

Growth inhibition induced by a disease resistance gene is uncoupled from plant defense activation.
Abstract
Activation of plant immune responses is often associated with an inhibition of plant growth. The molecular mechanisms underlying this fitness cost are unknown. Here, we utilize the autoimmune response mutant suppressor of npr1, constitutive1 (snc1) resulting from an activated form of the Disease Resistance (R) gene to dissect the genetic component mediating growth inhibition in Arabidopsis (Arabidopsis thaliana). The radical-induced cell death1 (rcd1) mutant defective in responses to reactive oxygen species (ROS) was isolated as an enhancer of the snc1 mutant in growth inhibition but not in defense response activation. Similarly, the vitamin C2 (vtc2) and vtc3 mutants defective in ROS detoxification enhanced the growth defects of snc1. Thus, perturbation of ROS status by R gene activation is responsible for the growth inhibition, and this effect is independent of defense response activation. This was further supported by the partial rescue of growth defects of rcd1 snc1 by the respiratory burst oxidase homolog D (rbohD) and rbohF mutations compromising the generation of ROS burst. Collectively, these findings indicate that perturbation of ROS homeostasis contributes to the fitness cost independent of defense activation.
It is commonly observed that plant growth is inhibited by elevated defense responses. Autoimmune mutants with constitutive defense responses often have reduced growth to different extents compared with the wild type, and these growth defects can be reduced or reverted by inhibition of defense responses (Clarke et al., 2001; Hua et al., 2001; Jambunathan et al., 2001; Shirano et al., 2002; Zhang et al., 2003; Yang and Hua, 2004). Cell expansion and sometimes cell division are reduced while more cell death occurs when defense responses are constitutively activated. Even in the absence of constitutive defense activation, there is a fitness cost associated with harboring disease resistance proteins in the genome (Tian et al., 2003). How defense responses repress plant growth at the molecular level is not well understood. The general assumption is that resources are channeled to defense responses and thus compromise plant growth. In addition, several molecules up-regulated in defense responses have adverse effects on plant growth. For instance, salicylic acid (SA), a signal for systemic acquired resistance (Durrant and Dong, 2004), inhibits plant growth, as revealed by a larger plant with the depletion of SA (Scott et al., 2004).
Reactive oxygen species (ROS) generated during defense may also contribute to the growth inhibition. ROS are produced from organelles with a highly oxidizing metabolic activity or with an intense rate of electron flow, including chloroplast, mitochondria, and peroxisomes (Mittler et al., 2004). Conditions that limit CO2 fixation, such as drought, salt, and temperature stress, particularly in combination with high light intensities, enhance ROS production (Mittler, 2002; Foyer and Noctor, 2005; Miller et al., 2008). ROS are also generated in plants during plant-microbe interaction, and the oxidative burst upon pathogen attack is thought to be part of the defense responses (Nanda et al., 2010). It is linked to papilla formation and the assembly of physical barriers in basal resistance. ROS are associated with the hypersensitive response, a form of programmed cell death, to restrict the spread of pathogens upon their recognition by plant Disease Resistance (R) genes. ROS play a dual role in plants as both toxic compounds and important signal transduction molecules (Apel and Hirt, 2004; Mittler et al., 2004; Foyer and Noctor, 2005). Hydroxyl radicals react instantaneously with proteins, lipids, and DNA, causing rapid cell damage. Superoxide (O2−) and hydrogen peroxide (H2O2) can inactivate various macromolecules directly or be converted to more toxic hydroxyl radicals. ROS can also cross-link Hyp-rich glycoproteins in the cell wall, likely contributing to the resistance of pearl millet (Pennisetum americanum; Deepak et al., 2007). Thus, cell growth inhibition by ROS might result from a combination of toxicity and direct physical limitation on cell expansion. On the other hand, ROS are also the positive regulators of the cell and organ growth under nonstress conditions (Foreman et al., 2003). H2O2 and O2− are thought to be involved in lignin formation in cell walls (Barceló et al., 2007). The Arabidopsis (Arabidopsis thaliana) RHD2 gene encoding the NADPH oxidase AtrbohC is required for ROS accumulation at the tip of the root hair and the elongation of the root hair (Schiefelbein and Somerville, 1990; Grierson et al., 1997). Exogenous application of diphenyleneiodonium, an inhibitor of flavoproteins such as NADPH oxidases, inhibits the growth of wild-type root hairs (Dunand et al., 2007; Jones et al., 2007), further supporting a positive role of ROS for root hair development. The distribution of ROS is also found to play a role in the transition from proliferation to differentiation in roots, with O2− accumulating primarily in the meristematic zone whereas H2O2 accumulates in the elongation zone (Tsukagoshi et al., 2010).
The Arabidopsis Radical-Induced Cell Death1 (RCD1) gene is involved in ROS signaling. It has been isolated from various genetic and biochemical screens. The rcd1 mutant is sensitive to ozone and has more expanded cell death compared with the wild type when exposed to ozone (Overmyer et al., 2000). However, it is more tolerant than the wild type to methyl viologen and UV-B that generate ROS in chloroplasts (Fujibe et al., 2004). It is more sensitive to salt but less sensitive to abscisic acid (ABA), ethylene, and methyl jasmonate (Ahlfors et al., 2004). RCD1, therefore, appears to serve as an integration point between hormone signaling and a coordinated ozone response. Under nonstressed growth conditions, the rcd1 mutant plant displays developmental defects, including reduced stature, early flowering, and altered leaf and rosette morphology (Ahlfors et al., 2004). The developmental defects become more severe in the double mutant of RCD1 and its homolog Similar to RCD-One1 (Teotia and Lamb, 2009). RCD1 encodes a protein with a WWE protein-protein interaction domain, three nuclear localization signals, and a poly(ADP-Rib) polymerase domain. RCD1 protein is found by yeast two-hybrid screens to interact with several transcription factors that are involved in both developmental and stress-related processes (Jaspers et al., 2009). It was recently shown to interact with DREB2A to influence senescence and stress responses (Vainonen et al., 2012). These data together indicate that RCD1 is required for ROS signaling and plant growth and development.
Here, we report the isolation of rcd1 as a mutant that enhances the growth defect but not disease resistance of an autoimmune mutant induced by an active form of the R protein. Further genetic characterization indicates that ROS have a major impact on growth defects induced by defense responses and that growth inhibition can be modulated independently from disease resistance.
RESULTS
Isolating an int51 Mutant as an Enhancer of snc1
To understand the molecular mechanisms underlying high-temperature suppression of growth defects in defense response mutants, we screened for temperature-insensitive mutants in the background of the temperature-sensitive autoimmune mutant suppressor of npr1, constitutive1 (snc1). The snc1 mutant has a missense mutation in a Nucleotide Binding-Leu Rich Repeat (NB-LRR) R protein resulting in constitutive activation of the SNC1 protein. It is dwarf at 22°C due to constitutive defense responses (Zhang et al., 2003). However, the mutant phenotype of snc1 exhibited at 22°C is rescued at 28°C due to a suppression of defense responses by the elevated temperature (Yang and Hua, 2004; Fig. 1A). Approximately 30,000 individual M2 plants were screened for mutants showing a dwarf phenotype at 28°C. One such mutant, insensitive to temperature51 (int51) snc1, exhibited a smaller stature than the snc1 single mutant (Fig. 1A). The single mutant of int51 was subsequently identified and confirmed (see below), and it exhibited a mild growth defect at both 22°C and 28°C (Fig. 1A). Rosette sizes were measured for the wild type, the single mutants, and the double mutant at 14 d after germination. At this developmental stage, when the phenotypes were not fully manifested, a synergistic interaction was already observed between int51 and snc1 (Fig. 1B). At 28°C, int51 was 57% of the wild type in size and int51 snc1 was approximately 34% of the wild type or snc1 (Fig. 1B). At 22°C, the snc1 and int51 single mutants were about 60% of the wild type in size and the double mutant was 25% of the wild type (Fig. 1B). The double mutant also showed root growth defects. At 14 d after germination, neither snc1-1 nor int51 single mutants showed drastic reduction of root growth. The double mutant had a much shorter root than either of the single mutants at 28°C but not at 22°C (Fig. 1C). These analyses indicate that int51 has a synergistic interaction with snc1 to affect both leaf and root growth.
Figure 1.

The int51 snc1 double mutant has reduced growth at both 28°C and 22°C. A, Growth phenotypes of wild-type Col-0, snc1 int51, and int51 snc1 at 22°C and 28°C before bolting. B, Diameter of rosettes of Col-0, snc1 int51, and int51 snc1 at 2 weeks after germination. C, Root length of the above plants grown on vertical plates 2 weeks after germination. A total of 20 and 25 seedlings were measured for A and B, respectively. Values represent means ± sd. Letters (a, b, c) indicate significant differences from the wild-type Col-0, and different letters indicate differences between different mutant genotypes (P < 0.05 according to Student’s t test). [See online article for color version of this figure.]
The int51 Mutant Is Defective in RCD1
The int51 phenotype is caused by a single nuclear recessive mutation. When int51 snc1 was backcrossed to snc1, the F1 plants exhibited wild-type morphology at 28°C and the F2 progeny segregated both dwarf and wild-type-looking plants at a ratio of approximately 1:3. To identify the int51 mutation, int51 snc1 in Columbia-0 (Col-0) was crossed to the wild-type Landsberg erecta (Ler), and their F2 progeny were used for map-based cloning. By genotyping 1,058 dwarf plants segregated at 28°C, the int51 mutation was positioned to a 28-kb region between markers Ch1.1161 and Ch1.1163 located at 11.614 and 11.642 Mb of chromosome 1, respectively (Fig. 2A; Supplemental Fig. S1A). Sequencing this region in int51 snc1 revealed a single G-to-A transition in the gene RCD1, resulting in a premature stop codon at Trp-431, and we named this mutant rcd1-5 (Fig. 2B). This allele, like the earlier allele rcd1-1 (Overmyer et al., 2000), showed a slightly dwarf phenotype, with 40% reduction in diameter at both temperatures (Fig. 1, A and B). The rcd1-5 defect could be rescued by transforming an RCD1 complementary DNA under the control of the 35S promoter (Supplemental Fig. S1B), indicating that the growth defect is indeed caused by the rcd1 mutation. RNA-blot analysis revealed a slightly higher expression of the RCD1 transcripts in rcd1-5 than in the wild type (Supplemental Fig. S1C), suggesting a possible feedback regulation of RCD1 itself.
Figure 2.

The int51 mutant is a new allele of RCD1. A, Map-based cloning of the INT51 gene. Shown are the molecular markers used for mapping, their positions on chromosome I (ChrI), and the number of recombinants (Rec #) at each molecular marker. B, A mutation was identified in the RCD1 gene in the int51 mutant. This G-to-A substitution leads to the change of Trp-431 to a stop codon. Exons are in black blocks, and introns are in gray blocks. The asterisks indicate different rcd1 alleles. int51 is named as rcd1-5. C, The rcd1-1 snc1 double mutant displays a dwarf phenotype similar to rcd1-5 snc1. Shown are 3-week-old plants at 28°C. [See online article for color version of this figure.]
To confirm that the rcd1-5 mutation indeed confers a dwarf phenotype to snc1 at 28°C, we generated a double mutant between rcd1-1 and snc1. The rcd1-1 snc1 double mutant showed the same phenotype to that of int51 snc1: dwarf at 28°C and 22°C, with a more severe phenotype at 22°C (Fig. 2C). Therefore, mutation in RCD1 indeed confers the int51 phenotype. For simplicity, we use rcd1 to refer to rcd1-5 (int51) and rcd1 snc1 to refer to int51 snc1 in the rest of this paper.
The Growth Defect Phenotype of rcd1 snc1 Is Not Due to Enhanced Disease Resistance
We determined whether growth defects of rcd1 snc1 at 28°C are due to enhanced disease resistance, as in a number of int mutants studied earlier (Zhu et al., 2010, 2012; Mang et al., 2012). As the rcd1 single mutant has a slightly dwarfed phenotype, we also asked if this growth defect is associated with enhanced disease resistance. A virulent bacterial pathogen Pseudomonas syringae pv tomato (Pst) DC3000, was used to infect the wild-type, snc1, rcd1, and rcd1 snc1 plants, and its growth in plants was measured at day 0 and day 3 after inoculation (Fig. 3A). At 22°C, wild-type and rcd1 plants supported the same amount of bacterial growth at 3 d after inoculation, while snc1 and rcd1 snc1 showed a similar extent of resistance to pathogen. At 28°C, rcd1 exhibited the same susceptibility to the pathogen as the wild type. Disease resistance in snc1 was reduced at 28°C than at 22°C. It had either no increase or a slight increase of resistance to pathogen compared with the wild type. The rcd1 snc1 double mutant was not more resistant than the snc1 single mutant at 28°C. Thus, rcd1 is as susceptible as the wild type to this bacterial pathogen, and it does not enhance the resistance of snc1 at 22°C or 28°C.
Figure 3.

rcd1 does not enhance disease resistance at 22°C or 28°C in Arabidopsis. A, Growth of Pst DC3000 in Col-0, rcd1, snc1, and rcd1 snc1 at 22°C and 28°C. Shown is the growth of bacterial strains at 0 and 3 d post inoculation. Each genotype at each temperature had three biological repeats in each experiment. Values represent means ± sd (n = 3). The letter a indicates a significant difference from the wild-type Col-0 but the same between the two genotypes (P < 0.05 according to Student’s t test). Similar results were obtained in three independent experiments, although snc1 and rcd1 snc1, while behaving the same, did not always show significant differences from Col-0 at 28°C at 3 d post inoculation. cfu, Colony-forming units. B, Expression of EDS1 and PR1 is not altered by the rcd1 mutation in snc1 at either 22°C or 28°C assayed by RNA blots. Ribosomal RNA was used as a loading control. C, Quantification of SA levels in Col-0, snc1, rcd1, and rcd1 snc1. Plants were grown on soil for 3 weeks at 22°C and 28°C. Data are means ± sd (n = 3) from three biological repeats. Asterisks indicate significant differences from the wild-type Col-0 (P < 0.05). FWT, Fresh weight. D, SA and PAD4 are not required for the dwarf phenotype of rcd1. Shown are the wild-type Col-0, rcd1, snc1, rcd1 snc1, snc1 NahG, rcd1 NahG, rcd1 snc1 NahG, snc1 pad4, rcd1 pad4, and rcd1 snc1 pad4 plants grown at 22°C and 28°C. [See online article for color version of this figure.]
Correlated with the disease resistance phenotype, the expression of defense response genes PR1 and EDS1 was not affected by the rcd1 mutation at either temperature (Fig. 3B). Similarly, no effect on the level of SA was observed for the rcd1 mutation (Fig. 3C). The rcd1 mutant accumulated the same amount of SA as the wild type, while rcd1 snc1 accumulated the same amount of SA as the snc1 single mutant at 22°C. At 28°C, the rcd1 single mutant appeared to accumulate a lower amount of SA, but rcd1 snc1 accumulated the same amount of SA as Col-0 and snc1, indicating that rcd1 does not enhance SA accumulation in the wild-type background or the snc1 background. The growth defects of rcd1 are not dependent on SA or PAD4, a mediator of defense responses (Wiermer et al., 2005). While nahG and pad4 largely suppressed the snc1 phenotype at 22°C, they did not alter the phenotype of rcd1 (Fig. 3D). The rcd1 snc1 nahG and rcd1 snc1 pad4 plants looked essentially the same as rcd1 plants (Fig. 3D), indicating that PAD4 and SA are required for R activation to inhibit growth. In sum, rcd1 enhances the growth defects of snc1 without enhancing its disease resistance, and the growth defects of rcd1 are not due to enhanced disease resistance.
rcd1 Enhances Growth Defects of the Autoimmune Mutant bon1
To investigate whether the rcd1 mutation could enhance the growth defects of other constitutive defense response mutants, we introduced the rcd1 mutation to the autoimmune mutant bonzai1-1 (bon1). The BON1 gene encodes an evolutionarily conserved calcium-binding protein localized to the plasma membrane (Hua et al., 2001; Li et al., 2010) and acts as a negative regulator of SNC1 (Yang and Hua, 2004). Similar to snc1, the bon1 mutant exhibits a temperature-dependent growth and defense phenotype (Yang and Hua, 2004). A double mutant of rcd1 and bon1 was generated, and it exhibited a more severe growth defect than either of the single mutants at 22°C (Supplemental Fig. S2A), indicating an enhancement of the growth defect of bon1 by rcd1. However, the double mutant had a growth phenotype similar to that of rcd1 at 28°C (Supplemental Fig. S2A), suggesting a more complete suppression of the bon1 defects than the snc1 defects at high temperatures.
Mutants vtc2 and vtc3 Enhance the Growth Defects of snc1
RCD1 was proposed to function in ROS signaling, and the enhancement of growth defects of snc1 by rcd1 independent of defense activation suggests a synergistic interaction of the defense response and the ROS response in growth regulation. Therefore, we tested the interaction of snc1 with several mutants defective in ROS generating or scavenging. l-Ascorbic acid (vitamin C) is an antioxidant that detoxifies ROS, in particular H2O2 (Smirnoff, 2000). Three vtc mutants (vtc1, vtc2, and vtc3) were identified as accumulating lower levels of l-ascorbic acid compared with the wild type (Conklin et al., 2000). Double mutants were generated between snc1 and the three vtc mutants, and their growth phenotypes were analyzed at 22°C and 28°C. While neither the snc1 nor the vtc mutants exhibited any growth defects at 28°C, the vtc2 snc1 and vtc3 snc1 double mutants, but not the vtc1 snc1 double mutant, showed more severe growth defects than either single mutant at both temperatures (Fig. 4, A and B; Supplemental Fig. S2).
Figure 4.

The vtc mutations enhance growth defects but not disease resistance of snc1. A, The vtc2 mutation enhances growth defects of snc1 at both 22°C and 28°C. Shown are Col-0, snc1, vtc2, and vtc2 snc1 plants grown on soil for 2 weeks. B, The vtc3 mutation enhances growth defects of snc1 at both 22°C and 28°C. Shown are Col-0, snc1, vtc3, and vtc3 snc1 plants grown on soil for 2 weeks. C, Disease resistance to Pst DC3000 is not enhanced in the vtc2 snc1 double mutant compared with the snc1 single mutant at 22°C or 28°C. Shown is the growth of bacterial strains at 0 and 3 d post inoculation. Each genotype at each temperature had three biological repeats in each experiment. Values represent means ± sd (n = 3). Asterisks indicate significant differences from the wild-type Col-0 (P < 0.05 according to Student’s t test). Similar results were obtained in three independent experiments. cfu, Colony-forming units. [See online article for color version of this figure.]
As the vtc2 mutant was reported to have elevated cell death and disease resistance (Pavet et al., 2005; Mukherjee et al., 2010), we tested if the growth defect in vtc2 snc1 at 28°C could be due to an enhanced disease resistance. The growth of the virulent pathogen Pst DC3000 was compared with the wild type, snc1, vtc2, and vtc2 snc1 (Fig. 4C). Both the snc1 and vtc2 mutants had enhanced disease resistance at 22°C but were as susceptible as the wild type at 28°C. However, the vtc2 snc1 double mutant did not exhibit an enhanced disease resistance compared with the single mutants at either temperature. Thus, similar to rcd1, the vtc2 and probably the vtc3 mutation enhances the growth defects of snc1 at 22°C and induces the growth defects of snc1 at 28°C without enhancing disease resistance.
High Accumulation of H2O2 in rcd1 snc1
The dissociation of the effects of defense and growth on snc1 by rcd1 and vtc2 suggests that perturbed ROS production or signaling might be the cause of growth defects in rcd1 snc1. To this end, we analyzed the levels of two ROS, H2O2 and O2−, in the wild type, snc1, rcd1, and rcd1 snc1 at 22°C and 28°C. As indicated by staining with 3,3′-diaminobenzidine (DAB), snc1 but not rcd1 accumulated a higher level of H2O2 than the wild type at 22°C, and rcd1 snc1 accumulated an even higher level of H2O2 (Fig. 5A). At 28°C, no obvious accumulation of H2O2 was observed in snc1, rcd1, or the wild type, but the rcd1 snc1 double mutant had a dramatic increase of H2O2 (Fig. 5A). As indicated by staining with nitroblue tetrazolium (NBT), the rcd1 single mutant had a higher accumulation of O2− than the wild type or snc1, while the rcd1 snc1 double mutant had a lower level of O2− than in rcd1 at both temperatures (Fig. 5B). Thus, the enhanced growth defects phenotype in rcd1 snc1 is correlated with a higher H2O2 level.
Figure 5.

The rcd1 snc1 mutant accumulates a higher level of H2O2. A, H2O2 is increased in the rcd1 snc1 double mutant at both 22°C and 28°C. Shown are DAB staining of the wild-type Col-0, rcd1, snc1, and rcd1 snc1 grown at two temperatures. B, O2− is decreased in the rcd1 snc1 double mutant at both 22°C and 28°C. Shown is NBT staining of the wild-type Col-0, rcd1, snc1, and rcd1 snc1 grown at two temperatures.
Partial Suppression of the Growth Defect of rcd1 snc1 by rboh Mutations
To determine if the high accumulation of H2O2 is the cause of growth defects, we introduced into rcd1 snc1 mutations that reduce the level of ROS. ROS are generated through metabolic processes as well as by RBOH (for respiration burst oxidase homolog) NADPH oxidases (Torres et al., 2005). Among the 10 RBOH members, AtRbohD and AtRbohF are the two expressed throughout most of the plant tissues, and they contribute to innate immune defense and ABA-dependent stomatal opening (Torres et al., 2002; Kwak et al., 2003). Loss-of-function mutants of rbohD and rbohF were introduced into rcd1 snc1. The rbohF mutation partially rescued the growth defect of rcd1 snc1 at 28°C but not 22°C (Fig. 6A). The level of H2O2 was greatly reduced in rcd1 snc1 rbohF compared with rcd1 snc1, correlating with the growth phenotype (Fig. 6B). The rbohD mutation suppressed the rcd1 snc1 defects at both 22°C and 28°C (Fig. 6C), and this was correlated with a reduction of H2O2 in the triple mutant. These data suggest that RbohD and RbohF are required for growth inhibition in the rcd1 snc1 double mutant and that this is likely through the production of H2O2.
Figure 6.

Partial suppression of the rcd1 snc1 phenotypes by rbohF and rbohD mutations. A and B, Growth phenotypes (top) and H2O2 accumulation (bottom) of the wild-type Col-0, snc1, rcd1, rbohF, rcd1 snc1, rcd1 rbohF, and rcd1 snc1 rbohF at 22°C (A) and 28°C (B). C and D, Growth phenotypes (top) and H2O2 accumulation (bottom) of the wild-type Col-0, snc1, rcd1, rbohD, rcd1 snc1, rcd1 rbohD, and rcd1 snc1 rbohD at 22°C (C) and 28°C (D). Accumulation of H2O2 in the above plants is indicated by DAB staining.
DISCUSSION
Plant growth is often inhibited by elevated defense responses, which is readily evident from the dwarf phenotypes exhibited by autoimmune mutants. Blocking defense responses by disrupting signaling components such as PAD4, EDS1, and SA often suppresses growth inhibition, indicating that early defense signaling molecules are responsible for both defense and growth. Whether and where the regulation of defense and growth become separated are not known. Here, we isolated an enhancer of autoimmune mutants only in growth inhibition but not in defense activation, revealing a bifurcation in the regulation of two processes after the core defense signaling involving PAD4 and SA (Fig. 7).
Figure 7.
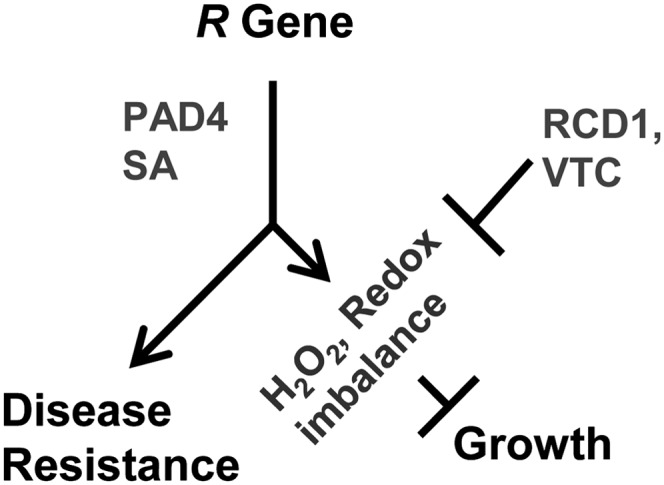
Model for growth inhibition induced by R genes.
Our study indicates that growth inhibition by defense activation is tightly linked to ROS homeostasis and responses and that this effect can be independent of defense responses (Fig. 7). It is striking that snc1 has no activation of defense responses at high temperature and yet snc1 still has a synergistic interaction with rcd1 on growth inhibition at high temperature. This indicates that the activated R gene snc1 has two effects: one is disease resistance induction, which can be suppressed by temperature, and the other is growth inhibition, which can be enhanced by other mutations independently of defense responses. The snc1 growth defect enhancer mutant rcd1 has altered ROS responses and stress responses, suggesting that the second effect of an autoimmune mutant is a change of redox status. This is supported by the enhancement of the growth defect of snc1 by vtc2 and vtc3 mutations, both of which cause lower levels of ROS-detoxifying ascorbic acids.
The level of H2O2 is correlated with the extent of growth inhibition: it is accumulated more in snc1 than in the wild type and even more in rcd1 snc1. The rcd1 snc1 phenotypes were suppressed by mutations in the ROS-generating genes RbohD and RbohF. These NADPH oxidases are responsible for oxidative bursts during pathogen invasion, and they often contribute to cell death but not necessarily disease resistance (Marino et al., 2012). The level of H2O2 was indeed reduced in rboh rcd1 snc1 compared with rcd1 snc1 when growth defects were inhibited, indicating that the high accumulation of H2O2 is at least partially responsible for the growth inhibition. We hypothesize that RBOH oxidases generate ROS in snc1, which leads to growth inhibition when the redox system is further perturbed. Mutation in RbohD but not RbohF suppressed the growth defects of snc1 rcd1 at 22°C, indicating that RbohD has a larger role than RbohF in mediating SNC1 function. This is consistent with earlier observations that the RbohD gene has a larger role than RbohF in most of the defense responses analyzed. We also observed a decrease of O2− in the rcd1 snc1 mutant compared with the single mutants. O2− has been implicated in promoting growth in roots, as it accumulates at the site of ectopic bulges in the suprecentipede1 mutant defective in the RhoGTPase GDP dissociation inhibitor AtRhoGDI1 (Carol et al., 2005). It has yet to be determined whether low O2− in rcd1 snc1 contributes to reduced growth and whether it results in high H2O2 through conversion.
In general, there is a synergistic interaction between defense response mutants and ROS mutants. More severe dwarf phenotypes were observed for rcd1 snc1, rcd1 bon1, vtc2 snc1, and vtc3 snc1 at 22°C. However, no enhancement was observed for rcd1 bon1 at 28°C. It is possible that high temperature has a more complete rescue of the bon1 defects than the snc1 defects and, therefore, that rcd1 bon1 behaves like rcd1. Although vtc1, vtc2, and vtc3 all accumulate less ascorbic acid and VTC1 and VTC2 both encode enzymes for vitamin biosynthesis, vtc1 does not enhance snc1 while vtc2 and vtc3 do. The VTC1-coded enzyme provides GDP-Man, which is used for cell wall carbohydrate biosynthesis and protein glycosylation as well as for ascorbic acid biosynthesis (Conklin et al., 1999). Therefore, the effect of vtc1 is pleiotropic and the interaction of vtc1 with snc1 might be more complex. It is also not yet determined whether these VTC genes have differential expression in response to temperature or defense activation and whether their mutants might perturb ROS homeostasis differently under different cellular or environmental conditions.
Moderately high temperatures such as 28°C can inhibit resistance mediated by R genes such as SNC1 due to its inhibition of R activities, and specific mutant forms of SNC1 can confer disease resistance at 28°C (Zhu et al., 2010). Therefore, potential moderate heat stress and/or perturbation of physiology induced by a moderate high temperature of 28°C are not responsible for the inhibition of disease resistance. However, the stress or perturbation could be enhanced by the loss of the ROS-regulating gene RCD1. This probably explains why the rcd1 snc1 double mutant had a great reduction in root elongation at 28°C but not at 22°C compared with the wild type and the single mutants (Fig. 1C).
This study further demonstrates a general tradeoff between growth and disease resistance. While biotic resistances are often associated with compromises in growth, growth defects, such as in a dwarfing mutant defective in growth hormone signaling, can be associated with resistance to some pathogens (Saville et al., 2012). Furthermore, ABA-deficient mutants can have enhanced disease resistance at high temperature (Mang et al., 2012; Zhu et al., 2012). Plants thus have complex systems to tune in to their environment and modulate their growth and responses to ensure best survival and reproduction.
MATERIALS AND METHODS
Plant Material and Growth Conditions
Arabidopsis (Arabidopsis thaliana) plants were grown on soil at 22°C or 28°C under constant light (approximately 100 μmol m−2 s−1) with a relative humidity between 40% and 60% for morphological phenotypic and gene expression analysis. Plants for pathogen tests were grown under a 12/12-h photoperiod. Arabidopsis seedlings used for protoplast transformation were grown on solid medium with one-half-strength Murashige and Skoog salts, 2% (w/v) Suc, and 0.8% (w/v) agar under a photoperiod of 8 h of light/16 h of dark.
Mutant Screen and Map-Based Cloning
The procedure was similar to one described previously (Mang et al., 2012). The snc1-1 seeds were treated with 0.25% ethane methylsulfonate for 12 h. Approximately 30,000 M2 plants (derived from 3,000 M1 plants) were screened at 28°C for int mutants with the 22°C snc1-1-like dwarf phenotype. The mapping populations for int51 were created by crossing the mutant in the Col-0 accession to the wild type of the Ler accession. Bulked segregation analysis was performed on pools of 40 plants with simple sequence length polymorphism, cleaved-amplified polymorphic sequence, and derived cleaved-amplified polymorphic sequence markers between Col-0 and Ler.
Transgenic Plant Generation
Agrobacterium tumefaciens stain GV3101 (Koncz and Schell, 1986) carrying the 35S:RCD1 construct was used to transform rcd1-5 plants via standard floral dipping. Primary transformants were selected on solid medium containing hygromycin.
RNA Analysis
The procedure was similar to one described previously (Zhu et al., 2010). Total RNAs were extracted using TRI Reagent (Molecular Research) from leaves of 3-week-old plants. Twenty micrograms of total RNAs per sample was used for RNA gel-blot analysis according to a standard procedure.
Pathogen Resistance Assay
Pseudomonas syringae pv tomato DC3000 was grown 2 to 3 d on King's B medium and resuspended at 105 colony-forming units mL−1 in a solution of 10 mm MgCl2 and 0.02% Silwet L-77. Two-week-old seedlings were dip inoculated with bacteria and kept covered for 1 h before being slowly uncovered. The amount of bacteria in plants was analyzed at 1 h after dipping (day 0) and 3 d after dipping (day 3). Three seedlings were pooled, and three pools were measured for bacteria growth in each genotype and condition.
DAB and NBT Staining
The Col-0, snc1, rcd1, and rcd1 snc1 plants were grown on soil at 22°C or 28°C for 2 or 3 weeks. Production of H2O2 was assayed by DAB staining. DAB was dissolved in 50 mm Tris-acetate (pH 5.0) at an concentration of 1 mg mL−1. Whole seedlings were placed in the DAB solution and vacuum infiltrated until the tissues were soaked. The seedlings were then incubated at room temperature in the dark for 24 h before the tissues were cleared by several rounds of ethanol (75%). Production of O2− was detected by soaking the whole plant in 50 mm Tris-acetate (pH 5.0) at a concentration of 500 mm NBT for 30 min.
Supplemental Data
The following materials are available in the online version of this article.
Supplemental Figure S1. Characterization of the rcd1-5 allele.
Supplemental Figure S2. Growth phenotypes of rcd1 bon1 and vtc1 snc1.
Acknowledgments
We thank the Arabidopsis Biological Resource Center for rcd1-1, rbohD, and rbohF mutant seeds, Tiina Blomster and Kirk Overmyer for the 35S::RCD1 construct, and Jed Spark, Kim Spark, Weiqiang Qian, and Zhilong Bao for assistance with mutant characterization.
Glossary
- SA
salicylic acid
- ROS
reactive oxygen species
- O2−
superoxide
- H2O2
hydrogen peroxide
- ABA
abscisic acid
- Col-0
Columbia-0
- Ler
Landsberg erecta
- Pst
Pseudomonas syringae pv tomato
- DAB
3,3′-diaminobenzidine
- NBT
nitroblue tetrazolium
References
- Ahlfors R, Lång S, Overmyer K, Jaspers P, Brosché M, Tauriainen A, Kollist H, Tuominen H, Belles-Boix E, Piippo M, et al. (2004) Arabidopsis RADICAL-INDUCED CELL DEATH1 belongs to the WWE protein-protein interaction domain protein family and modulates abscisic acid, ethylene, and methyl jasmonate responses. Plant Cell 16: 1925–1937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apel K, Hirt H. (2004) Reactive oxygen species: metabolism, oxidative stress, and signal transduction. Annu Rev Plant Biol 55: 373–399 [DOI] [PubMed] [Google Scholar]
- Barceló AR, Ros LVG, Carrasco AE. (2007) Looking for syringyl peroxidases. Trends Plant Sci 12: 486–491 [DOI] [PubMed] [Google Scholar]
- Carol RJ, Takeda S, Linstead P, Durrant MC, Kakesova H, Derbyshire P, Drea S, Zarsky V, Dolan L. (2005) A RhoGDP dissociation inhibitor spatially regulates growth in root hair cells. Nature 438: 1013–1016 [DOI] [PubMed] [Google Scholar]
- Clarke JD, Aarts N, Feys BJ, Dong X, Parker JE. (2001) Constitutive disease resistance requires EDS1 in the Arabidopsis mutants cpr1 and cpr6 and is partially EDS1-dependent in cpr5. Plant J 26: 409–420 [DOI] [PubMed] [Google Scholar]
- Conklin PL, Norris SR, Wheeler GL, Williams EH, Smirnoff N, Last RL. (1999) Genetic evidence for the role of GDP-mannose in plant ascorbic acid (vitamin C) biosynthesis. Proc Natl Acad Sci USA 96: 4198–4203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conklin PL, Saracco SA, Norris SR, Last RL. (2000) Identification of ascorbic acid-deficient Arabidopsis thaliana mutants. Genetics 154: 847–856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deepak S, Shailasree S, Kini RK, Hause B, Shetty SH, Mithöfer A. (2007) Role of hydroxyproline-rich glycoproteins in resistance of pearl millet against downy mildew pathogen Sclerospora graminicola. Planta 226: 323–333 [DOI] [PubMed] [Google Scholar]
- Dunand C, Crèvecoeur M, Penel C. (2007) Distribution of superoxide and hydrogen peroxide in Arabidopsis root and their influence on root development: possible interaction with peroxidases. New Phytol 174: 332–341 [DOI] [PubMed] [Google Scholar]
- Durrant WE, Dong X. (2004) Systemic acquired resistance. Annu Rev Phytopathol 42: 185–209 [DOI] [PubMed] [Google Scholar]
- Foreman J, Demidchik V, Bothwell JH, Mylona P, Miedema H, Torres MA, Linstead P, Costa S, Brownlee C, Jones JD, et al. (2003) Reactive oxygen species produced by NADPH oxidase regulate plant cell growth. Nature 422: 442–446 [DOI] [PubMed] [Google Scholar]
- Foyer CH, Noctor G. (2005) Redox homeostasis and antioxidant signaling: a metabolic interface between stress perception and physiological responses. Plant Cell 17: 1866–1875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujibe T, Saji H, Arakawa K, Yabe N, Takeuchi Y, Yamamoto KT. (2004) A methyl viologen-resistant mutant of Arabidopsis, which is allelic to ozone-sensitive rcd1, is tolerant to supplemental ultraviolet-B irradiation. Plant Physiol 134: 275–285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grierson CS, Roberts K, Feldmann KA, Dolan L. (1997) The COW1 locus of Arabidopsis acts after RHD2, and in parallel with RHD3 and TIP1, to determine the shape, rate of elongation, and number of root hairs produced from each site of hair formation. Plant Physiol 115: 981–990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua J, Grisafi P, Cheng SH, Fink GR. (2001) Plant growth homeostasis is controlled by the Arabidopsis BON1 and BAP1 genes. Genes Dev 15: 2263–2272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jambunathan N, Siani JM, McNellis TW. (2001) A humidity-sensitive Arabidopsis copine mutant exhibits precocious cell death and increased disease resistance. Plant Cell 13: 2225–2240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaspers P, Blomster T, Brosché M, Salojärvi J, Ahlfors R, Vainonen JP, Reddy RA, Immink R, Angenent G, Turck F, et al. (2009) Unequally redundant RCD1 and SRO1 mediate stress and developmental responses and interact with transcription factors. Plant J 60: 268–279 [DOI] [PubMed] [Google Scholar]
- Jones MA, Raymond MJ, Yang Z, Smirnoff N. (2007) NADPH oxidase-dependent reactive oxygen species formation required for root hair growth depends on ROP GTPase. J Exp Bot 58: 1261–1270 [DOI] [PubMed] [Google Scholar]
- Koncz C, Schell J. (1986) The promoter of the TL-DNA gene 5 controls the tissue-specific expression of chimeric genes carried by a novel type of Agrobacterium binary vector. Mol Gen Genet 204: 383–396 [Google Scholar]
- Kwak JM, Mori IC, Pei ZM, Leonhardt N, Torres MA, Dangl JL, Bloom RE, Bodde S, Jones JD, Schroeder JI. (2003) NADPH oxidase AtrbohD and AtrbohF genes function in ROS-dependent ABA signaling in Arabidopsis. EMBO J 22: 2623–2633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Gou M, Sun Q, Hua J. (2010) Requirement of calcium binding, myristoylation, and protein-protein interaction for the copine BON1 function in Arabidopsis. J Biol Chem 285: 29884–29891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mang HG, Qian W, Zhu Y, Qian J, Kang HG, Klessig DF, Hua J. (2012) Abscisic acid deficiency antagonizes high-temperature inhibition of disease resistance through enhancing nuclear accumulation of resistance proteins SNC1 and RPS4 in Arabidopsis. Plant Cell 24: 1271–1284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marino D, Dunand C, Puppo A, Pauly N. (2012) A burst of plant NADPH oxidases. Trends Plant Sci 17: 9–15 [DOI] [PubMed] [Google Scholar]
- Miller G, Shulaev V, Mittler R. (2008) Reactive oxygen signaling and abiotic stress. Physiol Plant 133: 481–489 [DOI] [PubMed] [Google Scholar]
- Mittler R. (2002) Oxidative stress, antioxidants and stress tolerance. Trends Plant Sci 7: 405–410 [DOI] [PubMed] [Google Scholar]
- Mittler R, Vanderauwera S, Gollery M, Van Breusegem F. (2004) Reactive oxygen gene network of plants. Trends Plant Sci 9: 490–498 [DOI] [PubMed] [Google Scholar]
- Mukherjee M, Larrimore KE, Ahmed NJ, Bedick TS, Barghouthi NT, Traw MB, Barth C. (2010) Ascorbic acid deficiency in Arabidopsis induces constitutive priming that is dependent on hydrogen peroxide, salicylic acid, and the NPR1 gene. Mol Plant Microbe Interact 23: 340–351 [DOI] [PubMed] [Google Scholar]
- Nanda AK, Andrio E, Marino D, Pauly N, Dunand C. (2010) Reactive oxygen species during plant-microorganism early interactions. J Integr Plant Biol 52: 195–204 [DOI] [PubMed] [Google Scholar]
- Overmyer K, Tuominen H, Kettunen R, Betz C, Langebartels C, Sandermann H, Jr, Kangasjärvi J. (2000) Ozone-sensitive Arabidopsis rcd1 mutant reveals opposite roles for ethylene and jasmonate signaling pathways in regulating superoxide-dependent cell death. Plant Cell 12: 1849–1862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavet V, Olmos E, Kiddle G, Mowla S, Kumar S, Antoniw J, Alvarez ME, Foyer CH. (2005) Ascorbic acid deficiency activates cell death and disease resistance responses in Arabidopsis. Plant Physiol 139: 1291–1303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saville RJ, Gosman N, Burt CJ, Makepeace J, Steed A, Corbitt M, Chandler E, Brown JK, Boulton MI, Nicholson P. (2012) The ‘Green Revolution’ dwarfing genes play a role in disease resistance in Triticum aestivum and Hordeum vulgare. J Exp Bot 63: 1271–1283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiefelbein JW, Somerville C. (1990) Genetic control of root hair development in Arabidopsis thaliana. Plant Cell 2: 235–243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott IM, Clarke SM, Wood JE, Mur LA. (2004) Salicylate accumulation inhibits growth at chilling temperature in Arabidopsis. Plant Physiol 135: 1040–1049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirano Y, Kachroo P, Shah J, Klessig DF. (2002) A gain-of-function mutation in an Arabidopsis Toll Interleukin1 receptor-nucleotide binding site-leucine-rich repeat type R gene triggers defense responses and results in enhanced disease resistance. Plant Cell 14: 3149–3162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smirnoff N. (2000) Ascorbic acid: metabolism and functions of a multi-facetted molecule. Curr Opin Plant Biol 3: 229–235 [PubMed] [Google Scholar]
- Teotia S, Lamb RS. (2009) The paralogous genes RADICAL-INDUCED CELL DEATH1 and SIMILAR TO RCD ONE1 have partially redundant functions during Arabidopsis development. Plant Physiol 151: 180–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian D, Traw MB, Chen JQ, Kreitman M, Bergelson J. (2003) Fitness costs of R-gene-mediated resistance in Arabidopsis thaliana. Nature 423: 74–77 [DOI] [PubMed] [Google Scholar]
- Torres MA, Dangl JL, Jones JD. (2002) Arabidopsis gp91phox homologues AtrbohD and AtrbohF are required for accumulation of reactive oxygen intermediates in the plant defense response. Proc Natl Acad Sci USA 99: 517–522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres MA, Jones JD, Dangl JL. (2005) Pathogen-induced, NADPH oxidase-derived reactive oxygen intermediates suppress spread of cell death in Arabidopsis thaliana. Nat Genet 37: 1130–1134 [DOI] [PubMed] [Google Scholar]
- Tsukagoshi H, Busch W, Benfey PN. (2010) Transcriptional regulation of ROS controls transition from proliferation to differentiation in the root. Cell 143: 606–616 [DOI] [PubMed] [Google Scholar]
- Vainonen JP, Jaspers P, Wrzaczek M, Lamminmäki A, Reddy RA, Vaahtera L, Brosché M, Kangasjärvi J. (2012) RCD1-DREB2A interaction in leaf senescence and stress responses in Arabidopsis thaliana. Biochem J 442: 573–581 [DOI] [PubMed] [Google Scholar]
- Wiermer M, Feys BJ, Parker JE. (2005) Plant immunity: the EDS1 regulatory node. Curr Opin Plant Biol 8: 383–389 [DOI] [PubMed] [Google Scholar]
- Yang S, Hua J. (2004) A haplotype-specific resistance gene regulated by BONZAI1 mediates temperature-dependent growth control in Arabidopsis. Plant Cell 16: 1060–1071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Goritschnig S, Dong X, Li X. (2003) A gain-of-function mutation in a plant disease resistance gene leads to constitutive activation of downstream signal transduction pathways in suppressor of npr1-1, constitutive 1. Plant Cell 15: 2636–2646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Mang HG, Sun Q, Qian J, Hipps A, Hua J. (2012) Gene discovery using mutagen-induced polymorphisms and deep sequencing: application to plant disease resistance. Genetics 192: 139–146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Qian W, Hua J. (2010) Temperature modulates plant defense responses through NB-LRR proteins. PLoS Pathog 6: e1000844. [DOI] [PMC free article] [PubMed] [Google Scholar]