar is essential for the calcium dependence of transmitter release
We investigated synaptic transmission (quantal content) as a function of extracellular calcium concentration. At 0.2 mM extracellular calcium, synaptic transmission in Adar5G1 mutant larvae (quantal content 24.03 +/- 3; n = 10) was indistinguishable from control animals (quantal content 20.4 +/- 2.1; n = 10; Fig. 4 A, B). However, when extracellular calcium was increased (0.3 to 0.5 mM Ca2+), synaptic transmission was significantly impaired in Adar5G1 mutants relative to controls (Fig. 4). At 0.5 mM Ca2+, Adar5G1 mutants release 48.4 +/- 2.1 quanta (n = 10) per evoked action potential while control animals release 66.2 +/- 3.3 (n = 10).
Figure 4. Adar is required to modulate the calciumdependence of vesicle release.
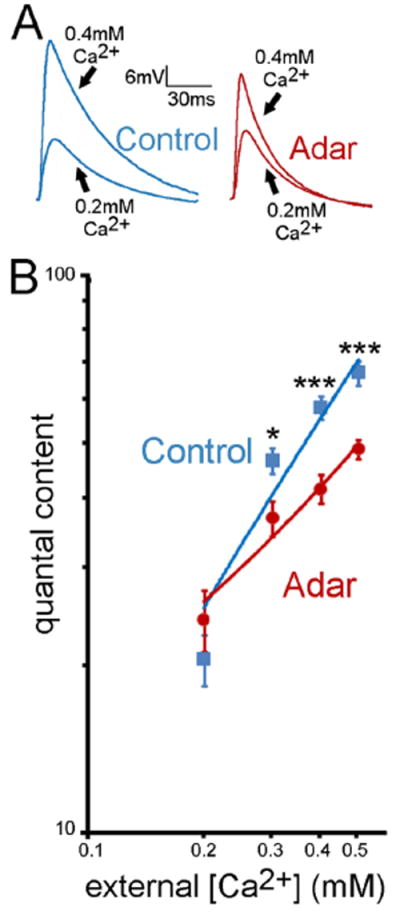
(A) Representative traces of control and Adar5G1 mutants at 0.2 and 0.4 mM extracellular Ca2+ concentration. (B) Quantification of average quantal content values at different extracellular Ca2+ concentrations for control (blue) and Adar5G1 mutants (red). At 0.2 mM extracellular Ca2+ quantal content of control and Adar5G1 mutants are not significantly different (p > 0.3; Student’s t-test). However, as the extracellular Ca2+ concentration increases, vesicle release between control and Adar5G1 becomes increasingly different (p < 0.03 at 0.3 mM extracellular Ca2+; p < 5 10-5 at 0.4 mM extracellular Ca2+; p < 4 10-4 at 0.5 mM extracellular Ca2+), behaving as if the Adar5G1 mutant synapses cannot appropriately sense the increasing extracellular Ca2+ concentration. Error bars represent average ± SEM.
While it is common knowledge that the entry of calcium into the presynaptic terminal is the key event triggering vesicle fusion (Llinas et al., 1976, Schneggenburger and Neher, 2000, Zucker and Regehr, 2002), the exact molecular mechanisms leading to vesicle fusion are still unclear. Nevertheless, abundant evidence links the synaptotagmin molecule to vesicle release (Pang and Sudhof, 2010). In particular, the synaptotagmin1 C2A and C2B domains are thought to act as Ca2+ sensors and have a pivotal role during exocytosis. Notably, the synaptotagmin C2B domain is edited at four different positions (Hoopengardner et al., 2003). One possibility, therefore, is that editing of the synaptotagmin C2B domain by Adar may modulate the calcium dependence of vesicle release. Importantly, Ca2+ dependence plotted at the log-log scale (Fig. 4) shows that at the Adar5G1 mutants the slope but not intercept is altered, suggesting that Ca2+ cooperativity is reduced (Rahamimoff, 1968). This finding is consistent with the hypothesis that editing the synaptotagmin C2B domain may alter the number of calcium ions bound to the calcium binding site. Indeed, one of the synaptotagmin editing sites is closely positioned to Ca2+- binding loops of the C2B domain (Hoopengardner et al., 2003).
Lack of Adar alters synapse ultrastructure
Among the proteins tested for their abundance at the Adar5G1 mutant NMJs, three out of four proteins associated with synaptic vesicles (Syn, Endo, Syt1, CSP) show increase abundance at the synapse. Dap160 and Brp are not associated with synaptic vesicles and do not show a change in abundance at the synapse. It is therefore possible that the synaptic up-regulation of Syn, Endo and Syt1 reflects a major change in numbers, size or membrane composition of synaptic vesicles. We therefore performed electron microscopy analysis to examine the effects of the lack of Adar on NMJ ultrastructure.
We found that ultrastructure of synaptic boutons in Adar5G1 mutants was dramatically modified. First, Adar5G1 boutons typically had irregular shapes, whereas control boutons were more round and smooth (Fig. 5A, E). Furthermore, unlike controls (Fig. 5A, B), the Adar5G1 mutant boutons were packed with vesicles very densely, showing no spaces between vesicles at areas where vesicles were present (Fig. 5E, F). Both control and Adar5G1 mutant boutons had areas devoid of vesicles (shown in Fig. 5C, G, respectively). However, the Adar5G1 mutant boutons had very distinct borders between areas packed with vesicles and areas devoid of vesicles (Fig. 5G), while in control boutons this transition was much smoother (Fig. 5C). It appeared that the Adar5G1 mutants had dense and tightly bound vesicle clusters while in controls the vesicle clusters were looser and not as dense (compare Fig. 5A-D to Fig.5 E-H).
Figure 5. Ultrastructural analysis shows that vesicle number, distribution and density are altered in Adar mutants.
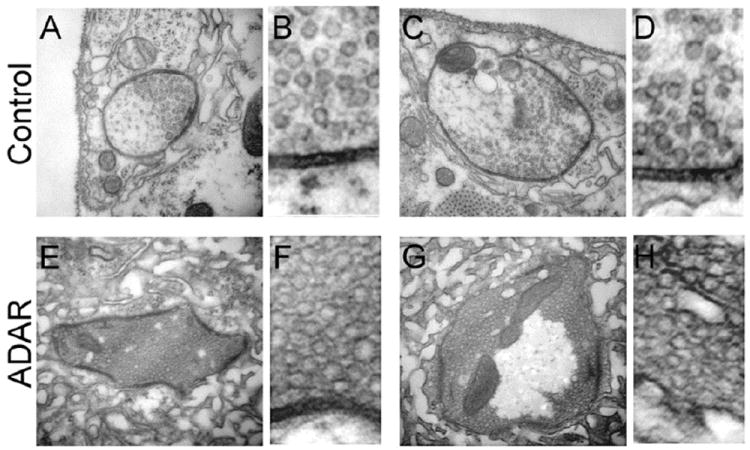
(A and C) Representative examples of control synaptic boutons. Note that parts of the bouton contain synaptic vesicles while some areas are devoid of vesicles.
(B and D) show a higher magnification of a portion of the bouton ultrastructure presented in A and C respectively.
(E and G) Representative examples of Adar5G1 mutant synaptic boutons. Note that, in E, the synaptic bouton is entirely covered by vesicles. In G, while the central part of the bouton is devoid of vesicles, its entire circumference is packed with vesicles. Both E and G show a large number of vesicles compared to controls. The density of the vesicles present in the mutant is increased compared to controls
(F and H) show a higher magnification of a portion of the bouton ultrastructure presented in E and G respectively. The vesicles present in F and H are much more densely packed than vesicles observed in B and D.
Interestingly, the phenotype observed in the Adar5G1 mutant boutons was opposite to the phenotype observed in synapsin null mutants (Akbergenova and Bykhovskaia, 2010), where vesicle density was reduced and areas devoid of vesicles were less distinct than in controls. Thus, hypothetically, the phenotype observed in the Adar5G1 boutons may result from excessive functioning of unedited synapsin. Interestingly, at both larval and adult stages, synapsin mRNA is edited by Adar. The edited site encodes a protein motif that is a site of phosphorylation and is conserved through several phyla; the unedited synapsin is phosphorylated by PKA while the edited version is not. Thus, it is possible that Adar may be able to modulate synapsin function (Diegelmann et al., 2006).
Lack of presynaptic Adar leads to a compensatory decrease in post-synaptic glutamate receptor subunit IIA
In an effort to determine whether the lack of Adar function could also lead to changes in the abundance of post-synaptic markers, we quantified the abundance of the glutamate receptor subunit IIA (GluRIIA), one of the two non-essential GluR subunits, at the NMJ (Petersen et al., 1997, DiAntonio et al., 1999, Qin et al., 2005). As control, we also quantified the abundance of the postsynaptic scaffolding protein Dlg, the homolog of PSD-95 that interacts at postsynaptic sites to form a multimeric scaffold for several receptors (Hough et al., 1997). We imaged synapses and the average staining intensity at the NMJ was measured for GluRIIA and Dlg as described above for the pre-synaptic markers. Using this method we find that while Dlg abundance does not vary between Adar5G1 mutants and controls, there is a 36.8% reduction in GluRIIA at Adar5G1 mutant synapses (n = 38; p < 3.10-11; Fig. 6A). Since reduction in GluRIIA levels is associated with a decrease in quantal size (Petersen et al., 1997), this result excludes the possibility that the increase in quantal size observed in the Adar5G1 mutants might be due to the variation in post-synaptic GluR at the synapse. Because the GluRIIA RNA is not subjected to editing we hypothesized that the decreased GluRIIA abundance might be a homeostatic compensation for an increase in quantal size provoked by a pre-synaptic change.
Figure 6. Adar mutants show a compensatory decrease in post-synaptic Glutamate Receptor Subunit IIA.
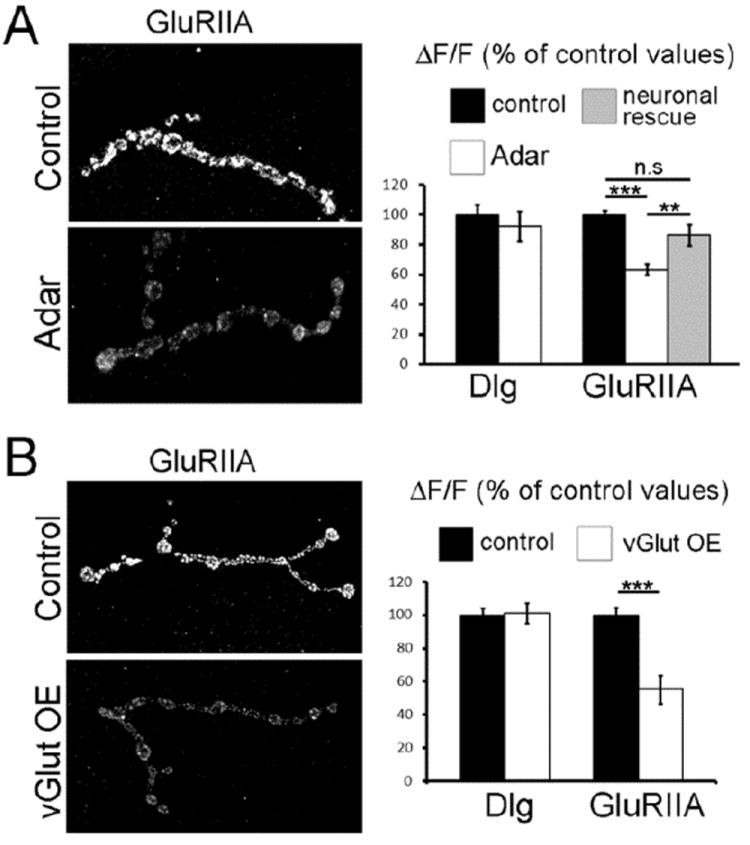
(A) (Left) Representative control and Adar5G1 mutant synaptic boutons from muscle 4 show that the postsynaptic abundance of GluRIIA subunit is decreased in Adar5G1 mutants. (Right) Quantification of fluorescence intensity as a measure of Dlg and GluRIIA abundance at the NMJ of muscle 4. While levels of Dlg do not vary between control and Adar5G1 mutant, GluRIIA levels are decreased by 37% in Adar5G1 mutant (n = 38) when compared to controls (n = 33; Student’s t-test; p < 2.9 10-11). Levels of expression are restored towards control values when Adar5G1 mutants are rescued by neuronal expression of Adar cDNA (neuronal rescue). Neuronal rescue values (n = 18) are significantly different from Adar5G1 mutants (p < 0.008) and not different from control values (p > 0.08).
(B) (Left) Representative control and vGlut over-expression (vGlutOE) synaptic boutons from muscle 4 show that the postsynaptic abundance of GluRIIA subunit is decreased in vGlut over-expression animals. (Right) Quantification of fluorescence intensity at the NMJ of muscle 4 shows that, while levels of Dlg do not vary, GluRIIA subunit is decreased in vGlut over-expression animals to 55.3 +/- 8.5% of control value (n = 25 and 26; p < 4.4 10-5). Each genotype is identified in the legend at the top of the chart. Error bars represent average ± SEM.
In order to test this hypothesis, we examined neuronal rescue flies expressing Adar cDNA only in the neurons of an otherwise Adar null animal. The GluRIIA synaptic staining intensity, that decreased to 63.2 +/- 3.6 % of control values in Adar5G1 mutant animals, recovers to 86.2 +/- 7.1 % (n = 18) of control values in neuronal rescue animals. The synaptic GluRIIA staining intensity observed in neuronal rescue animals is not significantly different from the wild type value (p > 0.07) but is significantly different from mutant data (p < 0.001; Fig. 6A). Because the synaptic GluRIIA intensity in the rescue animals is clearly modified towards control values (Fig. 6A), we conclude that the lack of Adar presynaptically is causing a decrease in postsynaptic GluRIIA. This is consistent with the fact that the editing of the Drosophila GluRIIA subunit has not been reported and that there is no evidence that ADAR is expressed in muscles at this stage. Because we showed that the postsynaptic decrease in GluRIIA levels is dependent on presynaptic adar and happens in conjunction with an increase in mEPSPs amplitude (fig.1), we propose that a compensatory mechanism is taking place that scales down GluRIIA expression in order to compensate for an increase in glutamate release.
We therefore decided to measure the abundance of the GluRIIA at the NMJ of flies releasing too much glutamate. To do so, we examined flies over-expressing the vesicular glutamate transporter (vGlut) in neurons. These flies show increased vesicle and quantal size and a compensatory decrease in vesicle release that maintain constant excitability (Daniels et al., 2004). We found that there is a 45% decrease of GluRIIA immunostaining in flies over expressing neuronal vGlut (vGlut OE; Fig. 6B; n = 25; p < 4.4 10-5) when compared to controls (n = 26). In contrast, the protein Dlg does not vary in vGlut over-expressors (n = 17) when compared to controls (n = 22). This shows that increasing quantal size through a presynaptic change leads to the selective decrease of GluRIIA abundance postsynaptically. These data are consistent with the model in which reduction of GluR abundance and muscle excitability is a compensatory response to increased glutamate release. Furthermore, it suggests that quantal scaling might be an important mechanism regulating the synaptic function at the Drosophila NMJ.
Discussion
Adar edits 597 RNAs from the Drosophila transcriptome (Graveley et al., 2011) and affects a large number of RNAs encoding proteins that regulate synaptic function and neuronal excitability (Hoopengardner et al., 2003, Stapleton et al., 2006, Graveley et al., 2011). Here, we studied the synaptic physiology and structure of Drosophila NMJ that do not contain any functional Adar. Our main finding is that the presynaptic loss of Adar modifies synaptic transmission and, more specifically, produces defects in quantal size, Ca2+ dependent transmitter release and synaptic ultrastructure. Interestingly, our findings raise the possibility that RNA editing is used by neurons to modulate synaptic release properties.
Notably, synaptic release is reduced in Adar mutant while the number of synaptic boutons is increased. This strongly suggests that probability of release is reduced in Adar mutant. Because Adar targets are known, it is tempting to associate the observed phenotype with the editing state of several synaptic proteins. The impairment of neurotransmitter release observed in Adar mutants is consistent with a difference in the activity of the presynaptic calcium channel cacophony or the transmembrane vesicle protein synaptotagmin, two known edited molecules (Hoopengardner et al., 2003, Keegan et al., 2005). Since Syt can modulate the release probability (Fernández-Chacón et al, 2001), the lack of Syt editing could be responsible for the decrease in the release probability. Interestingly, synaptotagmin is edited within the C2B calcium sensor domain (Hoopengardner et al., 2003), which is thought to be critical for Ca2+-dependent evoked release and Ca2+ cooperativity (Yoshihara and Littleton, 2002). The lack of editing of the syt C2B domain may alter the number of calcium ions bound to the calcium binding site and explain the reduced Ca2+ cooperativity observed in Adar mutants. Thus, it is possible that it is the lack of Syt editing that is responsible for the difference in calcium dependence of vesicle release observed in Adar mutants.
One of the most spectacular aspects of the adar loss of function phenotype is the effect on synapse ultrastructure; lack of RNA editing leads to altered distribution and density of synaptic vesicles. We also showed that a dramatic increase (200%) in the amount of synaptotagmin -a protein that accounts for 7-8% of the total vesicle protein (Chapman and Jahn, 1994)- was part of the Adar mutant phenotype. This increase is also notable with two other vesicle-associated proteins synapsin and endophilin. Taken together, these data suggest that RNA editing could affect vesicles number and distribution. This effect on synaptic vesicles could be a direct consequence of lack of editing or due to a compensatory mechanism aiming to counterbalance the lack of editing in another set of pre-synaptic molecules. The vesicle phenotype observed at the ultra-structural level could be explained by a gain of function of the unedited synapsin found in Adar mutants, since this phenotype is opposed to the one observed in the synapsin loss of function mutation (Akbergenova and Bykhovskaia, 2010). This possibility is particularly interesting since editing affects a protein motif that is phosphorylated by PKA. The unedited synapsin is phosphorylated by PKA while the edited version is not, raising the possibility that RNA editing modulates synapsin function (Diegelmann et al., 2006).
Interestingly, not all the vesicle-associated markers tested show an increased presence at the mutant synapse; indeed CSP levels stayed constant. Notably, the presynaptic markers showing increased amounts at the synapse in the Adar5G1 mutants are edited (Hoopengardner et al., 2003, Diegelmann et al., 2006, Stapleton et al., 2006, Graveley et al., 2011) while there is no evidence of editing of the synaptic markers whose abundance at the synapse did not change (Dap160, Brp and CSP). It is tempting to speculate that the increase in vesicle-associated markers in Adar mutants could be a consequence of a change in synaptic vesicle numbers and density. Alternatively, this increase could reflect a more global change in synaptic protein composition. For example, because RNA editing can affect RNA splicing (Rueter et al., 1999, Valente and Nishikura, 2005); changing Adar activity levels could affect the splicing profile of synaptic proteins. Similarly, there is increasing evidence demonstrating an interaction between the RNA editing and the RNA interference pathways (Nishikura, 2006, Wu et al., 2011). In this capacity, RNA editing could potentially influence the level of expression of many genes within neurons.
Importantly, we have also observed that quantal size is increased in Adar mutants. More specifically, the population of enlarged quanta is increased in the Adar mutants. Although the quantal size may be determined by many factors, presynaptic and postsynaptic, one possibility is that the observed effect is produced by changes in synaptic vesicles ultrastructure. In support of this hypothesis, we found that the effect on quantal size depends on presynaptic but not postsynaptic Adar. In addition, we did not detect any indication that post-synaptic GluR were up-regulated in the Adar mutants. On the contrary, we demonstrated that a postsynaptic compensatory mechanism is taking place in Adar mutants that decreases the amount of GluRIIA in response to the loss of presynaptic Adar (Fig. 6). We hypothesize that the excessive release of presynaptic glutamate is able to trigger this homeostatic response. It should to be noted that, consequentially, the increase in quantal size observed in Adar mutants is likely to be underestimated. Indeed, the measured quantal amplitude in Adar mutants should also reflect the decreased postsynaptic currents. We have also shown that the phenomenon of quantal downscaling is present in well studied transgenic flies that over-express vGlut (Daniels et al., 2004, 2011). This suggests that synaptic scaling, the process by which mEPSP amplitude is modified postsynaptically in order to compensate for a change in presynaptic activity (Turrigiano and Nelson, 2004, Turrigiano, 2008, Pozo and Goda, 2010), is likely to be a common mechanism of homeostatic regulation at the NMJ.
In summary, we present the evidence that Adar, a gene shown to modulate behaviors and maintain neuronal integrity, is essential for the normal presynaptic ultrastructure and physiology. Furthermore, we have shown that several presynaptic targets of Adar, including synapsin, synaptotagmin, and endophilin are abnormally expressed in mutant NMJs. Further experimentation is needed to determine the contribution of each of these RNA editing targets to the observed synaptic phenotype. The Drosophila model system allows the making of transgenic animals expressing either the edited or unedited version of any synaptic protein. This could allow the systematic characterization of the effects of editing on synaptic ultrastructure and physiology for every single edited synaptic protein.
Acknowledgments
We thank Mariam Vazquez for technical help. We thank Dr. Rosenthal and Dr. DiAntonio for providing fly strains, Dr. Kelly and Dr. Bellen for providing antibodies. We thank Dr. Blagburn for his comments on earlier versions of the manuscript. Supported by NINDS SC2NS077924 and RCMI RR-03051 to BM. CM is supported by NSF DBI-0932955; DA and BM are supported by NSF HRD-1137725; MB is supported by U54 NS039408; imaging equipment supported by NSF DBI 0115825 and DoD 52680-RT-ISP
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Akbergenova Y, Bykhovskaia M. Synapsin regulates vesicle organization and activity-dependent recycling at Drosophila motor boutons. Neuroscience. 2010;170:441–452. doi: 10.1016/j.neuroscience.2010.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basilio C, Wahba AJ, Lengyel P, Speyer JF, Ochoa S. Synthetic polynucleotides and the amino acid code. V Proc Natl Acad Sci U S A. 1962;48:613–616. doi: 10.1073/pnas.48.4.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bass BL. RNA editing by adenosine deaminases that act on RNA. Annu Rev Biochem. 2002;71:817–846. doi: 10.1146/annurev.biochem.71.110601.135501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bass BL, Weintraub H. An unwinding activity that covalently modifies its double-stranded RNA substrate. Cell. 1988;55:1089–1098. doi: 10.1016/0092-8674(88)90253-x. [DOI] [PubMed] [Google Scholar]
- Bhogal B, Jepson JE, Savva YA, Pepper AS, Reenan RA, Jongens TA. Modulation of dADAR-dependent RNA editing by the Drosophila fragile X mental retardation protein. Nat Neurosci. 2011;14:1517–1524. doi: 10.1038/nn.2950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bykhovskaia M. Synapsin regulation of vesicle organization and functional pools. Semin Cell Dev Biol. 2011;22:387–392. doi: 10.1016/j.semcdb.2011.07.003. [DOI] [PubMed] [Google Scholar]
- Chapman ER. How does synaptotagmin trigger neurotransmitter release? Annu Rev Biochem. 2008;77:615–641. doi: 10.1146/annurev.biochem.77.062005.101135. [DOI] [PubMed] [Google Scholar]
- Chapman ER, Jahn R. Calcium-dependent interaction of the cytoplasmic region of synaptotagmin with membranes. Autonomous function of a single C2-homologous domain. The J Biol Chem. 1994;269:5735–5741. [PubMed] [Google Scholar]
- Daniels RW, Collins CA, Gelfand MV, Dant J, Brooks ES, Krantz DE, DiAntonio A. Increased expression of the Drosophila vesicular glutamate transporter leads to excess glutamate release and a compensatory decrease in quantal content. J Neurosci. 2004;24:10466–10474. doi: 10.1523/JNEUROSCI.3001-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels RW, Miller BR, DiAntonio A. Increased vesicular glutamate transporter expression causes excitotoxic neurodegeneration. Neurobiol Dis. 2011;41:415–420. doi: 10.1016/j.nbd.2010.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis GW, DiAntonio A, Petersen SA, Goodman CS. Postsynaptic PKA controls quantal size and reveals a retrograde signal that regulates presynaptic transmitter release in Drosophila. Neuron. 1998;20:305–315. doi: 10.1016/s0896-6273(00)80458-4. [DOI] [PubMed] [Google Scholar]
- Davis GW, Goodman CS. Synapse-specific control of synaptic efficacy at the terminals of a single neuron. Nature. 1998;392:82–86. doi: 10.1038/32176. [DOI] [PubMed] [Google Scholar]
- DiAntonio A, Petersen SA, Heckmann M, Goodman CS. Glutamate receptor expression regulates quantal size and quantal content at the Drosophila neuromuscular junction. J Neurosci. 1999;19:3023–3032. doi: 10.1523/JNEUROSCI.19-08-03023.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diegelmann S, Nieratschker V, Werner U, Hoppe J, Zars T, Buchner E. The conserved protein kinase-A target motif in synapsin of Drosophila is effectively modified by pre-mRNA editing. BMC Neurosci. 2006;7:76. doi: 10.1186/1471-2202-7-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fergestad T, Broadie K. Interaction of stoned and synaptotagmin in synaptic vesicle endocytosis. J Neurosci. 2001;21:1218–1227. doi: 10.1523/JNEUROSCI.21-04-01218.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández-Chacón R, Königstorfer A, Gerber SH, García J, Matos MF, Stevens CF, Brose N, Rizo J, Rosenmund C, Südhof TC. Synaptotagmin I functions as a calcium regulator of release probability. Nature. 2001;410:41–49. doi: 10.1038/35065004. [DOI] [PubMed] [Google Scholar]
- Graveley BR, Brooks AN, Carlson JW, Duff MO, Landolin JM, Yang L, Artieri CG, van Baren MJ, Boley N, Booth BW, Brown JB, Cherbas L, Davis CA, Dobin A, Li R, Lin W, Malone JH, Mattiuzzo NR, Miller D, Sturgill D, Tuch BB, Zaleski C, Zhang D, Blanchette M, Dudoit S, Eads B, Green RE, Hammonds A, Jiang L, Kapranov P, Langton L, Perrimon N, Sandler JE, Wan KH, Willingham A, Zhang Y, Zou Y, Andrews J, Bickel PJ, Brenner SE, Brent MR, Cherbas P, Gingeras TR, Hoskins RA, Kaufman TC, Oliver B, Celniker SE. The developmental transcriptome of Drosophila melanogaster. Nature. 2011;471:473–479. doi: 10.1038/nature09715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greengard P, Valtorta F, Czernik AJ, Benfenati F. Synaptic vesicle phosphoproteins and regulation of synaptic function. Science. 1993;259:780–785. doi: 10.1126/science.8430330. [DOI] [PubMed] [Google Scholar]
- Higuchi M, Maas S, Single FN, Hartner J, Rozov A, Burnashev N, Feldmeyer D, Sprengel R, Seeburg PH. Point mutation in an AMPA receptor gene rescues lethality in mice deficient in the RNA-editing enzyme ADAR2. Nature. 2000;406:78–81. doi: 10.1038/35017558. [DOI] [PubMed] [Google Scholar]
- Hoopengardner B, Bhalla T, Staber C, Reenan R. Nervous system targets of RNA editing identified by comparative genomics. Science. 2003;301:832–836. doi: 10.1126/science.1086763. [DOI] [PubMed] [Google Scholar]
- Hough CD, Woods DF, Park S, Bryant PJ. Organizing a functional junctional complex requires specific domains of the Drosophila MAGUK Discs large. Genes Dev. 1997;11:3242–3253. doi: 10.1101/gad.11.23.3242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jepson JE, Reenan RA. Adenosine-to-inosine genetic recoding is required in the adult stage nervous system for coordinated behavior in Drosophila. J Biol Chem. 2009;284:31391–31400. doi: 10.1074/jbc.M109.035048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jepson JE, Savva YA, Yokose C, Sugden AU, Sahin A, Reenan RA. Engineered alterations in RNA editing modulate complex behavior in Drosophila: regulatory diversity of adenosine deaminase acting on RNA (ADAR) targets. J Biol Chem. 2011;286:8325–8337. doi: 10.1074/jbc.M110.186817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keegan LP, Brindle J, Gallo A, Leroy A, Reenan RA, O’Connell MA. Tuning of RNA editing by ADAR is required in Drosophila. EMBO J. 2005;24:2183–2193. doi: 10.1038/sj.emboj.7600691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keegan LP, Leroy A, Sproul D, O’Connell MA. Adenosine deaminases acting on RNA (ADARs): RNA-editing enzymes. Genome Biol. 2004;5:209. doi: 10.1186/gb-2004-5-2-209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kittel RJ, Wichmann C, Rasse TM, Fouquet W, Schmidt M, Schmid A, Wagh DA, Pawlu C, Kellner RR, Willig KI, Hell SW, Buchner E, Heckmann M, Sigrist SJ. Bruchpilot promotes active zone assembly, Ca2+ channel clustering, and vesicle release. Science. 2006;312:1051–1054. doi: 10.1126/science.1126308. [DOI] [PubMed] [Google Scholar]
- Klagges BR, Heimbeck G, Godenschwege TA, Hofbauer A, Pflugfelder GO, Reifegerste R, Reisch D, Schaupp M, Buchner S, Buchner E. Invertebrate synapsins: a single gene codes for several isoforms in Drosophila. J Neurosci. 1996;16:3154–3165. doi: 10.1523/JNEUROSCI.16-10-03154.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llinas R, Steinberg IZ, Walton K. Presynaptic calcium currents and their relation to synaptic transmission: voltage clamp study in squid giant synapse and theoretical model for the calcium gate. Proc Natl Acad Sci U S A. 1976;73:2918–2922. doi: 10.1073/pnas.73.8.2918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marie B, Sweeney ST, Poskanzer KE, Roos J, Kelly RB, Davis GW. Dap160/intersectin scaffolds the periactive zone to achieve high-fidelity endocytosis and normal synaptic growth. Neuron. 2004;43:207–219. doi: 10.1016/j.neuron.2004.07.001. [DOI] [PubMed] [Google Scholar]
- Marie B, Pym E, Bergquist S, Davis GW. Synaptic homeostasis is consolidated by the cell fate gene gooseberry, a Drosophila pax3/7 homolog. J Neurosci. 2010;30:8071–8082. doi: 10.1523/JNEUROSCI.5467-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martens S, McMahon HT. Mechanisms of membrane fusion: disparate players and common principles. Nat Rev Mol Cell Biol. 2008;9:543–556. doi: 10.1038/nrm2417. [DOI] [PubMed] [Google Scholar]
- Miech C, Pauer HU, He X, Schwarz TL. Presynaptic local signaling by a canonical wingless pathway regulates development of the Drosophila neuromuscular junction. J Neurosci. 2008;28:10875–10884. doi: 10.1523/JNEUROSCI.0164-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milosevic I, Giovedi S, Lou X, Raimondi A, Collesi C, Shen H, Paradise S, O’Toole E, Ferguson S, Cremona O, Camilli PD. Recruitment of endophilin to clathrin-coated pit necks is required for efficient vesicle uncoating after fission. Neuron. 2011;72:587–601. doi: 10.1016/j.neuron.2011.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishikura K. Editor meets silencer: crosstalk between RNA editing and RNA interference. Nat Rev Mol Cell Biol. 2006;7:919–931. doi: 10.1038/nrm2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palladino MJ, Keegan LP, O’Connell MA, Reenan RA. dADAR, a Drosophila double-stranded RNA-specific adenosine deaminase is highly developmentally regulated and is itself a target for RNA editing. RNA. 2000a;6:1004–1018. doi: 10.1017/s1355838200000248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palladino MJ, Keegan LP, O’Connell MA, Reenan RA. A-to-I pre-mRNA editing in Drosophila is primarily involved in adult nervous system function and integrity. Cell. 2000b;102:437–449. doi: 10.1016/s0092-8674(00)00049-0. [DOI] [PubMed] [Google Scholar]
- Pang ZP, Sudhof TC. Cell biology of Ca2+-triggered exocytosis. Curr Opin Cell Biol. 2010;22:496–505. doi: 10.1016/j.ceb.2010.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen SA, Fetter RD, Noordermeer JN, Goodman CS, DiAntonio A. Genetic analysis of glutamate receptors in Drosophila reveals a retrograde signal regulating presynaptic transmitter release. Neuron. 1997;19:1237–1248. doi: 10.1016/s0896-6273(00)80415-8. [DOI] [PubMed] [Google Scholar]
- Pozo K, Goda Y. Unraveling mechanisms of homeostatic synaptic plasticity. Neuron. 2010;66:337–351. doi: 10.1016/j.neuron.2010.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin G, Schwarz T, Kittel RJ, Schmid A, Rasse TM, Kappei D, Ponimaskin E, Heckmann M, Sigrist SJ. Four different subunits are essential for expressing the synaptic glutamate receptor at neuromuscular junctions of Drosophila. J Neurosci. 2005;25:3209–3218. doi: 10.1523/JNEUROSCI.4194-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahamimoff R. A dual effect of calcium ions on neuromuscular facilitation. J Physiol. 1968;195:471–480. doi: 10.1113/jphysiol.1968.sp008468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roos J, Kelly RB. Dap160, a neural-specific Eps15 homology and multiple SH3 domain-containing protein that interacts with Drosophila dynamin. J Biol Chem. 1998;273:19108–19119. doi: 10.1074/jbc.273.30.19108. [DOI] [PubMed] [Google Scholar]
- Rueter SM, Dawson TR, Emeson RB. Regulation of alternative splicing by RNA editing. Nature. 1999;399:75–80. doi: 10.1038/19992. [DOI] [PubMed] [Google Scholar]
- Ruiz-Canada C, Ashley J, Moeckel-Cole S, Drier E, Yin J, Budnik V. New synaptic bouton formation is disrupted by misregulation of microtubule stability in aPKC mutants. Neuron. 2004;42:567–580. doi: 10.1016/s0896-6273(04)00255-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneggenburger R, Neher E. Intracellular calcium dependence of transmitter release rates at a fast central synapse. Nature. 2000;406:889–893. doi: 10.1038/35022702. [DOI] [PubMed] [Google Scholar]
- Sharma M, Burre J, Sudhof TC. CSPalpha promotes SNARE-complex assembly by chaperoning SNAP-25 during synaptic activity. Nat Cell Biol. 2011;13:30–39. doi: 10.1038/ncb2131. [DOI] [PubMed] [Google Scholar]
- Stapleton M, Carlson JW, Celniker SE. RNA editing in Drosophila melanogaster: New targets and functional consequences. RNA. 2006;12:1922–1932. doi: 10.1261/rna.254306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweeney ST, Broadie K, Keane J, Niemann H, O’Kane CJ. Targeted expression of tetanus toxin light chain in Drosophila specifically eliminates synaptic transmission and causes behavioral defects. Neuron. 1995;14:341–351. doi: 10.1016/0896-6273(95)90290-2. [DOI] [PubMed] [Google Scholar]
- Tonkin LA, Saccomanno L, Morse DP, Brodigan T, Krause M, Bass BL. RNA editing by ADARs is important for normal behavior in Caenorhabditis elegans. EMBO J. 2002;21:6025–6035. doi: 10.1093/emboj/cdf607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrigiano GG. The self-tuning neuron: synaptic scaling of excitatory synapses. Cell. 2008;135:422–435. doi: 10.1016/j.cell.2008.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrigiano GG, Nelson SB. Homeostatic plasticity in the developing nervous system. Nat Rev Neurosci. 2004;5:97–107. doi: 10.1038/nrn1327. [DOI] [PubMed] [Google Scholar]
- Valente L, Nishikura K. ADAR gene family and A-to-I RNA editing: diverse roles in posttranscriptional gene regulation. Prog Nucleic Acid Res Mol Biol. 2005;79:299–338. doi: 10.1016/S0079-6603(04)79006-6. [DOI] [PubMed] [Google Scholar]
- Verstreken P, Kjaerulff O, Lloyd TE, Atkinson R, Zhou Y, Meinertzhagen IA, Bellen HJ. Endophilin mutations block clathrin-mediated endocytosis but not neurotransmitter release. Cell. 2002;109:101–112. doi: 10.1016/s0092-8674(02)00688-8. [DOI] [PubMed] [Google Scholar]
- Wagh DA, Rasse TM, Asan E, Hofbauer A, Schwenkert I, Durrbeck H, Buchner S, Dabauvalle MC, Schmidt M, Qin G, Wichmann C, Kittel R, Sigrist SJ, Buchner E. Bruchpilot, a protein with homology to ELKS/CAST, is required for structural integrity and function of synaptic active zones in Drosophila. Neuron. 2006;49:833–844. doi: 10.1016/j.neuron.2006.02.008. [DOI] [PubMed] [Google Scholar]
- Wagner RW, Smith JE, Cooperman BS, Nishikura K. A double-stranded RNA unwinding activity introduces structural alterations by means of adenosine to inosine conversions in mammalian cells and Xenopus eggs. Proc Natl Acad Sci U S A. 1989;86:2647–2651. doi: 10.1073/pnas.86.8.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu D, Lamm AT, Fire AZ. Competition between ADAR and RNAi pathways for an extensive class of RNA targets. Nat Struct Mol Biol. 2011;18:1094–1101. doi: 10.1038/nsmb.2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshihara M, Littleton JT. Synaptotagmin I functions as a calcium sensor to synchronize neurotransmitter release. Neuron. 2002;36:897–908. doi: 10.1016/s0896-6273(02)01065-6. [DOI] [PubMed] [Google Scholar]
- Zinsmaier KE. Cysteine-string protein’s neuroprotective role. J Neurogenet. 2010;24:120–132. doi: 10.3109/01677063.2010.489625. [DOI] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG. Short-term synaptic plasticity. Annu Rev Physiol. 2002;64:355–405. doi: 10.1146/annurev.physiol.64.092501.114547. [DOI] [PubMed] [Google Scholar]