

Abstract
With the completion of the zebrafish genome sequencing project, it becomes possible to analyze the function of zebrafish genes in a systematic way. The first step in such an analysis is to inactivate each protein-coding gene by targeted or random mutation. Here we describe a streamlined pipeline using proviral insertions coupled with high-throughput sequencing and mapping technologies to widely mutagenize genes in the zebrafish genome. We also report the first 6144 mutagenized and archived F1's predicted to carry up to 3776 mutations in annotated genes. Using in vitro fertilization, we have rescued and characterized ∼0.5% of the predicted mutations, showing mutation efficacy and a variety of phenotypes relevant to both developmental processes and human genetic diseases. Mutagenized fish lines are being made freely available to the public through the Zebrafish International Resource Center. These fish lines establish an important milestone for zebrafish genetics research and should greatly facilitate systematic functional studies of the vertebrate genome.
The zebrafish genome, along with the mouse and human genomes, are the only three vertebrate genomes that have been sequenced to a degree that they can be considered “finished” (Waterston et al. 2002; International Human Genome Sequencing Consortium 2004). Gene knockout remains the fundamental mechanism for deciphering protein function in vivo, and the first step in leveraging the full power of a model organism's genome project is a systematic mutation of all genes. In the last two decades, zebrafish has rapidly become a widely utilized model organism for studying vertebrate development and modeling human diseases. One of the primary reasons for the popularity of zebrafish is that they are particularly amenable to genetic studies, allowing the identification of mutations affecting both embryonic development and adult homeostasis. For zebrafish, most “forward” genetic studies have been conducted using the chemical mutagen ethylnitrosourea (ENU) (Solnica-Krezel et al. 1994) followed by screening for phenotypes of interest and positional cloning of the mutated genes (Talbot and Schier 1999; Bahary et al. 2004). However, for a systematic approach (e.g., one that allows testing gene function for entire classes of genes or even the entire genome in a nonredundant fashion), it is more effective to first create mutations in all genes and subsequently evaluate the effects of these mutations (i.e., “reverse” genetics). Because zebrafish are amenable to large-scale screening efforts (Mullins et al. 1994; Amsterdam and Hopkins 1999) and because they now have a completely sequenced genome (http://www.ensembl.org/Danio_rerio/Info/Index), they are an ideal organism for systematic reverse genetics in a vertebrate, and testing all protein coding genes in the zebrafish genome via reverse genetics is an achievable goal. As an alternative to ENU, Moloney murine leukemia virus ([M]MLV)-based insertional mutagenesis has been demonstrated to be an efficient approach for mutagenizing thousands of genes both in mouse embryonic stem cells (Friedel et al. 2005) and in large-scale zebrafish genetic screens (Amsterdam and Hopkins 1999). A major advantage of retroviral mutagenesis over ENU is that it allows for rapid identification of the mutated gene through the use of the proviral integration as a molecular “tag” at the site of insertion (Gaiano et al. 1996). Now that the zebrafish genome project is approaching completion, it is possible to isolate DNA fragments flanking the proviral integration on a large scale, sequence them and map the fragments to the proper location in the zebrafish genome, and then index the integration sites to cryo-preserved sperm samples. With this approach, a mutant line could be generated through in vitro fertilization of the frozen sperm sample containing an integration within the gene of interest (Wang et al. 2007). Here we have developed a new retroviral mutagenesis pipeline leveraging the power and cost efficiencies of a next-generation sequencing platform to isolate thousands of zebrafish gene mutations. We report the first 6144 mutagenized and archived F1 fish predicted to carry up to 3776 mutations in zebrafish genes. The mutagenesis is ongoing, and the mutagenized lines are being transferred to the Zebrafish International Resource Center (ZIRC) (Varga 2011) for open distribution to the research community. Large-scale mutagenesis of the zebrafish genome is the first step in defining the in vivo function of every gene in the zebrafish genome, and this retroviral mutagenesis resource complements other efforts in zebrafish to identify mutations using TILLING (targeting induced local lesions in genomes) and gene trap technologies.
Results
We infected zebrafish founder fish with pseudotyped [M]MLV as described earlier (Wang et al. 2007). We generated >3000 mosaic founder fish carrying multiple retroviral insertions. Each founder fish was then outcrossed with wild-type fish to obtain heterozygous F1 fish. An average of six male F1 fish per founder (ranging from four to 10 depending on the level of infection) were used to archive sperm samples, and tail-biopsies were collected for insertion site identification. The outline of the approach used in this manuscript is shown in Figure 1. In our previous strategy, the most cost-effective structure was to limit the sequences to four per fish (Wang et al. 2007). This maximized the number of unique sequences per fish, but capturing all integrations was unlikely because of PCR amplification biases and limited sampling. Next-generation sequencing platforms have the potential to overcome this limitation by making massive oversampling of sequences inexpensive and therefore cost effective. We redesigned our mapping pipeline to take advantage of the depth of sequence afforded by next-generation sequencing platforms.
Figure 1.

Overview of the retroviral mutagenesis pipeline. The pseudotyped murine leukemia virus (A) is injected into 1000–2000 cell stage blastula embryos (B). The infection rate is determined by quantitative PCR (qPCR), and founder fish with high infection rates are raised to adults. The founders are crossed to wild-type (T/AB) fish, and F1 male fish are used for sperm cryopreservation and fin biopsies. Integrations are amplified and mapped from gDNA isolated from the fin biopsies. Mapped integrations are assigned to the corresponding sperm samples, and desired mutations are recovered by in vitro fertilization.
Development of a high-throughput multiplexed mapping strategy
By adapting our mapping strategy to utilize Illumina HiSeq 2000 sequence data, we were able to significantly improve the efficiency of identifying the sequences flanking proviral integrations at a substantial reduction in cost. Each fish was receiving on average ≈390,000 sequences. This allowed us to use three frequently cutting restriction enzymes in parallel (MseI T/TAA, BfaI C/TAG, and Csp6I G/TAC) rather than just the single enzyme Mse1. Therefore if one restriction site was too close for mapping, the other sites provided additional chances for successful mapping.
Data generated using the new method captured a wider range of flanking sequences than did the original published single enzyme method. We adapted our linker-mediated PCR amplification protocol so that amplified genomic DNA adjacent to the retroviral integration sites could be directly sequenced on the HiSeq 2000 (Fig. 2). To link a particular amplified fragment to the F1 fish from which it originated, it was necessary to incorporate an index for the sequenced DNA fragments in the form of a six-base “barcode” sequence adjacent to the ligation site of the linker (Fig. 2). We synthesized 1024 nonredundant linkers each containing a unique barcode, differing by at least two nucleotides between each bar code to avoid incorrect assignment by sequencing miscalls. This allowed us to multiplex hundreds of samples in one sequencing lane.
Figure 2.

Overview of high-throughput strategy to identify retroviral integrations using a next-generation sequencing platform. Genomic DNAs corresponding to individual F1 fish were digested with three sets of restriction enzymes in parallel. After heat-inactivation of the restriction enzymes, the digested samples were then pooled together and ligated with DNA linkers, each containing a unique 6-bp barcode that indexes the F1 fish. The linker ligated DNA fragments were amplified by linker-mediated PCR using linker and viral LTR specific primers to amplify the adjacent genomic DNA sequences. The LTR/gDNA/linker amplicons are subsequently ligated to Illumina paired-end adapters and sequenced using the Illumina sequencing platform.
The Illumina HiSeq 2000 with “paired-end” reads provided up to 200 million paired sequencing reads per lane, with eight lanes on a chip. The platform routinely generated sequencing reads of 101 bp from each end of a paired-end sequence for a maximum of 202 bp of total sequence. Using the standard Illumina sequencing primers, we had to sequence through LTR and linker primers, which were both 25-bp long. We therefore obtained 76 bp of sequence from the viral LTR side and 70 bp of genomic sequence plus the 6-bp barcode index from the linker end. We had a maximum of 146 bp of sequence used to map integrations, larger than the average genomic fragment length sequenced (based on restriction enzyme site availability, it is ≈65 bp) with 46% of the sequences having overlap between the paired-end reads.
Insertion site mapping strategy
We developed a new customized bioinformatics pipeline to map retroviral insertions in the zebrafish genome. Each side of the raw sequence data was trimmed of LTR sequences or linker sequences. Barcodes for each fish were identified, indexed to the sequence but trimmed before alignment with the genome. We used two independent strategies to process the raw sequence data (contig construction or independent end mapping) followed by mapping retroviral integrations using the Bowtie algorithms (Fig. 3; see Supplemental Fig. 1 for details of mapping strategy; Langmead et al. 2009). A “consensus” list of unique integration coordinates based on the two different mapping strategies proved to significantly eliminate mismapped integrations, allowing a better recovery rate of correctly mapped integrations after in vitro fertilization.
Figure 3.
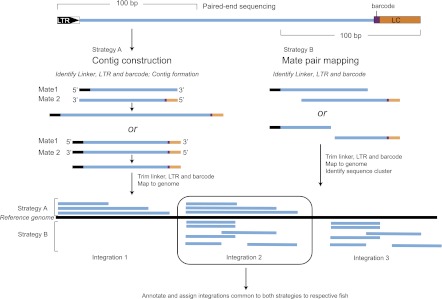
Strategies for mapping retroviral integrations. Paired-end sequencing was performed to capture the site of the retroviral integration (designated by LTR–retroviral 3′ long terminal repeat) and the linker cassette (LC) that contains the “barcode” identifier for the specific sample. Two strategies were used to map the integrations as this proved to be less error-prone than either strategy alone. In Strategy A, pairwise alignment of paired-end reads was performed to create contigs, and the resulting contigs were mapped to the zebrafish genome. Only contigs that mapped unambiguously were considered for identifying integrations. In Strategy B, each read from corresponding paired-ends was mapped independently, and colocalization in the correct orientation (pointing at each other) was used as the criterion for correct mapping. Integrations that mapped to the same genomic coordinates by both strategies were used for identification of integration events.
Zebrafish mutations generated from proviral integration
We utilized the new protocol to map retroviral insertions generated from founder injections. We processed F1 genomic DNA samples from 6144 male zebrafish (sixty-four 96-well plates) with matching cryo-preserved sperm using the overall approach described. Of the 6144 F1 fish, 15,223 unique integration sites (integrations with different genomic coordinates) were mapped using the consensus list from the two different mapping strategies. Among all insertions mapped to the genome, 52% of the integrations (7896/15,223) were in genes annotated by Ensembl. Insertions in exons generate a truncation at the site of integration, and our mapping data shows 12% of the gene hits were in exons (963/7896). 88% (6933/7896) of the gene “hits” were in introns. [M]MLV has a known bias to integrate near transcriptional start sites, and further analysis of the introns hits show 40% of the integrations (2813/6933) that occurred in introns were in the first intron (Table 1; Fig. 4A,B). This is consistent with the previous studies in mice (Mooslehner et al. 1990; Scherdin et al. 1990) and human tissue culture cells (Wu et al. 2003). Overall, 60% (9189/15,223) of all integrations landed either in genes or within 1 kb upstream of or downstream from genes. The mapped integrations showed roughly equal distribution across all chromosomes (Fig. 4D). In order to determine if we could expect a broad distribution of identified mutations, we looked at the distribution pattern for predicted mutations. As expected, the vast majority of identified mutations have been hit only once (3937), meaning we are still well below saturation for the technique. However, there are some clear hot spots for integration, with 97 instances of genes hit independently more than five times (based on uniquely mapped coordinates) and five examples of genes hit more than 10 times (Fig. 4C). The overall integration distribution profile is consistent with our previous studies (Wu et al. 2003; Wang et al. 2007).
Table 1.
Distribution profile of 15,223 retroviral integrations in 6,144 F1 fish
Figure 4.

Summary of proviral integrations from 6144 F1 fish. (A) Distribution of the 6933 retroviral integrations in introns; 40% of integrations (2813/6933) are in the first intron. (B) Distribution of 963 integrations in exons. (C) Number of hits per gene based on integrations with unique genomic coordinates; 72% of genes have only one integration. (D) Distribution of 15,223 integrations across all chromosomes.
We have previously shown that integrations both in exons and in the first intron of genes are highly mutagenic (Wang et al. 2007). In 80% of genes that contained integrations in their first intron, mRNA levels were reduced to <10% of wild type. Nearly one in five retroviral integrations is predicted to result in a disruption that reduces the gene expression level to 10% or less of the wild-type level (for the case of integrations in intron 1) or a truncation at the site of integration (for cases where the integration lands in an exon) (Wang et al. 2007). Extrapolating from our previous data, the number of potential null or severe hypomorph mutants in our data is up to 3776. The number of unique genes for these mutations is 3054.
Recovery of mapped insertions
Using more conservative mapping methods, we can recover alleles from frozen sperm stocks up to a 79% success rate. We attempted to recover 197 alleles from our frozen samples. We confirmed 156 (79%) using PCR primers designed from sequences adjacent to the site of insertion. Another four were recovered based on primers designed to the raw sequencing data, but they could not be confirmed to be in the correct genomic location, suggesting that gene is not correctly mapped to the genome despite supposedly having a unique mapping position.
Phenotypic characterization of recovered alleles
Genetic screens focused on zebrafish morphology have typically been performed during the first five days of embryonic development. As expected, our retroviral insertional lines produced early embryonic phenotypes (Fig. 5). To demonstrate the utility of this resource in producing biological information beyond embryonic development of zebrafish, we analyzed the nature of 41 mutations we recovered by in vitro fertilization through larval and adult lifespan (Supplemental Table 2). The F2 fish (counting generations from the founder fish) were raised to adults then genotyped to identify mutant carriers. F2 heterozygous mutant carriers were inbred, and the embryos were examined for early developmental defects. Twelve of the 41 showed obvious morphological defects in the developing embryo, and all were genetically linked to the predicted mutagenic insertion. We tested embryos from each mutant line by semiquantitative RT-PCR to determine if the mRNA for the predicted mutations were affected and found that all showed significant reduction in mRNA transcript levels.
Figure 5.

Representative embryonic, larval, and adult phenotypes from selected retroviral insertional alleles. (A) Insertion in the wee1 gene led to an early phenotype of cellular necrosis starting at the gastrulation stage. Images here show a wild-type and mutant embryo at the 12 somite stage. (B) Insertion in the eif3i gene led to a vascular defect in homozygous mutants. The upper panel shows bright field images and the lower panel shows the lack of intersegmental vessels labeled by the flk-gfp transgenic marker in the eif3s3−/− background at 1 day post-fertilization (dpf). (C) An insertion in the snapc1b gene causes embryonic phenotypes including jaw defects and a small liver visible at 5 dpf. Arrows point to the reduced jaw structures in the mutant, dashed lines demarcate the liver. (D) Homozygous rpa1 mutants at 2 dpf have small and necrotic heads, small eyes, and tails curling dorsally. These homozygous phenotypes are weaker but observable at 1 dpf. All homozygotes die at ∼5 dpf. (E) Insertion in a novel gene (zgc:194470) led to the larval phenotype of a larger body at day 12 of development. The mutant is homozygous viable, and the body sizes become the same as that of wild type when reaching the adult stage. One-hundred percent of the homozygous mutants show the larger larval phenotype (N = 200). (F) Slc7a5−/− fish showed no observable embryonic defects, but they are 40% smaller than their wild-type or heterozygous siblings at 4 mo of age. Slc7a5 is a small subunit of the L-type amino acid transporter 1. (G) 6-mo-old adult tg−/− (thyroglobulin) fish showed red swelling under the chins (black arrows), a phenotype reminiscent of human thyroid goiters. Tg−/− fish are fertile and showed no observable embryonic defects.
For all the mutations that did not have an early embryonic phenotype, we raised the F3 generations and genotyped the fish once they reached sexual maturity. In most of the lines, we could detect the normal Mendelian ratios of 1:2:1, suggesting that the mutated genes did not have a significant impact on viability in a laboratory setting.
Figure 5 shows several phenotypes identified in the tested alleles. We show four examples of genes that showed early embryonic phenotypes: wee1 mutants showed a very early cell death phenotype (Fig. 5A), eif3i (previously known as eif3s2) mutants showed defects in arterial patterning, snapc1b mutants showed liver and jaw defects, and rpa mutants showed a curled body axis and deficient brain and head structures. One allele (Zgc:194470) had a juvenile morphological phenotype of overgrowth, and two cases (Slc7A5 and Tg) had clear morphological phenotypes in adults. Zgc:194470−/− mutants were fully viable but had a larger body size by day 10–12 (Fig. 5E); although eventually, they became indistinguishable from their wild-type siblings. Slc7A5−/− adults were significantly smaller than their wild-type siblings, and Tg−/− siblings had an enlarged red growth under their chins resembling a thyroid hypertrophy (Fig. 5F,G; Jao et al. 2008). These data demonstrate that this resource will generate not only mutations that would be readily identified as early developmental defects in forward screening but can also identify genes that reduce viability, cause adult onset diseases, or alter adult morphology.
Distribution of mutants
After the integrations are mapped, the archived sperm samples are being deposited at the Zebrafish International Resource Center (ZIRC) for open distribution to the scientific community.
Discussion
We have mapped 15,223 [M]MLV proviral integrations onto the zebrafish genome resulting in 3776 predicted mutations in 3054 genes (see Supplemental Table 1). By adapting the mapping pipeline from capillary sequencing to the HiSeq 2000, the number of sequences per F1 fish increased from four sequences per fish to ≈390,000 sequences per fish. The extreme oversampling results in essentially saturating our ability to identify existing integrations (because of limited sampling and cost considerations, our previous approach only recovered ∼20% of the existing integrations). Approximately 24% of all integrations are either in exons (6%) or in the first intron (18%) (Table 1). There were multiple examples of genes with more than one unique integration, suggesting that as the number of identified integrations increases we will eventually reach a point at which we will need to shift to an insertional DNA element with a different integration bias to reach genomic saturation.
We successfully recovered ∼80% of the retroviral integrations from the frozen sperm samples. The failures to recover mutations could reflect mistakes in the genome assembly, gaps in the genomic sequence where the integration would have mapped, or polymorphisms that result in a misalignment. In addition, some rate of human error cannot be discounted. We expect the recovery rate to improve as updated versions of the zebrafish genome are released.
It is important to note the differences between our mapping approach compared to mutagenesis projects using [M]MLV previously undertaken in mouse embryonic stem cells. The mouse projects were very large-scale efforts; however, they relied on gene trap constructs and reporter expression (typically antibiotic resistance) to select for gene trap events. Thus, the number of genes ultimately trapped by this approach cannot exceed the number of genes expressed at the time of retroviral infection, and traps require both correct orientation and in-frame splicing events. Because of these limitations, typically gene traps do not end up trapping >50% of all genes in the genome. Because our approach does not rely on gene expression but only on identifying the exact site of integration, it is likely that we will be able to mutagenize a significantly larger number of genes before the approach reaches saturation. Based on the vast majority of mutations we have identified so far having only one integration event (Fig. 4C), we believe we are still very far from saturation for this approach and can continue to generate new mutations for several years. All frozen sperm samples are transferred to the Zebrafish International Resource Center. Each F1 fish has four frozen samples. If particular mutations are requested multiple times, ZIRC will use one sample to raise multiple fish and refreeze the samples, making this a durable mutant resource.
Recently, transcription activator-like effector nucleases (TALENs) are being used as an alternative method for knockout genes in zebrafish (Huang et al. 2011; Bedell et al. 2012). Although TALENs are an effective tool for targeted mutagenesis, scaling this method to mutagenize thousands of genes would be very difficult, and no genomewide resource utilizing TALENs is currently available.
One of the key aspects of generating a defined collection of mutations in a broad spectrum of zebrafish genes is the utility of these alleles for generating models for human disease. Approximately one-third of the integrations we predict to be mutagenic were in exons (Table 1; Fig. 4B); these are pure disruptions at the site of integration. The remaining alleles in the first intron are typically severe hypomorphs based on measured mRNA levels. Extensive literature in C. elegans, Drosophila, and even mouse have demonstrated that “weaker” alleles are often, if not in the majority of cases, superior to a null allele when analyzing gene effects in a multicellular organism. It is also worth mentioning that most human genetic diseases are hypomorphic mutations, and pathology is identified well after the embryonic stage.
The indexing technique and mapping pipeline we developed for pooling samples into a single lane of the Illumina HiSeq 2000 have important utility in a variety of research and clinical settings. The main advantages are: (1) a greater than tenfold reduced cost compared to capillary sequencing with an ≈58% improvement in identifying integrations; and (2) simple sample preparation that is amenable to scaling and automation. The technique does not require sonication of samples (Williams-Carrier et al. 2010) and has deeper sampling than 454 (Ciuffi et al. 2009). It is likely that the deeper sequencing is compensating for distortions caused by PCR amplification and other site cloning biases that may occur. The technique can be readily modified to map any DNA element being inserted into any sequenced genome. It has utility for mutagenesis using transposons, such as Tol2 or Piggyback, or in gene therapy experiments with any vectors that stably integrate into the genome.
Methods
Generation of virus-infected fish and cryopreservation
Founder production, F1 fish husbandry, and cryopreservation of sperm samples were performed as previously described (Wang et al. 2007). In brief, retroviral stocks were prepared according to Jao and Burgess (2009), and synchronized embryos were obtained from wild-type T/AB-5 fish. The concentrated viral stock was injected into blastula stage embryos (1000–2000 cell stage). Injected embryos were tested for the efficiency of proviral infection using qPCR-based assays to determine the copy number of the provirus (embryo assay values [EAV]). High-quality founder fish with high EAVs were raised, and F1 fish were generated by outcrossing with wild-type fish. Five to 10 F1 male fish per founder were selected for cryopreservation and to map the retroviral integrations. The sperm from each F1 fish line was collected and frozen, and the corresponding tail-cut was used to isolate genomic DNA for mapping.
Genomic DNA preparation and fragmentation
Genomic DNA was isolated from F1 fish tail biopsies. In 96-well plates, each F1 tail sample was lysed with 100 μL of lysis buffer (10 mM Tris/HCl pH 8.0, 1mM EDTA, 50mM KCl, 2mM MgCl2) 20 μg/μL proteinase K (Invitrogen, Inc.). After digestion for 3 h at 55°C, the DNA was precipitated with isopropanol and washed with ethanol. The DNA pellet was dissolved in 50 μL of distilled water. We used ∼500 ng of genomic DNA for fragmentation using three pairs of restriction enzymes (MseI/PstI, BfaI/BanII [New England Biolabs, Inc.] and Csp6I/ ECo24I [Fermentas, Inc.]) in parallel. The restriction digestion was done for 8 h at 37°C and heat inactivated for 10 min at 80°C.
Preparation of barcoded linkers
The barcode linkers followed a “splinkerette” design (Devon et al. 1995) with a 31-nucleotide-long upper strand and a 49-nt-long lower strand including a 6-nt barcode and a TA overhang. They were synthesized on a 10 nm scale by IDT DNA Inc. The synthesized oligonucleotides were reconstituted in TE buffer to a 200-μM concentration master stock. A 2-μM working concentration was prepared in STE buffer (TE with 50 mM NaCl). Barcode linkers were annealed for 3 min at 70°C and for 10 min 65°C. A final concentration of 0.2 μM was used to ligate onto the restriction enzyme-digested genomic fragments. The digested samples from each enzyme pair were pooled with prealiquoted barcoded linker in individual wells. The T4 DNA ligase (New England Biolabs, Inc.) was added, and the reaction mix was incubated for 6 h at 16°C.
Linker-mediated PCR
The linker-mediated PCR was performed in two steps. In the first step, PCR was done with one primer specific to the 3′- LTR (5′-GACTTGTGGTCTCGCTGTTCCTTGG-3′) and the other primer specific to linker sequences (5′-GTAATACGACTCACTATAGGGC-3′) using the following conditions: 2 min at 95°C, 7 cycles of 15 sec at 95°C, 1 min at 72°C, and then 32 cycles of 15 sec at 95°C, 1 min at 67°C, and a final step for 4 min at 67°C. The PCR products were diluted to 1:50 in dH2O, and a second round of PCR was performed using LTR (5′-GAGTGATTGACTACCCGTCAGCGGGGGTCTTTCA-3′) and Linker specific (5′ -ACT ATAGGGCACGCGTGGTCGACTGCGCAT-3′) nested primers to increase sensitivity and avoid nonspecific amplification. The nested PCR products from each 96-well plate were pooled together and processed for Illumina library preparation as per manufacturer's instructions.
Illumina library preparation
Illumina libraries were prepared from 1 μg of pooled PCR products. Illumina paired-end adaptors were ligated onto LTR-gDNA-linker amplicons generated from the nested PCR reactions following Illumina's sample preparation guide. In brief, the PE adaptor oligo mix was incubated with PCR amplicons using T4 DNA ligase for 20 min at room temperature. The ligation reaction was cleaned up using a QIAquick Min-elute column (QIAGEN) and eluted with EB buffer. The purified library was PCR-enriched with Phusion High-Fidelity polymerase in HF buffer. PCR was performed using primers (primer 1.0 and PE primer 2.0) supplied with the Illumina paired-end kit with the following conditions: 30 sec at 98°C, 15 cycles of 10 sec at 98°C, 30 sec at 65°C, 30 sec at 72°C, followed by a 5 min at 72°C, and a final hold at 4°C. The PCR enriched library was purified using a QIAquick Min-elute column and eluted in 20 μL of EB solution. An equimolar concentration of different barcoded libraries were pooled together, and the final concentration was determined using quantitative PCR prior to loading onto the Illumina sequencer flowcell.
Retroviral integration mapping
Paired-end sequencing of multiplexed samples was performed on the Illumina GAIIx or HiSeq 2000 platforms. Sequence reads were extracted from the ELAND or BAM files generated by the sequencer. Non-zebrafish sequences were trimmed from each read. The six-base nucleotide “barcode” sequence was then identified and compared to a database of indexed sequence codes. The resulting trimmed sequences were mapped to the zebrafish genome (Ensembl Zv9 assembly build e65) using Bowtie. In order to increase confidence in the ability to recover a specific integration, integration sites with ≥30 redundant mappable sequence reads were selected as higher confidence. Two bioinformatics methods for processing and mapping sequences were used.
Mapping method one
Preprocessing and alignment of proviral insertion sites
We assembled a curated, single-ended library from the original paired-end reads (Supplemental Fig. 1). Each of the single sequences is assembled in the form 5′-Linker-barcode-flanking genomic sequence-LTR-3′. A brute force exact alignment algorithm (Castruita et al. 2011) was used to align the paired reads along their overlapping regions and to find the location of both the linker and LTR sequences. All sequences are then stored in the form: 5′-linker-barcode/flanking genomic sequence/LTR-3′ for downstream analysis. Flanking sequences were extracted and aligned to the zebrafish genome assembly Zv9 using Bowtie (Langmead et al. 2009) with a tolerance of one mismatch. Only reads longer than 11 nt and with unambiguous alignments were used to pinpoint the insertion locus.
Some flanking sequences were sufficiently long that the paired reads did not overlap. In these cases, an oriented pseudo single-end sequence was generated in the form 5′-Linker-barcode/flanking genomic seq1/N-flanking genomic seq2/LTR-3′. The resulting flanking sequences were separately mapped to Zv9 using Bowtie (Langmead et al. 2009). Multiple hits were filtered to keep a maximum of 20 hits per read. In our model, the paired flanking sequences have a unique alignment if hits from both sequences are aligned to the same chromosomal region, same strand orientation, are at a distance of <1 kb between hits, and there is only one hit-pair that meets the above requirements.
Identifying insertion sites
All unique hits (i.e., unambiguously mapped to a single location in the genome) from the preprocessing step were pooled, and integration coordinates were extracted from the Bowtie mapping output. The integration site was defined as the genomic coordinate immediately adjacent to the portion of the read to which the 3′ LTR had been attached. The 3′-LTR end position was used to determine redundant sequences for each barcode or sample, and the longest fragment was used as representative to report and display the insertion locus.
For reporting and viewing the data, a bed-formatted file was produced, which gives the chromosome, flanking sequence start, flanking sequence end, barcode, frequency (number of reads per integration site), and orientation of each integration and is available as a download from http://research.nhgri.nih.gov/ZInC/ (Varshney et al. 2013).
Annotation of integration loci
The gene annotation file Ensembl ZV9 e65 was used to build a “One gene, One transcript” gene structure model (see Supplemental Fig. 2) as the exonic union of all the annotated transcripts. Finally, BEDTools was used to determine the overlap between the integration and the gene model, and integration sites were annotated as explained above.
Mapping method two
Trimming and orienting retroviral tag sequences and barcode identification
Prior to mapping the reads, a custom script was run through to trim off the 3′ retroviral LTR and linker cassette (LC) sequences and identify the barcodes. Sequence reads were discarded if they did not contain either the 3′ LTR or the LC primer sequence in their 5′ end. The six nucleotides directly adjacent to the LC primer sequence represented the barcode. Sequences were trimmed of the 3′ LTR and/or LC sequence as well as the barcode, the barcode was noted for sample identification, and the trimmed sequence was used for mapping integrations.
Mapping retroviral tags
Bowtie (Langmead et al. 2009) was again used to map the trimmed retroviral sequence tags to the Zv9 zebrafish genome assembly, allowing for one mismatch. Since the site of retroviral integration and the sample barcode could occur on separate mate pairs, it was important to perform paired-end sequencing. However, sequence ends were mapped separately due to wide variation in both the trimmed sequence lengths and the distances between the mate pairs. As above, the integration site was defined as the site of LTR insertion.
Pairing mapped sequence ends, collapsing redundant sequences, and identifying integration sites
After mapping, corresponding ends were paired and uniquely mapping read pairs were used to identify insertion sites. The following criteria were used for pairing and determining uniquely mapped insertion sites: both ends must map to the same chromosome within 1 kb of each other and with the correct orientation (the 3′ ends of the reads should point toward each other). Priority was given to the mapping that resulted in the smallest number of mismatches (Supplemental Fig. 1B). The number of redundant sequences was recorded. Integration sites with greater or equal to 60 redundant sequence reads were used for downstream analyses.
Annotation of retroviral integration sites
The genomic position of retroviral integrations was compared to those of zebrafish gene models obtained from Ensembl Zv9 e65. A custom perl script (Hu et al. 2008) was run to identify those retroviral insertions that occurred within a gene or 1 kb upstream or downstream from a gene.
Data access
All the sequence data from genomic DNA adjacent to the insertion sites used for mapping has been deposited in the public NCBI GSS database (http://www.ncbi.nlm.nih.gov/nucgss) (BioSample ID: LIBGSS_038780). The detailed insertion data are also incorporated into ZFIN (http://zfin.org), the zebrafish model organism database, as transgenic insertions. To help researchers from other fields, we created a database, ZInC (the Zebrafish Insertion Collection) that can be searched using different search inputs such as human, mouse gene symbols, or KEGG pathway terms (http://research.nhgri.nih.gov/ZInC) (Varshney et al. 2013). Insertion data can also be downloaded in bed file format to be used with the UCSC and Ensembl Genome Browser.
Acknowledgments
We would like to thank Colin Huck for superior animal care, Darryl Leja for illustrations and graphics work, and Robert Blakesley, Alice Young, and the National Intramural Sequencing Center (NISC) for providing sequence data. This research was supported by the Intramural Research Program of the National Human Genome Research Institute, National Institutes of Health (S.M.B. and T.G.W.), R01 GM085357 and R01 HL109525 (A.F.S.), and R01 DK084349 (S.L.) and NSFC 31110103904 (B.Z., S.L.).
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.151464.112.
References
- Amsterdam A, Hopkins N 1999. Retrovirus-mediated insertional mutagenesis in zebrafish. Methods Cell Biol 60: 87–98 [DOI] [PubMed] [Google Scholar]
- Bahary N, Davidson A, Ransom D, Shepard J, Stern H, Trede N, Zhou Y, Barut B, Zon LI 2004. The Zon laboratory guide to positional cloning in zebrafish. Methods Cell Biol 77: 305–329 [DOI] [PubMed] [Google Scholar]
- Bedell VM, Wang Y, Campbell JM, Poshusta TL, Starker CG, Krug Ii RG, Tan W, Penheiter SG, Ma AC, Leung AY, et al. 2012. In vivo genome editing using a high-efficiency TALEN system. Nature 491: 114–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castruita M, Casero D, Karpowicz SJ, Kropat J, Vieler A, Hsieh SI, Yan W, Cokus S, Loo JA, Benning C, et al. 2011. Systems biology approach in Chlamydomonas reveals connections between copper nutrition and multiple metabolic steps. Plant Cell 23: 1273–1292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciuffi A, Ronen K, Brady T, Malani N, Wang G, Berry CC, Bushman FD 2009. Methods for integration site distribution analyses in animal cell genomes. Methods 47: 261–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devon RS, Porteous DJ, Brookes AJ 1995. Splinkerettes–improved vectorettes for greater efficiency in PCR walking. Nucleic Acids Res 23: 1644–1645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedel RH, Plump A, Lu X, Spilker K, Jolicoeur C, Wong K, Venkatesh TR, Yaron A, Hynes M, Chen B, et al. 2005. Gene targeting using a promoterless gene trap vector (“targeted trapping”) is an efficient method to mutate a large fraction of genes. Proc Natl Acad Sci 102: 13188–13193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaiano N, Amsterdam A, Kawakami K, Allende M, Becker T, Hopkins N 1996. Insertional mutagenesis and rapid cloning of essential genes in zebrafish. Nature 383: 829–832 [DOI] [PubMed] [Google Scholar]
- Hu J, Renaud G, Gomes TJ, Ferris A, Hendrie PC, Donahue RE, Hughes SH, Wolfsberg TG, Russell DW, Dunbar CE 2008. Reduced genotoxicity of avian sarcoma leukosis virus vectors in rhesus long-term repopulating cells compared to standard murine retrovirus vectors. Mol Ther 16: 1617–1623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang P, Xiao A, Zhou M, Zhu Z, Lin S, Zhang B 2011. Heritable gene targeting in zebrafish using customized TALENs. Nat Biotechnol 29: 699–700 [DOI] [PubMed] [Google Scholar]
- International Human Genome Sequencing Consortium 2004. Finishing the euchromatic sequence of the human genome. Nature 431: 931–945 [DOI] [PubMed] [Google Scholar]
- Jao LE, Burgess SM 2009. Production of pseudotyped retrovirus and the generation of proviral transgenic zebrafish. Methods Mol Biol 546: 13–30 [DOI] [PubMed] [Google Scholar]
- Jao LE, Maddison L, Chen W, Burgess SM 2008. Using retroviruses as a mutagenesis tool to explore the zebrafish genome. Brief Funct Genomic Proteomic 7: 427–443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL 2009. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10: R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mooslehner K, Karls U, Harbers K 1990. Retroviral integration sites in transgenic Mov mice frequently map in the vicinity of transcribed DNA regions. J Virol 64: 3056–3058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullins MC, Hammerschmidt M, Haffter P, Nusslein-Volhard C 1994. Large-scale mutagenesis in the zebrafish: In search of genes controlling development in a vertebrate. Curr Biol 4: 189–202 [DOI] [PubMed] [Google Scholar]
- Scherdin U, Rhodes K, Breindl M 1990. Transcriptionally active genome regions are preferred targets for retrovirus integration. J Virol 64: 907–912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solnica-Krezel L, Schier AF, Driever W 1994. Efficient recovery of ENU-induced mutations from the zebrafish germline. Genetics 136: 1401–1420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talbot WS, Schier AF 1999. Positional cloning of mutated zebrafish genes. Methods Cell Biol 60: 259–286 [DOI] [PubMed] [Google Scholar]
- Varga ZM 2011. Aquaculture and husbandry at the Zebrafish International Resource Center. Methods Cell Biol 104: 453–478 [DOI] [PubMed] [Google Scholar]
- Varshney GK, Huang H, Zhang S, Lu J, Gildea DE, Yang Z, Wolfsberg TG, Lin S, Burgess SM 2013. The Zebrafish Insertion Collection (ZInC): A web based, searchable collection of zebrafish mutations generated by DNA insertion. Nucleic Acids Res 41: D861–D864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, Jao LE, Zheng N, Dolan K, Ivey J, Zonies S, Wu X, Wu K, Yang H, Meng Q, et al. 2007. Efficient genome-wide mutagenesis of zebrafish genes by retroviral insertions. Proc Natl Acad Sci 104: 12428–12433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterston RH, Lindblad-Toh K, Birney E, Rogers J, Abril JF, Agarwal P, Agarwala R, Ainscough R, Alexandersson M, An P, et al. 2002. Initial sequencing and comparative analysis of the mouse genome. Nature 420: 520–562 [DOI] [PubMed] [Google Scholar]
- Williams-Carrier R, Stiffler N, Belcher S, Kroeger T, Stern DB, Monde RA, Coalter R, Barkan A 2010. Use of Illumina sequencing to identify transposon insertions underlying mutant phenotypes in high-copy Mutator lines of maize. Plant J 63: 167–177 [DOI] [PubMed] [Google Scholar]
- Wu X, Li Y, Crise B, Burgess SM 2003. Transcription start regions in the human genome are favored targets for MLV integration. Science 300: 1749–1751 [DOI] [PubMed] [Google Scholar]