

Abstract
Small RNA-mediated regulation of chromatin structure is an important means of suppressing unwanted genetic activity in diverse plants, fungi, and animals. In plants specifically, 24-nt siRNAs direct de novo methylation to repetitive DNA, both foreign and endogenous, in a process known as RNA-directed DNA methylation (RdDM). Many components of the de novo methylation machinery have been identified recently, including multiple RNA polymerases, but specific genetic features that trigger methylation remain poorly understood. By applying whole-genome bisulfite sequencing to maize, we found that transposons close to cellular genes (particularly within 1 kb of either a gene start or end) are strongly associated with de novo methylation, as evidenced both by 24-nt siRNAs and by methylation specifically in the CHH sequence context. In addition, we found that the major classes of transposons exhibited a gradient of CHH methylation determined by proximity to genes. Our results further indicate that intergenic chromatin in maize exists in two major forms that are distinguished based on proximity to genes—one form marked by dense CG and CHG methylation and lack of transcription, and one marked by CHH methylation and activity of multiple forms of RNA polymerase. The existence of the latter, which we call CHH islands, may have implications for how cellular gene expression could be coordinated with immediately adjacent transposon repression in a large genome with a complex organization of genes interspersed in a landscape of transposons.
In a simplified view, eukaryotic genomes consist of two types of DNA, genic and intergenic, where the genic regions are associated with an open euchromatic state, and the inactive, intergenic regions are associated with a closed heterochromatic state. Segregation of the genome into active and repressed regions requires complex sets of enzymes to direct appropriate, context-specific activity of RNA polymerases. In theory, a conflict would emerge at the boundaries of intergenic and genic regions, where regions of genetic repression must cooperate or at least not interfere with the expression of genes. The plant Arabidopsis thaliana has provided a particularly fruitful system for investigating such conflicts, and multiple lines of evidence reveal spreading of repressive chromatin modifications from transposons into genes (e.g., Lippman et al. 2004; Henderson and Jacobsen 2008; Hollister and Gaut 2009; Ahmed et al. 2011).
In organisms with large genomes, the majority of the DNA is heterochromatic and comprised of tandem repeats and transposons, often one after another in nested arrangements, such that hundreds of kilobases of intergenic sequence may separate one gene or cluster of genes from the next. In these species, genes are the exception, special regions of genetic activity in a landscape of genetic repression. The largest contributor to the intergenic genome is typically the retroelement class of transposons (class I), which composes >75% of the maize genome (Baucom et al. 2009; Schnable et al. 2009). These transposons are found primarily in the deep intergenic spaces (multiple kb away from genes). In some cases, their locations can be explained by preference for insertion into heterochromatin because of transposon-encoded features such as chromodomains (Gao et al. 2008; Neumann et al. 2011). A smaller contribution to total genome size is made by the class II transposons, which are the DNA-type, cut-and-paste transposons first characterized by McClintock in maize (for review, see Feschotte et al. 2002). Certain class II transposons, such as mPING of the PIF/Harbinger superfamily in rice, have been observed to move frequently and tend to insert close to genes, affecting gene expression both positively and negatively (Naito et al. 2009). Others, such as members of the Mutator superfamily in maize, frequently insert within genes (for review, see Lisch 2002), and class II transposons are frequently implicated as being among the most important mutagens in many plants and animals (such as nematodes and insects).
Transposable elements and other repetitive regions are associated with DNA methylation, specific histone modifications, and other repressive chromatin features (for review, see Volpe and Martienssen 2011). Multiple regulatory mechanisms exist to direct these heterochromatic structural states, often overlapping with defense mechanisms against foreign nucleic acids. A prominent example is RNA interference (RNAi) (Fire et al. 1998), which can not only degrade RNA, but can also induce transcriptional gene silencing. In fact, short interfering RNA (siRNA)-mediated regulation of chromatin structure is widespread in both genic and intergenic regions and has been mechanistically linked to recruitment of heterochromatin modifications. The most thorough studies to date have been in plants and fungi (for review, see Volpe and Martienssen 2011); however, it is clear that similar mechanisms exist in animals, with recent examples including siRNA-induced trimethylation of Histone H3 Lysine 9 in Caenorhabditis elegans (Burkhart et al. 2011; Gu et al. 2012) and piRNA-induced recruitment of HP1 in Drosophila (Wang and Elgin 2011). Multiple observations indicate that small RNA-mediated chromatin modification mechanisms are also at work in mammals (for review, see Morris 2011).
The discovery of de novo DNA methylation in tobacco plants (Wassenegger et al. 1994) has provided a case study for how intergenic transcription coupled with siRNA production can induce chromatin modifications. This phenomenon, called RNA-directed DNA methylation (RdDM) establishes cytosine methylation in all sequence contexts. Separate pathways exist to maintain methylation depending on the sequence context (CG, CHG, and CHH; where H is A, C, or T); however, CHH methylation depends upon ongoing RdDM (for review, see Law and Jacobsen 2010). Whereas different proteins recognize methylation in CG and CHG contexts and induce other chromatin changes such as modification of nearby histones, consequences of CHH methylation remain unknown. RdDM has a clear role in repression of genetic activity, both for viral defense and transposon control, and cytosine methylation in all three sequence contexts is associated with repetitive DNA and gene silencing. The abundance of CHH methylation in plant repetitive elements is typically far less than either CHG or CG methylation (Feng et al. 2010; Zemach et al. 2010); hence the potency of RdDM is attributed to its recruitment of CG- and CHG-specific methyltransferases that amplify CG and CHG methylation independent of CHH.
To date, only plants with unusually small genomes and low repeat content have been selected for bisulfite sequencing-based, whole-genome methylation (Cokus et al. 2008; Feng et al. 2010; Zemach et al. 2010). We hypothesized that applying recent advances in bisulfite sequencing to maize, with its more representative genome size and rich repertoire of transposons (Baucom et al. 2009; Schnable et al. 2009), would reveal features of intergenic chromatin regulation and its relation to gene expression, transposon regulation, and intergenic chromatin structure that could be difficult to detect in the compact genomes already characterized. Despite being dominated by transposons and transposon relics, the maize genome has sufficient polymorphism in its repetitive sequences to align the majority of short reads (Schnable et al. 2009; Gent et al. 2012). Several studies have been carried out on whole-genome methylation analysis or at least on large parts of the genome. While each has made significant progress in elucidating the methylation distribution in the maize genome, all of these studies have been severely limited by weaknesses in experimental methods, i.e., antibody or other protein-binding biases (Schnable et al. 2009; Wang et al. 2009; Eichten et al. 2011; Gent et al. 2012), inability to measure methylation in repetitive regions (Eichten et al. 2011), or inability to distinguish methylation sequence context (Schnable et al. 2009; Wang et al. 2009; Eichten et al. 2011).
In order to overcome these limitations, we used Illumina technology and sequenced bisulfite-treated DNA to 7× coverage of the genome. Consistent with the abundance of repetitive elements in the maize genome, we found that a general feature of the genome is a dense background of methylation in all sequence contexts. Near genes, however, methylation in the CG and CHG contexts dropped dramatically, but methylation in the CHH context increased. We found that this CHH methylation could be explained by genes promoting de novo methylation on flanking intergenic chromatin (particularly within a kb of gene starts and ends).
Results
Enrichment for RdDM near genes
We recently reported that 24-nt siRNAs, the type that specifically guide de novo methylation, are enriched in gene-rich areas of chromosomes rather than repeat-rich areas (Gent et al. 2012). We examined this phenomenon further by looking at siRNA distributions around individual genes, and we noticed a strong trend for 24-nt siRNAs to be concentrated very close to gene ends, as illustrated by a cluster of genes in Figure 1A. To compare siRNA and genome-wide methylation patterns, we chose a tissue containing a diversity of cell types, the outer layer of mature maize ears prior to fertilization. We speculated that this complex tissue would allow us to detect general DNA methylation trends rather than ones that might be characteristic of a single cell type. We sequenced sodium bisulfite-treated DNA libraries using the Illumina HiSeq system. After trimming adapter sequences and aligning to the 10 chromosomes of the maize reference genome (version 2), we obtained 198,333,982 uniquely aligning reads with an average length of 72.8 bases, for a total coverage of 7.0× (2,058,582,553-base length of the 10 chromosomes divided by 14,442,902,870 bases in the aligned, trimmed reads). Despite the size and repetition of the genome, we obtained at least 1× coverage for 65% of the genome—in other words, at least 65% of the genome is effectively single copy for the purpose of aligning these short reads. The reason for this abundance of effectively single-copy sequence is that, despite transposons existing at copy numbers in the tens of thousands, their primary sequences are identical only in the case of recently and perfectly transposed copies, and active transposition is rare (for review, see Feschotte et al. 2002). This level of coverage allowed for high-confidence measurements of methylation levels across the genome. (See Supplemental Fig. S1 for comparisons of methylation and coverage for example transposon superfamilies).
Figure 1.

Example distributions of 24-nt siRNAs and methylation near genes. (A) An example of an ∼20-kb region of chromosome 2 showing clusters of 24-nt siRNAs near genes. Each blue segment represents a single siRNA. The figure was modified from a screenshot from the Genomaize Genome Browser (http://genomaize.org; released 15 March 2012) with the B73 reference genome, version 2. Genomaize is derived from the UCSC Genome Browser (Kent et al. 2002). (B) An example 2-mb region of the genome showing methylation frequency (methylcytosine per total cytosine in each specific sequence context, left axis) and RNA abundance (normalized read count, right axis) for each 10-kb interval. mRNA reads are from a previously published study (Wang et al. 2009). For mRNA in A and siRNA reads in B, repetitively mapping reads were excluded.
We measured methylation values for CG, CHG, and CHH individually. The genome averages for all three forms of methylation were substantially higher than previously reported in plants with compact genomes. The percent of methylcytosines over total cytosines in each specific sequence context was 86% for CG, 74% for CHG, and 5.4% for CHH. For comparison, values reported in rice are 59% for CG, 21% for CHG, and 2.2% for CHH, and in Arabidopsis are 22% for CG, 5.9% for CHG, and 1.5% for CHH (Feng et al. 2010). Also, unlike other species where CG and CHG are highly concentrated in specific domains of the chromosomes, in maize, CG and CHG methylation were abundant in intergenic regions throughout the genome. Even in areas of high gene density, the regions between genes often exceeded 90% CG methylation, as displayed for a 2-mb region of the short arm of chromosome 2 (Fig. 1B).
Surprisingly, CHH methylation did not correlate with either CG or CHG methylation. From a zoomed-out, low-resolution perspective, as depicted in the example 2-mb region of chromosome 2 split into 10-kb intervals, CHH methylation appeared to be slightly enriched near genes and corresponded to gaps in the background of dense CG and CHG methylation (Fig. 1B). A genome-wide analysis of 24-nt siRNAs and CHH methylation relative to genes revealed strong enrichments within ∼1 kb upstream of transcription start sites and 1 kb downstream from transcription termini (Fig. 2). The highest peak of CHH methylation occurred ∼400 bp upstream of the transcription start sites, a region where CG and CHG methylation was still relatively low (Fig. 2, cf. A and B with C–F). The 24-nt siRNAs exhibited a very similar spatial distribution to CHH methylation both in the same tissue (Fig. 2H) and in seedling root tips (Supplemental Fig. S2). To determine whether this unusual distribution of CHH methylation and siRNAs could be related to the frequencies of CG, CHG, or CHH motifs in the DNA, we plotted the frequency of each in these same regions. None of the three motifs exhibited a pattern that matched CHH methylation (Supplemental Fig. S3). We call these regions of high CHH methylation and 24-nt siRNA abundance “CHH islands.”
Figure 2.

Genome-wide summary of 24-nt siRNA and methylation distributions near genes. (A–F) Distributions of methylation in each sequence context near genes. Both relative frequency (5-methylcytosine over total cytosine in the specific sequence context) and absolute frequency (5-methylcytosine over total nucleotides) are shown. The genes were divided into four sets based on expression level. Methylation values were measured for each 100-bp interval in a 2-kb region upstream of and downstream from gene ends for all annotated genes in the filtered gene set (version 5b). Values were also measured for the first 600 bp inside genes on each end. (G) Comparison of gene expression vs. flanking CHH methylation. Genes were split into four quartiles based on the level of CHH methylation in either the upstream or downstream 1 kb. The average expression level for each quartile is shown in RPKM (reads per kilobase per million mapped reads). (Error bars) Standard errors of the means; (*) statistically significant difference between means (P-value < .005). (H) Distributions of 24-nt siRNAs near genes. The average number of 24-nt siRNAs that aligned within each 100-bp interval is displayed for each set of genes. Both uniquely aligning and repetitive siRNAs are included in this plot. For an analysis of just unique siRNAs, see Supplemental Figure S2A.
To test whether the enrichment for CHH methylation in CHH islands was significantly different from the genome as a whole, we compared the 1-kb regions upstream of genes with a set of randomly selected 1-kb controls. We divided both sets of loci into four quartiles based on their levels of CHH methylation and made pairwise comparisons for each quartile (Supplemental Fig. S4A). For each quartile, the upstream 1-kb loci had at least twofold higher CHH methylation than the control loci (all P-values << 0.005). These results also indicate that it was not a small number of extreme cases that dominate the curves in Figure 2. To estimate the number of genes exhibiting high CHH methylation upstream, we set an arbitrary threshold value of CHH methylation at 5.44% (the genome average for CHH methylation) and asked how many of the upstream 1-kb regions and controls exceeded this threshold. Approximately 30% of the control loci had higher CHH methylation than the genome average, whereas 67% of the 1-kb upstream loci did (2.2-fold enrichment; Supplemental Fig. S4B). More strikingly, while only 13% of the control regions had at least twofold higher CHH methylation than the genome average, 48% of the 1-kb upstream loci did (3.8-fold enrichment).
Given the connections between RdDM and repression of foreign or repetitive DNA, we asked whether CHH levels were associated with expression levels of the genes themselves. We sequenced poly(A)-enriched RNA (mRNA) from the same tissue type that we used for bisulfite sequencing. Consistent with what was originally discovered in Arabidopsis and later reported in other plants including rice (Zhang et al. 2006; Zilberman et al. 2007; Cokus et al. 2008; Feng et al. 2010; Zemach et al. 2010), CG and CHG methylation dipped at both 5′ and 3′ ends of genes and exhibited an inverse correlation with gene expression (Fig. 2C–F). CHH methylation was distinct in that it was stably low within genes independent of gene expression (Fig. 2A,B). CHH methylation flanking genes, however, exhibited a surprising positive correlation with gene expression level. To test the significance of this trend, we split genes into quartiles based on the level of CHH methylation in their upstream and downstream 1-kb regions and measured mRNA expression for each category (Fig. 2G). Each CHH quartile was associated with progressively higher average gene expression, and the differences between the first and second and between the second and third quartiles were highly significant (P-values < 0.005 for rejection of the null hypothesis of no difference between categories).
These unexpected distributions of CHH methylation suggest that de novo methylation has attributes that are distinct from both CG- or CHG-specific maintenance methylation, and that CHH islands are related to activity of cellular genes.
RdDM marks near-gene transposons
The 24-nt siRNAs tend to be derived from repetitive regions, and more than half of the 24-nt siRNAs in our data set could not be aligned uniquely (e.g., cf. Fig. 2H and Supplemental Fig. S2A). Furthermore, RdDM is known to repress the expression of repetitive DNA such as transposons. We wondered then what relation CHH islands would have with transposons. We first asked whether transposons that are enriched in genic areas would exhibit different levels of CHH methylation and siRNA accumulation from those that are enriched in deep intergenic regions. To test this, we aligned our siRNA reads to the set of exemplar transposon sequences available from the Maize Transposable Element database (excluding unclassified transposons). Table 1 shows the number of 24-nt siRNAs that aligned to each of the superfamilies represented. As a means of normalizing siRNA abundance both for transposon abundance and for potential procedural biases, we also aligned a set of randomly sheared DNA fragments (Tenaillon et al. 2011) that we trimmed to 24-nt, and aligned to the exemplars in parallel with the siRNAs. We then aligned the control reads for each superfamily to the genome and counted the number that specifically aligned within 1-kb upstream of a gene. For both class I (retrotransposons) and class II (DNA transposons) we found a strong correlation between enrichment in the 1 kb upstream of genes and siRNA accumulation. Exemplifying deep intergenic chromatin, long terminal-repeat (LTR) retrotransposons of the Gypsy or Copia superfamilies had less than a third of the number of 24-nt siRNAs that would be expected given a random distribution of siRNAs in the genome. In contrast, class II transposons that are found preferentially in near-gene intergenic chromatin had strong enrichments for siRNA accumulation. For example, the Tc1/Mariner superfamily had 26-fold the level of 24-nt siRNAs expected by a random sampling. Strikingly, the single class II superfamily that was not enriched for 24-nt siRNAs, CACTA, was also not enriched near genes. The CACTA, Gypsy, and Copia superfamilies occurred at median distances of 12–27 kb from the nearest gene, while the L1, Tc1/Mariner, PIF/Harbinger, and Mutator superfamilies were found at median distances of 1–6 kb from genes (Supplemental Fig. S1D). These data suggest that transposon superfamilies of both class I and class II that are concentrated near genes tend to engage RdDM, while those that are enriched in deep intergenic regions do not.
Table 1.
Correlations between abundance of 24-nt siRNAs and enrichment for locations within 1-kb upstream of genes across diverse transposon superfamilies
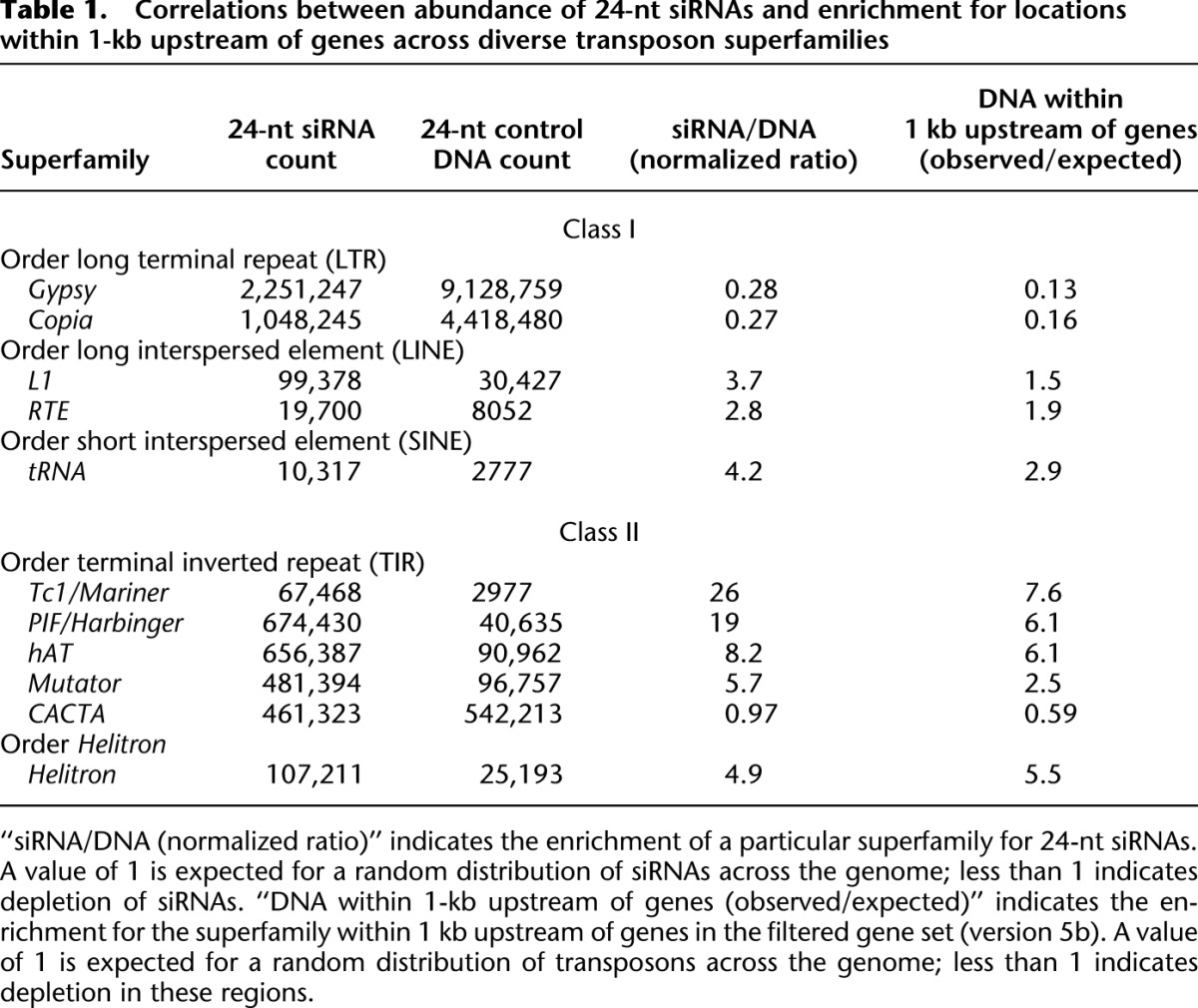
A question that immediately presents itself is whether these siRNAs have any impact on the expression of the transposons. These data predict that loss of 24-nt siRNAs would have strongest effects on transposons that are enriched near genes. Multiple genes required for accumulation of 24-nt siRNAs have been identified in maize (Alleman et al. 2006; Hale et al. 2007; Erhard et al. 2009; Sidorenko et al. 2009; Stonaker et al. 2009). One of these is an RNA-dependent RNA polymerase mutant homologous to rdr2 in Arabidopsis, called mop1 in maize (Alleman et al. 2006). Prior studies of the mop1 mutant found that Mutator transposons, which are enriched in genic areas (for review, see Lisch 2002) tend to transpose more frequently (Woodhouse et al. 2006), and Mutator and other class II transposons, with the notable exception of CACTA, exhibit increased RNA expression in the mop1 mutant (Jia et al. 2009). While effects of mop1 on whole-genome methylation patterns have not yet been characterized, the predicted consequence of reduced 24-nt siRNAs is reduced de novo methylation. Hence, the loss of 24-nt siRNAs and gain of mRNA expression for near-gene transposons in the mop1 mutant strongly supports the hypothesis that CHH islands repress near-gene transposons.
Near-gene transposons engage RdDM independent of transposon type
Two extreme possibilities could explain the association between near-gene transposons and RdDM. One is that certain types of transposons whose intrinsic features trigger RdDM are enriched near genes, either because of insertion preferences, selection, or other causes. The other extreme possibility is that intrinsic features of genes trigger RdDM on flanking transposons, independent of transposon type. To test these possibilities, we measured the average methylation for each transposon superfamily for the genome as a whole and compared it with the average methylation for the subset of transposon copies from the same superfamily that is located within 1 kb upstream of genes. In all cases, CHH methylation was higher upstream of genes relative to the genome as a whole (Fig. 3A). This trend was most striking for the retrotransposons that tended to be deeply intergenic—Gypsy and Copia—where CHH methylation increased greater than threefold close to genes (that is, within 1 kb). In contrast, CG and CHG methylation exhibited little variation for each transposon, regardless of location. Increased CHH methylation upstream of genes appeared to be independent of transposons, as even the regions that were not annotated as transposons exhibited high CHH methylation upstream of genes (though to a lesser degree than any of the eight transposon superfamilies examined).
Figure 3.

Methylation of transposons. (A) Comparison of transposon methylation in 1-kb regions upstream of genes with transposon methylation in the whole genome. In cases where a transposon copy extended beyond the 1-kb region, only the overlapping portion contributed to the analysis. For each superfamily, the difference in CHH methylation between the set of all copies (light blue, whole genome) and the subset upstream of genes (darker blue) was statistically significant (P-value < 0.005). Also shown are genome averages (both for the whole genome and for the regions within 1-kb upstream of genes) and transposon-free comparisons. Since excluding transposons enriched for genes, genes were also excluded. “Local” consists of portions of 1-kb regions that do not correspond to transposons or genes; “whole 1 KB” excludes entire 1-kb regions if they overlap at all with transposons or genes. (B) Comparison of methylation within single transposon copies relative to proximity of nearby genes. Copies that were contained in or overlapped with the region 1-kb upstream of genes were split into two halves, and the levels of methylation for the proximal and distal halves were measured separately. The transposons were also categorized by orientation relative to the genes, and the methylation averages for each orientation are shown separately. (Error bars) Standard errors of the means. For each of the superfamilies except for Tc1/Mariner and L1, the differences in CHH methylation between each half were statistically significant for both orientations (P-value < 0.005).
These data also suggest that proximity to genes is not the only determining factor in the extent to which transposons are subject to de novo methylation. Certain types of transposons are associated with higher levels of CHH methylation than others, even when close to genes. For example, PIF/Harbinger provides one extreme with an average of 33.2% CHH methylation at loci within 1 kb upstream of genes, while L1 LINEs are at the opposite extreme with 13.5% methylation (Fig. 3A). It is also possible that other features independent of transposon classification could contribute. For example, copy number and conservation of sequence identity among transposons could influence whether a particular transposon is targeted for de novo methylation. Copy number and identity are both reflected in the number of reads that can be uniquely aligned to a particular transposon; hence, to look for such effects on our measured CHH values we categorized transposons into quartiles based on how well they were covered by bisulfite reads, from no coverage (a coverage value of zero) to full-length coverage (reads spanning the entire copy, a coverage value of one). This analysis revealed a positive correlation between coverage and CHH methylation (Supplemental Fig. S1A). Mutator provided the most dramatic example, with 8.7% CHH methylation in the lowest coverage quartile and 13.4% CHH methylation in the highest coverage quartile. However, we found no consistent correlation between proximity to genes and transposon sequence coverage (Supplemental Fig. S1C,D). Therefore, while copy number and/or sequence identity do appear to contribute to CHH methylation within a transposon family, this trend does not detract from the larger point that transposons next to genes are more likely to experience higher CHH methylation that those of the same superfamily located elsewhere.
These data suggest that an interaction between genes and neighboring sequences is the major cause for CHH islands, where genes could induce methylation of neighboring sequences, though the magnitude of the effect could depend in part on other factors. To test whether proximity to genes is indeed a dominant factor, we selected all transposon copies with at least one edge that was within a kb from a gene, then split these copies into halves, and compared methylation of the proximal and distal halves. For each superfamily, the average proximal CHH methylation was higher than the distal CHH methylation, and the difference was evident regardless of the orientation of the copy relative to the gene (Fig. 3B). Taken together, the available data provide strong evidence that proximity to genes induces de novo methylation, regardless of transposon sequence or identity.
Discussion
The enrichment for 24-nt siRNAs and methylation specifically in the CHH context in regions immediately flanking genes is unexpected given the assumed primary role for de novo methylation: to give rise to high levels of CG and CHG methylation due to the activity of maintenance methyltransferases (for review, see Law and Jacobsen 2010). These forms of methylation are each associated with specific chromatin modifying enzymes; for example, the histone 3 lysine 9 methylase recognizes and binds specifically to methylated cytosines in the CHG context (Johnson et al. 2007). In contrast, factors that bind to methylated cytosine in the CHH context have not been identified. The disproportionally high frequency of CHH relative to CG and CHG that we observed near genes suggests a skewed ratio of de novo methylation over maintenance methylation near genes. Similarly, the types of transposons that were associated with the highest levels of both 24-nt siRNAs and CHH methylation are those that are enriched near genes.
Plant genomes exhibit a huge diversity of size and repeat content. Well-characterized plant genomes such as rice and Arabidopsis come from the small end of the genome size spectrum, while maize is more representative (for review, see Kelly and Leitch 2011). Maize is also distinct in that its repeats are not highly concentrated in the middle of the chromosomes, but also exist between genes even in gene-rich regions of chromosome arms (Schnable et al. 2009). However, a close look at methylation distributions in rice reveals that CHH methylation does not correlate with repeat density and exhibits a slight increase on both 5′ and 3′ ends of genes (Feng et al. 2010; Zemach et al. 2010). Arabidopsis is distinct from rice both in that it has a lower overall repeat content and in that its gene-rich areas are more clearly separated from its repeat-rich areas (for review, see Zhang 2008; Kelly and Leitch 2011). Hence, the lack of a strong CHH signal near genes in Arabidopsis, but the presence of one in rice makes sense in light of our observation that maize CHH islands reflect an interaction between genes and nearby sequences—in particular, an interaction that induces transposon silencing.
To summarize our results, maize intergenic chromatin exists in two general forms: deep intergenic chromatin that is densely methylated in CG and CHG contexts and is generally transcriptionally inactive, and near-gene intergenic chromatin that is enriched in methylation in the CHH context and is transcriptionally active. The evidence that the latter form, which we call CHH islands, is transcriptionally active comes from two sources: first, that we defined it by one form of a transcript (24-nt siRNAs); and second, that CHH methylation is known to require multiple forms of RNA polymerase activity. In fact, RdDM requires not only several unusual variants of RNA polymerase II (Pol II) and an RNA-dependent RNA polymerase (for review, see Haag and Pikaard 2011), but also RNA polymerase II itself (Zheng et al. 2009).
These observations bring up many intriguing possibilities as to the benefits of distinct modes of regulation of near-gene transposons and deep intergenic transposons. A form of densely methylated and potentially highly condensed deep intergenic heterochromatin makes good sense in terms of suppressing unwanted genetic activity, as the vast majority of the intergenic space is composed of transposons. Far away from genes, large-scale repressive chromatin structures could be tolerated without concern for inhibitory effects on gene expression. Genes, however, require windows of open chromatin for accessibility of Pol II and other components of transcription. These polymerase windows pose a challenge in that nearby transposons, particularly class II transposons—which can be dangerous mutagens because of their preference for insertion near or in genes—could take advantage of their proximity to Pol II to transpose. A nongenic form of transcription that occurs in CHH islands and its effects on chromatin structure may provide a mechanism for suppressing transposons without inhibiting gene expression.
The origin of transcripts 5′ and 3′ to genes and the precise function they provide is not clear; however, it may be related to the nature of gene transcription. While the vast majority of stable Pol II transcripts are derived from the enzyme's activity downstream from promoters, the production of unstable transcripts in the upstream orientation (bidirectional transcription) is well documented in budding yeast and human cells, and presumably present in plants as well (Core et al. 2008; Preker et al. 2008; Neil et al. 2009; Xu et al. 2009; Churchman and Weissman 2011). It is also conceivable that Pol II undergoes some form of scanning behavior in promoter areas, polymerizing transient RNAs in the process, and only initiating within the core promoter in response to appropriate cofactors associated with the classical TATA box or other transcriptional cis regulators. At gene 3′ ends, transcription by Pol II beyond the polyadenylation site is a well-established phenomenon (for review, see Mandel et al. 2008; Moore and Proudfoot 2009). On both ends of genes, even trace activity of Pol II could lead to engagement of RdDM, such that the very polymerase windows that could provide a dangerous opening for transposon expression could actually be used instead for transposon control.
These observations also raise the question of what restricts the activity of CG and CHG methyltransferases and potentially other heterochromatin factors in CHH islands. One intriguing possibility is that CHH islands also act as epigenetic insulators, using gene-intrinsic features such as the presence of Pol II to create flanking chromatin environments that help to inhibit the spread of dense heterochromatin into promoters and 3′ regulatory regions.
Methods
Tissue collection
Unfertilized ears from inbred B73 stock were harvested after silks had emerged. The inner cores (immature cobs) were removed and discarded and the kernel layer was flash-frozen in liquid nitrogen and preserved at −80°C.
Derivation of sequencing libraries
siRNAs
Small RNA-enriched RNA was extracted from frozen tissue using the mirVana miRNA Isolation Kit with Plant RNA Isolation Aid (Ambion), and Illumina sequencing libraries were prepared using oligos described by Gent et al. (2009) and a ligation scheme derived from the methods of Lau et al. ( 2001) and Pfeffer et al. ( 2005). This method selects for Dicer products and other small RNAs with both a 3′-OH and a 5′-monophosphate. The 3′ adapters were trimmed from the 50-nt, single-end reads using FAR (The Flexible Adapter Remover) Version 2.0 software (http://sourceforge.net/apps/mediawiki/theflexibleadap). Default parameters were used, except for the following: “format fastq–trim-end right–adaptive-overlap yes–min-readlength 22 max-uncalled 10”. The 5′-barcode, TAC, was also trimmed off the reads. To remove miRNAs from the data set, the reads were aligned to a list of maize miRNAs (http://www.mirbase.org) (Kozomara and Griffiths-Jones 2011). Blastall software was used for the alignment, with default parameters except for the expectation value (E) set to 1 × 10−4. After removal of miRNA matches, the resulting siRNA-enriched set was split up by length, and the 24-nt reads alone were included in subsequent analyses.
Control DNA
Control 24-nt reads were trimmed from longer reads derived from randomly sheared DNA (Tenaillon et al. 2011) as described previously (Gent et al. 2012).
mRNA
mRNA was extracted from frozen tissue using the Life Technologies Dynabeads mRNA DIRECT Kit, and Illumina sequencing libraries were prepared using the Epicentre ScriptSeq v2 RNA-Seq Library Preparation Kit. After sequencing, the 3′ adapters were trimmed from the reads using FAR with default parameters, except for the following: “format fastq–trim-end right–adaptive-overlap yes–min-readlength 53–max-uncalled 20.”
Bisulfite-treated DNA
Libraries were prepared for bisulfite sequencing using a method similar to that of Lister et al. (2009). The sample was Illumina sequenced in two parts: one lane of 50-nt, single-end, and a batch of seven lanes of 100-nt single-end. A substantial portion of the input DNA fragments were under 100 bp in length, so the 100-nt batch of reads was split into separate files based on length after trimming of 3′ adapter sequence (using cutadapt, http://code.google.com/p/cutadapt/): 30–39, 40–49, 50–59, 60–69, 70–79, 80–89, 90–99, and 100+ nt.
Whole-genome alignments, methylation calculations, and comparison with annotated genes
mRNA, siRNA, and control DNA reads were aligned to the maize genome version 2 using Bowtie software, version 2 (Langmead et al. 2009). For analysis of uniquely aligning reads, i.e., those with a single best alignment, only reads producing alignments with MAPQ values of greater than or equal to 30 were selected. Bisulfite reads were aligned using BS Seeker software (Chen et al. 2010). Depending on the trimmed read length, different numbers of mismatches were tolerated: 30–49, no mismatches; 50–69, one mismatch; 70–89, two mismatches; 80 and above, three mismatches. For the first set of reads (50-nt, single-end, default BS seeker parameters were used except “-t N -e 50 -m 1”. For the rest of the reads generated in the 100-nt, single-end run, -e was set to the lowest read length in the file, and -m to the appropriate number of mismatches.
Calculations of methylation frequency were obtained by counting the number of converted versus uncoverted cytosines in each aligned read for each cytosine (as indicated by BS Seeker). For loci containing multiple cytosines, the reported methylation value was calculated as the average of all the cytosines within a locus. For comparison of alignment counts or methylation frequencies near annotated genes, genome coordinates for the filtered gene set (version 5b) were obtained from http://ftp.maizesequence.org/current/filtered-set/. To split the genes into expression quartiles, we counted the number of mRNA reads that aligned to each exon, normalized by exon length, sorted by normalized read count to obtain RPKM values (reads per kilobase per million mapped reads), then divided into four categories, from lowest to highest RPKM. These categories are approximately equivalent to quartiles, but the lowest expression category has more genes (11,327 total) than each of the other three categories (9348 or 9349 total for each of the three). We categorized the genes in this way because 11,327 genes had zero corresponding reads, which would make it impossible to exactly separate the first and second quartiles (some of the second quartile genes would have zero reads and would be indistinguishable from the first quartile). Genome coordinates for transposon superfamilies were obtained from the set of MTEC repeats (version 5a; http://ftp.maizesequence.org/current/repeats/). Regions of overlap between genes and transposons were identified using Bedtools software (Quinlan and Hall 2010).
Determining transposon siRNA enrichments and distributions near genes
To identify reads that corresponded to particular transposons, both 24-nt control DNA and 24-nt siRNAs were aligned to the set of transposon sequences downloaded from the Maize Transposable Element Database (http://maizetedb.org/∼maize/ on 22 Sep 2011) using Blastall software with default parameters, except that the expectation value (E) was set to 1 × 10−4. siRNA/DNA normalized ratios in Table 1 were calculated by dividing the total number of siRNA and DNA reads for each superfamily of transposons by the total number of 24-nt siRNA reads and DNA reads that aligned to the 10 chromosomes (30,209,668 siRNA and 34,468,835 control). To estimate the abundance of transposons near genes, 24-nt control DNA reads corresponding to particular DNA superfamilies were aligned to the genome using Bowtie, version 1 (set to allow retention of repetitive alignments, “-n 0 -l 20 -M 1 –best”). The expected number of reads within 1-kb upstream of a gene was calculated for each transposon superfamily by multiplying the total number of control reads for that superfamily by the ratio of reads that aligned within 1-kb upstream of a gene over the total number that aligned to the 10 chromosomes (430,153/34,468,835).
Tests of statistical significance
To calculate P-values for the comparisons between gene expression and flanking CHH methylation, we used two-tailed Student's t-tests. Because we had prior expectations that CHH methylation would increase near genes, one-tailed Student's t-tests were used in the comparisons between CHH methylation relative to gene proximity. For the proximal-distal comparison of transposon halves, paired-sample t-tests were used. Welch's t-tests calculations were used in all cases because of the possibility of unequal variances.
Data access
All reads are available in the NCBI Sequence Read Archive (SRA) (http://www.ncbi.nlm.nih.gov/sra) under accession number SRA050144.
Acknowledgments
We thank Kathryn S. Hodges and Richard M. Gell for logistical support, Suhua Feng for help with preparing bisulfite sequencing libraries, Susan Wessler and Jeffrey Bennetzen for helpful discussions, and Yecheng Huang and the Georgia Advanced Computing Resource Center at the University of Georgia for computing and software support. This work was supported by NSF grants 0607123, 1118550, and 0922703, and by NIH grant F32GM095223.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.146985.112.
References
- Ahmed I, Sarazin A, Bowler C, Colot V, Quesneville H 2011. Genome-wide evidence for local DNA methylation spreading from small RNA-targeted sequences in Arabidopsis. Nucleic Acids Res 39: 6919–6931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alleman M, Sidorenko L, McGinnis K, Seshadri V, Dorweiler JE, White J, Sikkink K, Chandler VL 2006. An RNA-dependent RNA polymerase is required for paramutation in maize. Nature 442: 295–298 [DOI] [PubMed] [Google Scholar]
- Baucom RS, Estill JC, Chaparro C, Upshaw N, Jogi A, Deragon JM, Westerman RP, Sanmiguel PJ, Bennetzen JL 2009. Exceptional diversity, non-random distribution, and rapid evolution of retroelements in the B73 maize genome. PLoS Genet 5: e1000732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkhart KB, Guang S, Buckley BA, Wong L, Bochner AF, Kennedy S 2011. A pre-mRNA-associating factor links endogenous siRNAs to chromatin regulation. PLoS Genet 7: e1002249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen PY, Cokus SJ, Pellegrini M 2010. BS Seeker: Precise mapping for bisulfite sequencing. BMC Bioinformatics 11: 203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churchman LS, Weissman JS 2011. Nascent transcript sequencing visualizes transcription at nucleotide resolution. Nature 469: 368–373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cokus SJ, Feng S, Zhang X, Chen Z, Merriman B, Haudenschild CD, Pradhan S, Nelson SF, Pellegrini M, Jacobsen SE 2008. Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature 452: 215–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Core LJ, Waterfall JJ, Lis JT 2008. Nascent RNA sequencing reveals widespread pausing and divergent initiation at human promoters. Science 322: 1845–1848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichten SR, Swanson-Wagner RA, Schnable JC, Waters AJ, Hermanson PJ, Liu S, Yeh CT, Jia Y, Gendler K, Freeling M, et al. 2011. Heritable epigenetic variation among maize inbreds. PLoS Genet 7: e1002372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erhard KF, Stonaker JL, Parkinson SE, Lim JP, Hale CJ, Hollick JB 2009. RNA polymerase IV functions in paramutation in Zea mays. Science 323: 1201–1205 [DOI] [PubMed] [Google Scholar]
- Feng S, Cokus SJ, Zhang X, Chen PY, Bostick M, Goll MG, Hetzel J, Jain J, Strauss SH, Halpern ME, et al. 2010. Conservation and divergence of methylation patterning in plants and animals. Proc Natl Acad Sci 107: 8689–8694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feschotte C, Jiang N, Wessler SR 2002. Plant transposable elements: Where genetics meets genomics. Nat Rev Genet 3: 329–341 [DOI] [PubMed] [Google Scholar]
- Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC 1998. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 391: 806–811 [DOI] [PubMed] [Google Scholar]
- Gao X, Hou Y, Ebina H, Levin HL, Voytas DF 2008. Chromodomains direct integration of retrotransposons to heterochromatin. Genome Res 18: 359–369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gent JI, Schvarzstein M, Villeneuve AM, Gu SG, Jantsch V, Fire AZ, Baudrimont A 2009. A Caenorhabditis elegans RNA-directed RNA polymerase in sperm development and endogenous RNA interference. Genetics 183: 1297–1314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gent JI, Dong Y, Jiang J, Dawe RK 2012. Strong epigenetic similarity between maize centromeric and pericentromeric regions at the level of small RNAs, DNA methylation and H3 chromatin modifications. Nucleic Acids Res 40: 1550–1560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu SG, Pak J, Guang S, Maniar JM, Kennedy S, Fire A 2012. Amplification of siRNA in Caenorhabditis elegans generates a transgenerational sequence-targeted histone H3 lysine 9 methylation footprint. Nat Genet 44: 157–164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haag JR, Pikaard CS 2011. Multisubunit RNA polymerases IV and V: Purveyors of non-coding RNA for plant gene silencing. Nat Rev Mol Cell Biol 12: 483–492 [DOI] [PubMed] [Google Scholar]
- Hale CJ, Stonaker JL, Gross SM, Hollick JB 2007. A novel Snf2 protein maintains trans-generational regulatory states established by paramutation in maize. PLoS Biol 5: e275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson IR, Jacobsen SE 2008. Tandem repeats upstream of the Arabidopsis endogene SDC recruit non-CG DNA methylation and initiate siRNA spreading. Genes Dev 22: 1597–1606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollister JD, Gaut BS 2009. Epigenetic silencing of transposable elements: A trade-off between reduced transposition and deleterious effects on neighboring gene expression. Genome Res 19: 1419–1428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia Y, Lisch D, Ohtsu K, Scanlon M, Nettleton D, Schnable P 2009. Loss of RNA-dependent RNA polymerase 2 (RDR2) function causes widespread and unexpected changes in the expression of transposons, genes, and 24-nt small RNAs. PLoS Genet 5: e1000737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson LM, Bostick M, Zhang X, Kraft E, Henderson I, Callis J, Jacobsen SE 2007. The SRA methyl-cytosine-binding domain links DNA and histone methylation. Curr Biol 17: 379–384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly LJ, Leitch IJ 2011. Exploring giant plant genomes with next-generation sequencing technology. Chromosome Res 19: 939–953 [DOI] [PubMed] [Google Scholar]
- Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D 2002. The human genome browser at UCSC. Genome Res 12: 996–1006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozomara A, Griffiths-Jones S 2011. miRBase: Integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res 39: D152–D157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL 2009. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10: R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau NC, Lim LP, Weinstein EG, Bartel DP 2001. An abundant class of tiny RNAs with probable regulatory roles in Caenorhabditis elegans. Science 294: 858–862 [DOI] [PubMed] [Google Scholar]
- Law JA, Jacobsen SE 2010. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat Rev Genet 11: 204–220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippman Z, Gendrel AV, Black M, Vaughn MW, Dedhia N, McCombie WR, Lavine K, Mittal V, May B, Kasschau KD, et al. 2004. Role of transposable elements in heterochromatin and epigenetic control. Nature 430: 471–476 [DOI] [PubMed] [Google Scholar]
- Lisch D 2002. Mutator transposons. Trends Plant Sci 7: 498–504 [DOI] [PubMed] [Google Scholar]
- Lister R, Pelizzola M, Dowen RH, Hawkins RD, Hon G, Tonti-Filippini J, Nery JR, Lee L, Ye Z, Ngo QM, et al. 2009. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 462: 315–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandel CR, Bai Y, Tong L 2008. Protein factors in pre-mRNA 3′-end processing. Cell Mol Life Sci 65: 1099–1122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore MJ, Proudfoot NJ 2009. Pre-mRNA processing reaches back to transcription and ahead to translation. Cell 136: 688–700 [DOI] [PubMed] [Google Scholar]
- Morris KV 2011. The emerging role of RNA in the regulation of gene transcription in human cells. Semin Cell Dev Biol 22: 351–358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naito K, Zhang F, Tsukiyama T, Saito H, Hancock CN, Richardson AO, Okumoto Y, Tanisaka T, Wessler SR 2009. Unexpected consequences of a sudden and massive transposon amplification on rice gene expression. Nature 461: 1130–1134 [DOI] [PubMed] [Google Scholar]
- Neil H, Malabat C, d'Aubenton-Carafa Y, Xu Z, Steinmetz LM, Jacquier A 2009. Widespread bidirectional promoters are the major source of cryptic transcripts in yeast. Nature 457: 1038–1042 [DOI] [PubMed] [Google Scholar]
- Neumann P, Navrátilová A, Koblížková A, Kejnovský E, Hřibová E, Hobza R, Widmer A, Doležel J, Macas J 2011. Plant centromeric retrotransposons: A structural and cytogenetic perspective. Mob DNA 2: 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeffer S, Lagos-Quintana M, Tuschl T 2005. Cloning of small RNA molecules. Curr Protoc Mol Biol 72: 26.4.1–26.4.18 [DOI] [PubMed] [Google Scholar]
- Preker P, Nielsen J, Kammler S, Lykke-Andersen S, Christensen MS, Mapendano CK, Schierup MH, Jensen TH 2008. RNA exosome depletion reveals transcription upstream of active human promoters. Science 322: 1851–1854 [DOI] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM 2010. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 26: 841–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnable PS, Ware D, Fulton RS, Stein JC, Wei F, Pasternak S, Liang C, Zhang J, Fulton L, Graves TA, et al. 2009. The B73 maize genome: Complexity, diversity, and dynamics. Science 326: 1112–1115 [DOI] [PubMed] [Google Scholar]
- Sidorenko L, Dorweiler JE, Cigan AM, Arteaga-Vazquez M, Vyas M, Kermicle J, Jurcin D, Brzeski J, Cai Y, Chandler VL 2009. A dominant mutation in mediator of paramutation2, one of three second-largest subunits of a plant-specific RNA polymerase, disrupts multiple siRNA silencing processes. PLoS Genet 5: e1000725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stonaker JL, Lim JP, Erhard KF, Hollick JB 2009. Diversity of Pol IV function is defined by mutations at the maize rmr7 locus. PLoS Genet 5: e1000706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tenaillon MI, Hufford MB, Gaut BS, Ross-Ibarra J 2011. Genome size and transposable element content as determined by high-throughput sequencing in maize and Zea luxurians. Genome Biol Evol 3: 219–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volpe T, Martienssen RA 2011. RNA interference and heterochromatin assembly. Cold Spring Harb Perspect Biol 3: a003731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang SH, Elgin SC 2011. Drosophila Piwi functions downstream of piRNA production mediating a chromatin-based transposon silencing mechanism in female germ line. Proc Natl Acad Sci 108: 21164–21169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Elling AA, Li X, Li N, Peng Z, He G, Sun H, Qi Y, Liu XS, Deng XW 2009. Genome-wide and organ-specific landscapes of epigenetic modifications and their relationships to mRNA and small RNA transcriptomes in maize. Plant Cell 21: 1053–1069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wassenegger M, Heimes S, Riedel L, Sänger HL 1994. RNA-directed de novo methylation of genomic sequences in plants. Cell 76: 567–576 [DOI] [PubMed] [Google Scholar]
- Woodhouse MR, Freeling M, Lisch D 2006. Initiation, establishment, and maintenance of heritable MuDR transposon silencing in maize are mediated by distinct factors. PLoS Biol 4: e339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z, Wei W, Gagneur J, Perocchi F, Clauder-Münster S, Camblong J, Guffanti E, Stutz F, Huber W, Steinmetz LM 2009. Bidirectional promoters generate pervasive transcription in yeast. Nature 457: 1033–1037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zemach A, McDaniel IE, Silva P, Zilberman D 2010. Genome-wide evolutionary analysis of eukaryotic DNA methylation. Science 328: 916–919 [DOI] [PubMed] [Google Scholar]
- Zhang X 2008. The epigenetic landscape of plants. Science 320: 489–492 [DOI] [PubMed] [Google Scholar]
- Zhang X, Yazaki J, Sundaresan A, Cokus S, Chan SW, Chen H, Henderson IR, Shinn P, Pellegrini M, Jacobsen SE, et al. 2006. Genome-wide high-resolution mapping and functional analysis of DNA methylation in Arabidopsis. Cell 126: 1189–1201 [DOI] [PubMed] [Google Scholar]
- Zheng B, Wang Z, Li S, Yu B, Liu JY, Chen X 2009. Intergenic transcription by RNA polymerase II coordinates Pol IV and Pol V in siRNA-directed transcriptional gene silencing in Arabidopsis. Genes Dev 23: 2850–2860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zilberman D, Gehring M, Tran RK, Ballinger T, Henikoff S 2007. Genome-wide analysis of Arabidopsis thaliana DNA methylation uncovers an interdependence between methylation and transcription. Nat Genet 39: 61–69 [DOI] [PubMed] [Google Scholar]