

SUMMARY
Vascular endothelial growth factor inhibitors (VEGFi) are known to cause hypertension and renal injury that severely limits their use as an anticancer therapy. We hypothesized that the angiotensin-converting enzyme inhibitor captopril not only prevents hypertension, but also decreases renal injury caused by the VEGFi sorafenib.
Rats were administered sorafenib (20 mg/kg per day) alone or in combination with captopril (40 mg/kg per day) for 4 weeks. Sorafenib administration increased blood pressure, which plateaued by day 10.
Concurrent treatment with captopril for 4 weeks resulted in a 30 mmHg decrease in blood pressure compared with sorafenib alone (155 ± 5 vs 182 ± 6 mmHg, respectively; P < 0.05). Furthermore, concurrent captopril treatment reduced albuminuria by 50% compared with sorafenib alone (20 ± 8 vs 42 ± 9 mg/day, respectively; P < 0.05) and reduced nephrinuria by eightfold (280 ± 96 vs 2305 ± 665 μg/day, respectively; P < 0.05). Glomerular injury, thrombotic micro-angiopathy and tubular cast formation were also decreased in captopril-treated rats administered sorafenib. Renal autoregulatory efficiency was determined by evaluating the afferent arteriolar constrictor response to ATP. Sorafenib administration attenuated the vasoconstriction to ATP, whereas concurrent captopril treatment improved ATP reactivity.
In conclusion, captopril attenuated hypertension and renal injury and improved renal autoregulatory capacity in rats administered sorafenib. These findings indicate that captopril treatment, in addition to alleviating the detrimental side-effect of hypertension, decreases the renal injury associated with anticancer VEGFi therapies such as sorafenib.
Keywords: afferent arterioles, albuminuria, glomerular injury, hypertension.nephrinuria, vascular endothelial growth factor inhibitors
INTRODUCTION
Angiogenesis is a critical determinant of cancer progression and tumour metastasis. Thus, targeting angiogenesis has become an appealing therapeutic approach with broad applications in cancer therapy.1 It is known that receptor tyrosine kinases and their growth factor ligands, namely vascular endothelial growth factor (VEGF), play a fundamental role in tumour angiogenesis.2 Intense efforts have been directed at the development of efficient inhibitors of VEGF production and signal transduction for anti-tumourigenic purposes. These efforts include the development of humanized neutralizing anti-VEGF monoclonal antibodies, inhibitors of VEGF receptor kinases and small molecule VEGF receptor blockers.3,4 Of these agents, the US Food and Drug Administration (FDA) has approved bevacizumab (an anti-VEGF monoclonal antibody) and the tyrosine kinase receptor inhibitors sunitinib and sorafenib for anticancer therapy.1 Vascular endothelial growth factor signalling is important in normal cardiovascular physiology and its abrogation by anti-VEGF therapies causes vasoconstriction and microvascular rarefaction.5 These vascular changes finally lead to the development of hypertension and other complications, including renal injury. In line with this notion, it has been observed that VEGF-targeted chemotherapeutic agents provide substantial therapeutic benefit to cancer patients; however; hypertension and renal injury represent serious and limiting side-effects for the therapeutic efficacy of this class of compounds.6–10
Sorafenib is an orally active multikinase inhibitor of VEGF receptors with effects on tumour cell proliferation and tumour angiogenesis. It has been found to have broad spectrum activity against multiple tyrosine kinases of the VEGF receptor (VEGFR) family, including VEGFR1, VEGFR2 and VEGF3, RAF protein, platelet-derived growth factor receptor (PDGFR) family (PDGFR-β), stem-cell growth factor receptor (c-KIT), Fms-like tyrosine kinase 3 (Flt-3) and the receptors that are encoded by the ret proto-oncogene (RET). Sorafenib has strong anti-angiogenic actions mediated by its inhibitory effects on VEGFR and PDGFR.8 As with other multikinase inhibitor-based antineoplastic agents, the use of sorafenib in patients is associated with many side-effects, including hypertension, other cardiovascular events and renal injury.9,11,12
The management of hypertension in VEGF inhibitor (VEGFi)-treated patients is complex. The choice of treatment in the management of hypertension in these patients depends on several factors, including age, the presence of other comorbid conditions (type of cancer, renal insufficiency, diabetes, pre-existing hypertension) and patient lifestyle. Nonetheless, antihypertensive agents that target the renin–angiotensin system (RAS), particularly, angiotensin-converting enzyme inhibitors (ACEi), have so far been the most preferable choice to effectively combat the hypertension that occurs with VEGFi therapy.1,13–17
Although RAS inhibitors are given as antihypertensive agents to VEGFi-treated patients, their ability to decrease glomerular microangiopathy and improve renal vascular function remains unknown. With this background, in the present study we investigated the protective effect of the ACEi captopril on renal vascular injury caused by sorafenib administration to rats. Our results demonstrate that a preventative treatment approach with captopril not only decreases blood pressure, but also improves renal vascular and glomerular function in sorafenib-treated rats.
METHODS
Animal groups
The experiments were approved by and performed according to the guidelines of the Institutional Animal Care and Use Committee, Medical College of Wisconsin (Milwaukee, WI, USA). Male Sprague-Dawley rats weighing 180–200 g (Charles River, Wilmington, MA, USA) were fed an 8% NaCl diet. Rats were divided into three groups: (i) a control group, administered vehicle (pudding); (ii) a sorafenib-treated group given 20 mg/kg per day, p.o., sorafenib for 4 weeks; and (iii) a sorafenib + captopril-treated group given a combination of sorafenib (20 mg/kg per day, p.o.) plus captopril (40 mg/kg per day, p.o.) for 4 weeks. The doses of sorafenib and captopril were chosen based on previous studies in rats.18–21
Blood pressure measurements
Rats were anaesthetized with isoflurane and radiotelemetry transmitters (PA-C40; Data Science International, St Paul, MN, USA) were implanted. In implanting the radiotransmitter, the fluid-filled catheter of the transmitter was inserted into the right carotid artery and the body of the transmitter was placed subcutaneously at the back. Rats were allowed to recover from surgery and were returned to individual housing. Baseline arterial pressure was recorded for 5 days prior to the experimental period. Mean arterial pressure was recorded continuously throughout the experimental period. Radiotelemetry was used for the initial measurement of arterial blood pressure in rats administered sorafenib to assess the development of hypertension. Systolic blood pressure (SBP) was measured weekly for 4 weeks using tail-cuff plethysmography to compare changes in blood pressure in the different experimental groups.
Urinary albumin and nephrin measurement
During the treatment protocols, 24 h urine samples were collected on Days 14 and 28. Urinary albumin and nephrin excretion rates were determined by ELISA (Exocell, Philadelphia, PA, USA). Daily albumin and nephrin excretion rates were calculated from the volume of urine collected from each rat over the 24 h period.
Histological analysis
After the 4 week experimental period, rats were killed and kidney tissues collected. The left kidney was prepared for histopathological examination. The kidney was removed, decapsulated, cut into sections and fixed in 10% buffered formalin for 48 h. The fixed kidney tissues were embedded in paraffin and cut into 4 μm sections. Paraffin-embedded kidney slices were finally deparaffinized, rehydrated and stained with periodic acid-Schiff (PAS) and Masson’s trichrome. For semiquantitative evaluation, glomerular injury scores were obtained from the kidney sections that were stained with PAS. For each kidney specimen, approximately 100 glomeruli were examined to obtain a glomerular injury score. Based on an earlier described method,22 each glomerulus was graded from 1 to 4 as follows: Grade 1, normal glomerulus identified by light microscopy; Grade 2, involvement of sclerosis in up to one-third of the glomerulus; Grade 3, involvement of sclerosis in one- to two-thirds of the glomerulus; and Grade 4, two-thirds involvement or global sclerosis. The extent of glomerular injury is expressed as the glomerular injury index. Glomerular injury in the outer cortical and juxtamedullary areas of the kidney was also evaluated and compared. The tubular area that contains proteinaceous casts was determined at a magnification of × 100 to assess tubular damage using image-analysing software (NIS Elements AR version 3.0; Nikon Instruments, Melville, NY, USA). Renal tubular cast formation is characterized by proteinuria and is an important hallmark of a number of pathological conditions.23,24 The percentage area positive for casts was calculated from the mean of eight cortical and five medullary fields (× 100) for each animal.
Renal microvascular measurements
Renal microvascular experiments were conducted in vitro at the end of the 4 week experimental period using the juxtamedullary nephron preparation. As described previously,25–27 rats were anaesthetized with pentobarbital sodium (65 mg/kg, i.p.). The right kidney was isolated and after a midline laparotomy, the right renal artery was cannulated through the superior mesenteric artery and the kidney was perfused immediately with Tyrode’s solution containing 6% albumin and a mixture of L-amino acids. Once the microdissection procedures were completed, the renal artery perfusion pressure was set to 100 mmHg. The tissue surface was superfused continuously with Tyrode’s solution containing 1% albumin. After a 20 min equilibration period, an afferent arteriole was chosen for study and baseline diameter was measured. The afferent arteriole was exposed to increasing concentrations of ATP and changes in diameter were monitored for 5 min at each concentration. Arteriolar diameters were recorded at 20 s intervals and the steady state diameters were calculated from the average of all diameter measurements obtained during the final 2 min of each 5 min treatment period.
Statistical analysis
All data are presented as the mean ± SEM. Statistical significance was determined using one-way ANOVA followed by Tukey’s post hoc test to identify differences between the experimental groups. Differences between groups were considered significant when P < 0.05, where the critical value of P was two-sided. Analyses were performed using GRAPHPAD Prism Version 4.0 (GraphPad Software, La Jolla, CA, USA).
RESULTS
Blood pressure
Systolic blood pressure, as measured by radiotelemetry, increased over the first 2 weeks of sorafenib administration and averaged 168 ± 4 mmHg, compared with 119 ± 3 mmHg in the control group. The elevated blood pressure was maintained until the end of the 4 week sorafenib administration period (182 ± 6 vs 123 ± 3 mmHg in the sorafenib and control groups, respectively).
In a separate series of experiments in which blood pressure was measured by tail-cuff plethysmography, sorafenib administration significantly elevated SBP compared with that in the control group (P < 0.05). Captopril treatment significantly attenuated the sorafenib-induced increase in SBP (P < 0.05; Fig. 1).
Fig. 1.

Systolic blood pressure in control (vehicle-treated; △), sorafenib-treated (□) and sorafenib + captopril-treated (○) rats. Rats were treated over a period of 4 weeks. Systolic blood pressure was measured by tail-cuff plethysmography. Data are the mean ± SEM (n = 6 in each group). *P < 0.05 compared with vehicle control; †P < 0.05 compared with the sorafenib-treated group (one-way ANOVA followed by Tukey’s post hoc test).
Renal damage
Renal injury was determined by measuring urinary albumin and nephrin excretion in addition to histological evaluation. Sorafenib administration resulted in significant albuminuria at 2 weeks and albumin excretion increased fourfold at 4 weeks. Captopril treatment significantly attenuated the sorafenib-induced increase in albumin excretion by approximately 50% at 2 and 4 weeks (Fig. 2a). Sorafenib administration also resulted in significantly higher nephrin excretion at 4 weeks compared with the control group. Captopril treatment resulted in an eightfold reduction in nephrin excretion in sorafenib-treated rats (Fig. 2b).
Fig. 2.
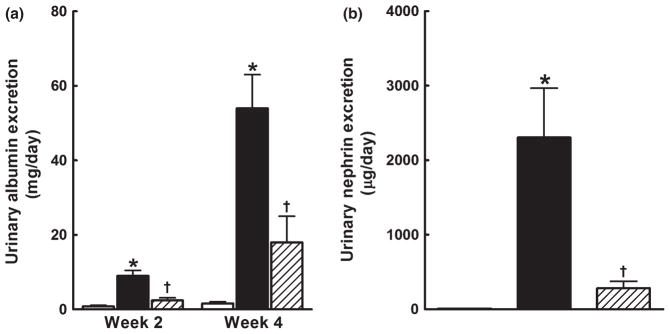
(a) Urinary albumin and (b) nephrin excretion in control (vehicle-treated;□), sorafenib-treated (■) and sorafenib + captopril-treated (▨) rats. Rats were treated over a period of 4 weeks. Progressively elevated albuminuria is seen in sorafenib-compared with vehicle-treated rats group (a). Captopril slowed the progression of albuminuria in the sorafenib-treated rats. Captopril treatment also significantly attenuated nephrinuria in sorafenib-treated rats (b). Data are the mean ± SEM (n = 5–6). *P < 0.05 compared with vehicle control; †P < 0.05 compared with the sorafenib-treated group (one-way ANOVA followed by Tukey’s post hoc test).
Renal injury in the groups was further assessed by histopathological examination. Sorafenib caused renal injury, as evident from the significantly higher glomerular injury index in the sorafenib group compared with that in the control group (P < 0.05). Captopril treatment protected the kidney against sorafenib-induced renal injury and decreased the glomerular injury index by 50% (Fig. 3a–g). Comparison of juxtamedullary and outer cortical glomeruli revealed greater sorafenib-induced injury in the juxtamedullary glomeruli (glomerular injury index 2.8 ± 0.2 vs 1.7 ± 0.3, respectively; P < 0.05). Compared with sorafenib treatment alone, concomitant captopril treatment significantly reduced glomerular injury to a similar extent in both the outer cortical (1.7 ± 0.3 vs 0.9 ± 0.2, respectively; P < 0.05) and juxtamedullary (2.8 ± 0.2 vs 1.8 ± 0.3, respectively; P < 0.05) regions. In addition, sorafenib administration resulted in the significant formation of intratubular proteinaceous casts in the cortical and medullary areas of the kidney. Captopril treatment protected the kidney against sorafenib-induced damage and significantly decreased cortical and medullary cast formation (Figs 4,5).
Fig. 3.

(a–f) Representative photomicrographs from (a, d) control (vehicle-treated), (b, e) sorafenib- and (c, f) sorafenib + captopril-treated rats showing that sorafenib-treated rats develop glomerular damage in the juxtamedullary area and that concurrent treatment with captopril attenuates this damage. (a–c) Periodic acid-Schiff (PAS) staining at low magnification (original magnification × 100); (d–f) PAS staining at higher magnification showing the glomeruli (original magnification × 400). Glomeruli from control group are almost intact (d), whereas those in the sorafenib-treated group (e) exhibit marked mesangial expansion and thrombosis (arrow), as well as fragmentation of erythrocytes (arrowhead). Glomeruli from the sorafenib + captopril-treated group exhibit decreased mesangial expansion (f). (g) Glomerular injury scores in the control (□), sorafenib-treated (■) and sorafenib + captopril-treated (▨) groups. Sorafenib-treated rats have a higher glomerular injury score than rats in the control group and this increased score was markedly attenuated by concomitant treatment with captopril. Data are the mean ± SEM (n = 4–6). *P < 0.05 compared with the vehicle control group; †P < 0.05 compared with the sorafenib-treated group (one-way ANOVA followed by Tukey’s post hoc test).
Fig. 4.

Kidney cortical cast formation in control (□), sorafenib-treated (■) and sorafenib + captopril-treated (▨) rats after 4 weeks treatment. The number of tubules containing proteinaceous casts was determined at × 100 magnification using Masson’s trichrome staining. The percentage area positive for casts was calculated from the mean of eight cortical fields (at × 100) for each animal. Data are the mean ± SEM (n = 5–6). *P < 0.05 compared with vehicle control; †P < 0.05 compared with the sorafenib-treated group (one-way ANOVA followed by Tukey’s post hoc test). (b–d) Representative photomicrographs from rats in the vehicle- (b), sorafenib- (c) and sorafenib + captopril (d)-treated groups.
Fig. 5.

Kidney medullary cast formation in control (□), sorafenib-treated (■) and sorafenib + captopril-treated (▨) rats after 4 weeks treatment. The number of tubules containing proteinaceous casts was determined at × 100 magnification using Masson’s trichrome staining. The percentage area positive for casts was calculated from the mean of five medullary fields (at × 100) for each animal. Data are the mean ± SEM (n = 5–6). *P < 0.05 compared with vehicle control; †P < 0.05 compared with the sorafenib-treated group (one-way ANOVA followed by Tukey’s post hoc test). (b–d) Representative photomicrographs from rats in the vehicle- (b), sorafenib- (c) and sorafenib + captopril (d)-treated groups.
Afferent arteriolar response to ATP
Afferent arterioles are the main vascular elements responsible for the pressure-mediated autoregulatory adjustments of pre-glomerular resistance that regulate renal blood flow and glomerular filtration rate; ATP is the important signalling molecule that regulates afferent arteriolar tone and mediates renal autoregulatorybehaviour.27 In the present study, we evaluated the renal afferent arteriolar response to ATP as an index of renal autoregulatory capacity. Basal arteriolar diameters were similar across the groups and averaged 16.4 ± 3.6 μm. As shown in Fig. 6, superfusion with ATP concentration-dependently reduced arteriolar diameter in the control group, with afferent arteriolar diameter decreasing by 22% in response to 100 μmol/L ATP. Afferent arteriolar responses to ATP were significantly blunted in rats administered sorafenib, with afferent arteriolar diameter decreasing by only 11% in response to 100 μmol/L ATP. These findings indicate that sorafenib administration impairs renal autoregulatory capacity. The afferent arteriolar response to ATP was partially restored in the sorafenib + captopril-treated group. These findings indicate that captopril treatment improves renal autoregulatory capacity in rats administered sorafenib.
Fig. 6.

Effect of chronic (4 weeks) sorafenib treatment alone (□) or in combination with captopril (△) on afferent arteriolar responses to ATP. (○), vehicle-treated group. The percentage change in afferent arteriolar diameter to ATP (0.1–100 μmol/L) is shown and is expressed as the percentage of the control diameter at 100 mmHg. Data are the mean ± SEM (n = 6 in each group). *P < 0.05 compared with vehicle control; †P < 0.05 compared with the sorafenib-treated group (one-way ANOVA followed by Tukey’s post hoc test).
DISCUSSION
The advent of angiogenesis inhibition in the treatment of cancer by blocking VEGF signalling has revolutionized the care of cancer patients.1,3,6,7 Although VEGFi offer excellent therapeutic options in treating a wide range of cancers, increased microvascular resistance and vascular regression leading to hypertension and other cardiovascular complications have been found to be associated with these drugs, seriously compromising their safe and effective use. Hypertension and renal injury are the most frequent comorbid conditions found in cancer patients treated with VEGFi-based therapies that cause systemic inhibition of VEGF signalling.1,5,6,8–14 The present study evaluated the VEGFi sorafenib, which also inhibits other tyrosine kinases and RAFs.1–4,11 In the present study, we demonstrated the development of hypertension, renal injury and impaired renal afferent arteriolar autoregulatory capacity during chronic administration of sorafenib, a widely used VEGFi, to rats.
The recognition and management of hypertension is an important issue in patients receiving anti-VEGF therapy because poorly controlled hypertension leads to serious cardiovascular consequences.6–12 The mechanism of action by which angiogenic inhibitors, including VEGFi, cause hypertension is not known.17,28 Recent experimental studies indicate a role for endothelin in VEGFi-related hypertension.29,30 A number of antihypertensive drugs are used in the management of hypertension that is frequently observed in VEGFi-treated cancer patients. Clinical trials on anti-VEGF-based cancer therapy have suggested that the use of drugs that inhibit the RAS, particularly ACEi, is the most effective and these agents are widely used in the management of VEGFi-related hypertension.13–17
Apart from the antihypertensive action of ACEi, numerous studies have demonstrated that ACEi also provide organ protection independent of their blood pressure-lowering effects in several different pathological conditions.31–34 Currently, the kidney-protective effects of ACEi in cancer patients receiving VEGFi have not been evaluated thoroughly. In this regard, a number of studies have demonstrated that cancer patients receiving anti-VEGF-based chemotherapy frequently develop renal injury.1,6–12 For instance, bevacizumab, which is a humanized monoclonal anti-VEGF antibody, causes renal injury in humans when used alone or in combination with other anti-VEGF, such as the tyro-sine kinase inhibitors sunitinib and sorafenib.1,6–12 Apart from the clinical studies, VEGFi-mediated renal injury has also been reported in animal studies. Rats treated with the strong VEGFi cediranib developed hypertension and proteinuria.35 In another study, the VEGFR inhibitor SU5416 enhanced dietary salt-induced hypertension and renal injury in rats.36 The renal injury in this model was characterized by marked proteinuria and albuminuria along with glomerulosclerosis.37 The findings of the present study demonstrate albuminuria, nephrinuria, microangiopathic and glomerular, as well as tubular damage evident from proteinaceous tubular cast formation, in rats administered sorafenib. We also observed greater glomerular injury in the juxtamedullary compared with outer cortical region in rats administered sorafenib. Indeed, there is experimental evidence that juxtamedullary glomerular injury occurs at an earlier time point and is more severe than superficial glomerular injury.38,39 Overall, the data obtained in the present study clearly demonstrate that captopril treatment markedly decreases the glomerular barrier injury, glomerular and tubular structural injury and renal vascular dysfunction that occur with sorafenib administration.
The mechanisms underlying the renal injury related to VEGF inhibition seen in the present study and in a large number of earlier clinical and animal studies are related to attenuated or dysfunctional VEGF signalling. In support of this notion, dysregulation of the VEGF signalling system has been identified in a wide variety of renal diseases, such as diabetic nephropathy, puromycin animo-nucleoside nephrosis and glomerulonephritis.40,41 In a mouse folic acid nephropathy model, peritubular capillary loss and tubular atrophy were correlated with the downregulation of VEGF signalling.42 It has also been reported that homozygous deletion of VEGF-A in the glomeruli results in perinatal lethality, whereas heterozygous deletion of renal VEGF-A causes end-stage kidney failure in mice.43 The findings of the present study provide additional evidence demonstrating that sorafenib treatment results in impaired renal autoregulatory capacity and glomerular and renal tubular injury.
The findings of the present study demonstrate that captopril treatment decreases sorafenib-induced glomerular and renal tubular injury. The ability of captopril to attenuate anti-VEGF therapy-related renal injury appears to be due to its protective effects on the glomerular filtration barrier. An integral component of this glomerular filtration barrier is a protein called nephrin. Nephrin is a transmembrane protein that is a structural component of the slit diaphragm. Recent evidence suggests that nephrin plays a key role in the function of the glomerular filtration barrier.44 Decreased glomerular nephrin levels are one of the mechanisms contributing to proteinuria and subsequent glomerulosclerosis in different pathological conditions, including hypertension.45,46 It has been demonstrated that ACEi induce re-expression of nephrin in diabetic nephropathy and improve endothelial function and microcirculatory density.47,48 In the present study, we observed marked nephrinuria associated with VEGFi administration that was attenuated by concomitant treatment with captopril.
Captopril treatment improved renal autoregulatory capacity during chronic VEGF inhibition and is one potential mechanism by which captopril decreases renal injury. Renal autoregulatory capacity is important for kidney function because pressure-mediated autoregulatory behaviour operates by recognizing a change in the renal perfusion pressure and initiates appropriate changes in the autoregulatory resistance to stabilize renal blood flow and glomerular filtration rate over a wide range of renal perfusion pressures.49 Renal autoregulatory capacity is impaired in pathological settings, such as hypertension, and the loss of autoregulatory efficiency could lead to glomerular stress, glomerular compensation and glomerular injury.50,51 Indeed, angiotensin II-induced hypertension exhibits attenuated renal autoregulatory capacity, as revealed by reduced pressure-mediated afferent arteriolar vasoconstriction followed by glomerular injury.52 Afferent arteriolar responses to purinoceptor stimulation by ATP were significantly attenuated in the kidneys of angiotensin II hypertensive animals.53 With this background, we studied the afferent arteriolar responses to purinoceptor stimulation by ATP as an index of renal autoregulatory capacity during VEGF inhibition. The impaired renal autoregulatory capacity caused by VEGF inhibition observed in the present study could result in enhanced transmission of the blood pressure to the glomerulus to increase glomerular pressure, resulting in injury. Thus, it can be suggested that improved renal autoregulatory capacity is an important mechanism contributing to the decrease in sorafenib-induced glomerular injury in rats treated with captopril.
In conclusion, we have demonstrated that administration of the anticancer VEGFi sorafenib to rats caused hypertension and renal injury. We further demonstrated that treatment with the ACEi captopril not only attenuated sorafenib-induced hypertension, but also decreased glomerular injury. Finally, we provide evidence that the mechanism by which captopril attenuated sorafenib-induced renal injury involved improvement of renal autoregulatory capacity.
References
- 1.Vaklavas C, Lenihan D, Kurzrock R, Tsimberidou AM. Anti-vascular endothelial growth factor therapies and cardiovascular toxicity: What are the important clinical markers to target? Oncologist. 2010;15:130–41. doi: 10.1634/theoncologist.2009-0252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Blume-Jensen P, Hunter T. Oncogenic kinase signaling. Nature. 2001;411:355–65. doi: 10.1038/35077225. [DOI] [PubMed] [Google Scholar]
- 3.Presta LG, Chen H, O’Connor SJ, et al. Humanization of an anti-vascular endothelial growth factor monoclonal antibody for the therapy of solid tumors and other disorders. Cancer Res. 1997;57:4593–9. [PubMed] [Google Scholar]
- 4.Neufeld G, Cohen T, Gengrinovitch S, Poltorak Z. Vascular endothelial growth factor (VEGF) and its receptors. FASEB J. 1999;13:9–22. [PubMed] [Google Scholar]
- 5.Mourad JJ, des Guetz G, Debbabi H, Levy BI. Blood pressure rise following angiogenesis inhibition by bevacizumab. A crucial role for microcirculation. Ann Oncol. 2008;19:927–34. doi: 10.1093/annonc/mdm550. [DOI] [PubMed] [Google Scholar]
- 6.Herbst RS. Toxicities of anti-angiogenic therapy in non-small-cell lung cancer. Clin Lung Cancer. 2006;8(Suppl):S23–30. doi: 10.3816/clc.2006.s.010. [DOI] [PubMed] [Google Scholar]
- 7.Faivre S, Sablin MP, Dreyer C, Raymond E. Novel anticancer agents in clinical trials for well-differentiated neuroendocrine tumors. Endocrinol Metab Clin North Am. 2010;39:811–26. doi: 10.1016/j.ecl.2010.09.006. [DOI] [PubMed] [Google Scholar]
- 8.Gurevich F, Perazella MA. Renal effects of anti-angiogenesis therapy: Update for the internist. Am J Med. 2009;122:322–8. doi: 10.1016/j.amjmed.2008.11.025. [DOI] [PubMed] [Google Scholar]
- 9.Okuno Y, Kume H, Hosoda C, Homma Y. Development of nephrotic syndrome after administration of sorafenib in a case of metastatic renal cell carcinoma. Case Rep Med. 2011;2011:710216. doi: 10.1155/2011/710216. Epub 5 October 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Van Heeckeren WJ, Ortiz J, Cooney MM, Remick SC. Hypertension, proteinuria, and antagonism of vascular endothelial growth factor signaling: Clinical toxicity, therapeutic target, or novel bio-marker? J Clin Oncol. 2007;25:2993–5. doi: 10.1200/JCO.2007.11.5113. [DOI] [PubMed] [Google Scholar]
- 11.Wilhelm S, Chien DS. BAY 43-9006: Preclinical data. Curr Pharm Des. 2002;8:2255–7. doi: 10.2174/1381612023393026. [DOI] [PubMed] [Google Scholar]
- 12.Chu D, Lacouture ME, Fillos T, Wu S. Risk of hand–foot skin reaction with sorafenib: A systematic review and meta-analysis. Acta Oncol. 2008;47:176–86. doi: 10.1080/02841860701765675. [DOI] [PubMed] [Google Scholar]
- 13.Gupta R, Maitland ML. Sunitinib, hypertension, and heart failure: A model for kinase inhibitor-mediated cardiotoxicity. Curr Hypertens Rep. 2011;13:430–5. doi: 10.1007/s11906-011-0229-4. [DOI] [PubMed] [Google Scholar]
- 14.Escalante CP, Zalpour A. Vascular endothelial growth factor inhibitor-induced hypertension: Basics for primary care providers. Cardiol Res Pract. 2011;2011:816897. doi: 10.4061/2011/816897. Epub 10 May 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Langenberg MH, Van Herpen CM, De Bono J, et al. Effective strategies for management of hypertension after vascular endothelial growth factor signaling inhibition therapy. Results from a Phase II randomized, factorial, double-blind study of Cediranib in patients with advanced solid tumors. J Clin Oncol. 2009;27:6152–9. doi: 10.1200/JCO.2009.22.2273. [DOI] [PubMed] [Google Scholar]
- 16.Pande A, Lombardo J, Spangenthal E, Javle M. Hypertension secondary to anti-angiogenic therapy: Experience with bevacizumab. Anticancer Res. 2007;27:3465–70. [PubMed] [Google Scholar]
- 17.Izzedine H, Ederhy S, Goldwasser F, et al. Management of hypertension in angiogenesis inhibitor-treated patients. Ann Oncol. 2009;20:807–15. doi: 10.1093/annonc/mdn713. [DOI] [PubMed] [Google Scholar]
- 18.Park YH, Roh SY, Lee YC. Effect of sorafenib on experimental choroidal neovascularization in the rat. Clin Exp Ophthalmol. 2010;38:718–26. doi: 10.1111/j.1442-9071.2010.02328.x. [DOI] [PubMed] [Google Scholar]
- 19.Ernsberger P, Johnson JL, Rosenthal T, Mirelman D, Koletsky RJ. Therapeutic actions of allylmercaptocaptopril and captopril in a rat model of metabolic syndrome. Am J Hypertens. 2007;20:866–74. doi: 10.1016/j.amjhyper.2007.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Swislocki AL, LaPier TL, Khuu DT, Fann KY, Tait M, Rodnick KJ. Metabolic, hemodynamic, and cardiac effects of captopril in young, spontaneously hypertensive rats. Am J Hypertens. 1999;12:581–9. doi: 10.1016/s0895-7061(99)00012-6. [DOI] [PubMed] [Google Scholar]
- 21.Gu FM, Li QL, Gao Q, et al. Sorafenib inhibits growth and metastasis of hepatocellular carcinoma by blocking STAT3. World J Gastroenterol. 2011;17:3922–32. doi: 10.3748/wjg.v17.i34.3922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pèrez De Lema G, De Wit C, Cohen CD, et al. Angiotensin inhibition reduces glomerular damage and renal chemokine expression in MRL/lpr mice. J Pharmacol Exp Ther. 2003;307:275–81. doi: 10.1124/jpet.103.053678. [DOI] [PubMed] [Google Scholar]
- 23.Turoni CM, Reynoso HA, Maraòûn RO, Coviello A, Peral de Bruno M. Structural changes in the renal vasculature in streptozotocin-induced diabetic rats without hypertension. Nephron Physiol. 2005;99:50–7. doi: 10.1159/000083136. [DOI] [PubMed] [Google Scholar]
- 24.Bowers MC, Katki KA, Rao A, et al. Role of calcitonin gene-related peptide in hypertension-induced renal damage. Hypertension. 2005;46:51–7. doi: 10.1161/01.HYP.0000168926.44648.ed. [DOI] [PubMed] [Google Scholar]
- 25.Imig JD, Dimitropoulou C, Reddy DS, White RE, Falck JR. Afferent arteriolar dilation to 11,12-EET analogs involves PP2A activity and Ca2+-activated K+ channels. Microcirculation. 2008;15:137–50. doi: 10.1080/10739680701456960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Imig JD, Pham BT, LeBlanc EA, Reddy KM, Falck JR, Inscho EW. Cytochrome P450 and cyclooxygenase metabolites contribute to the endothelin-1 afferent arteriolar vasoconstrictor and calcium responses. Hypertension. 2000;35:307–12. doi: 10.1161/01.hyp.35.1.307. [DOI] [PubMed] [Google Scholar]
- 27.Guan Z, Inscho EW. Role of adenosine 5′-triphosphate in regulating renal microvascular function and in hypertension. Hypertension. 2011;58:333–40. doi: 10.1161/HYPERTENSIONAHA.110.155952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Veronese ML, Mosenkis A, Flaherty KT, et al. Mechanisms of hypertension associated with BAY 43-9006. J Clin Oncol. 2006;24:1363–9. doi: 10.1200/JCO.2005.02.0503. [DOI] [PubMed] [Google Scholar]
- 29.Kappers MH, Smedts FM, Horn T, et al. The vascular endothelial growth factor receptor inhibitor sunitinib causes a preeclampsia-like syndrome with activation of the endothelin system. Hypertension. 2011;58:295–302. doi: 10.1161/HYPERTENSIONAHA.111.173559. [DOI] [PubMed] [Google Scholar]
- 30.Kappers MH, de Beer VJ, Zhou Z, et al. Sunitinib-induced systemic vasoconstriction in swine is endothelin mediated and does not involve nitric oxide or oxidative stress. Hypertension. 2012;59:151–7. doi: 10.1161/HYPERTENSIONAHA.111.182220. [DOI] [PubMed] [Google Scholar]
- 31.Chen SX, Song T, Zhou SH, Liu YH, Wu SJ, Liu LY. Protective effects of ACE inhibitors on vascular endothelial dysfunction induced by exogenous advanced oxidation protein products in rats. Eur J Pharmacol. 2008;584:368–75. doi: 10.1016/j.ejphar.2008.02.020. [DOI] [PubMed] [Google Scholar]
- 32.Liu XP, Pang YJ, Zhu WW, et al. Benazepril, an angiotensin-converting enzyme inhibitor, alleviates renal injury in spontaneously hypertensive rats by inhibiting advanced glycation end-product-mediated pathways. Clin Exp Pharmacol Physiol. 2009;36:287–96. doi: 10.1111/j.1440-1681.2008.05078.x. [DOI] [PubMed] [Google Scholar]
- 33.Brower GL, Levick SP, Janicki JS. Inhibition of matrix metalloproteinase activity by ACE inhibitors prevents left ventricular remodeling in a rat model of heart failure. Am J Physiol Heart Circ Physiol. 2007;292:H3057–64. doi: 10.1152/ajpheart.00447.2006. [DOI] [PubMed] [Google Scholar]
- 34.Yamamoto D, Takai S, Jin D, Inagaki S, Tanaka K, Miyazaki M. Molecular mechanism of imidapril for cardiovascular protection via inhibition of MMP-9. J Mol Cell Cardiol. 2007;43:670–6. doi: 10.1016/j.yjmcc.2007.08.002. [DOI] [PubMed] [Google Scholar]
- 35.Curwen JO, Musgrove HL, Kendrew J, Richmond GH, Ogilvie DJ, Wedge SR. Inhibition of vascular endothelial growth factor-A signaling induces hypertension: Examining the effect of cediranib (recentin; AZD2171) treatment on blood pressure in rat and the use of concomitant anti-hypertensive therapy. Clin Cancer Res. 2008;14:3124–31. doi: 10.1158/1078-0432.CCR-07-4783. [DOI] [PubMed] [Google Scholar]
- 36.Gu JW, Manning RD, Jr, Young E, Shparago M, Sartin B, Bailey AP. Vascular endothelial growth factor receptor inhibitor enhances dietary salt-induced hypertension in Sprague-Dawley rats. Am J Physiol Regul Integr Comp Physiol. 2009;297:R142–8. doi: 10.1152/ajpregu.90972.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mori T, Cowley AW., Jr Role of pressure in angiotensin II-induced renal injury: Chronic servo-control of renal perfusion pressure in rats. Hypertension. 2004;43:752–9. doi: 10.1161/01.HYP.0000120971.49659.6a. [DOI] [PubMed] [Google Scholar]
- 38.Ihara G, Kiyomoto H, Kobori H, et al. Regression of superficial glomerular podocyte injury in type 2 diabetic rats with overt albuminuria: Effect of angiotensin II blockade. J Hypertens. 2010;28:2289–98. doi: 10.1097/HJH.0b013e32833dfcda. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mori T, Cowley AW, Jr, Ito S. Molecular mechanisms and therapeutic strategies of chronic renal injury. Physiological role of angiotensin II-induced oxidative stress in renal medulla. J Pharmacol Sci. 2006;100:2–8. doi: 10.1254/jphs.fmj05003x2. [DOI] [PubMed] [Google Scholar]
- 40.Honkanen EO, Teppo AM, Gronhagen-Riska C. Decreased urinary excretion of vascular endothelial growth factor in idiopathic membranous glomerulo-nephritis. Kidney Int. 2000;57:2343–9. doi: 10.1046/j.1523-1755.2000.00094.x. [DOI] [PubMed] [Google Scholar]
- 41.Wolf G, Chen S, Ziyadeh F. From the periphery of the glomerular capillary wall toward the center of disease. Podocyte injury comes of age in diabetic nephropathy. Diabetes. 2005;54:1626–34. doi: 10.2337/diabetes.54.6.1626. [DOI] [PubMed] [Google Scholar]
- 42.Yuan HT, Li XZ, Pitera JE, Long DA, Woolf AS. Peritubular capillary loss after mouse acute nephrotoxicity correlates with down-regulation of vascular endothelial growth factor-A and hypoxia inducible factor-1. Am J Pathol. 2003;163:2289–301. doi: 10.1016/s0002-9440(10)63586-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Eremina V, Sood M, Haigh J, et al. Glomerular-specific alterations of VEGF-A expression lead to distinct congenital and acquired renal disease. J Clin Invest. 2003;111:707–16. doi: 10.1172/JCI17423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hauser PV, Collino F, Bussolati B, Camussi G. Nephrin and endothelial injury. Curr Opin Nephrol Hypertens. 2009;18:3–8. doi: 10.1097/MNH.0b013e32831a4713. [DOI] [PubMed] [Google Scholar]
- 45.Knight SF, Quigley JE, Yuan J, Roy SS, Elmarakby A, Imig JD. Endothelial dysfunction and the development of renal injury in spontaneously hypertensive rats fed a high-fat diet. Hypertension. 2008;51:352–9. doi: 10.1161/HYPERTENSIONAHA.107.099499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dai HY, Zheng M, Tang RN, et al. Effects of angiotensin receptor blocker on phenotypic alterations of podocytes in early diabetic nephropathy. Am J Med Sci. 2011;341:207–14. doi: 10.1097/MAJ.0b013e3182010da9. [DOI] [PubMed] [Google Scholar]
- 47.Agabiti-Rosei E. Structural and functional changes of the microcirculation in hypertension: Influence of pharmacological therapy. Drugs. 2003;63:19–29. [PubMed] [Google Scholar]
- 48.Sugimoto H, Hamano Y, Charytan D, et al. Neutralization of circulating vascular endothelial growth factor (VEGF) by anti-VEGF antibodies and soluble VEGF receptor 1 (sFlt-1) induces proteinuria. J Biol Chem. 2003;278:12605–8. doi: 10.1074/jbc.C300012200. [DOI] [PubMed] [Google Scholar]
- 49.Kitakaze M, Node K, Minamino T, et al. Inhibition of angiotensin-converting enzyme increases the nitric oxide levels in canine ischemic myocardium. J Mol Cell Cardiol. 1998;30:2461–6. doi: 10.1006/jmcc.1998.0806. [DOI] [PubMed] [Google Scholar]
- 50.Persson PB. Renal blood flow auto-regulation in blood pressure control. Curr Opin Nephrol Hypertens. 2002;11:67–72. doi: 10.1097/00041552-200201000-00010. [DOI] [PubMed] [Google Scholar]
- 51.Inscho EW, Carmines PK, Cook AK, Navar LG. Afferent arteriolar responsiveness to altered perfusion pressure in renal hypertension. Hypertension. 1990;15:748–52. doi: 10.1161/01.hyp.15.6.748. [DOI] [PubMed] [Google Scholar]
- 52.Inscho EW, Cook AK, Murzynowski JB, Imig JD. Elevated arterial pressure impairs autoregulation independently of AT(1) receptor activation. J Hypertens. 2004;22:811–8. doi: 10.1097/00004872-200404000-00025. [DOI] [PubMed] [Google Scholar]
- 53.Inscho EW, Imig JD, Deichmann PC, Cook AK. Candesartan cilexetil protects against loss of autoregulatory efficiency in angiotensin II-infused rats. J Am Soc Nephrol. 1999;10 (Suppl):S178–83. [PubMed] [Google Scholar]