

Abstract
Promyelocytic leukemia nuclear bodies (PML-NBs) are multi-protein complexes that include PML protein and localize in nuclear foci. PML-NBs are implicated in multiple stress responses, including apoptosis, DNA repair, and p53-dependent growth inhibition. ALT-associated PML bodies (APBs) are specialized PML-NBs that include telomere-repeat binding-factor TRF1 and are exclusively in telomerase-negative tumors where telomere length is maintained through alternative (ALT) recombination mechanisms. We compared cell cycle and p53 responses in ALT-positive cancer cells (U2OS) exposed to ionizing radiation (IR) or the p53 stabilizer Nutlin-3a. Both IR and Nutlin-3a caused growth arrest and comparable induction of p53. However, p21, whose gene p53 activates, displayed biphasic induction following IR and monophasic induction following Nutlin-3a. P53 was recruited to PML-NBs 3–4 days after IR, approximately coincident with the secondary p21 increase. These p53/PML-NBs marked sites of apparently unrepaired DNA double-strand breaks (DSBs), identified by colocalization with phosphorylated histone H2AX. Nutlin-3a and IR both caused a large increase in APBs that was dependent on p53 and p21 expression. Moreover, p21, and to a lesser extent p53, was recruited to APBs in a fraction of Nutlin-3a treated cells. These data indicate 1) p53 is recruited to PML-NBs after IR that likely mark unrepaired DSBs, suggesting p53 may either be further activated at these sites and/or function in their repair; 2) p53-p21 pathway activation increases the percentage of APB-positive cells, 3) p21 and p53 are recruited to ALT-associated PML-NBs after Nutlin-3a treatment, suggesting they may play a previously unrecognized role in telomere maintenance.
Keywords: P53, p21, PML nuclear bodies, irradiation, Nutlin
Introduction
The promyelocytic leukemia protein (PML) is a tumor suppressor implicated in multiple stress responses including apoptosis, senescence, and DNA repair [Bernardi and Pandolfi, 2007]. The PML gene was originally identified as a result of a reciprocal translocation t(15:17) associated with acute promyelocytic leukemia [de The et al., 1991; Goddard et al., 1991; Kakizuka et al., 1991; Pandolfi et al., 1991]. The t(15:17) translocation disrupts the PML gene on chromosome 15 and the retinoic acid receptor α (RARα) gene on chromosome 17 and is reciprocal in nature, resulting in the generation of novel fusion proteins PML-RARα and RARα-PML [Pandolfi, 2001]. The most striking feature of wild-type PML is its localization to distinct nuclear foci termed PML nuclear bodies (PML-NBs). These PML-NBs are multiprotein complexes and cells typically contain 10–30 PML-NBs/nucleus, although their number and size can vary during the cell cycle and following stress [Everett et al., 1999; Koken et al., 1995]. ALT-associated PML bodies (APBs) are specialized PML-NBs that are believed to function in telomere maintenance, and are found exclusively in telomerase-negative tumors in which telomere length is maintained through an alternative (ALT) recombination mechanism [Draskovic et al., 2009; Henson et al., 2002; Yu et al.] The PML protein does not have an intrinsic enzymatic activity but rather functions as a scaffold capable of interacting with multiple proteins simultaneously and bringing them in close proximity so they may interact with and regulate each other’s activity. Over 60 different proteins have been localized in PML-NBs to date [Dellaire et al., 2003], either spatially or temporally, thus implicating PML-NBs in multiple different cellular processes.
Wild-type p53 is a tumor suppressor protein and potent growth inhibitor. P53 is expressed at low levels in most normal cells due to a short protein half-life [Maki and Howley, 1997; Maltzman and Czyzyk, 1984]. However, p53 is stabilized and its levels increase in response to stresses, such as DNA damage and inappropriate oncogene signaling, that might otherwise predispose a normal cell toward carcinogenesis [Horn and Vousden, 2007; Maki and Howley, 1997; Maltzman and Czyzyk, 1984]. The majority of stabilized p53 accumulates in the nucleus where it functions as a transcription factor, activating expression of genes that cause either cell cycle arrest (P21) or apoptosis (PUMA, bax, Noxa) [Brown et al., 2007]. A smaller though significant portion of p53 also accumulates in mitochondria, where it interacts with pro- and anti-apoptotic members of the Bcl-2 family, resulting in release of factors from the mitochondria that drive apoptosis [Mihara et al., 2003; Vaseva and Moll, 2009]. Thus, p53 eliminates cells with potentially cancer-promoting lesions by either inhibiting their growth or causing them to die. A relationship between p53 and PML was first suggested by the finding that p53 could interact directly with PML and was recruited by PML into PML-NBs [Fogal et al., 2000]. Subsequent studies have shown that PML co-recruits p53 and the acetylase enzyme CBP into PML-NBs in response to DNA damaging stress (ionizing radiation) and oncogenic stress [Ferbeyre et al., 2000; Guo et al., 2000; Pearson et al., 2000]. CBP then promotes the acetylation of p53 C-terminal lysine residues, which increases p53’s DNA binding activity and thus leads to an activation of p53 responsive genes [Luo et al., 2004]. This ability to activate p53 likely contributes to the tumor suppressor function of PML. Still other studies have shown that p53 colocalizes with PML at sites of DNA damage in irradiated cells. For example, in irradiated normal human fibroblasts PML localized in nuclear foci with anti-phosphorylated histone γH2AX, an established marker of DNA double-strand breaks [Carbone et al., 2002]. P53 and the hMre11 DNA repair protein also associated with PML at these break sites, and irradiation promoted a stable association between PML, p53, and hMre11 that was revealed in co-immunoprecipitation experiments. The results of this study suggested a subset of PML-NBs function in the recognition and/or processing of DNA breaks and the recruitment of proteins (p53 and hMre11) required for both checkpoint activation and DNA repair.
P53 levels are controlled in part by MDM2, an ubiquitin E3 ligase enzyme that binds directly to p53 and promotes its degradation [Haupt et al., 1997; Kubbutat et al., 1997]. Nutlin-3a (Nutlin) is a preclinical drug that was specifically designed to inhibit the interaction between p53 and MDM2 [Vassilev et al., 2004]. Nutlin occupies the p53-binding pocket in MDM2, effectively blocking p53-MDM2 binding and stabilizing p53. Notably, phosphorylations in p53 that are commonly seen following DNA damage are not observed in Nutlin-treated cells, indicating that Nutlin stabilizes p53 in a non-genotoxic fashion [Thompson et al., 2004]. Multiple studies have reported that Nutlin can promote growth arrest and/or apoptosis in cancer cells that express wild-type p53, supporting its use in therapy against p53 wild-type cancers. In this study we compared the p53 response in an ALT-positive, p53 wild-type human cancer cell line (U2OS) exposed to either ionizing radiation (IR) or Nutlin. P53 was induced to comparable levels in response to both treatments. However, p21 displayed a biphasic induction in response to IR but not in response to Nutlin. P53 was first recruited into PML-NBs 3 days after IR treatment, roughly coincident with the secondary increase in p21. Further investigation revealed that these PML-NBs also include phosphorylated γH2AX and are likely sites of unrepaired DNA breaks. Both IR and Nutlin treatment caused a large increase in cells containing APBs, and this effect was dependent on p53 and p21 expression. Interestingly, p21, and to a lesser extent p53, was recruited to APBs in a percentage of Nutlin treated, but not IR treated, cells. In summary, the results demonstrate that p53 and p21 are recruited to distinct PML nuclear foci following either IR or Nutlin treatment. Specifically, p53 is recruited to PML-NBs after IR that likely mark unrepaired DSBs, suggesting p53 may either be activated at these sites and/or function in their repair. In contrast, p21 and p53 are recruited to APBs after non-genotoxic p53 stabilization by Nutlin treatment, suggesting p21 and p53 may play an as yet undetermined role in telomere maintenance.
MATERIALS AND METHODS
CELLS AND REAGENTS
U2OS cells were obtained from ATCC and were grown in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS), penicillin (100 U/mL) and streptomycin (100 μg/mL). Cells were plated 24h before being treated with irradiation (IR) or Nutlin-3 (Sigma, #N6287) at the indicated concentration. DMSO was used as vehicle control.
IMMUNOBLOTTING
Whole cell extracts were prepared by resuspending cell pellets in lysis buffer (160 mM NaCl, 5 mM EDTA, 0.5% NP40, 50 mM Tris, pH 7.5), resolved by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to polyvinylidene difluoride membranes (NEN Life Science Products). Antibodies to p53 (DO-1), p21 (187) were purchased from Santa Cruz Biotechnology. Primary antibodies were detected with goat anti-mouse secondary antibodies conjugated to horseradish peroxidase (Jackson ImmunoResearch), using enhanced chemiluminescence (Perkin-Elmer).
IMMUNOFLUORESCENCE
Cells were plated on glass coverslips in 6 well dishes and were treated either with IR (10Gy) or Nutlin-3 (5 μM) for indicated time periods. After treatment, cells were fixed in 4% paraformaldehyde. Coverslips were next blocked and permeabilized in 1% bovine serum albumin (BSA), 0.1% Triton X-100 in PBS and then incubated with primary antibodies (1:100 dilution in PBS/1% BSA for all antibodies) for 1.5 h. Cells were incubated with rhodamine conjugated goat anti-mouse IgG (1:2000 dilution in PBS/1%BSA; Jackson Immunoresearch, West Grove, PA) and/or Alexa 488 conjugated goat anti rabbit IgG (Invitrogen, Eugene, OR) for 1 h with DAPI. Primary antibodies used were specific against PML (PGM3), PML (H238), p21 (187), p21 (H168), p53 (FL-393) (Santa Cruz Biotechnology, Santa Cruz, CA); Anti-Histone H2A.X (Miliipore, Billerica, MA) and TRF1 (TRF-78) (Abcam Inc., Cambridge, MA). Fluorescent staining was visualized using an axiovert 200 fluorescence microscope (Zeiss, Gottingen, Germany).
CONFOCAL MICROSCOPY
Images were acquired from the Carl Zeiss LSM 510 META laser scanning confocal microscope equipped with a 63 X water-immersion objective. Beams of 488 nm and 542nm lasers were used for excitation. Green and red emissions were detected through 506nm and 568nm filters, respectively. The three different fluorochromes were scanned sequentially by using multi tracking function to avoid any bleed through between different dyes. Images were processed with Aim Image software. The thickness of optic section is around 1μM.
FLOW CYTOMETRY
For cell cycle analysis, cells were harvested and fixed in 25% ethanol overnight. The cells were then stained with propidium iodide (25 μg/mL, Calbiochem). Flow cytometry analysis was performed on FACS Canto (Becton Dickinson), analyzed with CellQuest (Becton Dickinson) and FlowJo 8.7 (Treestar Inc). For each sample, 10,000 events were collected. FSC-width vs. SSC-width was used to eliminate doublets.
SIRNA-MEDIATED TRANSIENT KNOCK-DOWN
p53, p21 RNAi (On-target plus smart pool) and Control RNAi (On-target plus siControl non-targeting pool) were purchased from Dharmacon and transfected according to the manufacturer’s guidelines using DharmaFECT I reagent.
RESULTS
P53 AND P21 INDUCTION IN IRRADIATED AND NUTLIN-3A TREATED CELLS
U2OS cells (p53 wild-type) were untreated, treated with IR (10 Gy), or continuously exposed to Nutlin (5 μM). Cell cycle analysis was performed by flow cytometry between 1–5 days after treatment. Wild-type p53 cells arrest transiently in G2 phase after IR treatment, before completing mitosis and becoming permanently arrested in the subsequent G1 phase [Di Leonardo et al., 1994; Nagasawa et al., 1998; Nagasawa et al., 1995]. Consistent with this, IR-treated U2OS cells were arrested in G2-phase 1 day after treatment, followed by a gradual decrease in G2-phase cells 2–5 days after treatment and a coincident increase in G1-phase cells (Fig 1). The percentage of cells in S-phase remained low up to 5 days after IR despite the accumulation of G1 cells (Fig 1), indicating the majority of these G1-phase cells were cell cycle arrested. In contrast, Nutlin treatment caused a cell cycle arrest in both 2N and 4N states that lasted the duration of the experiment (5 days). The percentage of cells with sub-G1 DNA content was used as a measure of apoptosis. Using this measure, both IR and Nutlin treatment caused comparable levels of apoptosis (20–26% at the 5 day time point for both treatments, Fig 1).
Figure 1. Flow Cytometry analysis of U2OS cells treated with IR or Nutlin.
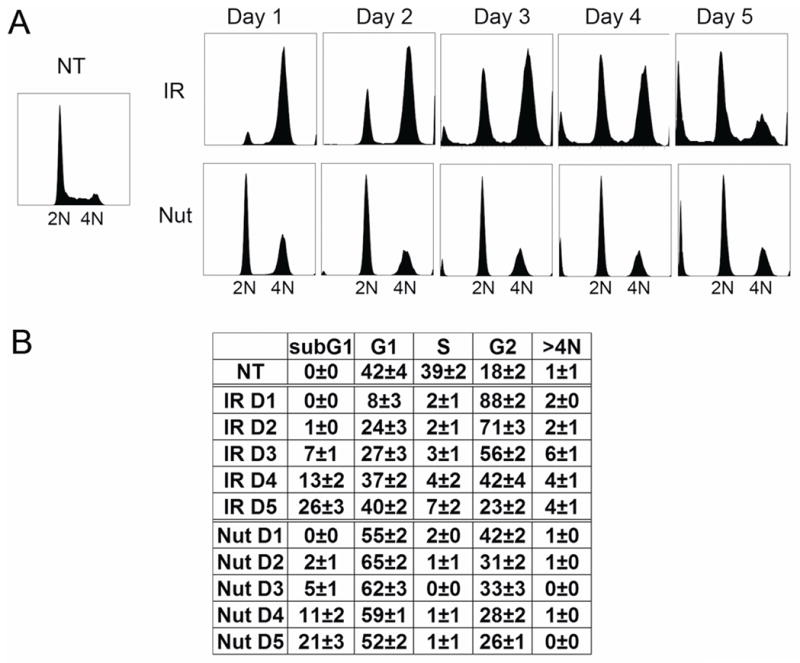
U2OS cells (asynchronously growing and ~50% confluent) were either untreated (NT), treated with irradiation (IR, 10Gy) or treated with Nutlin (Nut; 5μmol/L). The cells were harvested at the indicated time points. The culture medium was not changed during the course of the experiment. Fixed cells were stained with propidium iodide (25 μg/mL) and subjected to flow cytometry analysis. A. Representative DNA profile histograms were analyzed using FlowJo (cell count versus propidium iodide/DNA content). The position of 2N and 4N cells is indicated. B. Cell cycle distribution was determined from DNA profiles using FlowJo. The numbers represent averages from three independent experiments. (mean ± SE; n = 3)
Next, p53 and p21 protein levels were monitored in IR and Nutlin treated cells by immunoblotting at time points ranging from 6–120 hrs after treatment. As shown in Fig 2, p53 levels increased in response to both treatments. P53 was induced by the 6 hr time point after IR treatment and the 12 hr time point in Nutlin treated cells, and remained at elevated levels in response to both treatments for the duration of the experiment. P21 is a p53 gene target, and we monitored p21 levels as an indication of p53 activity. In the case of Nutlin, p21 induction followed that of p53, becoming elevated at the 18 hrs treatment time point and remaining at a high level for the duration of the experiment. Interestingly, we consistently observed a biphasic increase in p21 protein levels in IR treated cells. Specifically, p21 was first induced at the 24–30 hr time points after IR treatment, and plateaued at this level until the 84–90 hr time points, after which a second, further increase in p21 was observed (marked by an asterisk in Fig 2).
Figure 2. P53 and p21 induction in irradiated and Nutlin treated U2OS cells.
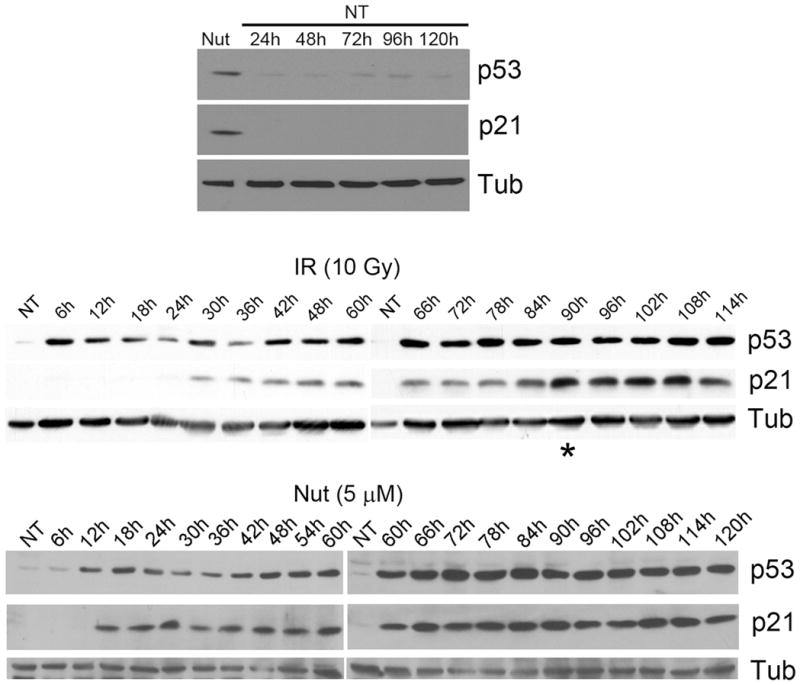
U2OS cells were either untreated (NT), treated with irradiation (IR, 10Gy) or treated with Nutlin (Nut; 5μmol/L), as in Figure 1. Cell lysates were harvested at indicated time points and analyzed by Immunoblotting with indicated antibodies. Tubulin (Tub) was loaded as loading control.
P53 LOCALIZES IN PML-NBs THAT MARK UNREPAIRED DNA DAMAGE SITES IN IRRADIATED CELLS
Recruitment of p53 to PML-NBs can lead to an increase in p53 activity as a transcription factor [Fogal et al., 2000]. We therefore asked whether the second increase in p21 levels 3–4 days after IR treatment coincided with recruitment of p53 to PML-NBs. First, p53 and PML localization was monitored in cells untreated (NT) or 5 days after IR treatment, a time point when both p53 and p21 were at high levels. P53 colocalization with PML was not detected in untreated (NT) cells (Fig 3A). In contrast, P53 and PML colocalization in nuclear foci was observed in irradiated cells at the 5 day time point (Fig 3B, white arrows). We used confocal microscopy looking at thin nuclear slices to confirm p53 and PML were localized in the same nuclear foci in IR treated cells (Fig 3C). Next, we quantified the percent of cells in which p53 and PML colocalization in nuclear foci was observed at multiple time points after IR. As shown in Fig 4, very few cells displayed p53 and PML colocalization in either the untreated condition or 2 days after IR. However, 3 days after IR treatment p53 and PML colocalization in nuclear foci was observed in over 10% of cells and this increased to over 20% of cells by the 5 day time point. Thus, recruitment of p53 to PML-NBs was first observed 3 days after IR, roughly coincident with the second increase in p21 protein expression. This is consistent with the possibility that the second increase in p21 results from p53 being recruited to PML-NBs and further activated as a transcription factor. Two things are important to point out: First, though p21 was highly expressed and displayed a nuclear localization 5 days after IR treatment (Fig 3B, 3C), we did not observe p21 localization in nuclear foci. Second, all (100%) of nuclear foci that contained p53 in IR treated cells also contained PML.
Figure 3. p53 colocalized with PML in irradiated U2OS cells.
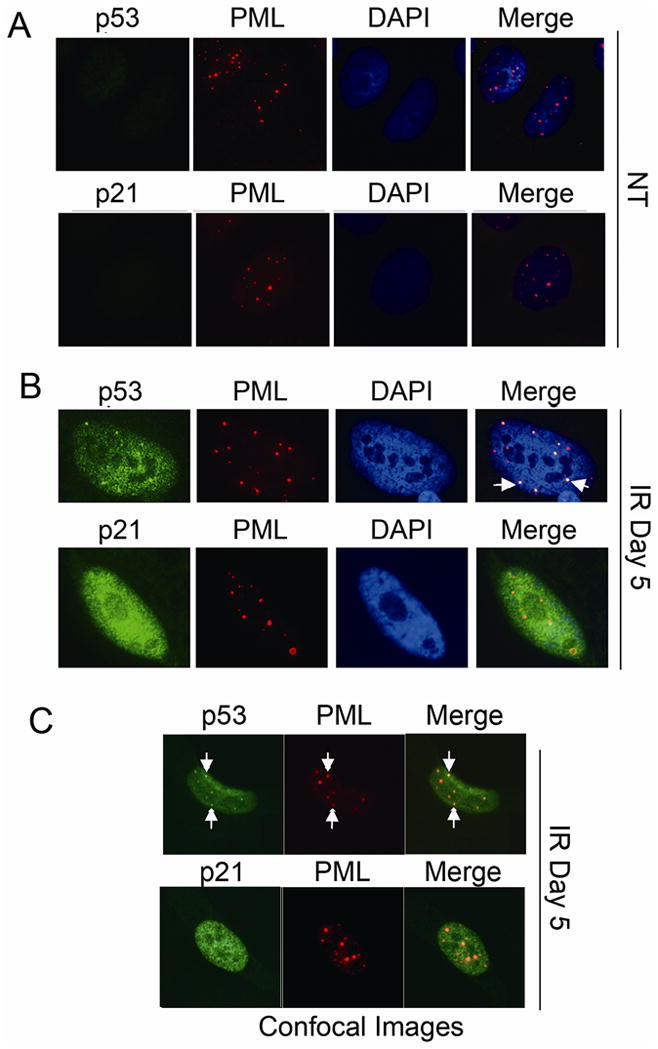
U2OS cells on glass coverslips were either untreated (NT, A) or treated with irradiation (IR, 10Gy, B,C). Cells were fixed with 4% formaldehyde at indicated time points. P53, p21 antibodies were detected using an Alexa-488 conjugated goat anti rabbit IgG. PML antibody were detected using a rhodamine-x conjugated goat anti mouse IgG. Cell nuclei were counterstained with DAPI (blue). Images were captured at 100× magnification. Representative images were shown. Representative foci with p53/PML colocalization were pointed with white arrows. Representative confocal images were shown in C.
Figure 4. Quantification of p53/PML colocalization in irradiated U2OS cells.
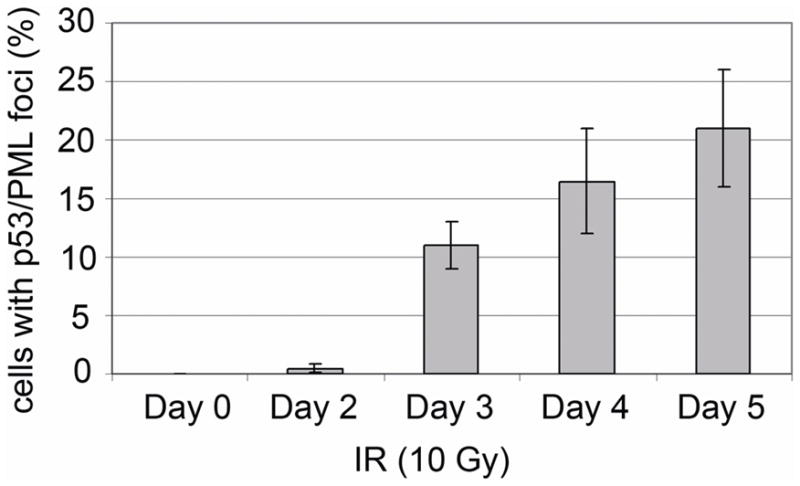
Percentage of cells with p53/PML colocalization was determined at indicated time points after treatment. Percentage represents Mean ± SEM of three independent experiments with 200 cells counted per experiment.
In a previous study it was reported that in IR treated normal human fibroblasts p53 and PML colocalized in nuclear foci that also contained the DNA repair protein hMre11 and phosphorylated histone H2AX (γH2AX), an established marker of DNA double strand breaks (DSBs) [Carbone et al., 2002]. This suggested a role for PML in DNA repair that involves the recruitment of DNA repair and cell cycle checkpoint factors to DNA break sites. We wished to examine whether the focal colocalization of p53 and PML we observed in IR treated U2OS cells corresponds to DNA break sites, and the potential relationship between formation of these p53/PML foci and DNA repair. Phosphorylated histone H2AX (γH2AX) localizes in nuclear foci in IR treated cells that mark DNA DSBs [Rogakou et al., 1999]. We first monitored γH2AX foci formation in U2OS cells between 0–5 days after IR (10 Gy) treatment. Most non-irradiated cells lacked γH2AX foci (Fig 5A), though a small number of foci was observed in some cells. In contrast, IR treated cells contained abundant γH2AX foci (Fig 5B), indicative of IR-induced DSBs. Some of these γH2AX foci were localized in PML-NBs (Fig 5B). We scored the percentage of cells with >10 γH2AX foci per nucleus between 0–5 days after IR treatment. Less than 10% of non-irradiated cell nuclei had >10 γH2AX foci, whereas over 90% of IR treated cell nuclei contained >10 γH2AX foci one day after IR (Fig 5A). A decrease in the number of γH2AX foci over time is indicative of DNA repair and a corresponding decreased number of DSBs [Taneja et al., 2004]. We observed the percentage of cells with >10 γH2AX foci per nucleus decreased to ~40% 2 days after IR and ~20% 3 days after IR, consistent with ongoing DNA repair, but did not decrease below 20% between 3 and 5 days (Fig 5A). These persistent γH2AX foci that remain up to 5 days after IR presumably mark unrepaired DSBs that are perhaps beyond the scope of the cells DNA repair capacity. Next we asked whether p53 and PML colocalized with γH2AX in these apparently unrepaired DSBs. U2OS cells were IR treated (10 Gy) and examined with p53 or PML and γH2AX antibodies after IR treatment. As shown in Fig 5B (white arrows), p53 colocalization with γH2AX in nuclear foci (DSBs) was first observed 3 days after IR treatment, and persisted up to 5 days after IR. Since all p53 containing nuclear foci we observed also contained PML, we can assume that PML is in the same γH2AX containing DSBs as is p53. Consistent with this, PML also localized in nuclear foci with γH2AX in IR treated cells (Fig 5B). The fact that p53 and PML were not recruited to these DSBs until 3 days after IR treatment suggests that p53 and PML are recruited specifically to unrepaired DSBs. We conclude p53 is recruited to PML-NBs after IR treatment that include γH2AX and likely mark persistently unrepaired IR induced DSBs.
Figure 5. p53 colocalized with γH2AX in irradiated U2OS cells.
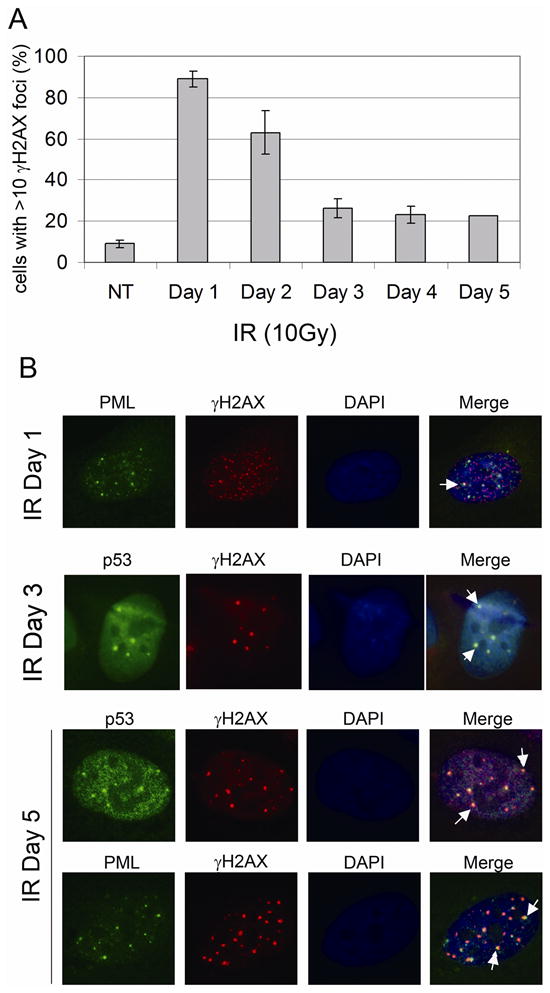
U2OS cells on glass coverslips were treated as described in Figure 2. A) Percentage of cells with >10 γH2AX foci was determined at indicated time points after treatment. Percentage represents Mean ± SEM of three independent experiments with 200 cells counted per experiment. B) γH2AX antibody were detected using a rhodamine-x conjugated goat anti mouse IgG. P53, PML antibody were detected using an Alexa-488 conjugated goat anti rabbit IgG. Representative foci with p53/γH2AX or γH2AX/PML colocalization were pointed with white arrows.
P21 AND P53 ARE RECRUITED TO ALT-ASSOCIATED PML-NBS IN NUTLIN-3A TREATED CELLS
Next, we asked whether the recruitment of p53 to PML containing nuclear foci was IR-specific or also occurred in U2OS cells exposed to Nutlin for 0–5 days. P53 was induced and accumulated in the nucleus of Nutlin treated U2OS cells (Fig 6A). Despite its high levels, however, p53 was only recruited to PML-containing nuclear foci in a small percentage (1–4%) of Nutlin treated cells between 2 and 5 days of treatment (Fig 6C). This indicates the recruitment of p53 to PML-NBs is largely IR-specific. Surprisingly, however, we observed p21 recruitment to PML-containing nuclear foci within the first 24 hrs of Nutlin treatment (Fig 6A, white arrows). We again used confocal microscopy looking at thin nuclear slices to confirm p21 and PML localized in the same nuclear foci in Nutlin treated cells (Fig 6B). As is the case with p53, all (100%) of p21-containing nuclear foci in Nutlin treated cells also included PML. The percentage of Nutlin treated cells in which p21 and PML focal colocalization was observed was quantified, peaking 2 days after Nutlin treatment (16% of cells), and then diminishing to less than 5% at the 4 and 5 day time points. Colocalization of p53, p21, and PML was observed in 1–3% of Nutlin-treated cells (Fig 6C).
Figure 6. p21 colocalized with PML in Nutlin treated U2OS cells.

U2OS cells on glass coverslips were continuously treated with Nutlin (5 μM). Cells were fixed with 4% formaldehyde at indicated time points. A) P53, p21 antibodies were detected using an Alexa-488 conjugated goat anti rabbit IgG. PML antibody were detected using a rhodamine-x conjugated goat anti mouse IgG. Representative foci with p21/PML colocalization were pointed with white arrows. B) Percentage of cells with p53/PML, p21/PML, p53/p21/PML colocalization was determined at indicated time points after treatment. Percentage represents Mean ± SEM of three independent experiments with 200 cells counted per experiment. C) Representative confocal images were shown.
Next, the percentage of cells with >10 γH2AX foci per nucleus was scored at different time points after Nutlin treatment. We observed that continuous Nutlin exposure for 0–5 days decreased the percentage of cells with >10 γH2AX foci per nucleus (Fig 7A). This decrease in γH2AX foci may result from Nutlin-induced cell cycle arrest [Ichijima et al., 2005]. Based on this, we speculated the p21- and PML-containing nuclear foci in Nutlin treated cells might not include γH2AX. Surprisingly, however, γH2AX was detected in the p21 and PML containing nuclear foci after Nutlin treatment (Fig 7B). Given that Nutlin does not induce DSBs, we considered the p21, PML, and γH2AX nuclear foci might represent novel PML-NBs in which DSBs or other chromosomal stresses are generated by an endogenous source. U2OS are telomerase-negative cancer celIs that maintain telomere length through an alternative (ALT) recombination mechanism [Scheel et al., 2001]. ALT-associated PML bodies (APBs) are specialized PML-NBs that include PML, telomere repeat binding protein-1 (TRF1), γH2AX, and multiple DNA repair/recombination proteins along with telomeric DNA [Henson et al., 2002][Tanaka et al., 2005]. We examined TRF1 localization in Nutlin treated U2OS cells to determine whether these p21-containing nuclear foci were APBs. TRF1 was detected in the p21 and PML containing nuclear foci 1–2 days after Nutlin treatment, identifying them as APBs (Fig 8A,B). We also observed that TRF1 colocalized with p53 in the small percentage (1–3%) of Nutlin treated cells that displayed p53/PML nuclear foci (Fig 8A), indicating p53 is also recruited to APBs after Nutlin treatment. In contrast, the overwhelming majority of PML-NBs in which p53 localized after IR treatment did not include TRF1 (Fig 8C), indicating they are not APBs. Based on these results we conclude that p21, and to a lesser extent p53, are recruited to APBs in Nutlin treated U2OS cells, while p53 is recruited to PML-NBs that mark unrepaired DNA damage sites after IR treatment.
Figure 7. p21 colocalized with γH2AX in Nutlin treated U2OS cells.
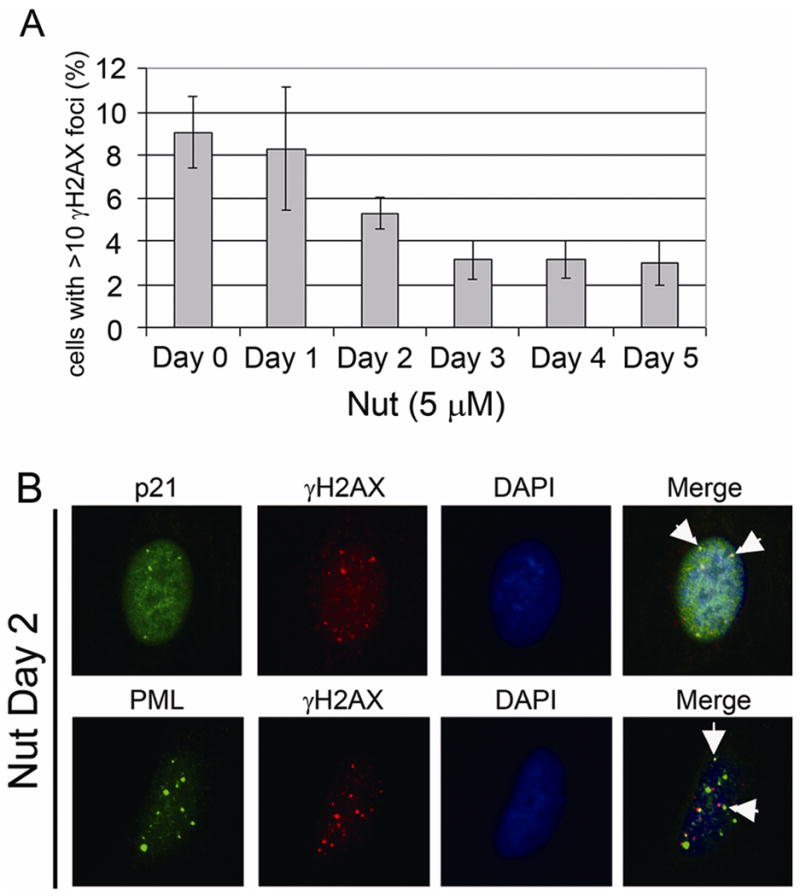
U2OS cells on glass coverslips were treated as described in Figure 4. A) Percentage of cells with >10 γH2AX foci was determined at indicated time points after treatment. Percentage represents Mean ± SEM of three independent experiments with 200 cells counted per experiment. B) p21, PML antibodies were detected using an Alexa-488 conjugated goat anti rabbit IgG. γH2AX antibody were detected using a rhodamine-x conjugated goat anti mouse IgG. Representative colocalization foci were pointed with white arrows.
Figure 8. p21 and p53 colocalized with TRF1 in PML-NB in Nutlin treated U2OS cells.

U2OS cells on glass coverslips were either untreated (NT), treated with irradiation (IR, 10Gy) or treated with Nutlin (Nut; 5μmol/L). P53, p21, PML antibodies were detected using an Alexa-488 conjugated goat anti rabbit IgG. TRF1 antibody were detected using a rhodamine-x conjugated goat anti mouse IgG. Representative colocalization foci were pointed with white arrows. Representative confocal images were shown in B.
It has been reported that within an asynchronously growing population of ALT cells, only 5–10% of the cells contain APBs [Wu et al., 2003; Yeager et al., 1999]. However, the proportion of APB-positive cells could be greatly increased by methionine starvation or by DNA damaging agents [Fasching et al., 2007; Jiang et al., 2007]. In each of these cases, the treatments that induced APBs also caused growth arrest. Both IR and Nutlin induce growth arrests that are largely (IR) or completely (Nutlin) dependent on the p53-p21 pathway. We therefore asked whether IR or Nutlin increase the percentage of APB-positive cells and, if yes, whether this increase requires p53 or p21. U2OS cells exposed to either IR or Nutlin were examined 1–5 days after treatment by immunofluorescence staining using antibodies against TRF1 and PML. APB positive cells were identified as those cells in which TRF1 and PML colocalized in large nuclear foci. Approximately 10% of untreated, asynchronously growing U2OS cells were APB-positive. In contrast, Nutlin or IR treatment for 2–5 days increased the proportion of APB positive cells to approximately 50% (Fig 9A). We next used an siRNA knockdown approach to ask whether this increase in APB cells was dependent of p53 and p21. As shown in Fig 9B, siRNA against either p53 or p21 blocked the increase in percent APB-positive cells caused by Nutlin, whereas control siRNA had no effect. We conclude that p53 and p21, and most likely growth arrest mediated by p53-p21 pathway activation, is responsible for the Nutlin induced increase in APB-positive cells.
Figure 9. APB positive cells increased in irradiated or Nutlin treated U2OS cells.
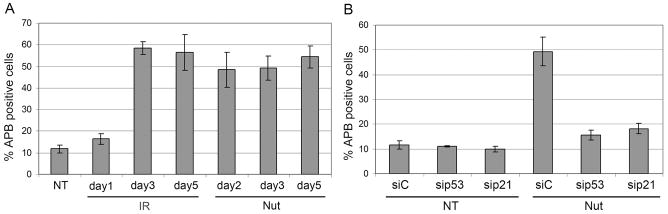
APB positive cells were identified as those cells in which TRF1 and PML colocalized in large nuclear foci. PML antibodies were detected using an Alexa-488 conjugated goat anti rabbit IgG. TRF1 antibody were detected using a rhodamine-x conjugated goat anti mouse IgG. A) Percentage of APB positive cells was determined at indicated time points after treatment. Percentage represents Mean ± SEM of three independent experiments with 200 cells counted per experiment. B) U2OS cells were either transfected with siControl (siC), sip53, or sip21. At 24hrs after transfection, the cells were untreated (NT) or treated with Nutlin (Nut; 5μmol/L). Cells were fixed with 4% formaldehyde 2 days after treatment and subjected to immunofluoresence staining.
DISCUSSION
PML localizes in distinct nuclear foci termed PML-NBs and can simultaneously bind and recruit multiple other proteins to these foci [Bernardi and Pandolfi, 2007]. At least 60 different proteins have been localized in PML-NBs, and subsets of PML-NBs that harbor different protein combinations are believed to affect different cellular activities [Bernardi and Pandolfi, 2007]. It was reported that a portion of p53 can be co-recruited to PML-NBs with the acetylase enzyme CBP in stressed cells [Ferbeyre et al., 2000; Guo et al., 2000; Pearson et al., 2000]. CBP then acetylates p53 C-terminal lysines, and this is believed to increase p53s ability to activate downstream targets such as p21 that inhibit cell growth [Ferbeyre et al., 2000; Luo et al., 2004; Pearson et al., 2000]. PML has also been shown to localize at DNA break sites in irradiated normal human fibroblasts and to recruit both p53 and the repair protein hMre11 to these sites [Carbone et al., 2002]. This suggests a subset of PML-NBs may play a role in the processing and/or repair of DNA breaks, perhaps by recruiting checkpoint proteins (p53) and repair proteins (hMre11) to break sites. ALT-associated PML bodies (APBs) are yet another subset of PML-NBs that include telomeric DNA, telomere repeat binding factors (TRFs), and multiple recombination and DNA repair proteins [Draskovic et al., 2009; Henson et al., 2002; Yu et al.]. These APBs are believed to play a role in telomere maintenance, and are found exclusively in telomerase-negative tumors in which telomere length is maintained through an alternative (ALT) recombination mechanism. Experiments in the current study were initiated to compare the response of U2OS cells when p53 was activated by either ionizing radiation (IR) or by treatment with the non-genotoxic p53 stabilizer Nutlin-3a. We found that p53 was induced to high levels and with monophasic induction kinetics following either IR or Nutlin treatment. P21, used as an indicator of p53 activity, was also induced to high levels by IR and Nutlin. However, while p21 displayed a monophasic induction after Nutlin treatment, it displayed a biphasic induction following IR characterized by an initial increase 24–30 hrs after IR treatment and a second, further increase in p21 approximately 3 days after IR. This prompted us to examine whether the second increase in p21 coincided with recruitment of p53 into PML-NBs. Indeed, p53 was recruited to PML-NBs beginning 3 days after IR treatment, approximately coincident with the second increase in p21 expression. Using γH2AX as a marker of DNA double strand breaks, we demonstrated these p53-containing PML-NBs are DNA break sites, and are likely unresolved or unrepaired DNA breaks that persist at late time points after IR. Both IR and Nutlin treatment caused cell cycle arrest, and our results showed that both treatments caused a large increase in the percentage of APB-positive cells. This effect was not observed in cells transfected with p53 or p21 siRNA, demonstrating the increase in APB-positive cells was p53- and p21-dependent. Interestingly, p21, and to a lesser extent p53, was recruited to APBs in a fraction of Nutlin treated, but not IR treated, cells. These data demonstrate that p53 and p21 are recruited to distinct PML-NBs in IR-treated and Nutlin treated cells. P53 is recruited to PML-NBs after IR that likely mark unrepaired DSBs, suggesting p53 may either be activated by PML at these sites and/or function in their repair. In contrast, p21 and p53 are recruited to ALT-associated PML-NBs after Nutlin treatment, suggesting p21 and p53 may play an as yet undetermined role in telomere maintenance.
Our findings raise two important questions. First, what is the mechanism for recruitment of p53 and p21 to these distinct PML-NBs, and second, what is the purpose of their recruitment to these PML-NBs? P53 is subject to various DNA damage-induced post-translational modifications that can stabilize the protein, increase its transcriptional activity, and cause it to accumulate in the nucleus or be directed to mitochondria [Boehme and Blattner, 2009]. We found that p53 was recruited to PML containing nuclear foci in IR-treated U2OS cells, and we confirmed these are sites of DNA double strand breaks by their colocalization with γH2AX. Our results are consistent with those of Carbone and colleagues (2002) who reported p53 and PML colocalization at DNA breaks sites in irradiated normal human fibroblasts [Carbone et al., 2002]. Nutlin stabilizes p53 through a completely non-genotoxic mechanism, and thus p53 induced by Nutlin is not subject to the same damage-induced post-translational modifications that it would be subject to in an IR-treated cell [Thompson et al., 2004]. Although p53 was induced by Nutlin to levels equally high or higher than that seen with IR, it retained a diffuse nuclear localization and only colocalized in nuclear foci with PML in a small percentage of cells (1–4%). This indicates increased levels of p53 alone are not sufficient for its efficient recruitment to PML-NBs. We considered that p53 may only associate with PML when PML is localized at DNA double strand breaks. However, we do not favor this possibility since purified p53 and PML bind each other strongly, and abundant colocalization of p53 with PML is seen in cells over-expressing RasV12, an oncogenic stress that does not cause overt DNA double strand breaks [Ferbeyre et al., 2000; Pearson et al., 2000]. Perhaps more likely is the possibility that DNA damage-induced modifications to p53 and/or PML promote their association and thus the recruitment of p53 to PML-NBs in IR treated cells. P21, and to a lesser extent p53, was recruited to PML-containing nuclear foci that also included γH2AX in Nutlin treated cells. We confirmed these were ALT-associated PML-NBs by their colocalization with TRF1. Consistent with our result, Jiang et al. reported that p21 was recruited to ALT-associated PML-NBs in cells in which an exogenously expressed p53-ER fusion protein was activated by tamoxifen [Jiang et al., 2009]. Since Nutlin is non-genotoxic and does not cause overt DNA damage, it seems likely that neither p21 nor p53 requires damage-induced post-translation modifications for recruitment to these PML-NBs. ALT-associated PML-NBs have been reported to include telomeric DNA and multiple factors including PML, telomere repeat binding factors 1 and 2, and different recombination/repair proteins including the MRN complex (Mre11, Rad50, Nbs1), RPA, Werners syndrome helicase (WRN) and Bloom’s syndrome helicase [Henson et al., 2002]. It is interesting that although p21 was induced to high levels after IR treatment it was not recruited to PML-NBs in IR treated cells. A trivial explanation for this would be if the ALT-associated PML-NBs to which p21 is recruited in Nutlin treated cells do not exist in cells exposed to IR. However, we do not favor this possibility since PML and TRF1-containing nuclear foci (ALT-associated PML-NBs) were readily detected 1–5 days after IR treatment (Fig 8C). Perhaps other proteins are recruited to ALT-associated PML-NBs after IR treatment that preclude the association or recruitment of p21. Alternatively, IR-induced modifications to p21, PML, or other factors might also preclude the recruitment of p21 to these ALT-associated PML-NBs.
Why are p53 and p21 recruited to PML-NBs in either IR or Nutlin treated cells? PML contains several protein-interaction domains and can co-recruit multiple proteins simultaneously. It has been suggested that PML’s function resides, at least in part, in its ability to bring proteins in close proximity so they may interact and regulate each others activity [Borden, 2002]. For example, co-recruitment of p53 and CBP into PML-NBs is thought to facilitate CBP-mediated acetylation of p53 and increase p53 transcriptional activity [Ferbeyre et al., 2000; Guo et al., 2000; Pearson et al., 2000]. In the case of IR, p53 was recruited to PML-NBs that were sites of DNA double strand breaks. Interestingly, the recruitment of p53 into these PML-NBs occurred 3 days after IR treatment, roughly coincident with the second increase in p21 expression. Thus, one possibility is that p53 is recruited to these sites to be brought in proximity with modifying enzymes (CBP) and kinases like ATM, ATR, and Chk2 that localize at DSBs and phosphorylate p53 to increase its activity. In this regard, it will be interesting to determine what other factors associate within foci with p53 and PML in IR- treated cells, and what if any post-translational modifications occur to p53 within these PML-NBs. A second possibility is that p53 is recruited to PML-NB/DNA break sites to directly participate in or regulate repair. For example, it has been known for several years that the p53 C-terminus has single-strand DNA binding activity. Wiman and colleagues (1994) reported purified p53 could bind single strand DNA ends and facilitate reannealing and strand transfer, potentially enhancing repair [Bakalkin et al., 1994]. More recently, Powell and colleagues (2000) reported that p53 could directly enhance rejoining of DNA double strand breaks with cohesive ends in IR-treated cells [Tang et al., 1999]. Thus, p53 might be recruited to PML-containing double strand break sites to bind and directly facilitate the rejoining and repair of DNA breaks. In contrast to IR, p21 and p53 were specifically recruited to APBs in Nutlin treated cells. APBs are believed to promote telomere maintenance in telomerase negative cancer cells, and have been reported to include telomeric DNA and binding proteins, and multiple recombination/repair proteins such as the MRN complex (Mre11, Rad50, Nbs1), RPA, Werners syndrome helicase (WRN) and Bloom’s syndrome helicase [Henson et al., 2002]. The ALT pathway maintains telomere length through a complex recombination and DNA synthesis mechanism [Henson et al., 2002]. The observation that p21 and p53 are recruited to APBs suggests they may play a previously unrecognized role in telomere maintenance, perhaps by interacting with and/or regulating the activity of factors within these APBs. It was interesting that p21 and p53 were only recruited to APBs in Nutlin treated cells and not IR treated cells, despite the fact that both treatments caused a large increase in the percentage of APB-positive cells. One possibility is that p21 and p53 are only recruited to and function in APBs in response to non-genotoxic stresses (such as Nutlin) but not in response to genotoxic stresses (such as IR). It was also interesting that the percentage of cells in which p21 localized in APBs peaked during the first 2–3 days of Nutlin treatment but then diminished by 4–5 days of treatment, despite p21 levels remaining at a steady, highly-elevated level. This suggests any potential role for p21 in telomere maintenance may be transient, occurring only within the first 1–3 days of Nutlin treatment.
A final consideration is what role p53 and p21 play in APB formation. Both IR and Nutlin treatment caused cell cycle arrest, and previous studies have shown these arrests are largely (IR) or completely (Nutlin) dependent on p53 and p21. IR and Nutlin treatment also both caused a pronounced increase in the percentage of APB positive cells. We showed in the case of Nutlin that this increase in APB-positive cells is p53 and p21 dependent. We believe the increase in APB positive cells is a secondary consequence of growth arrest resulting from p53-p21 pathway activation rather than a direct function of p53 or p21 within APBs. We believe this for three reasons: First, Nutlin caused a large increase in the percentage of APB-positive cells (increased from 15% in untreated cells to ~50% after Nutlin treatment), yet p21 was recruited to APBs in only a small percentage of Nutlin-treated cells (10–16%), and p53 was recruited to APBs in an even smaller percentage of cells (<5%). Thus, the vast majority of newly formed APBs in Nutlin treated cells do not include p21 or p53. Second, IR treatment caused a cell cycle arrest and large increase in APB positive cells, but without recruitment of p21 or p53 into APBs. Third, other studies have reported an increase in APB-positive cells when cell growth was arrested by either methionine starvation, exposure to DNA damaging agents, or treatment with tamoxifen in a cell line expressing a p53-ER fusion protein [Fasching et al., 2007; Jiang et al., 2007; Jiang et al., 2009]. In one report it was demonstrated that growth arrest and the increase in APB-positive cells required p53 and p21 [Jiang et al., 2009]. Our results are consistent with these findings and support the idea that the increase in APB-positive cells is a result of growth arrest mediated by the p53-p21 pathway. Nonetheless, the fact that p21 and p53 are recruited to APBs in a fraction of Nutlin treated cells suggests, as mentioned above, they may also have a direct function in these APBs.
Acknowledgments
The authors thank Dr. Jitesh Pratap for his advice and critical reading of the manuscript. This work was supported by NIH grants 1RO1CA137598-01A1 and RO1CA108843, both to CGM.
References
- Bakalkin G, Yakovleva T, Selivanova G, Magnusson KP, Szekely L, Kiseleva E, Klein G, Terenius L, Wiman KG. p53 binds single-stranded DNA ends and catalyzes DNA renaturation and strand transfer. Proc Natl Acad Sci U S A. 1994;91:413–7. doi: 10.1073/pnas.91.1.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardi R, Pandolfi PP. Structure, dynamics and functions of promyelocytic leukaemia nuclear bodies. Nat Rev Mol Cell Biol. 2007;8:1006–16. doi: 10.1038/nrm2277. [DOI] [PubMed] [Google Scholar]
- Boehme KA, Blattner C. Regulation of p53--insights into a complex process. Crit Rev Biochem Mol Biol. 2009;44:367–92. doi: 10.3109/10409230903401507. [DOI] [PubMed] [Google Scholar]
- Borden KL. Pondering the promyelocytic leukemia protein (PML) puzzle: possible functions for PML nuclear bodies. Mol Cell Biol. 2002;22:5259–69. doi: 10.1128/MCB.22.15.5259-5269.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown L, Boswell S, Raj L, Lee SW. Transcriptional targets of p53 that regulate cellular proliferation. Crit Rev Eukaryot Gene Expr. 2007;17:73–85. doi: 10.1615/critreveukargeneexpr.v17.i1.50. [DOI] [PubMed] [Google Scholar]
- Carbone R, Pearson M, Minucci S, Pelicci PG. PML NBs associate with the hMre11 complex and p53 at sites of irradiation induced DNA damage. Oncogene. 2002;21:1633–40. doi: 10.1038/sj.onc.1205227. [DOI] [PubMed] [Google Scholar]
- de The H, Lavau C, Marchio A, Chomienne C, Degos L, Dejean A. The PML-RAR alpha fusion mRNA generated by the t(15;17) translocation in acute promyelocytic leukemia encodes a functionally altered RAR. Cell. 1991;66:675–84. doi: 10.1016/0092-8674(91)90113-d. [DOI] [PubMed] [Google Scholar]
- Dellaire G, Farrall R, Bickmore WA. The Nuclear Protein Database (NPD) sub-nuclear localisation and functional annotation of the nuclear proteome. Nucleic Acids Res. 2003;31:328–30. doi: 10.1093/nar/gkg018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Leonardo A, Linke SP, Clarkin K, Wahl GM. DNA damage triggers a prolonged p53-dependent G1 arrest and long-term induction of Cip1 in normal human fibroblasts. Genes Dev. 1994;8:2540–51. doi: 10.1101/gad.8.21.2540. [DOI] [PubMed] [Google Scholar]
- Draskovic I, Arnoult N, Steiner V, Bacchetti S, Lomonte P, Londono-Vallejo A. Probing PML body function in ALT cells reveals spatiotemporal requirements for telomere recombination. Proc Natl Acad Sci U S A. 2009;106:15726–31. doi: 10.1073/pnas.0907689106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everett RD, Lomonte P, Sternsdorf T, van Driel R, Orr A. Cell cycle regulation of PML modification and ND10 composition. J Cell Sci. 1999;112(Pt 24):4581–8. doi: 10.1242/jcs.112.24.4581. [DOI] [PubMed] [Google Scholar]
- Fasching CL, Neumann AA, Muntoni A, Yeager TR, Reddel RR. DNA damage induces alternative lengthening of telomeres (ALT) associated promyelocytic leukemia bodies that preferentially associate with linear telomeric DNA. Cancer Res. 2007;67:7072–7. doi: 10.1158/0008-5472.CAN-07-1556. [DOI] [PubMed] [Google Scholar]
- Ferbeyre G, de Stanchina E, Querido E, Baptiste N, Prives C, Lowe SW. PML is induced by oncogenic ras and promotes premature senescence. Genes Dev. 2000;14:2015–27. [PMC free article] [PubMed] [Google Scholar]
- Fogal V, Gostissa M, Sandy P, Zacchi P, Sternsdorf T, Jensen K, Pandolfi PP, Will H, Schneider C, Del Sal G. Regulation of p53 activity in nuclear bodies by a specific PML isoform. Embo J. 2000;19:6185–95. doi: 10.1093/emboj/19.22.6185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goddard AD, Borrow J, Freemont PS, Solomon E. Characterization of a zinc finger gene disrupted by the t(15;17) in acute promyelocytic leukemia. Science. 1991;254:1371–4. doi: 10.1126/science.1720570. [DOI] [PubMed] [Google Scholar]
- Guo A, Salomoni P, Luo J, Shih A, Zhong S, Gu W, Pandolfi PP. The function of PML in p53-dependent apoptosis. Nat Cell Biol. 2000;2:730–6. doi: 10.1038/35036365. [DOI] [PubMed] [Google Scholar]
- Haupt Y, Maya R, Kazaz A, Oren M. Mdm2 promotes the rapid degradation of p53. Nature. 1997;387:296–9. doi: 10.1038/387296a0. [DOI] [PubMed] [Google Scholar]
- Henson JD, Neumann AA, Yeager TR, Reddel RR. Alternative lengthening of telomeres in mammalian cells. Oncogene. 2002;21:598–610. doi: 10.1038/sj.onc.1205058. [DOI] [PubMed] [Google Scholar]
- Horn HF, Vousden KH. Coping with stress: multiple ways to activate p53. Oncogene. 2007;26:1306–16. doi: 10.1038/sj.onc.1210263. [DOI] [PubMed] [Google Scholar]
- Ichijima Y, Sakasai R, Okita N, Asahina K, Mizutani S, Teraoka H. Phosphorylation of histone H2AX at M phase in human cells without DNA damage response. Biochem Biophys Res Commun. 2005;336:807–12. doi: 10.1016/j.bbrc.2005.08.164. [DOI] [PubMed] [Google Scholar]
- Jiang WQ, Zhong ZH, Henson JD, Reddel RR. Identification of candidate alternative lengthening of telomeres genes by methionine restriction and RNA interference. Oncogene. 2007;26:4635–47. doi: 10.1038/sj.onc.1210260. [DOI] [PubMed] [Google Scholar]
- Jiang WQ, Zhong ZH, Nguyen A, Henson JD, Toouli CD, Braithwaite AW, Reddel RR. Induction of alternative lengthening of telomeres-associated PML bodies by p53/p21 requires HP1 proteins. J Cell Biol. 2009;185:797–810. doi: 10.1083/jcb.200810084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakizuka A, Miller WH, Jr, Umesono K, Warrell RP, Jr, Frankel SR, Murty VV, Dmitrovsky E, Evans RM. Chromosomal translocation t(15;17) in human acute promyelocytic leukemia fuses RAR alpha with a novel putative transcription factor, PML. Cell. 1991;66:663–74. doi: 10.1016/0092-8674(91)90112-c. [DOI] [PubMed] [Google Scholar]
- Koken MH, Linares-Cruz G, Quignon F, Viron A, Chelbi-Alix MK, Sobczak-Thepot J, Juhlin L, Degos L, Calvo F, de The H. The PML growth-suppressor has an altered expression in human oncogenesis. Oncogene. 1995;10:1315–24. [PubMed] [Google Scholar]
- Kubbutat MH, Jones SN, Vousden KH. Regulation of p53 stability by Mdm2. Nature. 1997;387:299–303. doi: 10.1038/387299a0. [DOI] [PubMed] [Google Scholar]
- Luo J, Li M, Tang Y, Laszkowska M, Roeder RG, Gu W. Acetylation of p53 augments its site-specific DNA binding both in vitro and in vivo. Proc Natl Acad Sci U S A. 2004;101:2259–64. doi: 10.1073/pnas.0308762101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maki CG, Howley PM. Ubiquitination of p53 and p21 is differentially affected by ionizing and UV radiation. Mol Cell Biol. 1997;17:355–63. doi: 10.1128/mcb.17.1.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maltzman W, Czyzyk L. UV irradiation stimulates levels of p53 cellular tumor antigen in nontransformed mouse cells. Mol Cell Biol. 1984;4:1689–94. doi: 10.1128/mcb.4.9.1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mihara M, Erster S, Zaika A, Petrenko O, Chittenden T, Pancoska P, Moll UM. p53 has a direct apoptogenic role at the mitochondria. Mol Cell. 2003;11:577–90. doi: 10.1016/s1097-2765(03)00050-9. [DOI] [PubMed] [Google Scholar]
- Nagasawa H, Keng P, Maki C, Yu Y, Little JB. Absence of a radiation-induced first-cycle G1-S arrest in p53+ human tumor cells synchronized by mitotic selection. Cancer Res. 1998;58:2036–41. [PubMed] [Google Scholar]
- Nagasawa H, Li CY, Maki CG, Imrich AC, Little JB. Relationship between radiation-induced G1 phase arrest and p53 function in human tumor cells. Cancer Res. 1995;55:1842–6. [PubMed] [Google Scholar]
- Pandolfi PP. Oncogenes and tumor suppressors in the molecular pathogenesis of acute promyelocytic leukemia. Hum Mol Genet. 2001;10:769–75. doi: 10.1093/hmg/10.7.769. [DOI] [PubMed] [Google Scholar]
- Pandolfi PP, Grignani F, Alcalay M, Mencarelli A, Biondi A, LoCoco F, Grignani F, Pelicci PG. Structure and origin of the acute promyelocytic leukemia myl/RAR alpha cDNA and characterization of its retinoid-binding and transactivation properties. Oncogene. 1991;6:1285–92. [PubMed] [Google Scholar]
- Pearson M, Carbone R, Sebastiani C, Cioce M, Fagioli M, Saito S, Higashimoto Y, Appella E, Minucci S, Pandolfi PP, Pelicci PG. PML regulates p53 acetylation and premature senescence induced by oncogenic Ras. Nature. 2000;406:207–10. doi: 10.1038/35018127. [DOI] [PubMed] [Google Scholar]
- Rogakou EP, Boon C, Redon C, Bonner WM. Megabase chromatin domains involved in DNA double-strand breaks in vivo. J Cell Biol. 1999;146:905–16. doi: 10.1083/jcb.146.5.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheel C, Schaefer KL, Jauch A, Keller M, Wai D, Brinkschmidt C, van Valen F, Boecker W, Dockhorn-Dworniczak B, Poremba C. Alternative lengthening of telomeres is associated with chromosomal instability in osteosarcomas. Oncogene. 2001;20:3835–44. doi: 10.1038/sj.onc.1204493. [DOI] [PubMed] [Google Scholar]
- Tanaka H, Mendonca MS, Bradshaw PS, Hoelz DJ, Malkas LH, Meyn MS, Gilley D. DNA damage-induced phosphorylation of the human telomere-associated protein TRF2. Proc Natl Acad Sci U S A. 2005;102:15539–44. doi: 10.1073/pnas.0507915102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taneja N, Davis M, Choy JS, Beckett MA, Singh R, Kron SJ, Weichselbaum RR. Histone H2AX phosphorylation as a predictor of radiosensitivity and target for radiotherapy. J Biol Chem. 2004;279:2273–80. doi: 10.1074/jbc.M310030200. [DOI] [PubMed] [Google Scholar]
- Tang W, Willers H, Powell SN. p53 directly enhances rejoining of DNA double-strand breaks with cohesive ends in gamma-irradiated mouse fibroblasts. Cancer Res. 1999;59:2562–5. [PubMed] [Google Scholar]
- Thompson T, Tovar C, Yang H, Carvajal D, Vu BT, Xu Q, Wahl GM, Heimbrook DC, Vassilev LT. Phosphorylation of p53 on key serines is dispensable for transcriptional activation and apoptosis. J Biol Chem. 2004;279:53015–22. doi: 10.1074/jbc.M410233200. [DOI] [PubMed] [Google Scholar]
- Vaseva AV, Moll UM. The mitochondrial p53 pathway. Biochim Biophys Acta. 2009;1787:414–20. doi: 10.1016/j.bbabio.2008.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, Fotouhi N, Liu EA. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004;303:844–8. doi: 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]
- Wu G, Jiang X, Lee WH, Chen PL. Assembly of functional ALT-associated promyelocytic leukemia bodies requires Nijmegen Breakage Syndrome 1. Cancer Res. 2003;63:2589–95. [PubMed] [Google Scholar]
- Yeager TR, Neumann AA, Englezou A, Huschtscha LI, Noble JR, Reddel RR. Telomerase-negative immortalized human cells contain a novel type of promyelocytic leukemia (PML) body. Cancer Res. 1999;59:4175–9. [PubMed] [Google Scholar]
- Yu J, Lan J, Wang C, Wu Q, Zhu Y, Lai X, Sun J, Jin C, Huang H. PML3 interacts with TRF1 and is essential for ALT-associated PML bodies assembly in U2OS cells. Cancer Lett. 291:177–86. doi: 10.1016/j.canlet.2009.10.009. [DOI] [PubMed] [Google Scholar]