

Abstract
During craniofacial development, the Hedgehog (HH) signaling pathway is essential for mesodermal tissue patterning and differentiation. The HH family consists of three protein ligands: Sonic Hedgehog (SHH), Indian Hedgehog (IHH), and Desert Hedgehog (DHH), of which two are expressed in the craniofacial complex (IHH and SHH). Dysregulations in HH signaling are well documented to result in a wide range of craniofacial abnormalities, including holoprosencephaly (HPE), hypotelorism, and cleft lip/palate. Furthermore, mutations in HH effectors, co-receptors, and ciliary proteins result in skeletal and craniofacial deformities. Cranial suture morphogenesis is a delicate developmental process that requires control of cell commitment, proliferation and differentiation. This review focuses on both what is known and what remains unknown regarding HH signaling in cranial suture morphogenesis and intramembranous ossification. As demonstrated from murine studies, expression of both SHH and IHH is critical to the formation and fusion of the cranial sutures and calvarial ossification. SHH expression has been observed in the cranial suture mesenchyme and its precise function is not fully defined, although some postulate SHH to delay cranial suture fusion. IHH expression is mainly found on the osteogenic fronts of the calvarial bones, and functions to induce cell proliferation and differentiation. Unfortunately, neonatal lethality of IHH deficient mice precludes a detailed examination of their postnatal calvarial phenotype. In summary, a number of basic questions are yet to be answered regarding domains of expression, developmental role, and functional overlap of HH morphogens in the calvaria. Nevertheless, SHH and IHH ligands are integral to cranial suture development and regulation of calvarial ossification. When HH signaling goes awry, the resultant suite of morphologic abnormalities highlights the important roles of HH signaling in cranial development.
Keywords: craniofacial abnormalities, Sonic Hedgehog (SHH), Indian Hedgehog (IHH), cranial suture patterning/signaling, calvarial bone differentiation
Introduction
Craniofacial morphogenesis, an intricate developmental process, begins with the synchronized development of head primordia, which involves several organizing centers located in the neural ectoderm, axial mesendoderm, and the cranial neural crest. The differentiation and spatial patterning of these tissues must occur before they can be successfully integrated (Hu and Helms, 1999). The Sonic Hedgehog morphogen (SHH) is one of the signals involved in the axial and dorsoventral definition of craniofacial and limb development (Chiang et al., 1996; Capdevila and Johnson, 2000; Anderson et al., 2012). Dysregulation of the Hedgehog (HH) signaling pathway results in a wide array of craniofacial defects including holoprosencephaly (HPE), hypotelorism, and cyclopia, amongst others (Belloni et al., 1996). Both Indian Hedgehog (IHH) and SHH have been well studied in cartilage and bone patterning throughout the axial, appendicular and facial skeleton (Hammerschmidt et al., 1997; Capdevila and Johnson, 2000; Chai and Maxson, 2006). In contrast, the role of HH signaling in calvarial ossification and cranial suture morphogenesis is a relatively new and less examined area of scientific investigation.
The embryonic development of the cranium and cranial suture complex begins as far back as neural crest cell migration, a process starting on murine embryonic day 8 (E8) and completed within 2 days (Slavkin, 1979). In humans, this period lies approximately between E19 and E38 (Dixon et al., 1997). After migration of the neural crest, the calvarial mesenchyme originates from both the paraxial mesoderm and the migrated cranial neural crest (Noden, 1983). The demarcations between the neural crest and mesoderm-derived bone have undergone evolutionary changes resulting in species-specific differences. In avian skulls this boundary has been found on the caudal border of the frontal bone and that a portion of the calvarium was derived from neural crest cells (Noden, 1975; Le Lievre, 1978). Later studies have proposed that avian skull may even arise entirely from neural crest cells (Couly et al., 1993). Murine studies, using Wnt1-Cre (a reporter for neural crest origin) mice, suggest that the entire frontal bone is derived from neural crest origin, while other skull bones principally originate from the paraxial mesoderm (Chai et al., 2000; Jiang et al., 2000, 2002; Brault et al., 2001; Chai and Maxson, 2006; Ishii et al., 2012). Nevertheless, the process of cranial skeletogenesis occurs in concert with the expansion of the brain. Neural crest cells originate from the dorsal neural tube and as the cells migrate they are pulled by the expansion of the epidermis to form the frontonasal process. The frontal primordium mesenchyme then forms the frontal bones, which differentiates into the primary bone plates (Iseki et al., 1995; Ting et al., 2009). Subsequent growth of the bone plates occurs until they meet, resulting in formation of a suture.
Characterized as regions of fibrous tissue between cranial bones, sutures function as intramembranous osteogenic sites, permit cranium growth, inhibit bone separation, and absorb shock (Baer, 1954; Cohen, 1993; Wilkie, 1997; Lenton et al., 2005). Cranial sutures are distinguished with respect to their adjacent cranial bones (i.e., the metopic suture lies between frontal bones, etc.). It is crucial that sutures remain in an undifferentiated and proliferative state as the brain develops while permitting the growth of new bone at the suture margins until the they fuse (Opperman, 2000).
Cranial suture fusion is a process whereby the cells in the middle of the suture complex mature into osteoblasts that eventually fuse into midline sutures (i.e., the sagittal and metopic sutures), which join end-to-end. In humans all sutures close around the end of adolescence (Madeline and Elster, 1995). In contrast, in mice, the transversely situated sutures (lamboid and coronal sutures), do not undergo ossification and remain overlapping and patent. During the stages before physiologic suture fusion, an imbalance between cell proliferation and osteoblast differentiation can cause inappropriate suture fusion, or craniosynostosis. Suture synostosis prevents further bone formation and accommodation of neurocranial growth, leading to craniofacial dysmorphology and central nervous system (CNS) impairments. Potential CNS effects of craniosynostosis include elevated intracranial pressure (Bristol et al., 2004; Hayward and Gonsalez, 2005), high incidence of learning disabilities (Kapp-Simon, 1998; Panchal et al., 2001; Magge et al., 2002), and impaired eyesight (Macintosh et al., 2012). Therefore, regulation of cranial suture morphogenesis is crucial for maintenance of suture patency and proper physical and cognitive development.
Growth factor regulation is one of the main mechanisms that coordinate calvarial patterning and ossification. The most commonly studied signaling pathways regulating suture fusion include Fibroblast Growth Factor (FGF), Transforming Growth Factor (TGF-β), and Bone Morphogenetic Protein (BMP) signaling (Longaker, 2001; Warren et al., 2003; Nie et al., 2006a,b; Rawlins and Opperman, 2008). The dura mater (or outer layer of meninges) secretes a variety of growth factors in order to regulate cranial suture fusion and suture patency in a paracrine fashion (Opperman et al., 1993; Bradley et al., 1997; Cooper et al., 2012). FGF2 demonstrates a significant mitogenic effect and thus may stimulate proliferation of osteoprogenitors in the dura mater and overlying suture mesenchyme (Spector et al., 2002; Li et al., 2007). As a result, a majority of syndromic synostoses arise from Fibroblast growth factor receptor (FGFR) mutations (Cohen, 2009). Additionally, TGF-βs also serves to initiate suture fusion as there are high levels of TGF-β1 and 2 expression in fusing as compared to patent cranial sutures (Opperman et al., 1997, 1999; Ko et al., 2009; Slater et al., 2009). Studies dysregulating TGF-β signaling have also been shown to significantly alter suture ossification and fate (fusion vs. patency) (Derynck et al., 1994; Rawlins and Opperman, 2008; Chim et al., 2011). While the periosteum lies in close proximity in the suture, it does not contribute significantly to the regulation of suture fusion (Opperman et al., 1994, 1995). The significance of BMP signaling in cranial suture fusions was made clear by studies involving the BMP antagonist Noggin (Warren et al., 2003). While BMP is expressed in fusing sutures, the Noggin expression that it induces is counteracted by FGF2 signaling. In comparison to FGF, TGF-β, and BMP signaling, the role of HH signaling in cranial suture morphogenesis is a relatively new focus of investigation, yet one of clear importance.
This review will first provide an overview of HH signaling, including normal signal transduction and pertinent regulators of HH signaling. Next, a comprehensive review of the known functions of HH signaling in the cranial suture complex is presented. In addition, the role of HH in endochondral ossification and cranial base formation is examined. Finally, a discussion of what remains unknown or unclear regarding HH signaling in cranial suture biology is presented.
An overview of hedgehog signaling
The activity of the HH pathway was first identified in Drosophila, and its expression was later found in all vertebrates (Fietz et al., 1994). Three homologues of the Drosophila HH protein exist in vertebrates: SHH, IHH, and Desert hedgehog (DHH). DHH expression is primarily limited to the male reproductive tract, and the majority of DHH−/− mice do not exhibit mutant phenotypes in most tissues (Parmantier et al., 1999; Yao et al., 2002; Cohen, 2003; Kawai et al., 2010). In contrast, SHH and IHH are essential to embryonic development, as either SHH- or IHH-deficient mice demonstrate multiple severe congenital anomalies and neonatal lethality that will be further discussed below (Chiang et al., 1996; St-Jacques et al., 1999; Hayhurst and McConnell, 2003). SHH plays a diverse and key role in the development of the head process, notochord, ventrolateral midbrain, and ventral forebrain. SHH also plays an indispensible function in limb development, including limb budding, anterior-posterior patterning of limb skeleton and regulation of right/left asymmetry (Capdevila and Johnson, 2000). In regards to the craniomaxillofacial skeleton, SHH in the forebrain mediates the development of mid and upper face, frontonasal and maxillary processes (Byrnes et al., 2009). Closely related to SHH by a gene duplication event (Schlosser and Wagner, 2004), IHH regulates chondrocyte differentiation and stimulates endochondral bone formation (Cohen, 2003). IHH stimulates the proliferation of chondrocytes at the growth plate and further in development, osteoblast differentiation; in addition, it also regulates chondrocyte hypertrophic differentiation through a negative feedback loop involving Ihh-parathyroid hormone-related protein (PTHrP) (Bitgood and McMahon, 1995; St-Jacques et al., 1999; Long et al., 2004). As well, IHH promotes ossification and fusion of the cranial and palatine bones, discussed further below (Lenton et al., 2011; Levi et al., 2011). Both SHH and IHH are found to be crucial regulators of osteogenesis and therefore of importance in cranial suture biology.
All of the HH homologues undergo the same highly conserved HH signaling pathway that occurs through a three-step process. First, the insoluble HH morphogen is converted to a multimeric form, which renders it soluble and available for diffusion across cell membranes. This HH ligand is subsequently made active as HH ligands precursors undergo autocatalytic cleavage to form 19 kD proteins with a C-terminal cholesterol moiety (Porter et al., 1996; Cohen, 2003) (Figure 1). Palmitoyl acid then modifies the N-terminus, forming a palmitate (Pepinsky et al., 1998). After these two covalent lipid modifications, the HH ligand is now active and has an increased affinity for the cell membrane (Simpson et al., 2009). Second, a large transmembrane protein, Dispatched, releases the now lipid-anchored protein HH from the signaling cell, which allows HH binding to the receptor Patched (PTCH), a transcription inhibitor. After the HH ligand binds to PTCH, Smoothened (SMO), another transmembrane protein for downstream signaling, is freed from PTCH repression and transduces the signal intracellularly. In vertebrates, SMO leads to the transcription of target genes downstream through interaction with glioblastoma gene products (Gli) family of transcription factors (Gli1, Gli2, and Gli3). Recently, the so-called “non-canonical HH signaling pathway” has been discovered, which utilizes Gli independent pathways (Jenkins, 2009). This novel HH signaling has been studied during angiogenesis as well as cancer biology. However, this non-canonical signaling has not been sufficiently characterized for its operation in sutures, and will not be further addressed here (Chinchilla et al., 2010; Spek et al., 2010).
Figure 1.

Hedgehog pathway. The Hedgehog (HH) ligand precursor undergoes a series of modifications until reaching an active, multimeric form (shown in yellow). All three HH ligands then signal through the same pathway: following multimeric HH ligand release from Dispatched (DISP) and secretion from the signaling cell, the HH ligand binds to Patched (PTCH) on the receiving cell, releasing Smoothened (SMO) from constitutive inhibition (Cohen, 2003). Released SMO then shuttles through the cilia. This signals activation of the Gli2/3 complex (shown as Gli2/3A), which promotes gene expression via Gli1, while simultaneously inhibiting the Gli3 repressor form (shown as Gli3R). In the presence of HH activated SMO, the Kif3a motor complex promotes Gli2/3A expression and inhibits repression by Gli3R (Rohatgi and Scott, 2008). HHAT is necessary for post-translational palmitoylation of HH; in the absence of HHAT, HH secretion is decreased (Dennis et al., 2012). In the presence of HH, BOC, CDO, and GAS1 bind to PTCH to form complexes which repress SMO, allowing downstream signaling through Gli to continue (Izzi et al., 2011). RAB23 (shown in red) functions as a negative regulator of the HH pathway, most likely through interaction with the Gli2 activator and Gli3 repressor forms. Please note, the depiction of SMO as a 7-transmembrane protein and both PTCH and DISP as a 12-transmembrane protein correspond to their respective biological conformations (De Rivoyre et al., 2006; Cohen, 2010).
The Gli transcription factors are homologues of Cubitus interruptus (Ci), a transcription factor mediating HH signaling found in Drosophila (Huangfu and Anderson, 2006). Both Gli and Ci function similarly and act as key regulators of targeted gene expression. Gli proteins have been found to function as transcriptional activators, transcriptional repressors or both. Experiments have demonstrated that Gli2 and Gli3 both contain an amino-terminal repressor domain and a carboxyl-terminal activator domain that flanks the zinc fingers (Ruiz i Altaba, 1999; Sasaki et al., 1999; Wong and Reiter, 2008); however, Gli1 does not contain the repressor domain and so cannot be proteolytically processed (Ruiz i Altaba, 1998; Dai et al., 1999). Gene analysis has shown that Gli1 functions solely as a strong transcriptional activator (Hynes et al., 1997; Lee et al., 1997; Karlstrom et al., 2003). In mouse development, Gli1 does not appear to be essential since Gli1−/− mutants are viable and survive from birth to adulthood and exhibit a normal phenotype (Park et al., 2000; Bai et al., 2002). In contrast, the other members of the Gli family, Gli2, and Gli3, are required for organ patterning, such as lung development and spinal cord patterning (Brewster et al., 2000; Bai et al., 2004; Rutter et al., 2010). Both Gli2 and Gli3 can act as both activators and repressors. Their bifunctionality is determined by the presence of HH signaling. When there are high concentrations of HH ligands, proteolytic processing of Gli2 and Gli3 is inhibited, which allows for their activator function. Without HH presence, these transcription factors may undergo cleavage to become repressors (Wang et al., 2000; Bai et al., 2004; Pan et al., 2006). The deviations of the carboxyl-terminal amino acid sequences of Gli2 and Gli3 have been found to result in differential processing (Pan and Wang, 2007). Gli2 proteins have inefficient protein processing and therefore remain transcriptionally active in vivo (Fuccillo et al., 2006). In contrast, the majority of Gli3 proteins are partially degraded, thus mainly functioning as a transcription repressor (Theil et al., 1999; Persson et al., 2002; Rallu et al., 2002). In summary, the Gli protein family controls HH signaling through transcription, both in the cranial suture region and throughout the organism.
Craniofacial dysmorphism caused by hedgehog signaling
In the absence of an active HH ligand or interference with HH signal transduction, a transcriptional repression of HH target genes results in a slew of craniofacial anomalies (see Table 1). Aberrations in Gli3 are known to cause craniofacial dysmorphisms in both human and mice models. One result of altered Gli3 sequence is Greig cephalopolysyndactly syndrome, which causes metopic synostosis and is characterized by polydactyly and hypertelorism (Hui and Joyner, 1993; Quinn et al., 2012; Veistinen et al., 2012). Another caused by mutations in the Gli3 effector is Pallister-Hall Syndrome, with common craniofacial findings including disrupted midline development and abnormalities such as a short nose with flat nasal bridge, and cleft palate (Kuo et al., 1999; Naruse et al., 2010). In fact, the integral role of Gli3 as a transcriptional repressor is evident in studies with Gli3 null mice in which excessive osteoblastic proliferation and differentiation result in craniosynostotic phenotypes (Shimoyama et al., 2007). Interestingly, local application of recombinant FGF2 rescues loss of Gli3 as it stabilizes the increased osteoblastic proliferation observed in Gli3 deficient mice (Rice et al., 2010). HH signaling can also be effected by changes in the physical environment that transduces the signals of proteins in the HH pathway.
Table 1.
Genetic disorders in the hedgehog signaling network.
| Syndrome name | Mutated gene | MIM | Function of proteins* | Skeletal phenotypes* |
|---|---|---|---|---|
| Greig cephalopolysyndactyly syndrome | Gli3 | 175700 | Transcriptional repressor of HH signaling | Pre- and post-axial polydactyly and syndactyly of hands and feet, slight hypertelorism, high prominent forehead |
| Pallister-hall syndrome | Gli3 | 146510 | Transcriptional repressor of HH signaling | Disrupted midline development and craniofacial abnormalities including a short nose with flat nasal bridge and cleft palate |
| NBCCS | PTCH | 109400 | SHH receptor, inhibits SMO expression | Basal cell carcinomas, macroencephaly, cleft lip/palate, intracranial ectopic calcifications and facial dysmorphisms |
| Carpenter's syndrome | RAB23 | 201000 | Negative regulator of SHH | Syndactyly, brachydactyly with shortening or absence of middle phalanges, craniosynostosis of midline and coronal sutures |
| Acrocapitofemoral dysplasia | IHH | 607778 | HH ligand | Clinically short stature with short limbs, brachydactyly, shortening or loss of middle phalanges |
| Holoprosencephaly 1 | GAS1 | 236100 | Co-receptor of PTCH | Missing phalanges and anterior digit syndactyly |
| Holoprosencephaly 11 | CDO | 614226 | Co-receptor of PTCH | Lack of maxillary inscisors, primary palate, hypoplasia of the cartilage of the nasal septum |
| – | BOC | – | Co-receptor of PTCH | No abnormalities are observed in BOC−/− mutants alone. However, BOC and CDO mutants exhibit severe craniofacial midline abnormalities such as elongated nose, cleft lip, and hypotelorism. |
| Asphyxiating thoracic dystrophy 3, Short ribpolydactyly syndrome, type II and type III | DYNC2H1 | 613091, 263520, 263510 | IFT Protein, Ciliogenesis, signal transduction of HH pathway | Short rib polydactyly phenotype, shortened long bones, a narrow rib cage and polydactyly, variable malformations including cleft lip/palate |
| Asphyxiating thoracic dystrophy 5, Cranioectodermal dysplasia 4 | IFT144 | 614376, 614378 | IFT Protein, Ciliogenesis, signal transduction of HH pathway | Polydactyly, truncated ribs, craniosynostosis, exencephaly, reduced palatine bones and misshapen maxillary bones |
| – | HHAT | 206500, 202650 | Post-translational palmitoylation of HH proteins | Midface hypoplasia, agenesis of the jaw, loss of skeletal central bones, apoptosis in craniofacial mesenchyme |
Data from (Temtamy, 1966; Robinson et al., 1985; Shanley et al., 1994; Wild et al., 1997; Kuo et al., 1999; Lee et al., 2001; Liu et al., 2001, 2002; Cole and Krauss, 2003; Hellemans et al., 2003; Johnston et al., 2005; Zhang et al., 2006, 2011; Lo Muzio, 2008; Keaton et al., 2010; Naruse et al., 2010; Ashe et al., 2012; Dennis et al., 2012).
Abbreviations: GAS, growth arrest specific; HH, hedgehog; HHAT, hedgehog acyltransferase; IFT, intraflagellar transport; IHH, indian hedgehog; NBCCS, nevoid basal cell carcinoma syndrome; PTCH, patched; SHH, sonic hedgehog; SMO, smoothened.
Primary cilia serve an important and increasingly understood role in suture biology, as HH transduction initiates at the primary cilia (Tukachinsky et al., 2010). Studies involving cilial defects have shown that primary cilia are crucial to HH signaling. Currently, it is suggested that primary cilia provide an environment that facilitates interactions amongst the different pathway components in HH transduction (Ruat et al., 2012). During HH signaling, intraflagellar transport proteins (IFT), which are required for the production and preservation of cilia, have been found to affect the signal transduction of the HH pathway (Huangfu et al., 2003; Keady et al., 2012; Yang and Wang, 2012). IFT particles are formed by two complexes that use the Kif3 motor complex and the retrograde dynein motors to selectively import or export proteins between the cilium and cytoplasm (Ruat et al., 2012). Signal dependent transfer of PTCH, SMO, and Gli proteins requires ciliary transport in order to activate the HH pathway (Keady et al., 2012). Studies suggest that following PTCH regulation of SMO, SMO is consequently translocated to the cilium through the use of IFT proteins and interacts with Gli to promote Gli activation. Gli activators then move down the cilium to enter the nucleus and promote HH targeted genes (Huangfu and Anderson, 2006; Singla and Reiter, 2006). Interestingly, mutations that disrupt IFT proteins show phenotypes characteristic of SHH signaling defects. This observation extends to the craniofacial skeleton. For example, mutations in the IFT protein DYNC2H1, causes short rib polydactyly syndrome, a lethal autosomal recessive condition that features cerebral and skeletal abnormalities, such as HPE, in addition to other appendicular malformations (Dagoneau et al., 2009; Merrill et al., 2009; El Hokayem et al., 2012). Mutations in another IFT protein, IFT144, also result in craniofacial anomalies such as craniosynostosis and exencephaly, which results from deficient ciliogenesis and diminished response to upstream activation of HH signaling (Ashe et al., 2012). Gene analysis has found that IFT motor proteins such as Kif3a (Kinesin-like protein) are required for signaling of Gli transcription factors (Haycraft et al., 2005; Huangfu and Anderson, 2005; Liu et al., 2005). Kif3a conditional knockouts (Kif3a Wnt1-Cre) show a phenotype with severe cranial dysmorphisms including abnormal openings in the skull vault, associated with displacement of the neuroectodermal domains of SHH signaling (Brugmann et al., 2010). Interestingly, Kif3a deficient mouse skulls exhibit abnormalities partially overlapping with the SHH and IHH null phenotypes (see discussion below) (Koyama et al., 2007). In summary, cilia are involved in the regulation of HH signal transduction, although the precise mechanisms of this relationship are only partially elucidated.
Another potential cause of craniofacial abnormalities includes exposure to teratogens, which interfere with the HH signaling pathway. One of the main plant alkaloids that produce these deformities is the jervine family of alkaloids. Cyclopian deformities from alkaloids were first observed in the offspring of pregnant sheep grazing on Veratrum Californicum. Further experimentation found that these chemical compounds also trigger clefting and HPE in sheep and other animals by inhibiting the response of target tissues to HH ligands (Binns et al., 1963). The structural similarity of these alkaloids to cholesterol allows them to inhibit cholesterol's stimulatory effect on HH signaling (Cooper et al., 1998; Chen et al., 2002). For example, the steroidal alkaloid Cyclopamine (11-deoxojervine) causes teratogenic effects through direct binding of cyclopamine to the SMO heptahelical bundle thereby creating a PTCH independent pathway, thus resulting in HH pathway inhibition (Incardona et al., 2000; Chen et al., 2002). Cyclopamine is one of the causes of embryonic deficiency of midline and lower medial nasal prominence tissue, resulting in severe cranial defects including lateral cleft lip, cleft palate, and the cyclopia phenotype in many animal models including zebrafish and mice (Lipinski et al., 2010; Buttner et al., 2012).
Genetic mutations, rather than exposure to jervines, represent the major source of HH signaling abnormalities causing congenital dysmorphisms in humans. Currently, one third of all birth defects are craniofacial abnormalities, with HPE being the most common developmental disorder of the forebrain (Gorlin et al., 1990; Ming and Muenke, 1998; Ming et al., 1998). Despite the rare live-birth prevalence of 1 in 10,000 HPE infants, it may be as common as 1 in 250 conceptuses by some estimates (Vaz et al., 2012). In fact, SHH was the first gene identified to cause HPE in mice and humans from nonsense mutations or deletions that result in loss of function (Odent et al., 1999). In clinical studies, familial forms of HPE involve SHH gene mutations in up to 23% of affected families; much greater than the percentage of non-syndromic mutations, 1% (Roessler et al., 1997; Ming et al., 1998). SHH gene mutations are also responsible for lip and palatal defects as SHH signaling pathways regulate the epithelium and mesenchyme interactions that promote cell proliferation and palatal growth (Murray and Schutte, 2004). Additionally, IHH gene mutations, such as additional copies of the IHH locus, are associated malformations that result in syndactyly and craniosynostosis (Klopocki et al., 2011). Mutations in IHH can also cause acrocapitofermoral dysplasia, an autosomal recessive skeletal dysplasia resulting in shortening or loss of the middle phalanges (Gao et al., 2001; Hellemans et al., 2003; Byrnes et al., 2009).
Other gene mutations that induce HPE and result in HH pathway dysregulation involve PTCH, Growth arrest-specific 1 (GAS1), CDO, and BOC (Roessler and Muenke, 1998; Seppala et al., 2007; Allen et al., 2011; Bae et al., 2011; Zhang et al., 2012). CDO and BOC, the external binding domains of SHH, GAS1, the co-receptor of PTCH, and PTCH directly interact to activate the Gli transcription pathway (Izzi et al., 2011). GAS 1 has been found to promote SHH signaling in embryological development while being negatively regulated in response to SHH signaling (Allen et al., 2011). GAS 1 mutant mice have demonstrated various phenotypes similar to reduced SHH signaling (Liu et al., 2002). Co-existing CDO and GAS1 mutations in mice have resulted in HPE, therefore demonstrating these two co-receptors are essential to proper cranial patterning (Bae et al., 2011). Likewise, CDO and BOC double mutants also exhibit the HPE phenotype as well as extreme neural patterning defects. Mutations in BOC alone result in viable offspring with no HPE. Studies suggest that this absence may be compensated for by CDO, indicating that the two have similar functions (Zhang et al., 2012). Also, hedgehog acyltransferase (HHAT) loss of function decreases HH secretion and leads to the HPE phenotype; HHAT is necessary for modification of HH proteins (Chen et al., 2004; Dennis et al., 2012; Hardy and Resh, 2012). Yet another genetic mutation in the HH pathway is found in the mutation of the PTCH chromosome located on 9q22.3–q31 (Farndon et al., 1992). The loss of normal PTCH function leads to increased HH signaling and is thought to result in Nevoid Basal Cell Carcinoma Syndrome (NBCCS). This phenotype is characterized by basal cell carcinomas, macroencephaly, cleft lip and palate, intracranial ectopic calcifications, and facial dysmorphisms (Evans et al., 1993; Shanley et al., 1994; Lo Muzio, 2008; Zhang et al., 2011). Genetic mutations in regulator genes of HH signaling may also cause calvarial dysmorphisms. A mutation in the RAB23 gene leads to a dysfunctional repressor of HH signaling. This results in Carpenter's Syndrome, characterized by fusion of the midline sutures, obesity and syndactyly (Eggenschwiler et al., 2001; Jenkins et al., 2007). In summary, a substantial number of known human craniofacial anomalies in the HPE spectrum arise from HH and related genes mutations. See Table 1 for a summary of syndromes, gene mutations, and resultant craniofacial dysmorphisms.
Role of hedgehog signaling in cranial suture morphogenesis
Both SHH and IHH ligands have demonstrated importance in cranial suture morphogenesis. Studies have identified discrete expression patterns of SHH vs. IHH ligands in the developing skull, which are distinct and indicate probable differences in function (Figure 2). Murine studies have shown that SHH gene expression in the skull occurs at the end of embryonic development (from E18 and onwards) in a discontinuous pattern in the osteogenic fronts of the midline suture mesenchyme, but is absent in the coronal sutures (Kim et al., 1998). However, other investigators found contradicting evidence of weak SHH expression in the parietal bones of E16.5 mice, as well as absent SHH expression in the midline suture mesenchyme (Lenton et al., 2011). Thus, the true domains of SHH expression are relatively ill defined. IHH is present during calvarial osteoblastic development and is expressed during osteoblast proliferation at the osteogenic fronts in mice, (the leading edge of the ossifying calvarial bone; Jacob et al., 2007). In another study, IHH expression was also found in the sagittal suture mesenchyme albeit to a lesser degree than in ossifying bones and osteogenic fronts in E17.5 mice (Lenton et al., 2011). Thus, the expression patterns of SHH and IHH differ in the cranial suture complex, although some discrepancies regarding the precise domains of SHH expression exist in the literature.
Figure 2.
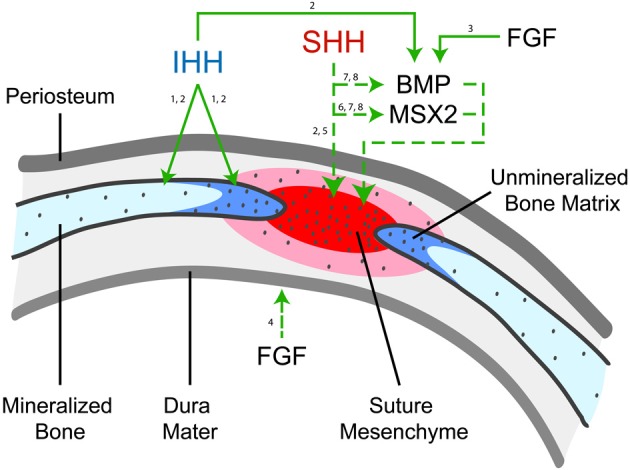
HH Ligand expression and function in cranial suture morphogenesis. Indian Hedgehog (IHH) is observed in the cranial bones (light blue), primarily at the osteogenic front (dark blue) (1, Jacob et al., 2007; 2, Lenton et al., 2011). In this figure, solid arrows represent consistent observations while dashed arrows represent postulated pathways. Studies have shown that IHH functions to increase new bone formation at the osteogenic fronts, likely through its upregulation of BMP2 and BMP4 (2, Lenton et al., 2011). Fibroblast growth factor (FGF) has been shown to promote BMP2/4 (3, Sahar et al., 2005) and is highly expressed in dura matter and is one of the main diffusible growth factors inducing sutural fusion (4, Li et al., 2007). In contrast, SHH has been observed to be expressed in a patched pattern in the midline suture mesenchyme (shown in red), although some disagreement regarding its expression pattern exists (2, Lenton et al., 2011). The function of SHH is less clear, although it has been postulated to function in maintaining suture patency (5, Kim et al., 1998). SHH may increase mesenchymal proliferation and suture mesenchyme thickness via promotion of MSX2 (6, Alappat et al., 2003), and similarities are present between the expression of SHH, MSX2, and BMP expression during neonatal craniofacial suture development (7, Liem et al., 2000; 8, Santagati and Rijli, 2003).
The domain-specific HH ligand expression has led investigators to examine different plausible functions for SHH and IHH ligands. The predominant presence of SHH in the suture mesenchyme and its overlapping expressing pattern with mesenchymal cell proliferation has led some to postulate that SHH has a role in maintaining suture patency (Kim et al., 1998). In fact, SHH may increase mesenchymal proliferation and suture mesenchyme thickness via promotion of MSX2, Muscle Segment Homeobox, a homeobox gene present in osteoblastic cells, in concert with BMP4 signaling (Alappat et al., 2003). Unlike other cytokines, which are concentrated in the dura mater of cranial sutures, SHH/IHH expression does not appear to be significantly derived from the underlying dura mater. In this way, HH signaling likely functions in a much different way than, for example, FGF2 or TGF-β (Mehrara et al., 1999; Moursi et al., 2002; Li et al., 2007; Gosain et al., 2009). In contrast, IHH is expressed by calvarial osteoblasts and disruption of IHH signaling impairs osteoblastogenesis (Murakami et al., 1997). IHH expression indicates that IHH induces PTCH and BMP2/4 expression, which results in intramembranous bone formation at the osteogenic fronts during intramembranous ossification and later suture fusion (Jacob et al., 2007; Lenton et al., 2011). Toward the completion of physiologic suture fusion, a decrease in osteoblast proliferation is observed, which coincides with a decrease in IHH expression. Another theory, although less frequently suggested, is that IHH functionally represses osteogenic lineage differentiation and that loss of IHH results in premature osteoprogenitor cell differentiation (Abzhanov et al., 2007). However, later studies by other research groups have not concurred with this observation (Lenton et al., 2011). Finally, IHH expression on the bony interfaces of synostotic rabbits may support the role of IHH in premature suture fusion (Nott et al., 2002a). Overall, the current available data suggests that SHH may prevent suture fusion and that IHH promotes calvarial ossification and sutural fusion. The idea that different, even opposing effects can result from two morphogens which use the same conserved pathway is a complex concept. Future research directly addressing this question may yield further insight.
Loss-of-function experiments involving either SHH or IHH results in profound craniofacial dysmorphisms. SHH−/− animals die before or shortly after birth with a variety of developmental defects, including absence of distinct forelimb and hindlimb structures, and a dysostoic calvarial phenotype (McMahon et al., 2003). Unfortunately, dorsal midline structures fail to form and therefore null animals demonstrate secondarily impeded calvarial ossification (Chiang et al., 1996). In addition, it has been found that when SHH signaling is blocked in the brain but SHH is also applied early to the frontonasal ectodermal zone, the formation of the face and upper jaw is significantly improved. This suggests that the SHH signaling, which mediates brain to face interaction, is time-dependent (Chong et al., 2012). Approximately half of IHH−/− mice die during mid-gestation due to yolk sac defects, while the remainder dies at birth, and is attributed to rib cage deformities and respiratory failure (Byrd et al., 2002). The IHH−/− mouse demonstrates a dysostotic phenotype with reduced calvarial bone size and ossification, and grossly widened cranial sutures (St-Jacques et al., 1999; Razzaque et al., 2005; Kolpakova-Hart et al., 2008). However, unlike the SHH null mouse, the IHH null mouse cranial phenotype is not secondary to underlying CNS dysgenesis. Further studies demonstrated that reduced ossification in the IHH−/− mouse is accompanied by global reduction of osteogenic markers and reduced BMP2/4 expression (Lenton et al., 2011). Overall, loss of function studies have suggested that IHH has a pro-osteogenic effect in calvarial ossification, while SHH studies are difficult to interpret due to widespread CNS malformations.
Both SHH and IHH ligands also interact with other signaling pathways in the developing skull, the most well studied of which are BMPs. BMPs, part of the TGF-β superfamily of growth factors, were first identified by their ability to induce ectopic ossification (Wozney et al., 1988). Currently, BMPs, such as BMP2, are in wide clinical use as a bone graft substitute for spinal fusion (Deyo et al., 2012; Hagen et al., 2012). The expression of the BMP signaling antagonist Noggin has been shown to be a critical regulator of cranial suture fusion (Warren et al., 2003; Jacob et al., 2007). In many studies, similarities were found between the patterning of SHH and BMP expression during neonatal craniofacial suture development (Liem et al., 2000; Santagati and Rijli, 2003) as well as with the transcription factor MSX2, a transcriptional repressor of neural crest-derived cells (Hodgkinson et al., 1993; Towler et al., 1994; Kim et al., 1998; Takahashi et al., 2001). However, the exact functional importance of SHH/BMP/MSX2 co-expression in the developing calvaria is not fully elucidated (Lallemand et al., 2009). Investigators have postulated that IHH also interacts with BMP signaling, and as previously mentioned, the IHH−/− mouse demonstrates reduced calvarial BMP2/4 expression (Lenton et al., 2011). In addition, in vitro studies in suture mesenchymal cells found that IHH positively regulated BMP2 and BMP4 transcript abundance, associated with upregulation of osteogenic markers (Lenton et al., 2011). In sum, this data suggests that IHH lies upstream of BMP2/4 signaling and may be a part of the regulatory network that controls BMP expression, thereby influencing cranial ossification. In addition, and although not thoroughly investigated, HH signaling likely interacts with FGF signaling (including FGF-2 and FGF-9). FGF2/9 expression peaks during physiologic suture fusion (Fakhry et al., 2005), and activating mutations in FGFR-2 are the main cause of syndromic synostosis (McGillivray et al., 2005; Marie et al., 2008). The interactions between FGF and HH signaling have been more thoroughly studied in facial development, including the coordinated development of the medial nasal prominence (Hu et al., 2003). Studies suggest that retinoid signaling mediates FGF-8 and SHH expression and synchronizes the development of the face (Schneider et al., 2001). While coordinate effects of HH/FGF signaling certainly exist in cranial suture, a precise characterization of this link has yet to be determined. In summary, strong data exist to support regulation of BMP signaling by both SHH and IHH in the calvaria.
Role of hedgehog signaling in endochondral processes
Another important aspect of HH signaling involves endochondral ossification, which is the process of bone development in the majority of bones in the axial and appendicular skeleton, as well as the cranial base (Nagayama et al., 2008). In contrast to intramembranous ossification (discussed previously), endochondral skeletogenesis occurs as mesenchymal cells differentiate into chondrocytes. As the cartilage enlarges, hypertrophic chondrocytes regulate matrix mineralization, vascularization and chondroclast attraction. As chondrocytes proliferate they are replaced by osteoblasts, which provide the scaffold for bone growth (Kronenberg, 2003). The cranial base is formed through endochondral ossification, in which the chondrocranium is formed and replaced by bones. These individual bones are connected by synchondroses, cartilaginous structures, which are similar to long bone growth plates (Nie, 2005). As with intramembranous bones, HH signaling is known to play an integral role in cranial base development (Figure 3).
Figure 3.
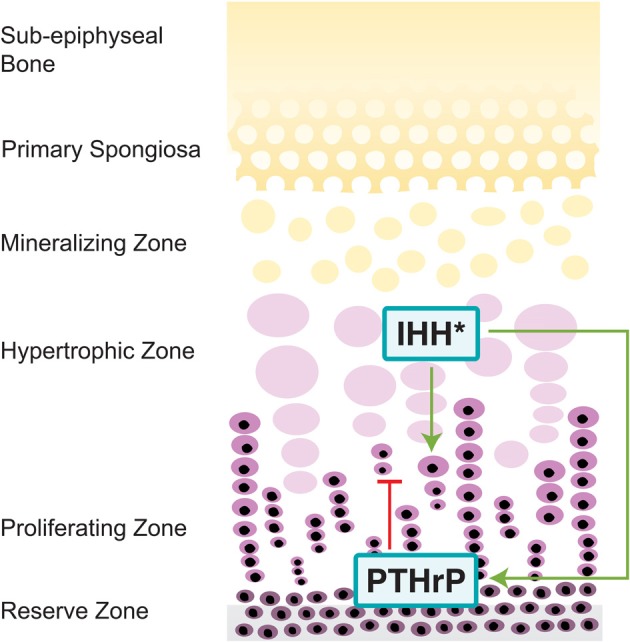
Hedgehog pathway in the cranial base. IHH, expressed in the pre-hypertrophic zones of growth plates, increases chondrocyte proliferation through increased expression of PTCH, a process mediated by intracellular component EVC (Long et al., 2001; Ruiz-Perez et al., 2007; Pacheco et al., 2012). Through a negative feedback loop, IHH also delays chondrocyte differentiation in order to sustain early chondrocyte production through synthesis of PTHrP, an IHH inhibitor (Lanske et al., 1996; Vortkamp et al., 1996; Kronenberg, 2003). *IHH also regulates craniofacial morphogenesis through altering the IHH/PTHrP negative feedback loop, though it is primarily expressed in the spheno-occipital synchondroses (Tavella et al., 2004).
SHH regulates craniofacial morphogenesis by altering the IHH/PTHrP negative feedback loop. Overexpression of SHH increases the amount of IHH inhibitor, PTHrP, thereby resulting in the failure of the cranial base to fully develop (Tavella et al., 2004). IHH works to delay the differentiation of chondrocytes through synthesis of PTHrP, which works on the PTH/PTHrP receptors in chondrocytes (Lanske et al., 1996). However, IHH is not present in the earliest stages of cranial base formation and patterning, as opposed to SHH, which indicates that their functions are not redundant (Nie et al., 2005). Increased SHH expression during later developmental period strongly correlates with IHH, and suggests that both IHH and SHH control subsequent bone formation in the cranial base. IHH, nonetheless, is the key regulator for endochondral ossification and is secreted by prehypertrophic chondrocytes and early hypertrophic chondrocytes (St-Jacques et al., 1999). IHH mainly functions to inhibit the progress of immature chondrocyte development in order to sustain early chondrocyte production (Vortkamp et al., 1996; Kronenberg, 2003). IHH in chondrocytes increases expression of PTCH, which then can activate SMO, allowing for increased chondrocyte proliferation (Long et al., 2001). Experiments with mice have shown that IHH regulates endochondral ossification in concert with a number of other proteins, including Ellis-van Creveld protein (EVC) and Kif3a (discussed below). IHH instigates the proliferation of chondrocytes and regulates maturation to promote cranial base elongation.
IHH is expressed in the pre-hypertrophic zones of growth plates while SHH expression is found in proximity to the spheno-occipital synchondroses. As the posterior portion of the cranial base is highly receptive to SHH, proper ossification occurs in both wild type and IHH−/− mice. However, the anterior intrasphenoidal synchondrosis appears to be less susceptive to SHH, resulting in IHH−/− mice with defective ossification in this area (Young et al., 2006). In addition IHH−/− mice show ectopic calcification in the cartilaginous synchondroses between the basioccipital and exoccipital bones in the cranial base and delayed hypertrophic chondrocyte differentiation (St-Jacques et al., 1999). This delayed differentiation also results in IHH−/− possessing disorganized synchrondosis growth plates and defects arise between IHH-producing pre-hypertrophic chondrocytes and PTHrP proliferating chondrocytes (Young et al., 2006). The many abnormalities produced from the IHH−/− phenotype demonstrate the integral part that IHH plays within cranial base synchondrosis and chondrogenesis.
Additionally, mutations in proteins involved in IHH signaling also contribute to anomalies in the cranial base. EVC mediates signaling in the IHH pathway and is required for control over the rate of chondrocyte hypertrophy (Ruiz-Perez et al., 2007; Pacheco et al., 2012). EVC −/− mice exhibit morphological defects of the cranial base including midline gaps and partially fused basisphenoid suture in the neonatal stage (Pacheco et al., 2012). EVC −/− mice demonstrate a decreased expression in IHH downstream genes, such as PTCH and Gli1, suggesting that EVC is an intracellular component of the IHH signaling pathway. The motor protein Kif3a, as discussed earlier with its function intramembranous bone defects, also regulates the development of the cranial base. In Kif3a-deficient mice, there is a delay in chondrocyte maturation and hypertrophy, as IHH has an increased gradient of expression throughout the upper growth plate and IHH receptors, such as PTCH, are reduced (Koyama et al., 2007). Thus, IHH is required for synchondrosis organization and function, and disrupted regulation of IHH signaling by EVC or Kif3a results in significantly abnormal cranial base phenotypes.
Unresolved concepts
While a number of studies have addressed the importance of HH signaling in cranial vault ossification and cranial suture biology, there are many questions that remain unanswered. Firstly, the precise expression pattern of SHH in the cranial suture is not yet agreed upon. Previous studies have found that SHH expression is principally observed in the ectodermal elements of the skull rather than in the bone or suture mesenchyme. In fact, using a reporter for HH activity, nearly complete loss of HH activity was observed in the skull and mesenchyme of IHH−/− null animals (Lenton et al., 2011). This suggests that SHH may not be as relevant to calvarial ossification as other researchers suggest. However, through the use of in situ hybridization for SHH, expression was found on the sagittal and metopic sutures (Kim et al., 1998). This ambiguity could be the result of different timepoints of analysis (E16 vs. E18) or differences in experimental methodology. Secondly, there exist a large number of in vitro studies that demonstrate the basic pro-osteogenic effects of SHH (Yuasa et al., 2002; van der Horst et al., 2003; James et al., 2010, 2012; Tian et al., 2012). These studies, which have used diverse cell types including pre-osteoblastic cell lines and primary mesenchymal stem cell sources, would contradict the hypothesis that SHH functions to prevent suture ossification. In addition, a study on craniosynostotic rabbits documented increased expression of SHH in craniosynostotic sutures, (Nott et al., 2002b) and SHH overexpression, which simulates loss of PTCH function, has resulted in the absence of calvarial bones in the NBCCS mouse phenotype (Hu and Helms, 1999; Cobourne et al., 2009), further complicating the role of SHH in suture morphogenesis. In summary, the exact endogenous function of SHH on calvarial ossification has yet to be fully elucidated, either as an inhibitor or inducer of calvarial ossification/suture fusion. Thirdly, it remains uncertain if IHH and SHH in fact have redundant or even opposing functions in regulating calvarial ossification. Indeed as IHH and SHH function through the same cell surface receptor, it is difficult to determine a manner in which there would have antagonistic effects. A potential method to clarify the specific functions of Indian vs. Sonic may be through the use of bone specific knockout mice, allowing for examination of postnatal bone phenotypes in the absence of specific HH ligands.
Conclusions
SHH and IHH have critical, sometimes synergistic, and currently debated biological roles in cranial suture development. Disruptions in either signaling ligand result in various defects in the development of cranial sutures. Examination of craniofacial defects arising from HH signaling abnormalities will provide new insights into the basic functions of the HH pathway in embryogenesis and patterning, and later bone and cartilage differentiation. Although much is known, there remain numerous questions for future study. Such questions include: (1) the precise function of SHH in cranial suture development, (2) whether IHH and SHH have different functions in the calvaria, and (3) improved understanding of the intersection of HH with other signaling pathways in craniofacial patterning, such as BMP signaling.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- Abzhanov A., Rodda S. J., McMahon A. P., Tabin C. J. (2007). Regulation of skeletogenic differentiation in cranial dermal bone. Development 134, 3133–3144 10.1242/dev.002709 [DOI] [PubMed] [Google Scholar]
- Alappat S., Zhang Z. Y., Chen Y. P. (2003). Msx homeobox gene family and craniofacial development. Cell Res. 13, 429–442 10.1038/sj.cr.7290185 [DOI] [PubMed] [Google Scholar]
- Allen B. L., Song J. Y., Izzi L., Althaus I. W., Kang J. S., Charron F., et al. (2011). Overlapping roles and collective requirement for the coreceptors GAS1, CDO, and BOC in SHH pathway function. Dev. Cell 20, 775–787 10.1016/j.devcel.2011.04.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson E., Peluso S., Lettice L. A., Hill R. E. (2012). Human limb abnormalities caused by disruption of hedgehog signaling. Trends Genet. 28, 364–373 10.1016/j.tig.2012.03.012 [DOI] [PubMed] [Google Scholar]
- Ashe A., Butterfield N. C., Town L., Courtney A. D., Cooper A. N., Ferguson C., et al. (2012). Mutations in mouse Ift144 model the craniofacial, limb and rib defects in skeletal ciliopathies. Hum. Mol. Genet. 21, 1808–1823 10.1093/hmg/ddr613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae G. U., Domene S., Roessler E., Schachter K., Kang J. S., Muenke M., et al. (2011). Mutations in CDON, encoding a hedgehog receptor, result in holoprosencephaly and defective interactions with other hedgehog receptors. Am. J. Hum. Genet. 89, 231–240 10.1016/j.ajhg.2011.07.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baer M. J. (1954). Patterns of growth of the skull as revealed by vital staining. Hum. Biol. 26, 80–126 [PubMed] [Google Scholar]
- Bai C. B., Auerbach W., Lee J. S., Stephen D., Joyner A. L. (2002). Gli2, but not Gli1, is required for initial Shh signaling and ectopic activation of the Shh pathway. Development 129, 4753–4761 [DOI] [PubMed] [Google Scholar]
- Bai C. B., Stephen D., Joyner A. L. (2004). All mouse ventral spinal cord patterning by hedgehog is Gli dependent and involves an activator function of Gli3. Dev. Cell 6, 103–115 10.1016/S1534-5807(03)00394-0 [DOI] [PubMed] [Google Scholar]
- Belloni E., Muenke M., Roessler E., Traverso G., Siegel-Bartelt J., Frumkin A., et al. (1996). Identification of Sonic hedgehog as a candidate gene responsible for holoprosencephaly. Nat. Genet. 14, 353–356 10.1038/ng1196-353 [DOI] [PubMed] [Google Scholar]
- Binns W., James L. F., Shupe J. L., Everett G. (1963). A Congenital cyclopian-type malformation in lambs induced by maternal ingestion of a range plant, veratrum californicum. Am. J. Vet. Res. 24, 1164–1175 [PubMed] [Google Scholar]
- Bitgood M. J., McMahon A. P. (1995). Hedgehog and Bmp genes are coexpressed at many diverse sites of cell-cell interaction in the mouse embryo. Dev. Biol. 172, 126–138 10.1006/dbio.1995.0010 [DOI] [PubMed] [Google Scholar]
- Bradley J. P., Levine J. P., McCarthy J. G., Longaker M. T. (1997). Studies in cranial suture biology: regional dura mater determines in vitro cranial suture fusion. Plast. Reconstr. Surg. 100, 1091–1099 discussion: 1100–1092. [DOI] [PubMed] [Google Scholar]
- Brault V., Moore R., Kutsch S., Ishibashi M., Rowitch D. H., McMahon A. P., et al. (2001). Inactivation of the beta-catenin gene by Wnt1-Cre-mediated deletion results in dramatic brain malformation and failure of craniofacial development. Development 128, 1253–1264 [DOI] [PubMed] [Google Scholar]
- Brewster R., Mullor J. L., Ruiz i Altaba A. (2000). Gli2 functions in FGF signaling during antero-posterior patterning. Development 127, 4395–4405 [DOI] [PubMed] [Google Scholar]
- Bristol R. E., Lekovic G. P., Rekate H. L. (2004). The effects of craniosynostosis on the brain with respect to intracranial pressure. Semin. Pediatr. Neurol. 11, 262–267 [DOI] [PubMed] [Google Scholar]
- Brugmann S. A., Allen N. C., James A. W., Mekonnen Z., Madan E., Helms J. A. (2010). A primary cilia-dependent etiology for midline facial disorders. Hum. Mol. Genet. 19, 1577–1592 10.1093/hmg/ddq030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buttner A., Busch W., Kluver N., Giannis A., Scholz S. (2012). Transcriptional responses of zebrafish embryos exposed to potential sonic hedgehog pathway interfering compounds deviate from expression profiles of cyclopamine. Reprod. Toxicol. 33, 254–263 10.1016/j.reprotox.2011.12.010 [DOI] [PubMed] [Google Scholar]
- Byrd N., Becker S., Maye P., Narasimhaiah R., St-Jacques B., Zhang X., et al. (2002). Hedgehog is required for murine yolk sac angiogenesis. Development 129, 361–372 [DOI] [PubMed] [Google Scholar]
- Byrnes A. M., Racacho L., Grimsey A., Hudgins L., Kwan A. C., Sangalli M., et al. (2009). Brachydactyly A-1 mutations restricted to the central region of the N-terminal active fragment of Indian Hedgehog. Eur. J. Hum. Genet. 17, 1112–1120 10.1038/ejhg.2009.18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capdevila J., Johnson R. L. (2000). Hedgehog signaling in vertebrate and invertebrate limb patterning. Cell. Mol. Life Sci. 57, 1682–1694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chai Y., Jiang X., Ito Y., Bringas P., Jr., Han J., Rowitch D. H., et al. (2000). Fate of the mammalian cranial neural crest during tooth and mandibular morphogenesis. Development 127, 1671–1679 [DOI] [PubMed] [Google Scholar]
- Chai Y., Maxson R. E., Jr. (2006). Recent advances in craniofacial morphogenesis. Dev. Dyn. 235, 2353–2375 10.1002/dvdy.20833 [DOI] [PubMed] [Google Scholar]
- Chen J. K., Taipale J., Cooper M. K., Beachy P. A. (2002). Inhibition of Hedgehog signaling by direct binding of cyclopamine to Smoothened. Genes Dev. 16, 2743–2748 10.1101/gad.1025302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M. H., Li Y. J., Kawakami T., Xu S. M., Chuang P. T. (2004). Palmitoylation is required for the production of a soluble multimeric Hedgehog protein complex and long-range signaling in vertebrates. Genes Dev. 18, 641–659 10.1101/gad.1185804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang C., Litingtung Y., Lee E., Young K. E., Corden J. L., Westphal H., et al. (1996). Cyclopia and defective axial patterning in mice lacking Sonic hedgehog gene function. Nature 383, 407–413 10.1038/383407a0 [DOI] [PubMed] [Google Scholar]
- Chim H., Manjila S., Cohen A. R., Gosain A. K. (2011). Molecular signaling in pathogenesis of craniosynostosis: the role of fibroblast growth factor and transforming growth factor-beta. Neurosurg. Focus 31, E7 10.3171/2011.5.FOCUS1197 [DOI] [PubMed] [Google Scholar]
- Chinchilla P., Xiao L., Kazanietz M. G., Riobo N. A. (2010). Hedgehog proteins activate pro-angiogenic responses in endothelial cells through non-canonical signaling pathways. Cell Cycle 9, 570–579 10.4161/cc.9.3.10591 [DOI] [PubMed] [Google Scholar]
- Chong H. J., Young N. M., Hu D., Jeong J., McMahon A. P., Hallgrimsson B., et al. (2012). Signaling by SHH rescues facial defects following blockade in the brain. Dev. Dyn. 241, 247–256 10.1002/dvdy.23726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobourne M. T., Xavier G. M., Depew M., Hagan L., Sealby J., Webster Z., et al. (2009). Sonic hedgehog signalling inhibits palatogenesis and arrests tooth development in a mouse model of the nevoid basal cell carcinoma syndrome. Dev. Biol. 331, 38–49 10.1016/j.ydbio.2009.04.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen M. M., Jr. (1993). Sutural biology and the correlates of craniosynostosis. Am. J. Med. Genet. 47, 581–616 10.1002/ajmg.1320470507 [DOI] [PubMed] [Google Scholar]
- Cohen M. M., Jr. (2003). The hedgehog signaling network. Am. J. Med. Genet. A 123A, 5–28 10.1002/ajmg.a.20495 [DOI] [PubMed] [Google Scholar]
- Cohen M. M., Jr. (2009). Perspectives on craniosynostosis: sutural biology, some well-known syndromes, and some unusual syndromes. J. Craniofac. Surg. 20Suppl. 1, 646–651 10.1097/SCS.0b013e318193d48d [DOI] [PubMed] [Google Scholar]
- Cohen M. M., Jr. (2010). Hedgehog signaling update. Am. J. Med. Genet. A 152A, 1875–1914 10.1002/ajmg.a.32909 [DOI] [PubMed] [Google Scholar]
- Cole F., Krauss R. S. (2003). Microform holoprosencephaly in mice that lack the Ig superfamily member CDO. Curr. Biol. 13, 411–415 10.1016/S0960-9822(03)00088-5 [DOI] [PubMed] [Google Scholar]
- Cooper G. M., Durham E. L., Cray J. J., Jr., Siegel M. I., Losee J. E., Mooney M. P. (2012). Tissue interactions between craniosynostotic dura mater and bone. J. Craniofac. Surg. 23, 919–924 10.1097/SCS.0b013e31824e645f [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper M. K., Porter J. A., Young K. E., Beachy P. A. (1998). Teratogen-mediated inhibition of target tissue response to Shh signaling. Science 280, 1603–1607 10.1126/science.280.5369.1603 [DOI] [PubMed] [Google Scholar]
- Couly G. F., Coltey P. M., Le Douarin N. M. (1993). The triple origin of skull in higher vertebrates: a study in quail-chick chimeras. Development 117, 409–429 [DOI] [PubMed] [Google Scholar]
- Dagoneau N., Goulet M., Genevieve D., Sznajer Y., Martinovic J., Smithson S., et al. (2009). DYNC2H1 mutations cause asphyxiating thoracic dystrophy and short rib-polydactyly syndrome, type III. Am. J. Hum. Genet. 84, 706–711 10.1016/j.ajhg.2009.04.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai P., Akimaru H., Tanaka Y., Maekawa T., Nakafuku M., Ishii S. (1999). Sonic Hedgehog-induced activation of the Gli1 promoter is mediated by GLI3. J. Biol. Chem. 274, 8143–8152 10.1074/jbc.274.12.8143 [DOI] [PubMed] [Google Scholar]
- De Rivoyre M., Ruel L., Varjosalo M., Loubat A., Bidet M., Therond P., et al. (2006). Human receptors patched and smoothened partially transduce hedgehog signal when expressed in Drosophila cells. J. Biol. Chem. 281, 28584–28595 10.1074/jbc.M512986200 [DOI] [PubMed] [Google Scholar]
- Dennis J. F., Kurosaka H., Iulianella A., Pace J., Thomas N., Beckham S., et al. (2012). Mutations in Hedgehog acyltransferase (Hhat) perturb Hedgehog signaling, resulting in severe acrania-holoprosencephaly-agnathia craniofacial defects. PLoS Genet. 8:e1002927 10.1371/journal.pgen.1002927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derynck R., Chen R. H., Ebner R., Filvaroff E. H., Lawler S. (1994). An emerging complexity of receptors for transforming growth factor-beta. Int. Symp. Princess Takamatsu Cancer Res. Fund 24, 264–275 [PubMed] [Google Scholar]
- Deyo R. A., Ching A., Matsen L., Martin B. I., Kreuter W., Jarvik J. G., et al. (2012). Use of bone morphogenetic proteins in spinal fusion surgery for older adults with lumbar stenosis: trends, complications, repeat surgery, and charges. Spine (Phila Pa 1976) 37, 222–230 10.1097/BRS.0b013e31821bfa3a [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon A. D., Hoyte D. A. N., Rönning O. (1997). Fundamentals of Craniofacial Growth. Boca Raton, FL: CRC Press; 497 [Google Scholar]
- Eggenschwiler J. T., Espinoza E., Anderson K. V. (2001). Rab23 is an essential negative regulator of the mouse Sonic hedgehog signalling pathway. Nature 412, 194–198 10.1038/35084089 [DOI] [PubMed] [Google Scholar]
- El Hokayem J., Huber C., Couve A., Aziza J., Baujat G., Bouvier R., et al. (2012). NEK1 and DYNC2H1 are both involved in short rib polydactyly Majewski type but not in Beemer Langer cases. J. Med. Genet. 49, 227–233 10.1136/jmedgenet-2011-100717 [DOI] [PubMed] [Google Scholar]
- Evans D. G., Ladusans E. J., Rimmer S., Burnell L. D., Thakker N., Farndon P. A. (1993). Complications of the naevoid basal cell carcinoma syndrome: results of a population based study. J. Med. Genet. 30, 460–464 10.1136/jmg.30.6.460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fakhry A., Ratisoontorn C., Vedhachalam C., Salhab I., Koyama E., Leboy P., et al. (2005). Effects of FGF-2/-9 in calvarial bone cell cultures: differentiation stage-dependent mitogenic effect, inverse regulation of BMP-2 and noggin, and enhancement of osteogenic potential. Bone 36, 254–266 10.1016/j.bone.2004.10.003 [DOI] [PubMed] [Google Scholar]
- Farndon P. A., Del Mastro R. G., Evans D. G., Kilpatrick M. W. (1992). Location of gene for Gorlin syndrome. Lancet 339, 581–582 [DOI] [PubMed] [Google Scholar]
- Fietz M. J., Concordet J. P., Barbosa R., Johnson R., Krauss S., McMahon A. P., et al. (1994). The hedgehog gene family in Drosophila and vertebrate development. Dev. Suppl. 43–51 [PubMed] [Google Scholar]
- Fuccillo M., Joyner A. L., Fishell G. (2006). Morphogen to mitogen: the multiple roles of hedgehog signalling in vertebrate neural development. Nat. Rev. Neurosci. 7, 772–783 10.1038/nrn1990 [DOI] [PubMed] [Google Scholar]
- Gao B., Guo J., She C., Shu A., Yang M., Tan Z., et al. (2001). Mutations in IHH, encoding Indian hedgehog, cause brachydactyly type A-1. Nat. Genet. 28, 386–388 10.1038/ng577 [DOI] [PubMed] [Google Scholar]
- Gorlin R. J., Cohen M. M., Levin L. S. (1990). Syndromes of the Head and Neck, 3rd Edn. New York, NY: Oxford University Press; xxi, 977 [Google Scholar]
- Gosain A. K., Machol J. A. T., Gliniak C., Halligan N. L. (2009). TGF-beta1 RNA interference in mouse primary dura cell culture: downstream effects on TGF receptors, FGF-2, and FGF-R1 mRNA levels. Plast. Reconstr. Surg. 124, 1466–1473 10.1097/PRS.0b013e3181b98947 [DOI] [PubMed] [Google Scholar]
- Hagen A., Gorenoi V., Schonermark M. P. (2012). Bone graft substitutes for the treatment of traumatic fractures of the extremities. GMS Health Technol. Assess. 8:Doc04 10.3205/hta000102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammerschmidt M., Brook A., McMahon A. P. (1997). The world according to hedgehog. Trends Genet. 13, 14–21 10.1016/S0168-9525(96)10051-2 [DOI] [PubMed] [Google Scholar]
- Hardy R. Y., Resh M. D. (2012). Identification of n-terminal residues of sonic hedgehog important for palmitoylation by hedgehog acyltransferase. J. Biol. Chem. 287, 42881–42889 10.1074/jbc.M112.426833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haycraft C. J., Banizs B., Aydin-Son Y., Zhang Q., Michaud E. J., Yoder B. K. (2005). Gli2 and Gli3 localize to cilia and require the intraflagellar transport protein polaris for processing and function. PLoS Genet. 1:e53 10.1371/journal.pgen.0010053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayhurst M., McConnell S. K. (2003). Mouse models of holoprosencephaly. Curr. Opin. Neurol. 16, 135–141 10.1097/01.wco.0000063761.15877.40 [DOI] [PubMed] [Google Scholar]
- Hayward R., Gonsalez S. (2005). How low can you go? Intracranial pressure, cerebral perfusion pressure, and respiratory obstruction in children with complex craniosynostosis. J. Neurosurg. 102, 16–22 10.3171/ped.2005.102.1.0016 [DOI] [PubMed] [Google Scholar]
- Hellemans J., Coucke P. J., Giedion A., De Paepe A., Kramer P., Beemer F., et al. (2003). Homozygous mutations in IHH cause acrocapitofemoral dysplasia, an autosomal recessive disorder with cone-shaped epiphyses in hands and hips. Am. J. Hum. Genet. 72, 1040–1046 10.1086/374318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgkinson J. E., Davidson C. L., Beresford J., Sharpe P. T. (1993). Expression of a human homeobox-containing gene is regulated by 1, 25(OH)2D3 in bone cells. Biochim. Biophys. Acta 1174, 11–16 10.1016/0167-4781(93)90086-S [DOI] [PubMed] [Google Scholar]
- Hu D., Helms J. A. (1999). The role of sonic hedgehog in normal and abnormal craniofacial morphogenesis. Development 126, 4873–4884 [DOI] [PubMed] [Google Scholar]
- Hu D., Marcucio R. S., Helms J. A. (2003). A zone of frontonasal ectoderm regulates patterning and growth in the face. Development 130, 1749–1758 10.1242/dev.00397 [DOI] [PubMed] [Google Scholar]
- Huangfu D., Anderson K. V. (2005). Cilia and Hedgehog responsiveness in the mouse. Proc. Natl. Acad. Sci. U.S.A. 102, 11325–11330 10.1073/pnas.0505328102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huangfu D., Anderson K. V. (2006). Signaling from Smo to Ci/Gli: conservation and divergence of Hedgehog pathways from Drosophila to vertebrates. Development 133, 3–14 10.1242/dev.02169 [DOI] [PubMed] [Google Scholar]
- Huangfu D., Liu A., Rakeman A. S., Murcia N. S., Niswander L., Anderson K. V. (2003). Hedgehog signalling in the mouse requires intraflagellar transport proteins. Nature 426, 83–87 10.1038/nature02061 [DOI] [PubMed] [Google Scholar]
- Hui C. C., Joyner A. L. (1993). A mouse model of greig cephalopolysyndactyly syndrome: the extra-toesJ mutation contains an intragenic deletion of the Gli3 gene. Nat. Genet. 3, 241–246 10.1038/ng0393-241 [DOI] [PubMed] [Google Scholar]
- Hynes M., Stone D. M., Dowd M., Pitts-Meek S., Goddard A., Gurney A., et al. (1997). Control of cell pattern in the neural tube by the zinc finger transcription factor and oncogene Gli-1. Neuron 19, 15–26 10.1016/S0896-6273(00)80344-X [DOI] [PubMed] [Google Scholar]
- Incardona J. P., Gaffield W., Lange Y., Cooney A., Pentchev P. G., Liu S., et al. (2000). Cyclopamine inhibition of Sonic hedgehog signal transduction is not mediated through effects on cholesterol transport. Dev. Biol. 224, 440–452 10.1006/dbio.2000.9775 [DOI] [PubMed] [Google Scholar]
- Iseki S., Osumi-Yamashita N., Miyazono K., Franzen P., Ichijo H., Ohtani H., et al. (1995). Localization of transforming growth factor-beta type I and type II receptors in mouse development. Exp. Cell Res. 219, 339–347 10.1006/excr.1995.1237 [DOI] [PubMed] [Google Scholar]
- Ishii M., Arias A. C., Liu L., Chen Y. B., Bronner M. E., Maxson R. E. (2012). A stable cranial neural crest cell line from mouse. Stem Cells Dev. 21, 3069–3080 10.1089/scd.2012.0155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izzi L., Levesque M., Morin S., Laniel D., Wilkes B. C., Mille F., et al. (2011). Boc and Gas1 each form distinct Shh receptor complexes with Ptch1 and are required for Shh-mediated cell proliferation. Dev. Cell 20, 788–801 10.1016/j.devcel.2011.04.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob S., Wu C., Freeman T. A., Koyama E., Kirschner R. E. (2007). Expression of Indian Hedgehog, BMP-4 and Noggin in craniosynostosis induced by fetal constraint. Ann. Plast. Surg. 58, 215–221 10.1097/01.sap.0000232833.41739.a5 [DOI] [PubMed] [Google Scholar]
- James A. W., Leucht P., Levi B., Carre A. L., Xu Y., Helms J. A., et al. (2010). Sonic Hedgehog influences the balance of osteogenesis and adipogenesis in mouse adipose-derived stromal cells. Tissue Eng. Part A 16, 2605–2616 10.1089/ten.TEA.2010.0048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- James A. W., Pang S., Askarinam A., Corselli M., Zara J. N., Goyal R., et al. (2012). Additive effects of sonic Hedgehog and Nell-1 signaling in osteogenic versus adipogenic differentiation of human adipose-derived stromal Cells. Stem Cells Dev. 21, 2170–2178 10.1089/scd.2011.0461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins D. (2009). Hedgehog signalling: emerging evidence for non-canonical pathways. Cell. Signal. 21, 1023–1034 [DOI] [PubMed] [Google Scholar]
- Jenkins D., Seelow D., Jehee F. S., Perlyn C. A., Alonso L. G., Bueno D. F., et al. (2007). RAB23 mutations in Carpenter syndrome imply an unexpected role for hedgehog signaling in cranial-suture development and obesity. Am. J. Hum. Genet. 80, 1162–1170 10.1086/518047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X., Iseki S., Maxson R. E., Sucov H. M., Morriss-Kay G. M. (2002). Tissue origins and interactions in the mammalian skull vault. Dev. Biol. 241, 106–116 10.1006/dbio.2001.0487 [DOI] [PubMed] [Google Scholar]
- Jiang X., Rowitch D. H., Soriano P., McMahon A. P., Sucov H. M. (2000). Fate of the mammalian cardiac neural crest. Development 127, 1607–1616 [DOI] [PubMed] [Google Scholar]
- Johnston J. J., Olivos-Glander I., Killoran C., Elson E., Turner J. T., Peters K. F., et al. (2005). Molecular and clinical analyses of Greig cephalopolysyndactyly and Pallister-Hall syndromes: robust phenotype prediction from the type and position of GLI3 mutations. Am. J. Hum. Genet. 76, 609–622 10.1086/429346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapp-Simon K. A. (1998). Mental development and learning disorders in children with single suture craniosynostosis. Cleft Palate Craniofac. J. 35, 197–203 10.1597/1545-1569(1998)035<0197:MDALDI>2.3.CO;2 [DOI] [PubMed] [Google Scholar]
- Karlstrom R. O., Tyurina O. V., Kawakami A., Nishioka N., Talbot W. S., Sasaki H., et al. (2003). Genetic analysis of zebrafish gli1 and gli2 reveals divergent requirements for gli genes in vertebrate development. Development 130, 1549–1564 10.1242/dev.00364 [DOI] [PubMed] [Google Scholar]
- Kawai Y., Noguchi J., Akiyama K., Takeno Y., Fujiwara Y., Kajita S., et al. (2010). A missense mutation of the Dhh gene is associated with male pseudohermaphroditic rats showing impaired Leydig cell development. Reproduction 141, 217–225 10.1530/REP-10-0006 [DOI] [PubMed] [Google Scholar]
- Keady B. T., Samtani R., Tobita K., Tsuchya M., San Agustin J. T., Follit J. A., et al. (2012). IFT25 links the signal-dependent movement of Hedgehog components to intraflagellar transport. Dev. Cell 22, 940–951 10.1016/j.devcel.2012.04.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keaton A. A., Solomon B. D., van Essen A. J., Pfleghaar K. M., Slama M. A., Martin J. A., et al. (2010). Holoprosencephaly and ectrodactyly: report of three new patients and review of the literature. Am. J. Med. Genet. C Semin. Med. Genet. 154, 170–175 10.1002/ajmg.c.30251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H. J., Rice D. P., Kettunen P. J., Thesleff I. (1998). FGF-, BMP- and Shh-mediated signalling pathways in the regulation of cranial suture morphogenesis and calvarial bone development. Development 125, 1241–1251 [DOI] [PubMed] [Google Scholar]
- Klopocki E., Lohan S., Brancati F., Koll R., Brehm A., Seemann P., et al. (2011). Copy-number variations involving the IHH locus are associated with syndactyly and craniosynostosis. Am. J. Hum. Genet. 88, 70–75 10.1016/j.ajhg.2010.11.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko S. H., Behr B., Longaker M. T. (2009). Discussion. TGF-beta1 RNA interference in mouse primary dura cell culture: downstream effects on TGF receptors, FGF-2, and FGF-R1 mRNA levels. Plast. Reconstr. Surg. 124, 1474–1476 10.1097/PRS.0b013e3181b989de [DOI] [PubMed] [Google Scholar]
- Kolpakova-Hart E., Nicolae C., Zhou J., Olsen B. R. (2008). Col2-Cre recombinase is co-expressed with endogenous type II collagen in embryonic renal epithelium and drives development of polycystic kidney disease following inactivation of ciliary genes. Matrix Biol. 27, 505–512 10.1016/j.matbio.2008.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koyama E., Young B., Nagayama M., Shibukawa Y., Enomoto-Iwamoto M., Iwamoto M., et al. (2007). Conditional Kif3a ablation causes abnormal hedgehog signaling topography, growth plate dysfunction, and excessive bone and cartilage formation during mouse skeletogenesis. Development 134, 2159–2169 10.1242/dev.001586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kronenberg H. M. (2003). Developmental regulation of the growth plate. Nature 423, 332–336 10.1038/nature01657 [DOI] [PubMed] [Google Scholar]
- Kuo J. S., Casey S. O., Thompson L., Truwit C. L. (1999). Pallister-Hall syndrome: clinical and MR features. AJNR Am. J. Neuroradiol. 20, 1839–1841 [PMC free article] [PubMed] [Google Scholar]
- Lallemand Y., Bensoussan V., Cloment C. S., Robert B. (2009). Msx genes are important apoptosis effectors downstream of the Shh/Gli3 pathway in the limb. Dev. Biol. 331, 189–198 10.1016/j.ydbio.2009.04.038 [DOI] [PubMed] [Google Scholar]
- Lanske B., Karaplis A. C., Lee K., Luz A., Vortkamp A., Pirro A., et al. (1996). PTH/PTHrP receptor in early development and Indian hedgehog-regulated bone growth. Science 273, 663–666 10.1126/science.273.5275.663 [DOI] [PubMed] [Google Scholar]
- Le Lievre C. S. (1978). Participation of neural crest-derived cells in the genesis of the skull in birds. J. Embryol. Exp. Morphol. 47, 17–37 [PubMed] [Google Scholar]
- Lee C. S., May N. R., Fan C. M. (2001). Transdifferentiation of the ventral retinal pigmented epithelium to neural retina in the growth arrest specific gene 1 mutant. Dev. Biol. 236, 17–29 10.1006/dbio.2001.0280 [DOI] [PubMed] [Google Scholar]
- Lee J., Platt K. A., Censullo P., Ruiz i Altaba A. (1997). Gli1 is a target of Sonic hedgehog that induces ventral neural tube development. Development 124, 2537–2552 [DOI] [PubMed] [Google Scholar]
- Lenton K., James A. W., Manu A., Brugmann S. A., Birker D., Nelson E. R., et al. (2011). Indian hedgehog positively regulates calvarial ossification and modulates bone morphogenetic protein signaling. Genesis 49, 784–796 10.1002/dvg.20768 [DOI] [PubMed] [Google Scholar]
- Lenton K. A., Nacamuli R. P., Wan D. C., Helms J. A., Longaker M. T. (2005). Cranial suture biology. Curr. Top. Dev. Biol. 66, 287–328 10.1016/S0070-2153(05)66009-7 [DOI] [PubMed] [Google Scholar]
- Levi B., James A. W., Nelson E. R., Brugmann S. A., Sorkin M., Manu A., et al. (2011). Role of Indian hedgehog signaling in palatal osteogenesis. Plast. Reconstr. Surg. 127, 1182–1190 10.1097/PRS.0b013e3182043a07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S., Quarto N., Longaker M. T. (2007). Dura mater-derived FGF-2 mediates mitogenic signaling in calvarial osteoblasts. Am. J. Physiol. Cell Physiol. 293, C1834–C1842 10.1152/ajpcell.00135.2007 [DOI] [PubMed] [Google Scholar]
- Liem K. F., Jr., Jessell T. M., Briscoe J. (2000). Regulation of the neural patterning activity of sonic hedgehog by secreted BMP inhibitors expressed by notochord and somites. Development 127, 4855–4866 [DOI] [PubMed] [Google Scholar]
- Lipinski R. J., Song C., Sulik K. K., Everson J. L., Gipp J. J., Yan D., et al. (2010). Cleft lip and palate results from Hedgehog signaling antagonism in the mouse: phenotypic characterization and clinical implications. Birth Defects Res. A. Clin. Mol. Teratol. 88, 232–240 10.1002/bdra.20656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu A., Wang B., Niswander L. A. (2005). Mouse intraflagellar transport proteins regulate both the activator and repressor functions of Gli transcription factors. Development 132, 3103–3111 10.1242/dev.01894 [DOI] [PubMed] [Google Scholar]
- Liu C., Yamada Y., Fan C. M. (2002). Growth arrest specific gene 1 acts as a region-specific mediator of Fgf10/Fgf8 regulatory loop in the limb. Development 129, 5289–5300 [DOI] [PubMed] [Google Scholar]
- Liu Y., May N. R., Fan C. M. (2001). Growth arrest specific gene 1 is a positive growth regulator for the cerebellum. Dev. Biol. 236, 30–45 10.1006/dbio.2000.0146 [DOI] [PubMed] [Google Scholar]
- Lo Muzio L. (2008). Nevoid basal cell carcinoma syndrome (Gorlin syndrome). Orphanet J. Rare Dis. 3:32 10.1186/1750-1172-3-32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long F., Chung U. I., Ohba S., McMahon J., Kronenberg H. M., McMahon A. P. (2004). Ihh signaling is directly required for the osteoblast lineage in the endochondral skeleton. Development 131, 1309–1318 10.1242/dev.01006 [DOI] [PubMed] [Google Scholar]
- Long F., Zhang X. M., Karp S., Yang Y., McMahon A. P. (2001). Genetic manipulation of hedgehog signaling in the endochondral skeleton reveals a direct role in the regulation of chondrocyte proliferation. Development 128, 5099–5108 [DOI] [PubMed] [Google Scholar]
- Longaker M. T. (2001). Role of TGF-beta signaling in the regulation of programmed cranial suture fusion. J. Craniofac. Surg. 12, 389–390 [DOI] [PubMed] [Google Scholar]
- Macintosh C., Wells R., Johnson D., Wall S. (2012). What are the effects of metopic synostosis on visual function? J. Craniofac. Surg. 22, 1280–1283 10.1097/SCS.0b013e31821c6a64 [DOI] [PubMed] [Google Scholar]
- Madeline L. A., Elster A. D. (1995). Suture closure in the human chondrocranium: CT assessment. Radiology 196, 747–756 [DOI] [PubMed] [Google Scholar]
- Magge S. N., Westerveld M., Pruzinsky T., Persing J. A. (2002). Long-term neuropsychological effects of sagittal craniosynostosis on child development. J. Craniofac. Surg. 13, 99–104 [DOI] [PubMed] [Google Scholar]
- Marie P. J., Kaabeche K., Guenou H. (2008). Roles of FGFR2 and twist in human craniosynostosis: insights from genetic mutations in cranial osteoblasts. Front. Oral Biol. 12, 144–159 10.1159/0000115036 [DOI] [PubMed] [Google Scholar]
- McGillivray G., Savarirayan R., Cox T. C., Stojkoski C., McNeil R., Bankier A., et al. (2005). Familial scaphocephaly syndrome caused by a novel mutation in the FGFR2 tyrosine kinase domain. J. Med. Genet. 42, 656–662 10.1136/jmg.2004.027888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon A. P., Ingham P. W., Tabin C. J. (2003). Developmental roles and clinical significance of hedgehog signaling. Curr. Top. Dev. Biol. 53, 1–114 [DOI] [PubMed] [Google Scholar]
- Mehrara B. J., Most D., Chang J., Bresnick S., Turk A., Schendel S. A., et al. (1999). Basic fibroblast growth factor and transforming growth factor beta-1 expression in the developing dura mater correlates with calvarial bone formation. Plast. Reconstr. Surg. 104, 435–444 [DOI] [PubMed] [Google Scholar]
- Merrill A. E., Merriman B., Farrington-Rock C., Camacho N., Sebald E. T., Funari V. A., et al. (2009). Ciliary abnormalities due to defects in the retrograde transport protein DYNC2H1 in short-rib polydactyly syndrome. Am. J. Hum. Genet. 84, 542–549 10.1016/j.ajhg.2009.03.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ming J. E., Muenke M. (1998). Holoprosencephaly: from Homer to Hedgehog. Clin. Genet. 53, 155–163 10.1111/j.1399-0004.1998.tb02666.x [DOI] [PubMed] [Google Scholar]
- Ming J. E., Roessler E., Muenke M. (1998). Human developmental disorders and the Sonic hedgehog pathway. Mol. Med. Today 4, 343–349 [DOI] [PubMed] [Google Scholar]
- Moursi A. M., Winnard P. L., Winnard A. V., Rubenstrunk J. M., Mooney M. P. (2002). Fibroblast growth factor 2 induces increased calvarial osteoblast proliferation and cranial suture fusion. Cleft Palate Craniofac. J. 39, 487–496 10.1597/1545-1569(2002)039<0487:FGFIIC>2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Murakami S., Nifuji A., Noda M. (1997). Expression of Indian hedgehog in osteoblasts and its posttranscriptional regulation by transforming growth factor-beta. Endocrinology 138, 1972–1978 10.1210/en.138.5.1972 [DOI] [PubMed] [Google Scholar]
- Murray J. C., Schutte B. C. (2004). Cleft palate: players, pathways, and pursuits. J. Clin. Invest. 113, 1676–1678 10.1172/JCI22154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagayama M., Iwamoto M., Hargett A., Kamiya N., Tamamura Y., Young B., et al. (2008). Wnt/beta-catenin signaling regulates cranial base development and growth. J. Dent. Res. 87, 244–249 10.1177/154405910808700309 [DOI] [PubMed] [Google Scholar]
- Naruse I., Ueta E., Sumino Y., Ogawa M., Ishikiriyama S. (2010). Birth defects caused by mutations in human GLI3 and mouse Gli3 genes. Congenit. Anom. (Kyoto) 50, 1–7 10.1111/j.1741-4520.2009.00266.x [DOI] [PubMed] [Google Scholar]
- Nie X. (2005). Cranial base in craniofacial development: developmental features, influence on facial growth, anomaly, and molecular basis. Acta Odontol. Scand. 63, 127–135 10.1080/00016350510019847 [DOI] [PubMed] [Google Scholar]
- Nie X., Luukko K., Kettunen P. (2006a). BMP signalling in craniofacial development. Int. J. Dev. Biol. 50, 511–521 10.1387/ijdb.052101xn [DOI] [PubMed] [Google Scholar]
- Nie X., Luukko K., Kettunen P. (2006b). FGF signalling in craniofacial development and developmental disorders. Oral Dis. 12, 102–111 10.1111/j.1601-0825.2005.01176.x [DOI] [PubMed] [Google Scholar]
- Nie X., Luukko K., Kvinnsland I. H., Kettunen P. (2005). Developmentally regulated expression of Shh and Ihh in the developing mouse cranial base: comparison with Sox9 expression. Anat. Rec. A. Discov. Mol. Cell. Evol. Biol. 286, 891–898 10.1002/ar.a.20231 [DOI] [PubMed] [Google Scholar]
- Noden D. M. (1975). An analysis of migratory behavior of avian cephalic neural crest cells. Dev. Biol. 42, 106–130 [DOI] [PubMed] [Google Scholar]
- Noden D. M. (1983). The role of the neural crest in patterning of avian cranial skeletal, connective, and muscle tissues. Dev. Biol. 96, 144–165 10.1016/0012-1606(83)90318-4 [DOI] [PubMed] [Google Scholar]
- Nott R. L., Stelnicki E. J., Mack J. A., Ben Y., Mitchell R., Mooney M. P. (2002a). Changes in the protein expression of hedgehog and patched-1 in perisutural tissues induced by cranial distraction. Plast. Reconstr. Surg. 110, 523–532 [DOI] [PubMed] [Google Scholar]
- Nott R. L., Stelnicki E. J., Mack J. A., Ben Y., Mitchell R., Mooney M. P. (2002b). Comparison of hedgehog and patched-1 protein expression in the cranial sutures of craniosynostotic and wild-type rabbits. Plast. Reconstr. Surg. 110, 515–522 [DOI] [PubMed] [Google Scholar]
- Odent S., Atti-Bitach T., Blayau M., Mathieu M., Aug J., Delezo de A. L., et al. (1999). Expression of the Sonic hedgehog (SHH) gene during early human development and phenotypic expression of new mutations causing holoprosencephaly. Hum. Mol. Genet. 8, 1683–1689 10.1093/hmg/8.9.1683 [DOI] [PubMed] [Google Scholar]
- Opperman L. A. (2000). Cranial sutures as intramembranous bone growth sites. Dev. Dyn. 219, 472–485 10.1002/1097-0177(2000)9999:9999<::AID-DVDY1073>3.0.CO;2-F [DOI] [PubMed] [Google Scholar]
- Opperman L. A., Chhabra A., Cho R. W., Ogle R. C. (1999). Cranial suture obliteration is induced by removal of transforming growth factor (TGF)-beta 3 activity and prevented by removal of TGF-beta 2 activity from fetal rat calvaria in vitro. J. Craniofac. Genet. Dev. Biol. 19, 164–173 [PubMed] [Google Scholar]
- Opperman L. A., Nolen A. A., Ogle R. C. (1997). TGF-beta 1, TGF-beta 2, and TGF-beta 3 exhibit distinct patterns of expression during cranial suture formation and obliteration in vivo and in vitro. J. Bone Miner. Res. 12, 301–310 10.1359/jbmr.1997.12.3.301 [DOI] [PubMed] [Google Scholar]
- Opperman L. A., Passarelli R. W., Morgan E. P., Reintjes M., Ogle R. C. (1995). Cranial sutures require tissue interactions with dura mater to resist osseous obliteration in vitro. J. Bone Miner. Res. 10, 1978–1987 10.1002/jbmr.5650101218 [DOI] [PubMed] [Google Scholar]
- Opperman L. A., Persing J. A., Sheen R., Ogle R. C. (1994). In the absence of periosteum, transplanted fetal and neonatal rat coronal sutures resist osseous obliteration. J. Craniofac. Surg. 5, 327–332 [DOI] [PubMed] [Google Scholar]
- Opperman L. A., Sweeney T. M., Redmon J., Persing J. A., Ogle R. C. (1993). Tissue interactions with underlying dura mater inhibit osseous obliteration of developing cranial sutures. Dev. Dyn. 198, 312–322 10.1002/aja.1001980408 [DOI] [PubMed] [Google Scholar]
- Pacheco M., Valencia M., Caparros-Martin J. A., Mulero F., Goodship J. A., Ruiz-Perez V. L. (2012). Evc works in chondrocytes and osteoblasts to regulate multiple aspects of growth plate development in the appendicular skeleton and cranial base. Bone 50, 28–41 10.1016/j.bone.2011.08.025 [DOI] [PubMed] [Google Scholar]
- Pan Y., Bai C. B., Joyner A. L., Wang B. (2006). Sonic hedgehog signaling regulates Gli2 transcriptional activity by suppressing its processing and degradation. Mol. Cell. Biol. 26, 3365–3377 10.1128/MCB.26.9.3365-3377.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Y., Wang B. (2007). A novel protein-processing domain in Gli2 and Gli3 differentially blocks complete protein degradation by the proteasome. J. Biol. Chem. 282, 10846–10852 10.1074/jbc.M608599200 [DOI] [PubMed] [Google Scholar]
- Panchal J., Amirsheybani H., Gurwitch R., Cook V., Francel P., Neas B., et al. (2001). Neurodevelopment in children with single-suture craniosynostosis and plagiocephaly without synostosis. Plast. Reconstr. Surg. 108, 1492–1498 discussion: 1499–1500. [DOI] [PubMed] [Google Scholar]
- Park H. L., Bai C., Platt K. A., Matise M. P., Beeghly A., Hui C. C., et al. (2000). Mouse Gli1 mutants are viable but have defects in SHH signaling in combination with a Gli2 mutation. Development 127, 1593–1605 [DOI] [PubMed] [Google Scholar]
- Parmantier E., Lynn B., Lawson D., Turmaine M., Namini S. S., Chakrabarti L., et al. (1999). Schwann cell-derived Desert hedgehog controls the development of peripheral nerve sheaths. Neuron 23, 713–724 10.1016/S0896-6273(01)80030-1 [DOI] [PubMed] [Google Scholar]
- Pepinsky R. B., Zeng C., Wen D., Rayhorn P., Baker D. P., Williams K. P., et al. (1998). Identification of a palmitic acid-modified form of human Sonic hedgehog. J. Biol. Chem. 273, 14037–14045 10.1074/jbc.273.22.14037 [DOI] [PubMed] [Google Scholar]
- Persson M., Stamataki D., te Welscher P., Andersson E., Bose J., Ruther U., et al. (2002). Dorsal-ventral patterning of the spinal cord requires Gli3 transcriptional repressor activity. Genes Dev. 16, 2865–2878 10.1101/gad.243402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter J. A., Ekker S. C., Park W. J., von Kessler D. P., Young K. E., Chen C. H., et al. (1996). Hedgehog patterning activity: role of a lipophilic modification mediated by the carboxy-terminal autoprocessing domain. Cell 86, 21–34 10.1016/S0092-8674(00)80074-4 [DOI] [PubMed] [Google Scholar]
- Quinn M. E., Haaning A., Ware S. M. (2012). Preaxial polydactyly caused by Gli3 haploinsufficiency is rescued by Zic3 loss of function in mice. Hum. Mol. Genet. 21, 1888–1896 10.1093/hmg/dds002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rallu M., Machold R., Gaiano N., Corbin J. G., McMahon A. P., Fishell G. (2002). Dorsoventral patterning is established in the telencephalon of mutants lacking both Gli3 and Hedgehog signaling. Development 129, 4963–4974 [DOI] [PubMed] [Google Scholar]
- Rawlins J. T., Opperman L. A. (2008). Tgf-beta regulation of suture morphogenesis and growth. Front. Oral Biol. 12, 178–196 10.1159/0000115038 [DOI] [PubMed] [Google Scholar]
- Razzaque M. S., Soegiarto D. W., Chang D., Long F., Lanske B. (2005). Conditional deletion of Indian hedgehog from collagen type 2alpha1-expressing cells results in abnormal endochondral bone formation. J. Pathol. 207, 453–461 10.1002/path.1870 [DOI] [PubMed] [Google Scholar]
- Rice D. P., Connor E. C., Veltmaat J. M., Lana-Elola E., Veistinen L., Tanimoto Y., et al. (2010). Gli3Xt-J/Xt-J mice exhibit lambdoid suture craniosynostosis which results from altered osteoprogenitor proliferation and differentiation. Hum. Mol. Genet. 19, 3457–3467 10.1093/hmg/ddq258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson L. K., James H. E., Mubarak S. J., Allen E. J., Jones K. L. (1985). Carpenter syndrome: natural history and clinical spectrum. Am. J. Med. Genet. 20, 461–469 10.1002/ajmg.1320200307 [DOI] [PubMed] [Google Scholar]
- Roessler E., Muenke M. (1998). Holoprosencephaly: a paradigm for the complex genetics of brain development. J. Inherit. Metab. Dis. 21, 481–497 [DOI] [PubMed] [Google Scholar]
- Roessler E., Ward D. E., Gaudenz K., Belloni E., Scherer S. W., Donnai D., et al. (1997). Cytogenetic rearrangements involving the loss of the Sonic Hedgehog gene at 7q36 cause holoprosencephaly. Hum. Genet. 100, 172–181 10.1007/s004390050486 [DOI] [PubMed] [Google Scholar]
- Rohatgi R., Scott M. P. (2008). Cell biology. Arrestin' movement in cilia. Science 320, 1726–1727 10.1126/science.1160448 [DOI] [PubMed] [Google Scholar]
- Ruat M., Roudaut H., Ferent J., Traiffort E. (2012). Hedgehog trafficking, cilia and brain functions. Differentiation 83, S97–S104 10.1016/j.diff.2011.11.011 [DOI] [PubMed] [Google Scholar]
- Ruiz i Altaba A. (1998). Combinatorial Gli gene function in floor plate and neuronal inductions by Sonic hedgehog. Development 125, 2203–2212 [DOI] [PubMed] [Google Scholar]
- Ruiz i Altaba A. (1999). Gli proteins encode context-dependent positive and negative functions: implications for development and disease. Development 126, 3205–3216 [DOI] [PubMed] [Google Scholar]
- Ruiz-Perez V. L., Blair H. J., Rodriguez-Andres M. E., Blanco M. J., Wilson A., Liu Y. N., et al. (2007). Evc is a positive mediator of Ihh-regulated bone growth that localises at the base of chondrocyte cilia. Development 134, 2903–2912 10.1242/dev.007542 [DOI] [PubMed] [Google Scholar]
- Rutter M., Wang J., Huang Z., Kuliszewski M., Post M. (2010). Gli2 influences proliferation in the developing lung through regulation of cyclin expression. Am. J. Respir. Cell Mol. Biol. 42, 615–625 10.1165/rcmb.2008-0390OC [DOI] [PubMed] [Google Scholar]
- Sahar D. E., Longaker M. T., Quarto N. (2005). Sox9 neural crest determinant gene controls patterning and closure of the posterior frontal cranial suture. Dev. Biol. 280, 344–361 10.1016/j.ydbio.2005.01.022 [DOI] [PubMed] [Google Scholar]
- Santagati F., Rijli F. M. (2003). Cranial neural crest and the building of the vertebrate head. Nat. Rev. Neurosci. 4, 806–818 10.1038/nrn1221 [DOI] [PubMed] [Google Scholar]
- Sasaki H., Nishizaki Y., Hui C., Nakafuku M., Kondoh H. (1999). Regulation of Gli2 and Gli3 activities by an amino-terminal repression domain: implication of Gli2 and Gli3 as primary mediators of Shh signaling. Development 126, 3915–3924 [DOI] [PubMed] [Google Scholar]
- Schlosser G., Wagner G. P. (2004). Modularity in Development and Evolution. Chicago, IL: University of Chicago Press; x, 600 [Google Scholar]
- Schneider R. A., Hu D., Rubenstein J. L., Maden M., Helms J. A. (2001). Local retinoid signaling coordinates forebrain and facial morphogenesis by maintaining FGF8 and SHH. Development 128, 2755–2767 [DOI] [PubMed] [Google Scholar]
- Seppala M., Depew M. J., Martinelli D. C., Fan C. M., Sharpe P. T., Cobourne M. T. (2007). Gas1 is a modifier for holoprosencephaly and genetically interacts with sonic hedgehog. J. Clin. Invest. 117, 1575–1584 10.1172/JCI32032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shanley S., Ratcliffe J., Hockey A., Haan E., Oley C., Ravine D., et al. (1994). Nevoid basal cell carcinoma syndrome: review of 118 affected individuals. Am. J. Med. Genet. 50, 282–290 10.1002/ajmg.1320500312 [DOI] [PubMed] [Google Scholar]
- Shimoyama A., Wada M., Ikeda F., Hata K., Matsubara T., Nifuji A., et al. (2007). Ihh/Gli2 signaling promotes osteoblast differentiation by regulating Runx2 expression and function. Mol. Biol. Cell 18, 2411–2418 10.1091/mbc.E06-08-0743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson F., Kerr M. C., Wicking C. (2009). Trafficking, development and hedgehog. Mech. Dev. 126, 279–288 10.1016/j.mod.2009.01.007 [DOI] [PubMed] [Google Scholar]
- Singla V., Reiter J. F. (2006). The primary cilium as the cell's antenna: signaling at a sensory organelle. Science 313, 629–633 10.1126/science.1124534 [DOI] [PubMed] [Google Scholar]
- Slater B. J., Kwan M. D., Gupta D. M., Lee J. K., Longaker M. T. (2009). The role of regional posterior frontal dura mater in the overlying suture morphology. Plast. Reconstr. Surg. 123, 463–469 10.1097/PRS.0b013e3181954d21 [DOI] [PubMed] [Google Scholar]
- Slavkin H. C. (1979). Developmental Craniofacial Biology. Philadelphia, PA: Lea and Febiger; xv, 464 [Google Scholar]
- Spector J. A., Greenwald J. A., Warren S. M., Bouletreau P. J., Detch R. C., Fagenholz P. J., et al. (2002). Dura mater biology: autocrine and paracrine effects of fibroblast growth factor 2. Plast. Reconstr. Surg. 109, 645–654 [DOI] [PubMed] [Google Scholar]
- Spek C. A., Bijlsma M. F., Queiroz K. C. (2010). Canonical Hedgehog signaling drives proangiogenic responses in endothelial cells. Cell Cycle 9, 1683 10.4161/cc.9.9.11653 [DOI] [PubMed] [Google Scholar]
- St-Jacques B., Hammerschmidt M., McMahon A. P. (1999). Indian hedgehog signaling regulates proliferation and differentiation of chondrocytes and is essential for bone formation. Genes Dev. 13, 2072–2086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K., Nuckolls G. H., Takahashi I., Nonaka K., Nagata M., Ikura T., et al. (2001). Msx2 is a repressor of chondrogenic differentiation in migratory cranial neural crest cells. Dev. Dyn. 222, 252–262 10.1002/dvdy.1185 [DOI] [PubMed] [Google Scholar]
- Tavella S., Biticchi R., Schito A., Minina E., Di Martino D., Pagano A., et al. (2004). Targeted expression of SHH affects chondrocyte differentiation, growth plate organization, and Sox9 expression. J. Bone Miner. Res. 19, 1678–1688 10.1359/JBMR.040706 [DOI] [PubMed] [Google Scholar]
- Temtamy S. A. (1966). Carpenter's syndrome: acrocephalopolysyndactyly. An autosomal recessive syndrome. J. Pediatr. 69, 111–120 [DOI] [PubMed] [Google Scholar]
- Theil T., Alvarez-Bolado G., Walter A., Ruther U. (1999). Gli3 is required for Emx gene expression during dorsal telencephalon development. Development 126, 3561–3571 [DOI] [PubMed] [Google Scholar]
- Tian Y., Xu Y., Fu Q., Dong Y. (2012). Osterix is required for sonic Hedgehog-induced osteoblastic MC3T3-E1 cell differentiation. Cell Biochem. Biophys. 64, 169–176 10.1007/s12013-012-9369-7 [DOI] [PubMed] [Google Scholar]
- Ting M. C., Wu N. L., Roybal P. G., Sun J., Liu L., Yen Y., et al. (2009). EphA4 as an effector of Twist1 in the guidance of osteogenic precursor cells during calvarial bone growth and in craniosynostosis. Development 136, 855–864 10.1242/dev.028605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Towler D. A., Bennett C. D., Rodan G. A. (1994). Activity of the rat osteocalcin basal promoter in osteoblastic cells is dependent upon homeodomain and CP1 binding motifs. Mol. Endocrinol. 8, 614–624 10.1210/me.8.5.614 [DOI] [PubMed] [Google Scholar]
- Tukachinsky H., Lopez L. V., Salic A. (2010). A mechanism for vertebrate Hedgehog signaling: recruitment to cilia and dissociation of SuFu-Gli protein complexes. J. Cell Biol. 191, 415–428 10.1083/jcb.201004108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Horst G., Farih-Sips H., Lowik C. W., Karperien M. (2003). Hedgehog stimulates only osteoblastic differentiation of undifferentiated KS483 cells. Bone 33, 899–910 10.1016/j.bone.2003.07.004 [DOI] [PubMed] [Google Scholar]
- Vaz S. S., Chodirker B., Prasad C., Seabrook J. A., Chudley A. E., Prasad A. N. (2012). Risk factors for nonsyndromic holoprosencephaly: a Manitoba case-control study. Am. J. Med. Genet. A 158A, 751–758 10.1002/ajmg.a.35240 [DOI] [PubMed] [Google Scholar]
- Veistinen L., Takatalo M., Tanimoto Y., Kesper D. A., Vortkamp A., Rice D. P. (2012). loss-of-function of gli3 in mice causes abnormal frontal bone morphology and premature synostosis of the interfrontal suture. Front. Physiol. 3:121 10.3389/fphys.2012.00121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vortkamp A., Lee K., Lanske B., Segre G. V., Kronenberg H. M., Tabin C. J. (1996). Regulation of rate of cartilage differentiation by Indian hedgehog and PTH-related protein. Science 273, 613–622 10.1126/science.273.5275.613 [DOI] [PubMed] [Google Scholar]
- Wang B., Fallon J. F., Beachy P. A. (2000). Hedgehog-regulated processing of Gli3 produces an anterior/posterior repressor gradient in the developing vertebrate limb. Cell 100, 423–434 10.1016/S0092-8674(00)80678-9 [DOI] [PubMed] [Google Scholar]
- Warren S. M., Brunet L. J., Harland R. M., Economides A. N., Longaker M. T. (2003). The BMP antagonist noggin regulates cranial suture fusion. Nature 422, 625–629 10.1038/nature01545 [DOI] [PubMed] [Google Scholar]
- Wild A., Kalff-Suske M., Vortkamp A., Bornholdt D., Konig R., Grzeschik K. H. (1997). Point mutations in human GLI3 cause Greig syndrome. Hum. Mol. Genet. 6, 1979–1984 10.1093/hmg/6.11.1979 [DOI] [PubMed] [Google Scholar]
- Wilkie A. O. (1997). Craniosynostosis: genes and mechanisms. Hum. Mol. Genet. 6, 1647–1656 10.1093/hmg/6.10.1647 [DOI] [PubMed] [Google Scholar]
- Wong S. Y., Reiter J. F. (2008). The primary cilium at the crossroads of mammalian hedgehog signaling. Curr. Top. Dev. Biol. 85, 225–260 10.1016/S0070-2153(08)00809-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wozney J. M., Rosen V., Celeste A. J., Mitsock L. M., Whitters M. J., Kriz R. W., et al. (1988). Novel regulators of bone formation: molecular clones and activities. Science 242, 1528–1534 10.1126/science.3201241 [DOI] [PubMed] [Google Scholar]
- Yang S., Wang C. (2012). The intraflagellar transport protein IFT80 is required for cilia formation and osteogenesis. Bone 51, 407–417 10.1016/j.bone.2012.06.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao H. H., Whoriskey W., Capel B. (2002). Desert Hedgehog/Patched 1 signaling specifies fetal Leydig cell fate in testis organogenesis. Genes Dev. 16, 1433–1440 10.1101/gad.981202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young B., Minugh-Purvis N., Shimo T., St-Jacques B., Iwamoto M., Enomoto-Iwamoto M., et al. (2006). Indian and sonic hedgehogs regulate synchondrosis growth plate and cranial base development and function. Dev. Biol. 299, 272–282 10.1016/j.ydbio.2006.07.028 [DOI] [PubMed] [Google Scholar]
- Yuasa T., Kataoka H., Kinto N., Iwamoto M., Enomoto-Iwamoto M., Iemura S., et al. (2002). Sonic hedgehog is involved in osteoblast differentiation by cooperating with BMP-2. J. Cell. Physiol. 193, 225–232 10.1002/jcp.10166 [DOI] [PubMed] [Google Scholar]
- Zhang T., Chen M., Lu Y., Xing Q., Chen W. (2011). A novel mutation of the PTCH1 gene activates the Shh/Gli signaling pathway in a Chinese family with nevoid basal cell carcinoma syndrome. Biochem. Biophys. Res. Commun. 409, 166–170 10.1016/j.bbrc.2011.04.047 [DOI] [PubMed] [Google Scholar]
- Zhang W., Hong M., Bae G. U., Kang J. S., Krauss R. S. (2012). Boc modifies the holoprosencephaly spectrum of Cdo mutant mice. Dis. Model Mech. 4, 368–380 10.1242/dmm.005744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W., Kang J. S., Cole F., Yi M. J., Krauss R. S. (2006). Cdo functions at multiple points in the Sonic Hedgehog pathway, and Cdo-deficient mice accurately model human holoprosencephaly. Dev. Cell 10, 657–665 10.1016/j.devcel.2006.04.005 [DOI] [PubMed] [Google Scholar]