

Abstract
Although glutamate receptor 1 (GluR1)-containing α-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate receptors (GluR1-AMPARs) are implicated in synaptic plasticity, it has yet to be demonstrated whether endogenous GluR1-AMPARs undergo activity-dependent trafficking in vivo to synapses to support short-term memory (STM) formation. The paradigm of pavlovian fear conditioning (FC) can be used to address this question, because a discrete region—the lateral amygdala (LA)—has been shown unambiguously to be necessary for the formation of the associative memory between a neutral stimulus (tone [CS]) and a noxious stimulus (foot shock [US]). Acquisition of STM for FC can occur even in the presence of protein synthesis inhibitors, indicating that redistribution of pre-existing molecules to synaptic junctions underlies STM. We employed electron microscopic immunocytochemistry to evaluate alterations in the distribution of endogenous AMPAR subunits at LA synapses during the STM phase of FC. Rats were sacrificed 40 minutes following three CS-US pairings. In the LA of paired animals, relative to naïve animals, the proportion of GluR1-AMPAR-labeled synapses increased 99% at spines and 167% in shafts. In the LA of unpaired rats, for which the CS was never associated with the US, GluR1 immunoreactivity decreased 84% at excitatory shaft synapses. GluR2/3 immunoreactivity at excitatory synapses did not change detectably following paired or unpaired conditioning. Thus, the early phase of FC involves rapid redistribution specifically of the GluR1-AMPARs to the postsynaptic membranes in the LA, together with the rapid translocation of GluR1-AMPARs from remote sites into the spine head cytoplasm, yielding behavior changes that are specific to stimulus contingencies.
INDEXING TERMS: receptor trafficking, synaptic plasticity, pavlovian, fear conditioning, auditory cue, conditioned inhibition, endogenous gene products, associative memory, STM, LTM, stimulus contingencies
Fear conditioning is a robust learning paradigm that provides an excellent ingress toward understanding the cellular and molecular mechanisms underlying memory formation. In the paradigm of fear conditioning, presentation of an originally neutral conditioned stimulus (CS) elicits a defensive response after being paired with an aversive stimulus (unconditioned stimulus [US]) (Blan-chard and Blanchard, 1969; Fanselow and Bolles, 1979). Although converging evidence over the past few decades suggests that the association between the CS and US occurs in the amygdala (Cain and LeDoux, 2008), descriptions of the subcellular and molecular events underlying the acquisition of fearful memory remains incomplete.
The amygdala, located deep within the medial temporal lobes of the brain, is comprised of a dozen or so nuclei (Pitkanen et al., 1997), of which the lateral (LA), basal, and central nuclei have been most convincingly linked to conditioned fear behaviors (Davis and Shi, 1999; Fendt and Fanselow, 1999; LeDoux, 2000; Maren, 2005; Sah et al., 2008). The dorsal tip of the LA is the first site where sensory information representing the US and the CS from cortical and thalamic regions converge (Quirk et al., 1996). Both of these inputs are excitatory in nature, utilizing α-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate receptors (AMPARs) and N-methyl-D-aspartic acid receptors (NMDARs) expressed by LA neurons (Farb et al., 1995; Farb and LeDoux, 1997, 1999; Mahanty and Sah, 1998; Radley et al., 2007; Woodson et al., 2000)
There is compelling evidence that synaptic plasticity in the LA involves NMDAR activation (Blair et al., 2001), followed by the insertion of AMPARs into synapses. The molecular mechanism underlying fear conditioning may be similar to that of hippocampal slice long-term potentiation (LTP), in that the exogenous green fluorescent protein (GFP)-tagged glutamate receptor 1 (GluR1)-containing AMPARs increase at thalamo-LA synapses following fear conditioning (Rumpel et al., 2005; Yeh et al., 2006), as do the hippocampal synapses that receive the LTP-inducing tetanus (Shi et al., 1999). Seminal studies on hippocampal neurons have shown that the mechanism regulating the trafficking of GluR1-containing AMPARs to the synapses is distinct from and overrides the presence of GluR2 subunits in the heteromer. Specifically, the GluR1-containing AMPARs (but not the GluR2-containing/GluR1-lacking) AMPARs require an LTP-inducing tetanus or activation of protein kinases as well as the interactions of the C-terminal tail of AMPARs with the PDZ domains (“PDZ” derives from the proteins PSD-95, Dlg, and ZO1, which contain the domain) of anchoring proteins at excitatory postsynaptic membranes (Hayashi et al., 2000; Passafaro et al., 2001).
Although these studies used hippocampal neurons that were dissociated (Passafaro et al., 2001; Shi et al., 1999) or maintained within organotypic slices (Hayashi et al., 2000), the importance of GluR1-containing AMPARs in synaptic plasticity of the LA has been confirmed by demonstrating that knockout of the GluR1 subunit leads to the loss of LTP at thalamic inputs to the LA and auditory fear conditioning (Humeau et al., 2007). Additional support for the idea that an LTP-like process underlies fear conditioning in the LA comes from the observation that both in vitro and in vivo synaptic plasticity in the LA are sensitive to the same stimulus contingencies (Bauer et al., 2001) and similar biochemical mechanisms (Huang et al., 2000; Rodrigues et al., 2004b).
The features described above for the GluR1-containing AMPARs is likely to be the basis for the early rise of GluR1-containing AMPARs at tetanized synapses (Shi et al., 1999). However, it is not yet known whether the redistribution of endogenous GluR1-containing AMPARs occurs during the formation of short-term memory (STM). In order to examine this point, we used electron microscopic (EM) immunocytochemistry to analyze the synaptic and nonsynaptic distributions of endogenous GluR1 subunits at LA synapses during the STM phase of fear memory formation. Our results provide the first ultrastructural evidence that fear memory formation involves the rapid (<40 minutes after fear conditioning) redistribution of endogenous GluR1-containing AMPARs to the LA synapses and that this redistribution is sensitive to stimulus contingencies. Results from the pilot studies of this project were first presented at the Annual Meeting of the Society for Neuroscience in 2006 (Lázaro-Muñoz et al., 2006).
MATERIALS AND METHODS
Antibody information and chemicals
The rabbit anti-GluR1 subunit was purchased from Chemicon (now Millipore, Temecula, CA, cat. no. AB1504). The antibody is directed against the carboxy-terminus of the rat GluR1 subunit, corresponding to the amino acid sequence SHSSGMPLGATGL, and was generated by immunizing rabbits. The antibody has been shown to be compatible with the glutaraldehyde fixation condition required for ultrastructural preservation. Specificity of this antibody has been demonstrated previously by Western blot (manufacturer’s data sheet and Petralia and Wenthold, 1992; Wenthold et al., 1992), which shows that this antibody recognizes a single band corresponding to the molecular weight, ~108 kDa, of the GluR1 subunit and neither the GluR2/3 nor GluR4 subunits of AMPARs expressed by COS-7 cells transfected with the respective GluR-subunit cDNA.
The antibody directed against the GluR2/3 subunits of AMPARs was also purchased from Chemicon (now Millipore, cat. no. AB1506). This antibody is directed against the carboxy-terminus of the rat GluR2 and -3 subunits, corresponding to the amino acid sequence EGYNVYGIESVKI (amino acids 850–862), and was generated by immunizing rabbits. Specificity of this antibody was demonstrated by the same studies that characterized the GluR1 antibody, by using Western blot (manufacturer’s data sheet and Petralia and Wenthold, 1992; Wenthold et al., 1992) to show that this antibody recognizes a single band corresponding to the molecular weight, ~108 kDa, of the GluR2/3 subunits and neither the GluR1 nor the GluR4 subunits of AMPARs expressed by COS-7 cells transfected with the respective GluR-subunit cDNA. Previous EM studies (Petralia and Wenthold, 1992), including those from this laboratory (Farb et al., 1995; Farb and LeDoux, 1997, 1999; Levy and Aoki, 2002), had determined that these antibodies recognize asymmetric synapses but not the symmetric synapses of the amygdala and cortex. Asymmetric synapses, in turn, have been shown to be associated with excitatory synapses (Gray, 1959).
The secondary antibody was gold-conjugated goat anti-rabbit IgG; the colloidal gold particles were 10 nm in diameter (Aurion, EMS, Hatfield, PA, cat. no. 810.011).
The control peptides used to perform preadsorption control of the GluR2/3 antibody were purchased from Millipore (cat. no. AG305). The control peptides used to perform preadsorption control of the GluR1 antibody was also purchased from Millipore (cat. no. AG360).
Chemicals required for the osmium-free processing of tissue for EM (Phend et al., 1995) included glutaraldehyde, paraformaldehyde, tannic acid, p-phenylenediamine (PPND), uranyl acetate, and EMBED-812. These were purchased from EMS. Iridium tetrabromide (IrB4), another chemical required for osmium-free tissue processing, was purchased from Premion (Wardhill, MA, cat. no. 39493). Maleate buffer salts, Tris buffer salts, and Triton X-100 were purchased from Sigma (St. Louis, MO).
Behavioral training
All procedures for the care of animals were according to the NIH Guide for the Care and Use of Laboratory Animals, and also the guidelines approved by New York University’s Animal Care and Use Committee (Animal Welfare Assurance Number A3317-01).
All animals were adult male Sprague-Dawley rats weighing 300–350 g. Rats were fear conditioned according to the paradigm schematized in Figure 1. Training consisted of a 5-minute acclimation to the novel fear conditioning chamber (Rat Test Cage, Coulbourn Instruments, Allen-town, PA), followed by three presentations of the CS (tone, 30 seconds, 75dB, 5 kHz), paired with an aversive US (foot shock, 1 second, 0.8 mA, co-terminating with the CS) that were separated by 5-minute intertrial intervals. In contrast to these fear-conditioned animals that received the CS and US in a paired fashion, the control animals received the same CSs and USs but in an explicitly unpaired fashion (unpaired): tone CSs were delivered on the same schedule as the paired group (5-minute inter-tone intervals), but the shock USs were delivered, on average, 120 seconds before the CSs. Unpaired controls using similar protocols have repeatedly been shown to produce little or no associative fear of the CS (Cain and LeDoux, 2007).
Figure 1.
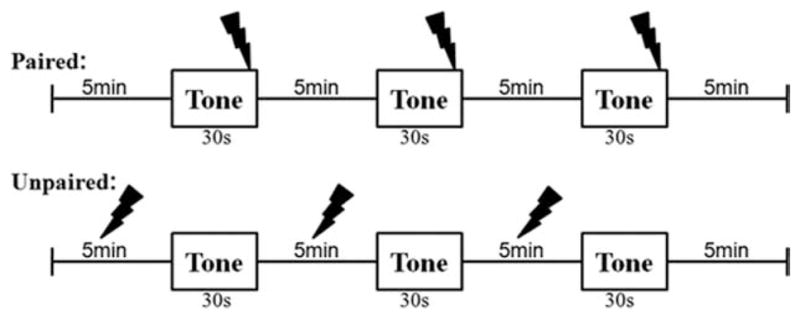
Schematic of the fear conditioning paradigm for the paired and unpaired rats.
Both treatment groups were returned to their home cage 5 minutes following the last CS presentation, and then anesthetized 40 minutes later, using Nembutal (50 mg/kg i.p.). A second group of controls, the naïves, received no stimuli and no experience in the chamber and were anesthetized within their home cage. Four animals were allocated to each treatment group. Forty minutes following the last presentation of the CS and US, rats were anesthetized by i.p. injection with Nembutal (50 mg/kg; Abbott Laboratories, North Chicago, IL), in preparation for euthanasia by transcardial perfusion with fixatives.
Preparation of brain tissue for electron microscopy
Once the depth of anesthesia was confirmed by the lack of reflex to a tail pinch or light touching of the cornea, animals were perfused with 0.1 M phosphate buffer (PB; pH 7.4) containing 4% paraformaldehyde and 0.1% glutaraldehyde at a rate of 50 ml/min during the first 3 minutes, followed by perfusion with the identical solution but at a rate of 20 ml/min for the succeeding 7 minutes. Brains were left in the skull for 1 hour post perfusion to allow in situ fixation to continue.
Forty-micrometer-thick Vibratome sections were collected from the dorsal LA corresponding to coronal planes of Bregma −2.25 to −3.75 mm. To prevent loss of antigenicity due to excess fixation, sections were treated with sodium borohydride (1%, in 0.1 M PB) at the 5th hour following transcardial perfusion. From this step on, brain sections were stored at 4°C, suspended in 0.01 M PB with 0.9% NaCl and 0.05% sodium azide (PBS-azide). Four days later, the Vibratome sections were postfixed with 2% glutaraldehyde for 10 minutes, then processed osmium-free (Phend et al., 1995), dehydrated, infiltrated with 100% EPON (EMBED-812 from EMS), and then placed between two sheets of Aclar plastic and incubated at 60°C overnight (24 hours) to cure the resin. All Vibratome sections were processed in parallel, so as to reduce interanimal and intertissue variability resulting from treatments.
Region analyzed for electron microscopy
All of the EM analyses examined the dorsal portion of the LA, where thalamic afferents innervate LA neurons most densely (Quirk et al., 1996). Prior to ultrathin sectioning, the position of the dorsal LA within Vibratome sections was identified based on landmarks visualized by light microscopy, by using the external capsule as the lateral border and the diagonally coursing lateral association bundle to delineate the dorsal and medial borders (Rodrigues et al., 2004b) (Fig. 2).
Figure 2.

Light micrographs depicting the dorsal tip of the lateral amygdala (LAd) within adult rat brain sections. B,D: Position of trapezoidal mesas chosen for ultrathin sectioning; photographed with a 5′ objective. A,B: Section immunolabeled for the GluR1 subunit of AMPA receptors (AMPARs). C,D: Section immunolabeled for the GluR2/3 subunits of AMPARs. GluR1 labeling is relatively weak within the LAd, compared with the immediately surrounding regions, which are the caudate-putamen nucleus (CP) and cortex (Ctx) dorsally, the amygdalostriatal transition area (AST) and central amygdala (CeA) medially, and the basal nucleus of the amygdala (BA) ventrally. Scale bar = 0.5 mm in A–D.
Electron microscopic immunocytochemistry
EM immunocytochemistry was performed to discriminate the synaptic versus nonsynaptic localization of GluR1 subunits forming the GluR1-containing AMPARs (summarized in Fig. 3), by using the postembedding immunogold labeling (PEG) procedure (Aoki et al., 2000). The same procedure was used to detect the synaptic versus nonsynaptic localization of GluR2 and -3 subunits forming AMPARs. The PEG procedure was as described by Phend et al. (1995). Ultrathin sections were collected on grids from all animals within a single 3-hour period, and then processed in parallel, so as to reduce interanimal and intertissue variability resulting from the immunocytochemical procedure. Seventy to 90-nm-thick ultrathin sections were collected on 200-mesh Formvar-coated nickel grids. We collected one to three ultrathin sections per grid and at least three grids per sample for GluR1 immunocytochemistry and at least three additional grids for GluR2/3 immunocytochemistry. Of these, at least one underwent a no-primary-antibody control of the PEG procedure (described in the next section), and the remaining grids served as duplicates to immunolabel for the GluR1 or the GluR2/3 subunits of AMPARs.
Figure 3.

Categorization of postembedding immunogold labeling (PEG) in relation to the postsynaptic density (PSD; gray fill) of asymmetric synapses. PEG particles positioned at the cleft, over the PSD or near the PSD (<60 nm from the postsynaptic membrane surface) were categorized as “postsynaptic.” PEG particles at positions removed from the postsynaptic membrane surface by more than 60 nm were all categorized as “nonsynaptic.” The “nonsynaptic” PEG particles included those residing in the spine neck, sometimes as much as a micrometer removed from the PSD. PEG particles residing along the plasma membrane or in the intracellular cytoplasm were included in the nonsynaptic category. The total number of PEG particles residing postsynaptically and nonsynaptically was categorized as “total spinous” for spine profiles.
The anti-GluR1 antibody or the anti-GluR2/3 antibody was used at a concentration of 1–14 μg/ml, diluted in saline buffered with 0.1 M Tris and containing 0.1% Triton X-100 and 0.3% NaCl, adjusted to pH 7.4 (TBST-7.4). The ultrathin sections mounted on Formvar-coated 200-mesh grids were incubated in the primary antibody within 5–8 hours after ultrathin sectioning. The incubation in the primary antibody was overnight (~14 hours) at room temperature. Grids were immersed in droplets of TBST-7.4 containing either the anti-GluR1 or the anti-GluR2/3 antibody. On the following day, the grids were rinsed in TBST-7.4, and then rinsed in TBST adjusted to pH 8.4 and with 0.9% NaCl (TBST-8.4) before being incubated for 1 hour at room temperature in TBST-8.4 and containing the secondary antibody at a dilution of 1:40. At the end of this incubation in the secondary antibody, grids were rinsed first in TBST-7.4 in 0.9% NaCl and then in water, and postfixed for 10 minutes at room temperature, by using 1% glutaraldehyde in water. At the end of the PEG procedure, grids were briefly counterstained with Reynold’s lead citrate (15 seconds).
Control experiments of electron microscopic immunocytochemistry
Two types of control experiments were conducted. One was the “no-primary-antibody” control, in which a subset of grids holding adjacent or nearly adjacent ultra-thin sections to those that were processed for GluR1 or GluR2/3 immunocytochemistry was incubated overnight in the identical TBST buffer (pH 7.4; NaCl concentration at 0.3%) but containing no primary antibody. Tissue from every animal underwent this control, every time the PEG experiment was run.
A subset of tissue obtained from naïve, paired, and unpaired conditions were processed for preadsorption control. For these, the GluR1 primary antibody was preadsorbed with a control peptide corresponding to the antigen used to generate the antibody. The original Western blot report on the nature of the AMPAR antibodies indicated that preadsorption of antibodies was successful at a control peptide concentration of 1 mg/ml (Wenthold et al., 1992). Therefore, the control peptide concentration was set to a value as close as possible to this original condition, namely, 800 μg/ml; lower concentrations of the control peptide were also tried. For all conditions, pre-adsorption was achieved by preincubating the antibody with the control peptide, together with 0.05% sodium azide, overnight at room temperature and for an additional hour at 30°C, before the antibody-peptide mixture was applied to grids.
Quantitative electron microscopic analysis
For all parts of the quantitative analysis, the researcher was kept blind to the treatment of the animal. Digital images were captured at a magnification of 50,000×, by making systematic sweeps within grid squares to avoid resampling of synaptic profiles. Within the two-dimensional digitized images, synapses were identified as asymmetric and thus presumably excitatory, based on the presence of thick postsynaptic densities (PSDs) aligned within the intracellular surfaces of plasma membranes juxtaposed by vesicle-filled axon terminals. So as to maximize randomness, we evaluated all synapses encountered, strictly in the order that they were encountered. Usually, at the magnification of 50,000×, we could not resolve PEG particles until the digitized images were averaged across multiple frames. This feature of image capturing allowed us to ensure that we did not preselect synapses to analyze or not analyze, based on immunoreactivity.
Immunolabels were categorized according to their position, relative to the PSD at asymmetric synaptic junctions. Those PEG particles occurring within the cleft were categorized as being in Zone I (“at cleft” in Fig. 3). The position of the immunolabel was categorized as Zone II (“at PSD” in Fig. 3), if the immunolabel was directly over the PSD and as Zone III (“near PSD” in Fig. 3), if the immunolabel was within a distance equal to the thickness of the PSD in its plane of section (<60 nm from the postsynaptic membrane surface) but not directly over the PSD. Zones I, II, and III were combined and considered “postsynaptic.” Labeling that occurred removed from the postsynaptic membrane surface by >60 nm but still within the cytoplasm or along the plasma membrane of the spine head or neck was categorized as Zone IV and considered to be nonsynaptic. PEG particles occurring across Zones I–IV of spines were considered “total” spinous labeling, determined by counting PEG particles at “synaptic” plus “nonsynaptic” zones. Figure 3 shows our schematic of the zones used for PEG categorization. Our synaptic Zones I–III correspond closely to Bins 1–3 and 5 of Yildirim et al. (2008), whereas our Zone IV overlaps mostly with Yildirim’s Bin 4.
PSD length measurements
In order to determine whether the changes in GluR1 or GluR2/3 immunoreactivity could result from the increased rate of detection due to the enlargement of PSD lengths, randomly encountered PSDs from each animal were subjected to PSD length measurements. The segmented line tool of ImageJ (1.37J, NIH) was used to measure PSD lengths. All images were digital, captured at a magnification of 50,000×, by using the same source of tissue that underwent GluR1- and GluR2/3-immunore-activity analysis.
Statistical analyses
Our design consisted of three treatment groups—paired, unpaired, and naïve—and four animals were assigned to each treatment group.
From each animal’s brain tissue, data were pooled from EM images captured across two grids, so as to avoid over-representation of data from any one grid. A minimum of 200 asymmetric synapses was sampled from the dorsal tip of the LA of each animal, and these were separated into two groups: axo-spinous synapses versus axo-shaft synapses. Typically, about 180 of the encountered synapses were on spines, and 12 or more of the asymmetric synapses were along the dendritic shafts. Because the number of asymmetric synapses occurring along dendritic shafts was small, the number of axo-shaft synapses analyzed per animal was kept constant at 12, by randomly sampling 6 of the synapses from one grid and randomly sampling 6 of the synapses from the second grid.
For each synapse on a spine, the number of PEG particles occurring synaptically and nonsynaptically was tallied. For shafts, only the PEG particles at postsynaptic sites were tallied, because nonsynaptic sites lacked clear boundaries. Synapses (axo-spinous or axo-shaft) and spines (spine head and neck) exhibiting one or more PEG particle were categorized as immunolabeled. Four mean values were derived for each animal: the proportion of spines immunolabeled anywhere (synaptic or nonsynaptic); the proportion of axo-spinous synapses immunolabeled at nonsynaptic sites; the proportion of spines labeled at synapses; and the proportion of axo-shaft synapses immunolabeled at the synapse. Treatment effects on the proportion of immunolabeling were analyzed by one-way ANOVA with planned post hoc comparisons (Fisher’s LSD).
In order to determine whether fear conditioning altered levels of receptors containing specific subunits of AMPARs, the mean number of PEG particles at synaptic and nonsynaptic sites was determined for each animal, and the mean values of each animal were compared across treatments. To avoid oversampling of data from any single animal’s tissue, the number of spines sampled from each animal was equalized to 164 for the GluR1 samples and 126 for the GluR2/3 samples. A one-way ANOVA and Fisher’s LSD post hoc comparisons were performed upon these data as well.
All data from the EM immunocytochemisty quantification were analyzed by using the software SPSS (version 13.0) and Statistica (version 6.0, released from StatSoft), while the analysts were kept blind to the treatment condition of each animal. Differences were accepted as significant for P < 0.05.
Light microscopic immunocytochemistry
In order to confirm that immunoreactivity of the amygdala to the GluR1 and GluR2/3 subunits was as observed previously (Farb et al., 1995), Vibratome sections of naïve animals were used to immunolabel the tissue by the avidin-biotin complex, by using Vector’s Elite kit for rabbit IgGs (Vector, Burlingame, CA, cat. no. PK-6200). Substrate for peroxidase was 0.3% 3,3′-diaminobenzidine HCl with 0.03% H2O2, and the buffer was 0.01 M phosphate buffer (pH 7.4) containing 0.9% NaCl.
Image production procedures
The digital EM images, first captured by using a Hamamatsu CCD camera and software produced by AMT, were adjusted with Adobe Photoshop (version 7.0; Adobe Systems, San Jose, CA). Light microscopic images were captured by using the Scientific Image Processing software (version 3.070, Scanalytics, Fairfax, VA). Contrast adjustments were performed for the purpose of matching the contrast and brightness of one digital image to another within a single figure. Adobe Photoshop was also used for cropping the image and for adding text and arrows. No dodging or copy-pasting of digital images was performed.
RESULTS
GluR1 and GluR2/3 immunoreactivities were considered synaptic when PEG particles were detected precisely over the PSD or within 60 nm from the postsynaptic membrane surface (black arrows in Fig. 4 and schematized in Fig. 3). PEG particles occurring within the synaptic cleft of asymmetric synaptic junctions were also included in the tally of “synaptic” immunolabeling. Immunoreactivity was considered to be nonsynaptic, although it was potentially and functionally linked to the synapse, when the PEG particles occurred at a position >60 nm from the postsynaptic membrane surface of the spine but still within the cytoplasm of the spine (white arrows in Fig. 4 and schematized in Fig. 3).
Figure 4.
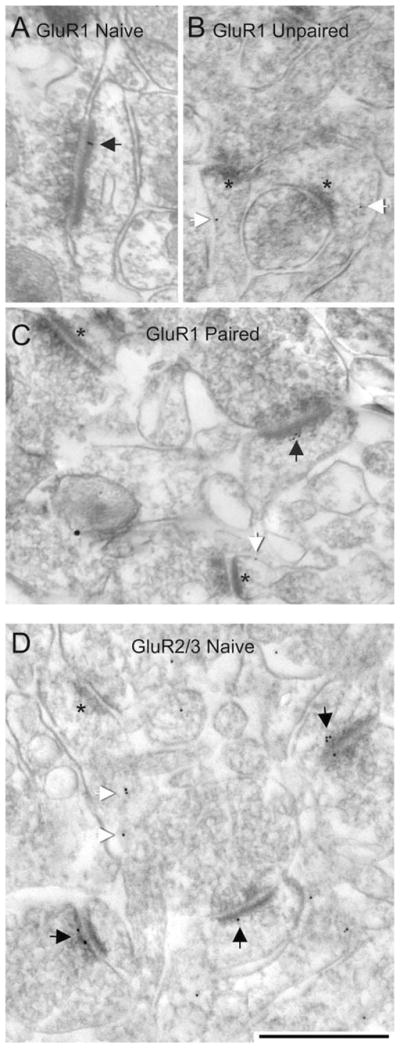
Representative electron micrographs from the dorsal LA of rats that received no stimuli (i.e., naïve), paired, and unpaired presentation of stimuli.A–C: Postembedding gold (PEG) labeling for the GluR1 subunit of AMPARs. D: PEG labeling for the GluR2/3 subunits of AMPARs. Black arrows across all panels indicate synaptic PEG labeling; white arrows indicate nonsynaptic labeling within spine heads (B,C) and spine neck (D). Asterisks point to PSDs of unlabeled synapses. Scale bar = 500 nm in D (applies to A–D).
Results of control experiments
Initial survey of grids prepared from tissue containing the LA of naïve rats indicated that the procedure yielded GluR1 immunolabeling of approximately 20% of spines and none of the symmetric (presumably inhibitory) synapses, with a substantial portion of the PEGs occurring non-synaptically (white arrows in Fig. 4). In contrast, the identical PEG procedure but using the GluR2/3 antibody, in lieu of the GluR1 antibody, yielded immunolabeling of approximately 70% of spines, with the majority of them exhibiting PEG directly over the PSD (black arrows in Fig. 4). These differences indicated that a larger portion of asymmetric synapses utilize AMPARs that contain the GluR2/3 subunits than the GluR1 subunits. This pattern was recaptured by light microscopy, which showed that GluR2/3 immunolabeling of the neuropil is robust and evenly distributed across subregions of the amygdala and at levels comparable to those found in the cortex and caudate-putamen nucleus, whereas the GluR1 immunoreactivity is relatively sparse, especially within the LA (Fig. 2).
Omission of the primary antibody in the incubation buffer resulted in labeling only 1 asymmetric synapse out of over 200 asymmetric synapses encountered from multiple brains of the paired, unpaired, and naïve animals, indicating that the PEG procedure was specific. This elimination of immunolabeling at asymmetric synapses was confirmed for every LA tissue examined in the current study (n = 12).
Preadsorption greatly reduced GluR1 immunolabeling at asymmetric synapses. At the dilution of 400 μg/ml of the control peptide, 93% of the synapses were devoid of even a single particle of PEG labeling (Fig. 5A). In contrast, ultrathin sections from paired animals’ LA that were incubated with the antibody but in the absence of the control peptide exhibited immunolabeling of approximately 30% of the synapses, and of these, 88% were labeled by just one PEG particle (Fig. 5C). Preadsorption reduced the proportion of asymmetric synapses labeled for GluR2/3 as well. In the presence of the control peptide, 95% of the synapses were devoid of PEG particles (Fig. 5B). In contrast, when the ultrathin section was incubated in the presence of the antibody but in the absence of the control peptide, only 30% of the synapses lacked PEG particles and approximately 70% of the synapses were immunolabeled with one, two, three, or more PEG particles. Of the GluR2/3-immunolabeled synapses, 48% exhibited one PEG particle (Fig. 5D). Based on these observations, we concluded that the association of one PEG particle with a spine or shaft synapse could be considered the threshold for GluR1 or GluR2/3 immunolabeling.
Figure 5.

Preadsorption control reduces background GluR1 and GluR2/3- immunolabeling in the vicinity of synapses. Preadsorption control was performed to determine the degree to which the postembedding gold (PEG) labeling results from nonspecific binding of the primary antibody to ultrathin sections. A: Preincubation of the control peptide (CP) with the GluR1 antibody decreased the frequency of PEG occurring in the vicinity of spines to 7%. B: Similarly, preincubation of the control peptide with the GluR2/3 antibody also reduced the frequency of PEG labeling within spines down to 5%. C: The number of PEG particles occurring within spines was predominantly zero for grids incubated with the GluR1 antibody, even in the absence of the control peptide, but the frequency increased further in the presence of the control peptide. D: Following preincubation of the GluR2/3 antibody with the control peptide, the frequency of spines containing no PEG particle rose threefold, leaving 5% of the spines labeled by one PEG particle and none labeled by more than one PEG particle.
The proportion of LA spines immunoreactive for GluR1 at the synaptic junction increases after fear conditioning
Ultrastructural analysis revealed that the great majority of asymmetric (presumably excitatory) synapses in the LA occur on spines. In order to determine whether fear conditioning altered the proportion of axo-spinous synapses immunoreactive for GluR1, we captured electron microscopic images from the LA until more than 200 synapses were encountered per animal. Data were collected from three treatment groups: paired, naïve control, and unpaired. The unpaired group served as a second control, in that these animals were exposed to the same stimuli of tone and foot shock, but the tone followed the shock. This sequence of stimuli does not yield the conditioned response of freezing (Cain and LeDoux, 2007).
A spine was considered immunolabeled if a PEG particle occurred anywhere within the spine. One-way ANOVA indicated no effect of the treatment upon the proportion of spines labeled generally (Fig. 6A). The proportions of spines immunolabeled at nonsynaptic sites were also not significantly different across the treatment groups (Fig. 6B). However, rats in the paired group showed a 99% increase in the proportion of spines immunolabeled at the synaptic junction (Zones I–III), compared with the spines of naïve rats, and this effect was statistically significant (F(2,8) = 5.5694, P < 0.05) (Fig. 6C). Spines of the LA from the unpaired group were also elevated at the synaptic junction, relative to the spines of naïve rats, but this change did not reach statistical significance (P = 0.10) (Fig. 6C).
Figure 6.

The proportion of spines immunoreactive for GluR1 increases following fear conditioning, whereas GluR2/3 immunoreactivity remains unchanged. GluR1 immunoreactivity was analyzed by sampling approximately 200 spines from the dorsal tip of the LA of each of four naïve animals, together with four animals that received the paired presentation of a tone with foot-shock (paired) and three animals that received the foot shock preceding the tone (unpaired). A: The proportion of spines immunolabeled for GluR1 at the synaptic junction or nonsynaptically was not significantly different across the three treatment groups. B: The proportion of spines immunolabeled for GluR1 at nonsynaptic sites within spines was also not significantly different. C: However, analysis of those spines immunolabeled specifically at the synaptic junction was significantly different among the treatment groups (P < 0.05). Fisher’s LSD post hoc comparisons revealed a significant difference between the paired and naïve groups. GluR2/3 immunoreactivity was analyzed by sampling 120 synapses per animal from the same region of the LA of four naïve, four paired, and four unpaired animals. D–F: In contrast to the GluR1 pattern, neither the paired nor unpaired conditioning evoked an increase in the proportion of spines labeled (D), the proportion of spines labeled at nonsynaptic sites (E) or the proportion of spines labeled specifically at synaptic sites (F). Bars represent mean and SEM. *, P < 0.05.
In order to determine whether GluR1-immunoreactivity levels within single spines were altered by fear conditioning, we also compared the mean PEG counts at the synaptic junction and at nonsynaptic portions of spine heads of each animal. This comparison indicated no effect of fear conditioning at the synaptic junction (F(2,7) = 2.1948, P = 0.18) or at nonsynaptic portions of spine heads (F(2,7) = 1.4597, P = 0.30).
Paired presentation of stimuli elevates GluR1 immunoreactivity at excitatory synapses formed on dendritic shafts
The frequency of asymmetric synapses occurring along dendritic shafts in the LA was much less than at spines, ranging from 7 to 20% of all asymmetric synapses encountered. Although less frequent, these asymmetric synapses on shafts would still be expected to affect neuronal excitability, because the absence of the spine neck makes these shaft synapses electrotonically and biochemically “closer” to the axon initial segment and soma. In order to determine whether axo-shaft excitatory synapses also undergo changes evoked by the paired presentation of stimuli, we analyzed this population of synapses separately from the axo-spinous asymmetric synapses. Results of one-way ANOVA revealed a robust increase in the proportion of axo-shafts synapses immunolabeled for GluR1 at the synaptic junction (F(2,7) = 23.013; P < 0.001), reflecting a 167% change in the proportion of synapses from the LA of the paired group that were immuno-labeled, relative to those of the Naives (P < 0.005) (Fig. 7A). PEG counts at axo-shaft synapses were also strongly altered by the fear conditioning treatment (F(2,7) = 102.81; P < 0.00001), due to the 167% elevation among synapses of the paired group, relative to those of the naïves (P < 0.00005) (Fig. 7B).
Figure 7.

GluR1 immunoreactivity is increased by fear conditioning at axo-shaft synapses, but GluR2/3 immunoreactivity is unchanged. A: The proportion of axo-shaft synapses immunolabeled for GluR1 increased for the paired group of animals, relative to axo-shaft synapses of the naïve and unpaired groups of animals (P < 0.001). B: The increase was also evident for GluR1 immunoreactivity at the axo-shaft synapses (P < 0.0001), reflected by the increase of PEG counts at the axo-shaft synaptic junction of the paired group, relative to the PEG counts of naïves. PEG counts at the synaptic junction of the unpaired group decreased, relative to the PEG counts of naïves. For both graphs, N indicates the number of animals analyzed. C,D: Identical analysis of sections immunolabeled for GluR2/3 revealed no effect of the treatment on the proportion of synaptic synapses labeled (C) or the PEG counts at shaft synapses (D). **, P < 0.005; ***, P < 0.0005 using Fisher’s post hoc LSD comparisons.
The unpaired presentation of stimuli lowers GluR1 immunoreactivity at axo-shaft asymmetric synapses
Not only did the axo-shaft synapses exhibit a robust increase of GluR1 immunoreactivity among the paired group, but they also exhibited a significant decrease in GluR1 immunoreactivity among the unpaired group (Fig. 7). Although the decline in the proportion of axo-shaft synapses labeled among the unpaired group did not reach statistical significance (P = 0.09; Fig. 7A), the PEG counts per synapse did (Fig. 7B). Specifically, PEG counts at the synaptic junction decreased by 84%, relative to the that of the naïves, and this difference was significant (P < 0.005). This decrease at the axo-shaft synapses contrast sharply with the pattern observed at spines of the unpaired group, namely, the absence of change of PEG counts at axo-spinous synapses and a slight trend toward an increase in the proportion of spines immunolabeled at the synaptic junction, relative to that of the naïves (P = 0.10; Fig. 6C).
Taken together, these results show that fear conditioning increases both the levels of GluR1 immunoreactivity and the proportion of synapses immunoreactive for GluR1 subunits in the LA, but only when presented with the stimuli in a sequence that leads to the acquisition of predictive value for the CS and the conditioned response of freezing.
PSD lengths are not altered by fear conditioning
In order to determine whether the increased encounter with GluR1-immunoreactive asymmetric LA synapses from the paired group was due to an increase in their synaptic size, we measured the lengths of the PSDs of asymmetric synapses that we encountered randomly from each animal and were analyzed for GluR1 immunoreactivity within single two-dimensional digitized images. A one-way ANOVA indicated no effect of the treatment. The mean PSD lengths were 258 ± 9 nm for the paired group, 279 ± 11 nm for the unpaired group, and 265 ± 9 nm for the naïve group. Although these measurements do not reflect true sizes of synapses (because we did not attempt three-dimensional reconstructions), the similarity in the PSD lengths across the treatment groups indicate that the rate of encounter with synaptic profiles within two-dimensional images is not likely to have been affected by differences in their sizes.
GluR2/3 immunoreactivity at synapses is unchanged 40 minutes following fear conditioning
GluR1 subunits can occur as homomers or as heteromers containing GluR2 subunits (Greger et al., 2003). Because GluR1 immunoreactivity rises at synaptic junctions following paired conditioning, a correlative rise of GluR2/3 immunoreactivity might also be expected. In order to test this possibility, we analyzed synapses of the same LA tissue for GluR2/3 immunoreactivity. The analysis indicated that no change was induced by the paired conditioning with regard to the proportion of synapses labeled for GluR2/3 at the synaptic junction of spines (F(2,9) = 0.126, P = 0.88; Fig. 6F), the proportion of spines labeled at nonsynaptic portions (F(2,9) = 0.098; P = 0.91; Fig. 6E), or the proportion of dendritic shaft synapses labeled (F(2,9) = 0.05884, P = 0.94; Fig. 7C). PEG levels at dendritic shaft synapses also were not detectably different (F(2,9) = 0.19031; P = 0.83; Fig. 7D), nor were PEG levels at spine synapses (F(2,9) = 0.32604; P = 0.73).
DISCUSSION
Synaptic plasticity likely involves a plethora of synaptic molecules (Malenka and Bear, 2004). Of these, previous studies highlighted GluR1 as one synaptic protein whose level at synapses becomes augmented following fear conditioning (Rumpel et al., 2005; Yeh et al., 2006). These previous studies detected increases in synaptic AMPAR currents carried by exogenous recombinant genes by the third hour after fear onditioning (Rumpel et al., 2005) and of endogenous surface AMPAR, as assessed by Western blotting, by the second hour following fear conditioning (Yeh et al., 2006). To date, our study is the first to focus on the formation of short-term memory (STM) of fear by analyzing the distribution of receptor subunits at synapses within 40 minutes following fear conditioning. Our results support and extend previous findings by confirming that endogenous GluR1-containing AMPARs, like the GFP-tagged GluR1-containing AMPARs (Rumpel et al., 2005), increase at LA synapses following fear conditioning. The ultrastructural data reveal that GluR1-containing AMPARs rise earlier than reported previously (Rumpel et al., 2005; Yeh et al., 2006). This difference is likely to be due to the greater sensitivity with which ultrastructural data can detect changes in the number of synapses expressing GluR1-containing AMPARs precisely at the synaptic junction.
Methodological considerations
In the light microscopic portion of the present study, we noted that GluR1 immunoreactivity in the LA is much lower than is the GluR2/3 immunoreactivity. This pattern is predicted from earlier studies, indicating that GluR1 immunoreactivity occurs predominantly in inhibitory aspiny interneurons of the basolateral amygdala, whereas the GluR2/3 immunoreactivity occurs much more prevalently within the spiny, pyramidal-like neurons (McDonald, 1996), combined with the fact that many more of the neurons in the LA are excitatory than are inhibitory (Johnson et al., 2001). Based on this observation, the lower level of GluR1 immunoreactivity that we detected by the PEG procedure (~20%), relative to the GluR2/3 immunoreactivity that we detected by using the same procedure (~70%), is likely to reflect, at least in part, differences in GluR1 versus GluR2/3 antigen levels, rather than the failure to detect the GluR1 antigen.
Previous studies showed that approximately 60–70% of the LA spines forming synapses with thalamic or cortical afferents were GluR1-immunoreactive (Farb and LeDoux, 1997, 1999). This difference in proportion of GluR1 immunoreactivity raises the possibility that PEG, as opposed to the horseradish peroxidase-diaminobenzidine (HRP-DAB) that was used in previous studies, resulted in greater failure to detect GluR1 immunoreactivity. However, the rate of detection of GluR2/3 within spines by the two procedures is comparable (60–70% for both) (Farb and LeDoux, 1997, 1999; Radley et al., 2007), so the idea that PEG is less efficient than HRP-DAB for detecting synaptic proteins is certainly not generalizable. Another possibility is that the lower level of GluR1 detected in the current study reflects differential sensitivity of the two GluR subunits to the tissue fixation condition or occurred because the other studies included the intercalated, GABAergic neurons that express high levels of GluR1.
Yet another more interesting explanation for the difference observed among the studies is that a substantial proportion of spines in the LA receive inputs from sources other than the thalamus or cortex, the two sources for which GluR1 immunoreactivity at synapses had been assessed by the HRP-DAB method (Farb and LeDoux, 1997, 1999). If spines that receive input from sources other than thalamus or cortex express only low levels of GluR1, then our PEG procedure, which did not preselect for glutamergic axons of any particular source, would yield a lower proportion of GluR1-immunoreactive spines (~20%).
For the proportion of postsynaptic GluR1 immunoreactivity to drop from 60% to 20%, a large portion of the pre-synaptic axons would have to be noncortical and nonthalamic. That the proportion of spines targeted by the thalamus or cortex may be small is not an unreasonable assumption. Another, thoroughly analyzed, brain region such as layer 4 of cortex receives as little as 6% (Ahmed et al., 1994) and no more than 20% (White, 1986) of its excitatory synaptic input from the thalamus. The current PEG result would be expected, if the proportion of spines receiving thalamic or cortical axons is about a third of all spines in the LA, and only those spines postsynaptic to the thalamic-plus-cortical axons express GluR1. What might be the source of nonthalamic/noncortical afferents to spines of the LA? One possibility is the spiny, projecting neurons of the LA, which presumably form axon collaterals to innervate other spiny neurons in the LA. The proportion of LA spines targeted by other LA neurons is a measurement that has not yet been examined, nor has it been determined whether excitatory axons originating from LA neurons target spines lacking GluR1 immunoreactivity. The idea that spines express different levels of GluR1, depending on their afferent partner, is novel but not impossible, because a substantial amount of evidence indicates that spines can undergo extensive biophysical and biochemical changes locally, independent of the parent dendritic shaft or soma (Carter et al., 2007; Matsuzaki et al., 2004; Yuste and Denk, 1995).
GluR1 trafficking to support the formation of short-term fear memory
Although the proportion of synapses immunoreactive to the GluR1 subunit was small, our findings were still able to demonstrate that the increase in the number of GluR1-containing AMPARs at the synaptic junction occurs rapidly enough to support short-term fear memory. Earlier studies showed that STM for fear conditioning is measurable 1 hour after training, even in the presence of protein synthesis inhibitors (Schafe and LeDoux, 2000). Because the time point that we analyzed overlaps with the reported phase that is resistant to protein synthesis inhibitors, synapse strengthening via GluR1-containing AMPAR accumulation at the LA synapses is likely to proceed mainly, although by no means exclusively, by phosphorylation and trafficking of reserve pools of GluR1-containing AMPARs from nonsynaptic sites to the postsynaptic sites.
Did the rise in the proportion of labeled axo-spinous synaptic junctions result from local translocation, e.g., the relocation from the nonsynaptic pool of GluR1-containing AMPARs within spines? Alternatively, did the rise in the proportion of labeled synapses involve the influx of GluR1-containing AMPARs from the parent dendritic shafts into spine heads? If the rise resulted from the local, intraspinous translocation only, without the concomitant influx from the parent shaft into spine heads, we would expect to have detected depletion of GluR1 immunoreactivity at nonsynaptic sites within spines. Because we observed no decrease in the proportion of spines labeled at nonsynaptic sites (Fig. 6B), the rise of GluR1 immunoreactivity at synaptic junctions following fear conditioning is likely to have been supported by the influx of GluR1-containing AMPARs from remote shaft sites into nonsynaptic positions within spine heads, to replenish the non-synaptic GluR1-containing AMPAR pool within spine heads that repositioned to the postsynaptic membrane.
Previous studies, using in vitro hippocampal slices to measure LTP, also point to the rapidity, e.g., within 15 minutes, with which GluR1-containing AMPARs can be delivered to synapses to mediate postsynaptic potentiation (Shi, 2001; Shi et al., 1999). Whether LA synapses of our in vivo preparation would also have shown increased levels of endogenous GluR1 immunoreactivity at the synaptic junction within 15 minutes and whether these arise through lateral diffusion from nonsynaptic plasma membrane or through exocytosis from the cytoplasmic pool is a question that can be addressed in the future.
Cellular mechanisms associated with trafficking of GluR1-containing AMPARs toward synaptic junctions
Besides the GluR1-containing AMPARs, previous studies have revealed changes within intact LA of at least five other synaptic proteins following fear conditioning: Ca2+/calmodulin-dependent protein kinase IIα (CaMKIIα) (Rodrigues et al., 2004a); myosin light chain kinase (Lamprecht et al., 2006b); the activity-regulated cytoskeletal-associated protein Arc/Arg3.1 (Ploski et al., 2008); profilin (Lamprecht et al., 2006a); and spinofilin (Radley et al., 2006).
CaMKIIα is autophosphorylated at the Thr286 site during the acquisition phase of fear conditioning, yielding persistent activity of this kinase (Rodrigues et al., 2004a). Because inhibition of CaMKIIα prevents the NMDAR-dependent form of LTP at thalamic input synapses in the LA, CaMKIIα activation is undoubtedly downstream from NMDAR activation. Both the NMDARs and GluR1-AMPARs lacking GluR2 subunits, as well as the voltage-gated calcium channels, can support the calcium influx necessary to autophosphorylate CaMKIIα. It is possible that some portion of the GluR1-containing AMPARs lack GluR2 subunits. If so, then those GluR1-containing AMPARs that arrive newly to synapses of the paired animals during the earliest phase of STM may contribute, in part, to the calcium influx needed for CaMKIIα activation and LTP in the LA.
The increase in the number of spines immunoreactive for the two F-actin binding proteins, profilin (Lamprecht et al., 2006a) and spinofilin (Radley et al., 2006), within the LA was detected by using an approach similar to ours. Of the two, the increase of spinophilin-immunoreactive spines was observed 24 hours after fear conditioning, e.g., during the period that is sensitive to protein synthesis inhibitors and after STM is converted to LTM. Because these events extend beyond the time point that we measured, the mechanisms underlying the increased GluR1-containing AMPAR that we observed may not have interacted with the mechanisms underlying the increase of spinophilin in the LA.
In contrast, the elevation of profilin was detected within the early time window, like this study—within the first hour following the first pairing of CS and US (Lamprecht et al., 2006a). Therefore, it is plausible to consider that the GluR1-containing AMPARs and profilin interact with one another during the early phase of fear memory formation, as they are both translocated into dendritic spines immediately following fear conditioning. Profilin promotes F-actin elongation and stabilization of spine morphology (Witke, 2004), and is translocated into spine heads of cultured hippocampal neurons in an activity-dependent manner (Ackermann and Matus, 2003; Neuhoff et al., 2005). One possibility is that the enhancement of synaptic strength through the increase of GluR1-containing AMPARs in the LA facilitates profilin influx into spines and this influx of profilin, in turn, stabilizes the F-actin matrix that ramifies the spine head. Stabilized F-actin matrix within spine heads may be one step during STM maintenance and conversion of STM to LTM that facilitates the subsequent trafficking of GluR2/3-containing AMPARs into and out of the spines that have been tagged by the elevated levels of GluR1-containing AMPARs.
The observation that activity-regulated cytoskeletal-associated protein (Arc/Arg3.1) is altered following fear conditioning (Ploski et al., 2008) is relevant to the current study on AMPAR subunit changes, because it is widely accepted that Arc/Arg3.1 regulates AMPAR trafficking (Chowdhury et al., 2006; Rial Verde et al., 2006; Shepherd et al., 2006; Bramham et al., 2008). Rial Verde et al. (2006 showed that upregulation of Arc/Arg3.1 leads to the removal of AMPARs containing GluR2/3-subunits, with less of an effect on recently potentiated synapses containing AMPARs with GluR1/2 subunits. Timed manipulation of Arc/Arg3.1 protein levels through administration of antisense mRNA into the LA indicates that the synaptic plasticity underlying STM is not dependent on the presence of this protein, but Arc/Arg3.1 protein is important for the conversion of STM to LTM (Ploski et al., 2008). When these data are taken together, it is likely that the rise of the GluR1-containing AMPARs that we observed does not depend on Arc/Arg3.1. Instead, the delay in the rise of this protein may be suitable for removing GluR2/3-AMPARs from nonactivated pathways and for re-establishing homeostasis of synaptic activity in the LA, after synapses of the activated pathways become strengthened by fear conditioning.
Absence of change in GluR2/3 immunoreactivity
Because the GluR1-containing AMPARs are likely to also contain GluR2/3 subunits (Greger et al., 2003), it is somewhat surprising that we did not detect a rise in GluR2/3 immunoreactivity, as we did for the GluR1 immunoreactivity. If, as hypothesized above, the GluR1/2 heteromers occur predominantly postsynaptic to the thalamic-plus-cortical axons and these are the predominant pathways undergoing synapse strengthening during the first hour after fear conditioning, then the change occurring at these pathways may have been masked by the GluR2/3-containing AMPARs present at the remaining LA-to-LA synapses.
It may also be that synapse strengthening via the increase of GluR1/2-containing AMPARs at synapses formed with the thalamic-plus-cortical axons are counterbalanced by the Arc3.1-mediated efflux of GluR2/3-containing AMPARs at the LA-to-LA synapses or other pathways that remained inactive during fear conditioning. Pathway-specific strengthening, against a background of unchanging or weakened synapses, may be desirable for maintaining homeostasis of LA’s overall excitability. This study focused on measuring changes during STM formation and did not measure changes occurring during the conversion from STM to LTM. The postsynaptic changes that ensue during LTM formation may involve changes in the levels of GluR2/3-containing AMPARs. In particular, it is possible that synapses that have undergone an increase of GluR1-containing AMPARs become strengthened through the insertion of GluR2/3-containing AMPARs during the LTM formation phase. Future studies that quantify GluR2/3 immunoreactivity within spines that are positively identified to be postsynaptic to thalamic and cortical axons would help with testing these working models.
Responses differ across axo-spinous versus axo-shaft synapses
Following the paired presentation of stimuli, asymmetric synapses responded with an increase in GluR1 immunoreactivity, regardless of the location of the synapse (spine or shaft). However, following the unpaired presentation of stimuli, the asymmetric synapses at the two types of locations responded oppositely: synapses formed along the spine remained unchanged or slightly elevated in GluR1 immunoreactivity, whereas synapses formed along shafts decreased GluR1 immunoreactivity. Although the number of synapses analyzed along the dendritic shaft was smaller than that for the spines, the magnitude of the change at axo-shaft synapses was sufficiently large to be detectable.
It is not easy to attribute the changes in GluR1 at axo-shaft synapses to any specific pathway within the LA, because we did not perform experiments to determine whether the postsynaptic cells were excitatory or inhibitory or whether the population of synapses showing increased GluR1 with paired or unpaired training overlap. These experiments are planned for the future. However, it is most likely that cells postsynaptic to asymmetric axo-shaft synapses are inhibitory interneurons. This supposition is based on the synaptic pattern described for the cortex, where aspiny and sparsely spiny neurons receive excitatory inputs directly on dendritic shafts, whereas spiny neurons receive most of their excitatory inputs at spine heads (White, 1989). Moreover, previous anatomical studies indicate that spines in the lateral and basal nuclei of amygdala are more likely to belong to pyramidal-like cells, because these are very spiny and also more numerous, whereas the aspiny and sparsely spiny neurons belong to the inhibitory interneurons (Millhouse and DeOlmos, 1983; McDonald, 1996; McDonald et al., 2002; Muller et al., 2007, 2009; Pinard et al., 2008). In contrast, excitatory neurons, defined by their immunoreactivity to CaMKII or by their spininess, as visualized through three-dimensional reconstruction, have been observed to receive excitatory inputs along dendritic shafts (L. Ostroff and C. Farb, personal communication).
Inhibitory neurons have been reported to utilize AMPARs that lack GluR2 subunits (McDonald, 1996) and thus are calcium-permeable (Seeburg, 1993). If the shafts exhibiting augmented GluR1 immunoreactivity following the paired presentation of stimuli do belong to inhibitory interneurons, then this change is likely to exert a potent influence upon the postsynaptic membrane potential and allow influx of calcium, independent of NMDAR activity. In both the cortex (Hull et al., 2009) and LA (Rainnie et al., 1991; Bissiere et al., 2003; Kodirov et al., 2006; Shaban et al., 2006), the excitatory afferents targeting inhibitory interneurons exert potent feed-forward inhibition upon the spiny excitatory neurons. In the LA, this pathway is active, even in the naïve state, but even more, following paired conditioning, according to the results of the present study. Following the paired presentation of stimuli, an increase in the feed-forward inhibition may occur specifically upon the pathways in the LA that were not simultaneously activated by CS and US inputs, and, in this way, increase the signal-to-noise ratio for the pathway subserving the CS and also contribute toward maintaining an overall homeostasis of LA excitability.
The unpaired group was chosen as an associative control, because these animals receive the same number of CSs and USs but in a non-overlapping manner. Indeed, unpaired controls in fear conditioning experiments commonly show no signs of CS-elicited fear following training, indicating that no positive association was formed between the CS and US (e.g., Cain and LeDoux, 2007). However, it is important to note that unpaired controls can learn. Like their paired counterparts, they can undergo nonassociative learning from exposure to the aversive US (i.e., sensitization), associative context conditioning, or even conditioned inhibition (safety signal learning).
The decline of GluR1-containing AMPARs at axo-shaft synapses with unpaired training does not seem consistent with a cellular mechanism for either context conditioning or conditioned inhibition. However, this change is consistent with a nonassociative sensitization process resulting from US exposure. A variety of stressors can lead to hyperexcitability in the amygdala, often manifested as impairments in local γ-aminobutyric acid (GABA)ergic inhibition (Braga et al., 2004; Rainnie et al., 2004; Rodriguez Manzanares et al., 2005). Thus, USs that are not predicted by any explicit CS may prime the amygdala for future associative conditioning and/or lower the threshold for amygdala activation by other fear-eliciting stimuli. Recent studies indicate that suppression of feed-forward inhibition is important for induction of LTP in the LA (Bissiere et al., 2003; Kodirov et al., 2006), consistent with this priming hypothesis.
CONCLUSIONS
The present EM immunocytochemical study revealed two contrasting types of cellular responses, distinguished by the location of asymmetric synapses, e.g., dendritic spine versus shaft. Following the paired presentation of CS and US, GluR1 immunoreactivity undergoes augmentation at both the spine and the shaft. In contrast, the unpaired presentation of CS and US elicits a decrease in GluR1 immunoreactivity, only at the asymmetric synapses on shafts, leaving the asymmetric synapses on spines relatively unchanged. This difference in the cellular response may be critical for eliciting the fear response to the CS following the paired presentation of stimuli, but not after the unpaired presentation of stimuli. The identification of such diversity points to the importance of conducting anatomical studies that have the resolving power of differentiating synaptic from nonsynaptic domains, and of distinguishing synapses that form on spines from those that form directly on dendritic shafts. Moreover, only by performing analysis upon intact tissue were we able to identify synaptic changes in the LA that link sensory experience to fear learning within the first hour post conditioning.
In this way, our study extends beyond preceding studies that led us to examine trafficking of GluR1-containing AMPARs. Our analysis shows that trafficking of GluR1-containing AMPARs is evoked not only after LTP induction within hippocampal slices or following sensory experience during development (Takahashi et al., 2003) but also within intact LA following fear learning in adulthood. Moreover, we show that this can be achieved through the rapid trafficking of endogenous GluR1-AMPARs, without depleting the nonsynaptic reserve pool of GluR1-AMPAR, leading to behavior, emotion, and arousal that are robustly plastic, yet also specific and protected from the overgeneralizations associated with anxiety disorders.
Acknowledgments
We thank Veeravan Mahadomrongkul, Sho Fujisawa, and Robert Levy for helping with tissue processing of pilot studies and Hilda M. Fernandez and Tunazzina Ahmed for their technical assistance with tissue preparation. We thank Claudia Farb for many hours of discussions regarding the circuitry of LA, critical reading of the manuscript, advice regarding identification of the dorsal tip of LA, and providing the light microscopic slides used for this paper. We thank Beth Bauer, PhD, Barry Cohen, PhD, Marjorie McMeniman, PhD (New York University), and Diana T. Gheorghiu, MA (Cluj-Napoca University) for their critical input on the statistical analysis.
Grant sponsor: New York University’s Dean’s Undergraduate Research Fund (to C.M.K.); Grant sponsor: National Institutes of Mental Health; Grant numbers: R01-MH046516, P50-MH058911, and R37-MH038774 (to J.E.L.) and F32 MH077458-03 (to C.K.C.); Grant Sponsor: National Eye Institute; Grant number: EY13079 (to C.A.).
LITERATURE CITED
- Ackermann M, Matus A. Activity-induced targeting of profilin and stabilization of dendritic spine morphology. Nat Neurosci. 2003;6:1194–1200. doi: 10.1038/nn1135. [DOI] [PubMed] [Google Scholar]
- Ahmed B, Anderson JC, Douglas RJ, Martin KA, Nelson JC. Polyneuronal innervation of spiny stellate neurons in cat visual cortex. J Comp Neurol. 1994;341:39–49. doi: 10.1002/cne.903410105. [DOI] [PubMed] [Google Scholar]
- Aoki C, Rodrigues S, Kurose H. Use of electron microscopy in the detection of adrenergic receptors. Methods Mol Biol. 2000;126:535–563. doi: 10.1385/1-59259-684-3:535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer EP, LeDoux JE, Nader K. Fear conditioning and LTP in the lateral amygdala are sensitive to the same stimulus contingencies. Nat Neurosci. 2001;4:687–688. doi: 10.1038/89465. [DOI] [PubMed] [Google Scholar]
- Bissiere S, Humeau Y, Luthi A. Dopamine gates LTP induction in lateral amygdala by suppressing feedforward inhibition. Nat Neurosci. 2003;6:587–592. doi: 10.1038/nn1058. [DOI] [PubMed] [Google Scholar]
- Blair HT, Schafe GE, Bauer EP, Rodrigues SM, LeDoux JE. Synaptic plasticity in the lateral amygdala: a cellular hypothesis of fear conditioning. Learn Mem. 2001;8:229–242. doi: 10.1101/lm.30901. [DOI] [PubMed] [Google Scholar]
- Blanchard RJ, Blanchard DC. Passive and active reactions to fear-eliciting stimuli. J Comp Physiol Psychol. 1969;68:129–135. doi: 10.1037/h0027676. [DOI] [PubMed] [Google Scholar]
- Braga MF, Aroniadou-Anderjaska V, Manion ST, Hough CJ, Li H. Stress impairs alpha(1A) adrenoceptor-mediated noradrenergic facilitation of GABAergic transmission in the basolateral amygdala. Neuropsychopharmacology. 2004;29:45–58. doi: 10.1038/sj.npp.1300297. [DOI] [PubMed] [Google Scholar]
- Bramham CR, Worley PF, Moore MJ, Guzowski JF. The immediate early gene arc/arg3.1: regulation, mechanisms, and function. J Neurosci. 2008;28:11760–11767. doi: 10.1523/JNEUROSCI.3864-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cain CK, LeDoux JE. Escape from fear: a detailed behavioral analysis of two atypical responses reinforced by CS termination. J Exp Psychol Anim Behav Process. 2007;33:451–463. doi: 10.1037/0097-7403.33.4.451. [DOI] [PubMed] [Google Scholar]
- Cain CK, LeDoux JE. Brain mechanisms of Pavlovian and instrumental aversive conditioning. In: Nutt DJ, Blanchard RJ, Blanchard DC, Griebel G, editors. Handbook of anxiety and fFear. Amsterdam: Elsevier Academic Press; 2008. pp. 103–125. [Google Scholar]
- Carter AG, Soler-Llavina GJ, Sabatini BL. Timing and location of synaptic inputs determine modes of subthreshold integration in striatal medium spiny neurons. J Neurosci. 2007;27:8967–8977. doi: 10.1523/JNEUROSCI.2798-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chowdhury S, Shepherd JD, Okuno H, Lyford G, Petralia RS, Plath N, Kuhl D, Huganir RL, Worley PF. Arc/Arg3.1 interacts with the endocytic machinery to regulate AMPA receptor trafficking. Neuron. 2006;52:445–459. doi: 10.1016/j.neuron.2006.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis M, Shi C. The extended amygdala: are the central nucleus of the amygdala and the bed nucleus of the stria terminalis differentially involved in fear versus anxiety? Ann N Y Acad Sci. 1999;877:281–291. doi: 10.1111/j.1749-6632.1999.tb09273.x. [DOI] [PubMed] [Google Scholar]
- Fanselow MS, Bolles RC. Naloxone and shock-elicited freezing in the rat. J Comp Physiol Psychol. 1979;93:736–744. doi: 10.1037/h0077609. [DOI] [PubMed] [Google Scholar]
- Farb CR, LeDoux JE. NMDA and AMPA receptors in the lateral nucleus of the amygdala are postsynaptic to auditory thalamic afferents. Synapse. 1997;27:106–121. doi: 10.1002/(SICI)1098-2396(199710)27:2<106::AID-SYN2>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- Farb CR, Ledoux JE. Afferents from rat temporal cortex synapse on lateral amygdala neurons that express NMDA and AMPA receptors. Synapse. 1999;33:218–229. doi: 10.1002/(SICI)1098-2396(19990901)33:3<218::AID-SYN6>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- Farb CR, Aoki C, Ledoux JE. Differential localization of NMDA and AMPA receptor subunits in the lateral and basal nuclei of the amygdala: a light and electron microscopic study. J Comp Neurol. 1995;362:86–108. doi: 10.1002/cne.903620106. [DOI] [PubMed] [Google Scholar]
- Fendt M, Fanselow MS. The neuroanatomical and neurochemical basis of conditioned fear. Neurosci Biobehav Rev. 1999;23:743–760. doi: 10.1016/s0149-7634(99)00016-0. [DOI] [PubMed] [Google Scholar]
- Gray EG. Axo-somatic and axo-dendritic synapses of the cerebral cortex: an electron microscope study. J Anat. 1959;93:420–433. [PMC free article] [PubMed] [Google Scholar]
- Greger IH, Khatri L, Kong X, Ziff EB. AMPA receptor tetramerization is mediated by Q/R editing. Neuron. 2003;40:763–774. doi: 10.1016/s0896-6273(03)00668-8. [DOI] [PubMed] [Google Scholar]
- Hayashi Y, Shi SH, Esteban JA, Piccini A, Poncer JC, Malinow R. Driving AMPA receptors into synapses by LTP and CaMKII: requirement for GluR1 and PDZ domain interaction. Science. 2000;287:2262–2267. doi: 10.1126/science.287.5461.2262. [DOI] [PubMed] [Google Scholar]
- Huang YY, Martin KC, Kandel ER. Both protein kinase A and mitogen-activated protein kinase are required in the amygdala for the macromolecular synthesis-dependent late phase of long-term potentiation. J Neurosci. 2000;20:6317–6325. doi: 10.1523/JNEUROSCI.20-17-06317.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hull C, Isaacson JS, Scanziani M. Postsynaptic mechanisms govern the differential excitation of cortical neurons by thalamic inputs. J Neurosci. 2009;29:9127–9136. doi: 10.1523/JNEUROSCI.5971-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humeau Y, Reisel D, Johnson AW, Borchardt T, Jensen V, Gebhardt C, Bosch V, Gass P, Bannerman DM, Good MA, Hvalby O, Sprengel R, Luthi A. A pathway-specific function for different AMPA receptor subunits in amygdala long-term potentiation and fear conditioning. J Neurosci. 2007;27:10947–10956. doi: 10.1523/JNEUROSCI.2603-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson LR, Hou M, Albert JL, Farb CR, Hof PR, LeDoux J. Quantification of the total neuronal structure of the fear conditioning circuit of the lateral amygdala of the rat. Annual Meeting of the Society for Neuroscience Society for Neuroscience.2001. [Google Scholar]
- Kodirov SA, Takizawa S, Joseph J, Kandel ER, Shumyatsky GP, Bolshakov VY. Synaptically released zinc gates long-term potentiation in fear conditioning pathways. Proc Natl Acad Sci U S A. 2006;103:15218–15223. doi: 10.1073/pnas.0607131103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamprecht R, Farb CR, Rodrigues SM, LeDoux JE. Fear conditioning drives profilin into amygdala dendritic spines. Nat Neurosci. 2006a;9:481–483. doi: 10.1038/nn1672. [DOI] [PubMed] [Google Scholar]
- Lamprecht R, Margulies DS, Farb CR, Hou M, Johnson LR, LeDoux JE. Myosin light chain kinase regulates synaptic plasticity and fear learning in the lateral amygdala. Neuroscience. 2006b;139:821–829. doi: 10.1016/j.neuroscience.2005.12.055. [DOI] [PubMed] [Google Scholar]
- Lázaro-Muñoz G, Mahadomrongkul V, Cain C, Aoki C. Fear conditioning increases GluR1 subunit of AMPA receptors at postsynaptic densities of lateral amygdala spines. Vol. 2006. Atlanta, Georgia: Society for Neuroscience; 2006. [Google Scholar]
- LeDoux JE. Emotion circuits in the brain. Annu Rev Neurosci. 2000;23:155–184. doi: 10.1146/annurev.neuro.23.1.155. [DOI] [PubMed] [Google Scholar]
- Levy RB, Aoki C. Alpha7 nicotinic acetylcholine receptors occur at postsynaptic densities of AMPA receptor-positive and -negative excitatory synapses in rat sensory cortex. J Neurosci. 2002;22:5001–5015. doi: 10.1523/JNEUROSCI.22-12-05001.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahanty NK, Sah P. Calcium-permeable AMPA receptors mediate long-term potentiation in interneurons in the amygdala. Nature. 1998;394:683–687. doi: 10.1038/29312. [DOI] [PubMed] [Google Scholar]
- Malenka RC, Bear MF. LTP and LTD: an embarrassment of riches. Neuron. 2004;44:5–21. doi: 10.1016/j.neuron.2004.09.012. [DOI] [PubMed] [Google Scholar]
- Maren S. Synaptic mechanisms of associative memory in the amygdala. Neuron. 2005;47:783–786. doi: 10.1016/j.neuron.2005.08.009. [DOI] [PubMed] [Google Scholar]
- Matsuzaki M, Honkura N, Ellis-Davies GC, Kasai H. Structural basis of long-term potentiation in single dendritic spines. Nature. 2004;429:761–766. doi: 10.1038/nature02617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald AJ. Localization of AMPA glutamate receptor subunits in subpopulations of non-pyramidal neurons in the rat basolateral amygdala. Neurosci Lett. 1996;208:175–178. doi: 10.1016/0304-3940(96)12585-4. [DOI] [PubMed] [Google Scholar]
- McDonald AJ, Muller JF, Mascagni F. GABAergic innervation of alpha type II calcium/calmodulin-dependent protein kinase immunoreactive pyramidal neurons in the rat basolateral amygdala. J Comp Neurol. 2002;446:199–218. doi: 10.1002/cne.10204. [DOI] [PubMed] [Google Scholar]
- Millhouse OE, DeOlmos J. Neuronal configurations in lateral and basolateral amygdala. Neuroscience. 1983;10:1269–1300. doi: 10.1016/0306-4522(83)90112-4. [DOI] [PubMed] [Google Scholar]
- Muller JF, Mascagni F, McDonald AJ. Serotonin-immunoreactive axon terminals innervate pyramidal cells and inter-neurons in the rat basolateral amygdala. J Comp Neurol. 2007;505:314–335. doi: 10.1002/cne.21486. [DOI] [PubMed] [Google Scholar]
- Muller JF, Mascagni F, McDonald AJ. Dopaminergic innervation of pyramidal cells in the rat basolateral amygdala. Brain Struct Funct. 2009;213:275–288. doi: 10.1007/s00429-008-0196-y. [DOI] [PubMed] [Google Scholar]
- Neuhoff H, Sassoe-Pognetto M, Panzanelli P, Maas C, Witke W, Kneussel M. The actin-binding protein profilin I is localized at synaptic sites in an activity-regulated manner. Eur J Neurosci. 2005;21:15–25. doi: 10.1111/j.1460-9568.2004.03814.x. [DOI] [PubMed] [Google Scholar]
- Passafaro M, Piech V, Sheng M. Subunit-specific temporal and spatial patterns of AMPA receptor exocytosis in hippocampal neurons. Nat Neurosci. 2001;4:917–926. doi: 10.1038/nn0901-917. [DOI] [PubMed] [Google Scholar]
- Petralia RS, Wenthold RJ. Light and electron immunocytochemical localization of AMPA-selective glutamate receptors in the rat brain. J Comp Neurol. 1992;318:329–354. doi: 10.1002/cne.903180309. [DOI] [PubMed] [Google Scholar]
- Phend KD, Rustioni A, Weinberg RJ. An osmium-free method of Epon embedment that preserves both ultra-structure and antigenicity for post-embedding immunocytochemistry. J Histochem Cytochem. 1995;43:283–292. doi: 10.1177/43.3.7532656. [DOI] [PubMed] [Google Scholar]
- Pinard CR, Muller JF, Mascagni F, McDonald AJ. Dopaminergic innervation of interneurons in the rat basolateral amygdala. Neuroscience. 2008;157:850–863. doi: 10.1016/j.neuroscience.2008.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitkanen A, Savander V, LeDoux JE. Organization of intra-amygdaloid circuitries in the rat: an emerging framework for understanding functions of the amygdala. Trends Neurosci. 1997;20:517–523. doi: 10.1016/s0166-2236(97)01125-9. [DOI] [PubMed] [Google Scholar]
- Ploski JE, Pierre VJ, Smucny J, Park K, Monsey MS, Overeem KA, Schafe GE. The activity-regulated cytoskeletal-associated protein (Arc/Arg3.1) is required for memory consolidation of pavlovian fear conditioning in the lateral amygdala. J Neurosci. 2008;28:12383–12395. doi: 10.1523/JNEUROSCI.1662-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quirk GJ, Armony JL, Repa JC, Li XF, LeDoux JE. Emotional memory: a search for sites of plasticity. Cold Spring Harb Symp Quant Biol. 1996;61:247–257. [PubMed] [Google Scholar]
- Radley JJ, Johnson LR, Janssen WG, Martino J, Lamprecht R, Hof PR, LeDoux JE, Morrison JH. Associative Pavlovian conditioning leads to an increase in spinophilin-immunoreactive dendritic spines in the lateral amygdala. Eur J Neurosci. 2006;24:876–884. doi: 10.1111/j.1460-9568.2006.04962.x. [DOI] [PubMed] [Google Scholar]
- Radley JJ, Farb CR, He Y, Janssen WG, Rodrigues SM, Johnson LR, Hof PR, LeDoux JE, Morrison JH. Distribution of NMDA and AMPA receptor subunits at thalamo-amygdaloid dendritic spines. Brain Res. 2007;1134:87–94. doi: 10.1016/j.brainres.2006.11.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rainnie DG, Asprodini EK, Shinnick-Gallagher P. Inhibitory transmission in the basolateral amygdala. J Neurophysiol. 1991;66:999–1009. doi: 10.1152/jn.1991.66.3.999. [DOI] [PubMed] [Google Scholar]
- Rainnie DG, Bergeron R, Sajdyk TJ, Patil M, Gehlert DR, Shekhar A. Corticotrophin releasing factor-induced synaptic plasticity in the amygdala translates stress into emotional disorders. J Neurosci. 2004;24:3471–3479. doi: 10.1523/JNEUROSCI.5740-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rial Verde EM, Lee-Osbourne J, Worley PF, Malinow R, Cline HT. Increased expression of the immediate-early gene arc/arg3.1 reduces AMPA receptor-mediated synaptic transmission. Neuron. 2006;52:461–474. doi: 10.1016/j.neuron.2006.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigues SM, Farb CR, Bauer EP, LeDoux JE, Schafe GE. Pavlovian fear conditioning regulates Thr286 auto-phosphorylation of Ca2+/calmodulin-dependent protein kinase II at lateral amygdala synapses. J Neurosci. 2004a;24:3281–3288. doi: 10.1523/JNEUROSCI.5303-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigues SM, Schafe GE, LeDoux JE. Molecular mechanisms underlying emotional learning and memory in the lateral amygdala. Neuron. 2004b;44:75–91. doi: 10.1016/j.neuron.2004.09.014. [DOI] [PubMed] [Google Scholar]
- Rodriguez Manzanares PA, Isoardi NA, Carrer HF, Molina VA. Previous stress facilitates fear memory, attenuates GABAergic inhibition, and increases synaptic plasticity in the rat basolateral amygdala. J Neurosci. 2005;25:8725–8734. doi: 10.1523/JNEUROSCI.2260-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rumpel S, LeDoux J, Zador A, Malinow R. Postsynaptic receptor trafficking underlying a form of associative learning. Science. 2005;308:83–88. doi: 10.1126/science.1103944. [DOI] [PubMed] [Google Scholar]
- Sah P, Westbrook RF, Luthi A. Fear conditioning and long-term potentiation in the amygdala: what really is the connection? Ann N Y Acad Sci. 2008;1129:88–95. doi: 10.1196/annals.1417.020. [DOI] [PubMed] [Google Scholar]
- Schafe GE, LeDoux JE. Memory consolidation of auditory pavlovian fear conditioning requires protein synthesis and protein kinase A in the amygdala. J Neurosci. 2000;20:RC96. doi: 10.1523/JNEUROSCI.20-18-j0003.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeburg PH. The TINS/TiPS Lecture. The molecular biology of mammalian glutamate receptor channels. Trends Neurosci. 1993;16:359–365. doi: 10.1016/0166-2236(93)90093-2. [DOI] [PubMed] [Google Scholar]
- Shaban H, Humeau Y, Herry C, Cassasus G, Shigemoto R, Ciocchi S, Barbieri S, van der Putten H, Kaupmann K, Bettler B, Luthi A. Generalization of amygdala LTP and conditioned fear in the absence of presynaptic inhibition. Nat Neurosci. 2006;9:1028–1035. doi: 10.1038/nn1732. [DOI] [PubMed] [Google Scholar]
- Shepherd JD, Rumbaugh G, Wu J, Chowdhury S, Plath N, Kuhl D, Huganir RL, Worley PF. Arc/Arg3.1 mediates homeostatic synaptic scaling of AMPA receptors. Neuron. 2006;52:475–484. doi: 10.1016/j.neuron.2006.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi SH. Amersham Biosciences & Science Prize. AMPA receptor dynamics and synaptic plasticity. Science. 2001;294:1851–1852. doi: 10.1126/science.1067844. [DOI] [PubMed] [Google Scholar]
- Shi SH, Hayashi Y, Petralia RS, Zaman SH, Wenthold RJ, Svoboda K, Malinow R. Rapid spine delivery and redistribution of AMPA receptors after synaptic NMDA receptor activation. Science. 1999;284:1811–1816. doi: 10.1126/science.284.5421.1811. [DOI] [PubMed] [Google Scholar]
- Takahashi T, Svoboda K, Malinow R. Experience strengthening transmission by driving AMPA receptors into synapses. Science. 2003;299:1585–1588. doi: 10.1126/science.1079886. [DOI] [PubMed] [Google Scholar]
- Wenthold RJ, Yokotani N, Doi K, Wada K. Immunochemical characterization of the non-NMDA glutamate receptor using subunit-specific antibodies. Evidence for a hetero-oligomeric structure in rat brain. J Biol Chem. 1992;267:501–507. [PubMed] [Google Scholar]
- White EL. Terminations of thalamic afferents. I. In: Jones EG, Peters A, editors. The cerebral cortex sensory-motor areas and aspects of cortical connectivity. New York: Plenum; 1986. pp. 271–289. [Google Scholar]
- White EL. Cortical circuits: synaptic organization of the cerebral cortex: structure, function and theory. Boston: Birkhauser; 1989. [Google Scholar]
- Witke W. The role of profilin complexes in cell motility and other cellular processes. Trends Cell Biol. 2004;14:461–469. doi: 10.1016/j.tcb.2004.07.003. [DOI] [PubMed] [Google Scholar]
- Woodson W, Farb CR, Ledoux JE. Afferents from the auditory thalamus synapse on inhibitory interneurons in the lateral nucleus of the amygdala. Synapse. 2000;38:124–137. doi: 10.1002/1098-2396(200011)38:2<124::AID-SYN3>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- Yeh SH, Mao SC, Lin HC, Gean PW. Synaptic expression of glutamate receptor after encoding of fear memory in the rat amygdala. Mol Pharmacol. 2006;69:299–308. doi: 10.1124/mol.105.017194. [DOI] [PubMed] [Google Scholar]
- Yildirim M, Janssen WG, Tabori NE, Adams MM, Yuen GS, Akama KT, McEwen BS, Milner TA, Morrison JH. Estrogen and aging affect synaptic distribution of phosphorylated LIM kinase (pLIMK) in CA1 region of female rat hippocampus. Neuroscience. 2008;152:360–370. doi: 10.1016/j.neuroscience.2008.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuste R, Denk W. Dendritic spines as basic functional units of neuronal integration. Nature. 1995;375:682–684. doi: 10.1038/375682a0. [DOI] [PubMed] [Google Scholar]