

In this study, Kannan et al. investigate spliceosome function in telomerase TER1 RNA processing in fission yeast. The mechanism of spliceosome-mediated 3′ end processing has been unclear. The authors identify a strong RNA branch site and a weak polypyrimidine tract within the TER1 intron that attenuate the transition from the first to the second step of splicing, This work thus provides new insight into the roles of the branch site and pyrimidine tract during the second step of splicing and sheds new light on RNA splicing mechanisms.
Keywords: telomerase, fission yeast, splicing, branch site, 3′ end processing
Abstract
The fission yeast telomerase RNA (TER1) precursor harbors an intron immediately downstream from its mature 3′ end. Unlike most introns, which are removed from precursor RNAs by the spliceosome in two sequential but tightly coupled transesterification reactions, TER1 only undergoes the first cleavage reaction during telomerase RNA maturation. The mechanism underlying spliceosome-mediated 3′ end processing has remained unclear. We now demonstrate that a strong branch site (BS), a long distance to the 3′ splice site (3′ SS), and a weak polypyrimidine (Py) tract act synergistically to attenuate the transition from the first to the second step of splicing. The observation that a strong BS antagonizes the second step of splicing in the context of TER1 suggests that the BS–U2 snRNA interaction is disrupted after the first step and thus much earlier than previously thought. The slow transition from first to second step triggers the Prp22 DExD/H-box helicase-dependent rejection of the cleaved products and Prp43-dependent “discard” of the splicing intermediates. Our findings explain how the spliceosome can function in 3′ end processing and provide new insights into the mechanism of splicing.
The enzyme telomerase replenishes repetitive DNA sequences at the ends of eukaryotic chromosomes that otherwise shorten with each round of replication (Blackburn and Collins 2011). At its core, telomerase is comprised of a catalytic protein subunit and a noncoding RNA component. The RNA serves as a scaffold for the assembly of the holoenzyme and provides the template for telomeric repeat synthesis by the catalytic subunit telomerase reverse transcriptase (TERT).
The gene encoding the telomerase RNA subunit from the fission yeast Schizosaccharomyces pombe has been identified (Leonardi et al. 2008; Webb and Zakian 2008), and examination of the maturation pathway revealed that the RNA is first transcribed as a longer polyadenylated precursor containing an intron immediately downstream from the 3′ end of the mature form (Box et al. 2008). Interestingly, the mature 3′ end of TER1 is generated by a single cleavage reaction at the 5′ splice site (5′ SS) akin to the first step of splicing (Fig. 1A).
Figure 1.

The TER1 intron contains all elements required for spliceosomal cleavage. (A) Schematic of TER1 3′ end processing by the first step of splicing. The BS is shown as a circle, and poly(A) tails are indicated by dashed lines. (B) Northern blot on RNA isolated from cells expressing reporter constructs as indicated. Ura4 sequence is shown in blue, TER1 sequence is in green, and mutations at the 5′ SS and 3′ SS are symbolized by red crosses. (Top panel) A 32P-labeled oligonucleotide complementary to the second exon (position indicated by purple line above lane 3) was used for detection of precursor and spliced forms. (Middle panel) The Northern was reprobed with a radiolabeled oligo complementary to the first exon to also visualize the cleaved form (position of probe is indicated by red line above lane 3). A probe against snoRNA snR101 was used as loading control (LC). (C) Schematic of RNaseH cleavage assay. Cleavage sites are indicated by clusters of vertical arrows. Those cleavage products that are homogenous in size and visualized on Northern blots are underlined. The probe used for detection of all three forms is indicated by a red line. (D) RNaseH cleavage followed by Northern blot analysis of the indicated constructs. TER1 sequences are in green, URA4 sequences are in blue, and a red arrow points to the cleaved form. Products were detected using a probe complementary to the first exon as indicated in C. (Lane 1) The ratios of cleaved form to LC for each lane were normalized to wild type, with the data being represented as mean ± SEM from three independent experiments. (E) As in D for additional constructs, including one that places the TER1 intron in an entirely heterologous context (shown in lane 4). Differences in mobility for each form between lanes reflect differences in the length and sequence of constructs used. Quantification as described in E, with n = 2.
Prior to cleavage, a ring of seven Sm proteins binds the TER1 precursor directly upstream of the intron and recruits the methylase Tgs1, which converts the monomethyl guanosine cap at the 5′ end of TER1 into the trimethyl guanosine form (Tang et al. 2012). After cleavage by the spliceosome has occurred, Sm proteins dissociate from the RNA and are replaced by a related protein complex comprised of seven Like-Sm proteins: Lsm2 to Lsm8 (Wilusz and Wilusz 2005). The Lsm complex protects the 3′ end of telomerase RNA against degradation and promotes assembly of the RNA with the catalytic subunit of telomerase (Tang et al. 2012). The identification of several key events that convert the telomerase RNA precursor into the mature form has laid the foundation for mechanistic studies. A question of particular interest is how the spliceosome carries out a single cleavage reaction in 3′ end processing when its normal role is to remove introns in two tightly coupled steps.
In the first step of intron excision, the 2′ hydroxyl of the branch point (BP) adenosine, located within the branch site (BS) sequence, attacks the sugar–phosphate bond at the 5′ SS, forming a 2′–5′ linkage and producing a free 5′ exon and a branched species, termed the lariat intermediate (Wahl et al. 2009). In the second reaction, the now exposed 3′ hydroxyl of the upstream exon attacks the phosphate at the 3′ splice site (3′ SS), yielding ligated mRNA and the lariat form of the intron. Although the TER1 intron contains all RNA elements required for complete splicing, the level of spliced TER1 accounts for <1% of TER1 isoforms in actively growing cells (Box et al. 2008).
Consistent with telomerase RNA 3′ end processing involving only the first step of splicing, nucleotide changes at the 5′ SS and the BP adenosine blocked TER1 maturation (Box et al. 2008). In contrast, mutations at the 3′ SS did not impair the accumulation of the cleaved form but eliminated the small amount of spliced TER1 that is normally observed (Box et al. 2008). Direct evidence for spliceosome-mediated 3′ end processing of TER1 came from experiments in which 5′ SS mutations were rescued by compensatory changes in the U1 snRNA, which base-pairs with the 5′ SS during spliceosome assembly. Furthermore, replacement of the TER1 intron with a heterologous intron from a protein-encoding gene resulted in the production of spliced TER1 RNA, which failed to support telomerase activity. When the 3′ SS was mutated in this context, some cleaved TER1 was produced, and telomere maintenance was partially restored (Box et al. 2008). Blocking the completion of splicing is thus of pivotal importance for producing functional telomerase enzyme.
The mechanism of splicing has been studied primarily in the context of protein-encoding genes. There, release of the 5′ exon after the first cleavage reaction would block gene expression, as released splicing intermediates will be degraded rather than translated. It is thus no surprise that the two steps of splicing are tightly coupled, and at least for some introns, the first step of splicing is dependent on the presence of an intact 3′ SS (Reed 1989; Romfo and Wise 1997; Hilleren and Parker 2003). It is presently unclear whether fission yeast telomerase RNA is unique in using the first step of splicing for 3′ end processing or whether it is the first example of a class of noncoding RNAs that are processed in this manner. The observation that introns are found near the 3′ ends of telomerase RNAs from several other fungi indicates that spliceosomal 3′ end processing is at least not limited to S. pombe (Gooding et al. 2006; Gunisova et al. 2009; R Kannan and P Baumann, unpubl.).
Here we show that the ability of the spliceosome to function in 3′ end processing is intrinsic to the kinetic quality control mechanism that permits discard of suboptimal intermediates (Hilleren and Parker 2003; Mayas et al. 2006, 2010; Harigaya and Parker 2012). The uncoupling of the first and second step of splicing that occurs naturally in the context of TER1 processing provides new insights into the roles of the BS in splicing.
Results
RNA elements within the intron inhibit the second step of splicing
Many RNA processing events are initiated by the recruitment of processing factors to the C-terminal domain (CTD) of RNA polymerase II (Hernandez and Weiner 1986; Baillat et al. 2005; Moldon et al. 2008). These factors travel with the polymerase during transcription and act on the nascent RNA transcript upon recognition of a processing signal (Bentley 2005). To test whether the ter1 promoter and first exon are required for 3′ end processing by the spliceosome, we replaced them with the promoter and ORF of the ura4 gene. As the binding of Sm proteins directly upstream of the 5′ splice site may be responsible for inhibiting the completion of splicing, 14 nucleotides (nt) of TER1 sequence, including the Sm site, were included in the chimeric construct. A radiolabeled oligonucleotide probe complementary to TER1 exon 2 was used to detect the precursor and spliced forms (Fig. 1B, top panel). Reprobing the Northern with a probe complementary to the first exon (Ura4 ORF) revealed an additional faster-migrating form corresponding in size to the first exon, the expected product of the first step of splicing (Fig. 1B, middle panel). This form was insensitive to a mutation of the 3′ SS (Fig. 1B, middle panel, lane 4) but was abolished when the 5′ SS was mutated (Fig. 1B, middle panel, lane 5). Its identity as the product of spliceosomal cleavage was further confirmed by cloning and sequencing its 3′ terminus (Supplemental Fig. 1). Consistent with previous findings in the context of endogenous TER1 (Box et al. 2008), reducing the distance between the BP and 3′ SS to 9 nt promoted complete splicing (Fig. 1B, lane 6).
The similarity in sizes and length heterogeneity arising from multiple polyadenylation sites and variable poly(A) tail length complicated quantification of the different RNA forms on Northern blots. This issue was resolved by treating the RNA with RNaseH in the presence of two DNA oligonucleotides complementary to sites in exons 1 and 2, respectively (Fig. 1C). Following RNaseH cleavage, precursor, spliced, and cleaved forms resolved into three well-separated bands (Fig. 1D, lane 1). A 2.3-fold increase in the cleaved product was observed when the second step of splicing was blocked by a 3′ SS mutation (Fig. 1D, lane 2). A 5′ SS mutation reduced the first step by >10-fold (Fig. 1D, lane 3), and shortening the BP-to-3′ SS distance promoted complete splicing (Fig. 1D, lane 4).
The observed cleavage of a reporter containing a heterologous promoter and first exon indicated that the TER1 Sm site, intron, and second exon contain all elements required to inhibit the second step of splicing. To further define the essential elements, we deleted the Sm site (Fig. 1E, lane 3) and replaced the TER1 second exon with the 3′ untranslated region (UTR) of URA4 (Fig. 1E, lane 4), neither of which reduced the levels of the cleaved form. The spliced form was more abundant in the context of the URA4 3′ UTR than in the presence of the TER1 second exon. This may reflect differences in stability between the two products or a context dependency of splicing efficiency. In either case, these results show that the TER1 intron is sufficient to promote spliceosomal cleavage of a heterologous RNA and thus harbors the elements required to uncouple the two transesterification reactions.
The strength and spacing of the 3′ intronic elements control uncoupling
Since deletion of 14 nt between the BP and 3′ SS promoted complete splicing, we asked whether inhibition of the second step of splicing was due to the long distance or a specific sequence motif. Starting with the ter1-55 mutant (Fig. 2A) we increased the distance between the BP and 3′ SS in a stepwise manner by insertion of adenines. Adenines were chosen, as increasing pyrimidine content between the BP and 3′ SS has been shown to promote splicing in fission yeast (Romfo and Wise 1997) and metazoans (Reed 1989). As the BP-to-3′ SS distance was increased from 9 to 12 and 16 nt, we observed a stepwise increase in the cleaved form (Fig. 2B). However, the spliced product was also readily detected by Northern blot for all three constructs and dominated over the precursor in RT–PCR analysis (Fig. 2C). Further increasing the distance from 16 to 20 nt resulted in an eightfold increase in the cleaved product, indicating that the second step of splicing was specifically inhibited by the increased distance between the BP and 3′ SS. The first step appeared unaffected by the replacement of endogenous TER1 sequence with adenosines, as the 9+11A and 9+14A constructs yielded as much or more cleaved product as wild-type TER1 (Fig. 2B, cf. lanes 1 and 4,5).
Figure 2.

A long distance between the BP and 3′ SS inhibits the second step of splicing. (A) Intronic sequences of TER1 constructs used in B and C. Splice sites and BSs are in capital letters for emphasis. Inserted adenosines are underlined. (B) Northern blot analysis for ter1 constructs expressed from plasmids under the control of the ter1 promoter. RNaseH cleavage was used to improve resolution of precursor, spliced, and cleaved forms. The cleaved form was quantified relative to loading control snRNA101 (LC) and normalized to wild-type levels. The data are represented as mean ± SEM, with n = 2 for “9+3A” construct and n = 3 for all other constructs. An arrow points to the cleaved form. (C) RT–PCR across the TER1 intron to visualize the relative abundance of precursor and spliced forms.
The observation that the first step but not the second step of splicing proceeded efficiently in the 9+14A mutant was surprising, as the replacement of endogenous TER1 sequence with 14 adenosines eliminated the polypyrimidine (Py) tract. Biochemical analysis in fission yeast cell-free extracts has identified the Py tract as an important element in spliceosome assembly at a step preceding U2 recruitment (Huang et al. 2002). In humans, Py tract strength predominantly affects spliceosome assembly and efficiency of the first step (Reed 1989; Coolidge et al. 1997), but effects of length and sequence on the second step have been observed as well (Reed 1989; Fu et al. 1991; Umen and Guthrie 1995).
To further investigate the effect of pyrimidine content in the context of telomerase RNA 3′ end processing, additional constructs with weak and strong Py tracts were synthesized and integrated at the TER1 locus (Fig. 3A). TER1 itself has a moderately strong Py tract with an uninterrupted run of five pyrimidines and an overall pyrimidine content of 65% between the BS and 3′ SS. Increasing the pyrimidine content favored completion of the second step, as indicated by a twofold reduction in the cleaved form and now readily detectable levels of fully spliced product for the Py+ construct (Fig. 3B). In contrast, loss of the Py tract in the context of the TER1 intron had little effect on the first or second step of splicing (Fig. 3B, lane 3).
Figure 3.

A strong Py tract and U2AF59 favor completion of the second step of splicing. (A) TER1 mutants with strengthened (Py+) or weakened (Py−) pyrimidine tract or cytosines replaced with uridines (C2U) were integrated at the ter1 locus. (B) Detection of precursor, spliced, and cleaved forms by Northern blot. Loading control (LC) and quantification are as in Figure 2B, with n = 2. (C) Northern analysis of RNA from cells containing the ura4 reporter with TER1 intron in the context of prp2+ (U2AF59) and prp2D307N mutant, respectively. Cells were grown at 25°C to 5 × 106 cells per milliliter, shifted to 36°C, and harvested after 0 or 30 min. (Lane 1) Quantification of cleaved and spliced forms relative to LC (snR101) for each lane and normalized to wild type. Quantification was performed as in Figure 2B, with n = 2.
A role for U2AF in promoting the second step of splicing
During spliceosome assembly, the Py tract is bound by the large subunit of U2AF (U2AF65 in mammals; U2AF59 in fission yeast) (Zamore and Green 1989, 1991; Potashkin et al. 1993). Py tract strength correlates with U2AF65-binding affinity, and selection experiments have shown that U2AF65 prefers binding sites containing uridines over cytosines (Singh et al. 1995). Replacement of the five cytosines in the TER1 Py tract with uridines (C2U mutant) resulted in an increase in the spliced form at the expense of the first-step product (Fig. 3B, lane 4). This result further supports that a strong Py tract favors completion of splicing and indicates that the effect may be mediated by U2AF.
We thus examined processing of the TER1 reporter construct in the context of a temperature-sensitive allele (ts) of U2AF59 (Haraguchi et al. 2007). When RNA was isolated from cells immediately after shifting to the restrictive temperature, a 2.5-fold lower level of cleaved TER1 was observed in U2AF59ts compared with wild-type cells (Fig. 3C). This is consistent with a modest assembly and first-step defect even at the permissive temperature. Interestingly, no further reduction in the first-step product was observed in the mutant after 30 min at the restrictive temperature. However, the second step of splicing was now impaired, as evidenced by a twofold reduction in the level of spliced form in the mutant. Taken together, these observations reveal that in the context of the TER1 intron, the first step of splicing occurs largely independent of the presence of a Py tract. Notably though, completion of the second step of TER1 splicing was favored by a strong Py tract and functional U2AF59.
Unexpected role for the BS in inhibiting completion of splicing
Although the BP-to-3′ SS distance is about twice as long in the TER1 intron than the median distance for all annotated introns in the S. pombe genome (see below), well over 100 introns in protein-encoding genes have a similar or greater BP-to-3′ SS distance. We thus wondered whether these introns are efficiently spliced. The first intron in the fcp1 gene encoding for a serine phosphatase (Meyer et al. 2011) resembles the TER1 intron in overall length and architecture, with a BP-to-3′ SS distance of 24 nt (Supplemental Fig. 2A,B). However, when this intron was inserted into the reporter, robust completion of splicing was observed (Fig. 4A, lane 2). The intron was also efficiently spliced in the context of the endogenous fcp1 gene (Supplemental Fig. 2C). A longer distance between the BP and 3′ SS is therefore not sufficient to account for the uncoupling of the first and second step of splicing in TER1.
Figure 4.

A strong BS inhibits the second step of splicing. (A) Northern blot analysis of reporter constructs that contain the TER1 intron (green) or FCP1 intron (pink) between the ura4 ORF and 3′ UTR (blue). Chimeric reporters contain the TER1 or FCP1 BS, indicated by green or pink filled circles, respectively. A probe against snR101 was used as loading control (LC) for normalization of cleaved form relative to lane 1, with the data being represented as mean ± SEM (n = 2). (B) Schematic of base pair interactions between U2 snRNA and BSs of FCP1 and variations, including the TER1 BS sequence. A vertical line is used to represent Watson-Crick base-pairing, and two dots represent G:U interactions. The bulged adenosine at the BP is drawn above the paired sequence. Nucleotide changes from the wild-type FCP1 sequence are highlighted in green. (C) Northern blot analysis for reporter constructs with the FCP1 intron containing the indicated BS sequences. Quantification is as in A, with n = 2 for UAUUAAC and AACUAAC mutants, and n = 3 for other mutants. (D) Distance between the BP and 3′ SS was varied in the context of the FCP1 intron with a strong BS (for schematic, see Supplemental Fig. 2E). The cleaved forms were quantified relative to loading control (LC) and normalized to lane 1 (n = 2).
To identify sequence elements responsible for spliceosomal cleavage, several chimeric introns were generated and tested in the context of the reporter assay. Chimeras containing TER1 sequences upstream of the BS and FCP1-derived sequences between the BS and 3′ SS were spliced well, whereas reciprocal chimeras were processed poorly (Fig. 4A; Supplemental Fig. 2D). Most notable, in otherwise identical constructs, spliceosomal cleavage was observed when the BS sequence was derived from TER1 but not with the BS sequence from FCP1 (Fig. 4A, lanes 3,4; Supplemental Fig. 2D). This observation was surprising, as cross-linking between U2 and the BS in the pre-mRNA and in the excised intron lariat had led to the view that the U2–BS association is maintained through both steps of splicing (Wassarman and Steitz 1992). In contrast, our observation suggests that a tight U2–BS interaction promotes accumulation of the 5′ cleavage product, although in the context of the reporter, the spliced form still predominates.
The TER1 BS is fully complementary to the BS-binding sequence in U2 snRNA (Fig. 4B), whereas the BS in FCP1 intron 1 deviates by two nucleotides from the U2 complement, resulting in a mismatch and the replacement of a G-C base pair with a G:U wobble (Fig. 4B). These two nucleotide differences alone reduced the level of cleavage product by 10-fold (Fig. 4C). Restoring one or the other of the Watson-Crick base pairs resulted in spliceosomal cleavage at 16% and 27% of the level observed with the TER1 intron, respectively (Fig. 4C, lanes 3,4). When both nucleotides were changed together, spliceosomal cleavage products accumulated to 56% of the TER1 control (Fig. 4C, lane 5). Further increasing the distance between the BS and 3′ SS to 38 nt stimulated spliceosomal cleavage at the expense of splicing, whereas reducing the distance promoted the second step of splicing (Fig. 4D).
These observations demonstrate that, at least in the context examined here, the U2–BS interaction has opposing effects on the first and second step of splicing. High U2 complementarity promotes spliceosome assembly and the first cleavage reaction but is inhibitory to the completion of splicing. The most parsimonious explanation for these findings is that U2 must dissociate at least partially from the BS for splicing to proceed beyond the first step.
Our findings are reminiscent of prior observations revealing that U6 snRNA has to dissociate from the 5′ SS of the Saccharomyces cerevisiae actin intron between the first and second step and that hyperstabilization of this interaction inhibits the second step (Konarska et al. 2006). The 5′ SSs in TER1 and FCP1 differ only at position +4, with a uridine in TER1 contributing an additional base pair with U6 as opposed to a C/A mismatch in FCP1 (Supplemental Fig. 3A). We speculated that more extensive base-pairing between U6 snRNA and the 5′ SS in TER1 also contributes to slowing the transition from the first to the second step and thus favors the release of the first-step products. However, replacing the 5′ SS in FCP1 with that of TER1 or two other sequences with increased U6 complementarity did not have the expected effect (Supplemental Fig. 3B). Instead, splicing went to completion for GUAUGU in the context of the FCP1 intron, and GUCUGU and GUCAGU inhibited splicing altogether.
The ‘discard’ pathway promotes spliceosomal cleavage
The transition from the first to the second step of splicing involves major conformational changes within the spliceosome. Biochemical experiments using budding yeast cell-free extracts have revealed a quality control mechanism that monitors the success of exon ligation (Mayas et al. 2006, 2010). When the DExD/H-box helicase Prp22 hydrolyzes ATP prior to the exon ligation step, the splicing intermediates are rejected. This process has been suggested to permit recycling of spliceosomes bound to mutant RNA substrates that arise due to RNA polymerase errors or mutations in the DNA template (Semlow and Staley 2012). It also allows recovery of spliceosomes that have selected suboptimal 3′ SSs.
Here we identified several elements in the TER1 intron that encumber the first-to-second-step transition and may thus trigger Prp22-dependent “discard” of the first-step products. Based on sequence similarity with S. cerevisiae PRP22 and other DExD/H-box helicases (Xu and Query 2007), we designed point mutations in S. pombe prp22. T665A and H634A displayed weak temperature sensitivity, whereas S663A was strongly cold-sensitive (Supplemental Fig. 4A). As Prp22 promotes the release of first- and second-step products in an ATPase-dependent manner and also stabilizes the exon ligation conformation in an ATPase-independent manner, it was important to distinguish between these effects. Mutations that affect the ATPase activity and thereby impair “rejection” are expected to permit increased completion of splicing, whereas mutations that compromise the Prp22 function in exon ligation will reduce the amount of spliced product. We therefore calculated what fraction of RNA had undergone the first and second step, respectively, and normalized to the wild-type sample at 32°C, for which both values were set to 100. Although such quantification does not directly measure the actual percentage of RNA molecules that are released after the first step, it is nevertheless an established measure for first- and second-step efficiencies (Konarska et al. 2006). All three mutations in the Prp22 helicase domain shifted the ratio of first to second step product in favor of spliced RNA, supporting a role for the “discard pathway” in TER1 biogenesis (Fig. 5A,B; Supplemental Fig. 4B).
Figure 5.

Prp22 and Prp43 promote spliceosomal cleavage. (A) Northern blot analysis of reporter containing wild-type TER1 intron. RNA was isolated from prp22+ and prp22T665A cells initially grown at 32°C to 5 × 106 cells per milliliter, shifted to 22°C, and harvested immediately. First-step efficiencies were calculated as cleaved plus spliced form over all forms normalized to wild type at 32°C multiplied by 100. Second-step efficiencies were calculated as spliced form over cleaved plus spliced form normalized to wild type at 32°C multiplied by 100. Data are represented as mean ± SEM (n = 3). (B) Northern blot analysis of a second Prp22 mutant prp22S663A. Analysis of TER1 processing is as in A, with n = 3. (C) Analysis for prp43S229A and prp43R406N by Northern blotting. Quantification is as described in A, with n = 2.
The Prp43 helicase functions downstream from Prp22 in the release of spliced mRNA as well as the discard of suboptimal splicing intermediates (Mayas et al. 2010). A mutation in budding yeast PRP43 further revealed a role in enhancing the specificity of exon ligation by repressing the use of a cryptic 3′ SS. Consistent with a role for S. pombe prp43 in TER1 processing, two mutations (Supplemental Fig. 4C) also reduced first-step and increased second-step efficiencies for the TER1 reporter (Fig. 5C).
In summary, several RNA elements in the TER1 intron act cooperatively to attenuate the second step of splicing, thus creating an opportunity for the rejection and discard of the splicing intermediates in a Prp22- and Prp43-dependent manner.
Global selection against spliceosomal cleavage
We next examined what fraction of annotated fission yeast introns share the combination of a strong BS, long distance, and weak Py tract and are therefore likely candidates for spliceosomal cleavage. While the use of the spliceosome in 3′ end processing may be limited to noncoding RNAs, spliceosomal cleavage could also serve a function in regulating gene expression by reducing the amount of spliced mRNA that is produced (Harigaya and Parker 2012).
To identify putative BSs, we first used a Gibb's sampler algorithm to identify motifs that are overrepresented in S. pombe introns (excluding the 5′ and 3′ SSs) (see the Materials and Methods for details). The positional weight matrix (PWM) generated by this approach strongly resembled known BSs (Fig. 6A). We then used the PWM to score every position in the intron data set. This score (referred to as SBS) ranges from 1.0 to 7.86. Plotting the SBS values versus their positions relative to the acceptor site revealed that sites with scores >6 clustered near the 3′ SS (Supplemental Fig. 5A) Only 189 annotated introns (4%) returned no match for a site with SBS > 6.0 and were excluded from further analysis. Forty-two percent had exactly one site with a score ≥6, and the remaining 54% had more than one such site (Supplemental Fig. 5B). For introns with more than one site ≥6, the site closest to the 3′ SS was chosen as the putative BS.
Figure 6.

Global analysis indicates selection against the intron structure that favors release after the first step of splicing. (A) Sequence logo representation of the S. pombe BS PWM constructed as described in the Materials and Methods. (B) Histogram showing the locations of BSs relative to the 3′ SS. (C) Pie chart representation of introns with long and short distances between the BP and 3′ SS, respectively. (D) Graphical representation of the relationship between BS strength and distance to the 3′ SS. (E) Scatter plot illustrating Py tract strength for introns that resemble TER1 in the BS sequence and distance to the 3′ SS. (F) Northern blot for the reporter RNA containing the indicated introns. A probe against snR101 was used as loading control (LC). Data are represented as mean ± SEM, with n = 2.
With putative BSs mapped in this manner, the mean distance from BP to 3′ SS was 11 nt (Fig. 6B), a finding that is in good agreement with the positions of predicted BSs based on a subset of fission yeast introns (Zhang and Marr 1994). For 96% of introns, the distance was <20 nt, and only 176 introns had a distance between the predicted BP and 3′ SS in the range of where we detect uncoupling of splicing for TER1 (Fig. 6C). We next compared the strength of the BSs between these introns and those with a shorter distance. Whereas 60% of introns with a BP located within 20 nt from the acceptor have strong predicted BSs (defined as a score >7.0), only 30% of introns with a distance >20 nt have strong BSs (Fig. 6D). This statistically significant difference (χ2 P-value = 9.1 × 10−15) between these categories demonstrates that the combination of a strong BS and a long BP-to- 3′ SS distance is underrepresented in introns of protein-encoding genes.
We next plotted the uridine to pyrimidine ratio over the pyrimidine content as a measure of Py tract strength for the 10 introns that share the consensus BS sequence with TER1 and have a BP-to-3′ SS distance of ≥20 nt (Fig. 6E). With the exception of MBX1 intron 3, all candidates have stronger Py tracts than TER1. The two introns that resembled TER1 most closely were cloned into the reporter construct and examined by Northern. No cleavage was observed for MBX1, but the free 5′ exon was readily detected for ATP23 (Fig. 6F). To assess whether the cleavage of the ATP23 RNA was a consequence of placing the intron into a heterologous context, we examined the endogenous message in the context of wild-type fission yeast and an rrp6 deletion. The latter compromises the activity of the nuclear exosome and is thus expected to stabilize a released 5′ exon. We indeed observed the spliceosomal cleavage product for ATP23 in both strain backgrounds (Supplemental Fig. 5C). However, unlike TER1, the spliced form of ATP23 RNA dominated over the cleaved form in steady state. In summary, the combination of RNA elements that promote the uncoupling of the two steps of splicing is largely absent from the vast majority of annotated introns, but at least in one case, cleaved pre-mRNA is detected. Importantly, the global analysis exposed a fervent anti-correlation between a strong BS and a long BS-to-3′ SS distance.
Discussion
The realization that fission yeast telomerase RNA 3′ end processing is carried out by the first step of splicing was surprising at first. The need to accurately remove thousands of introns from pre-mRNAs has resulted in tight coupling of the two steps of splicing so as to not inadvertently release the intermediates. Utilization of the first step of splicing as a means for 3′ end formation necessitates that splicing is aborted after the first cleavage reaction. Here we show how the spliceosome can be converted into a 3′ end processing machinery in the context of TER1 RNA: A combination of RNA elements that promote spliceosome assembly and the first step but encumber the transition to the second step is sufficient to uncouple the two reactions and promote the release of the free 5′ exon via the discard pathway. In addition to providing further insights into the mechanism of telomerase biogenesis, these studies shed new light onto aspects of the splicing reaction that are still only poorly understood.
A conserved role for distance and the Py tract in regulating the second step
The most obvious feature that distinguishes the TER1 intron from the majority of introns in fission yeast is the distance between the BP and 3′ SS. At 23 nt, TER1 is in the top three percentile of introns with respect to BP-to-3′ SS distance. Although the BP-to-3′ SS distance varies considerably between introns, there is a clear species-specific optimum into which the majority of introns fall. In budding yeast, the mean distance between the BP and 3′ SS is ∼39 (Spingola et al. 1999; Meyer et al. 2011). Increasing the distance in the context of a specific intron hampers progression through the second step (Cellini et al. 1986; Luukkonen and Seraphin 1997). In humans, BP-to-3′ SS distances also cluster in a fairly narrow window ∼25 nt (Gao et al. 2008; Corvelo et al. 2010), with the exception of a small number of introns that contain distant BSs that can be >150 nt from the 3′ SS (Gooding et al. 2006). Altering the distance between the BP and 3′ SS in the context of in vitro splicing substrates results in accumulation of splicing intermediates (Reed 1989; Chua and Reed 2001). So, despite differences during spliceosome assembly between yeasts and metazoans, uncoupling of the two steps of splicing as a consequence of a long BP-to-3′ SS distance has been observed in diverse systems.
In vertebrates, binding of U2AF to the Py tract promotes U2 recruitment (Zamore and Green 1989, 1991), and mutations that shorten the Py tract or replace pyrimidines with purines interfere with spliceosome assembly (Roscigno et al. 1993; Coolidge et al. 1997). Similarly, in fission yeast, the Py tract has been shown to function prior to the first step of splicing (Romfo and Wise 1997). Here, a preformed complex of SF1 and the small and large subunits of U2AF bind cooperatively to the BS, Py tract, and 3′ SS (Huang et al. 2002). The prominent role of a Py tract during the early stages of splicing has complicated the analysis of its roles during the second step. However, synthetic splicing substrates containing competing splice sites revealed a function of the Py tract in 3′ SS choice and second-step kinetics (Reed 1989). Notably, completion of the second step was only dependent on a Py tract in the context of a long distance between the BP and 3′ SS. Similarly, in budding yeast, where introns were long thought not to contain functionally important Py tracts, examination of model substrates revealed a critical function for uridine-rich motifs in the identification and utilization of distant splice acceptor sites (Patterson and Guthrie 1991). A role for Py tracts in promoting the second step therefore appears to be widely conserved as well.
What is more surprising is that a temperature-sensitive allele of U2AF59 would compromise completion of the second step at the restrictive temperature (Fig. 3C). At least in mammalian splicing extracts, the large subunit of U2AF is replaced by three proteins of the U5 snRNP prior to the second step of splicing (Chiara et al. 1997) and is not detected in the C complex (Agafonov et al. 2011). Fission yeast may differ in this regard, or U2AF may remain transiently associated with the spliceosome and promote the second step even after direct interactions with the Py tract has ceased.
A strong BS can impede completion of splicing
Binding of the U2 snRNP to the BS is one of the earliest—and in some cases, the earliest—events in spliceosome assembly (Hoskins et al. 2011), and the extent of sequence complementarity correlates positively with the rate of assembly and first cleavage reaction (Zhuang et al. 1989). Pioneering cross-linking studies in the early 1990s suggested that the U2–BS association is maintained through both steps of splicing (Wassarman and Steitz 1992), and consistent with this assertion, the U2 snRNP remains part of the spliceosome after the first step (Bessonov et al. 2008). What has remained puzzling in this regard is that BS mutations to either G or U inhibit the second step of splicing in yeast and human cell extract (Fouser and Friesen 1986; Horning et al. 1986; Jacquier and Rosbash 1986; Tseng et al. 2011). Using an orthogonal splicing system involving a highly substituted and reporter-specific second copy of U2, it has been shown that a change in the position of the bulged nucleophile imposes a block on the second step (Smith et al. 2009). One interpretation of these results is that the nucleotide change affects the flexibility of the RNA, and this impairs the conformational rearrangements that position the RNA correctly for the second transesterification reaction.
An indication that the U2–BS interaction may unwind prior to 5′ SS cleavage came from the observation of trans-splicing products between the 5′ end of U2 and the 3′ exon of a reporter gene in budding yeast (Smith et al. 2007). While those results indicated that U2–BS base-pairing is more dynamic than previously thought, it did not suggest that maintenance of the U2–BS interaction is detrimental to the completion of splicing. Our analysis showed that in the appropriate context, sequence complementarity between the BS and U2 antagonizes the completion of splicing and favors uncoupling of the two steps. In light of these observations, it seems likely that at least partial or transient disruption of U2–BS base-pairing is part of the transition from the first to the second step of a normal splicing reaction. The molecular function of this rearrangement will need to be defined by future studies.
A ‘proofreading pathway’ generates functional telomerase RNA
Our results show that a combination of intronic elements hampers the transition from the first to the second step of splicing and is critical for the release of the TER1 5′ exon after the first step of splicing. The implication of Prp22 and Prp43 in TER1 processing further suggests that the “discard pathway” is not merely acting in the recovery of spliceosomes from defective or suboptimal substrates, but in the case of TER1, promotes the release of a functional product. As the mechanism for attenuating the transition from the first to the second step as well as the molecular machinery are widely conserved, additional targets for this pathway are likely to be discovered. The observation that a partial block to the second step occurs for at least one intron in a protein-encoding gene in S. pombe, together with the recent identification of a substantial number of splicing intermediates in S. cerevisiae (Harigaya and Parker 2012), indicate that the discard pathway may also function in the regulation of gene expression.
Materials and methods
Yeast strains and constructs
Genotypes of the strains used in this study are listed in Supplemental Table I. The basic reporter was generated by replacing the ura4 ORF in pDblet (Brun et al. 1995) with a synthetic DNA fragment comprised of the ura4 ORF followed by 14 nt of TER1 exon 1 sequence (GGGCCCAUUUUUUG), the TER1 intron, and the TER1 second exon using AvrII and StuI restriction sites. Derivatives of this construct were generated by site-directed mutagenesis or subcloning. Plasmids were introduced into S. pombe strains PP138 and PP407 by electroporation, and transformants were selected on Edinburgh minimal medium (EMM) lacking uracil. For genomic integration, ter1∷ura4 spores were generated from PP433 (Tang et al. 2012), germinated on YEA, and selected on EMM lacking uracil. A DNA fragment containing the mutations in the context of a genomic copy of TER1 was amplified from plasmid DNA by PCR using primers PBoli923 (GTAAACGGAATATCCGCGATG) and BLoli1122 (TTCCATATAGTCGATGCTCG). The PCR product was purified, and 1 μg was used for lithium acetate transformations as described (Baumann and Cech 2000). Transformants were selected on medium containing 5-fluoroorotic acid (5′ FOA), and strains containing the correct integration were identified by colony PCR and sequencing.
The prp22 knockout strain was generated by replacing one copy of prp22+ in a diploid strain with the kanamycin resistance marker. The heterozygous prp22+/prp22∷kanR strain was transformed with plasmid pSH3 containing wild-type prp22+ and the ura4 gene. The diploid transformants were sporulated, and prp22∷kanR cells containing pSH3 were selected. The resultant strain was then transformed with a plasmid derived from pBG1 (Burke and Gould 1994) containing prp22+ or point mutants and the his3 gene as a selection marker. Medium containing 5′ FOA and lacking histidine was used to select for cells that have lost pSH3 and now harbor a prp22 mutant or prp22+ on the pBG1-derived plasmid. The presence of each prp22 mutant allele was confirmed by PCR and sequencing. Temperature and cold sensitivity were examined by incubation at 36°C and 22°C, respectively. The same procedure was employed to generate temperature-sensitive prp43 strains, which contained prp43 mutants on a plasmid derived from pBG1 in a prp43∷kanR background.
RNA analysis
RNA isolation was performed as described previously (Box et al. 2008) with the following modifications for temperature-sensitive strains. Cultures (400 mL) of U2AF59ts strains were grown at 25°C to a density of 5 × 106 cells per milliliter. The culture was shifted rapidly to 36°C by immersion and agitation in warm water. Once the culture medium had reached 36°C, a 100-mL sample was taken for RNA isolation. Cultures were then transferred into a shaker at 36°C, and additional samples were harvested at the indicated time points. Similarly, prp22 and prp43 mutant strains were grown into log phase at 32°C and shifted rapidly to 22°C. The point at which the culture medium reached 22°C is referred to as 0 min. To isolate RNA, three or five rounds of phenol-chloroform extraction were performed, depending on whether the sample volume was 100 mL or 400 mL. After the final chloroform isoamyl alcohol extraction, the supernatant containing RNA was ethanol-precipitated at −20°C for at least 1 h. Total RNA was precipitated by centrifugation, and the pellet was dissolved in 50 mM sodium acetate (pH 5.2). Except for the Northern shown in Figure 1B, all samples were subjected to RNaseH cleavage prior to Northern analysis to improve resolution of precursor, spliced, and cleaved forms. Briefly, 15 μg of DNase-treated RNA were combined with two DNA oligos (600 pmol) complementary to sites in the first and second exon, respectively. BLoli2269 (AACATCCAAGCCGATACCAG) and BLoli2326 (GCAAACAAGGCATCGACTTTTTCAATAACCAACCAAAA) were used for constructs containing the ORF and 3′ UTR of ura4. BLoli2326 was replaced with BLoli1275 (CGGAAACGGAATTCAGCATGT) for constructs containing TER1 sequences downstream from the intron. Oligonucleotides BLoli3695 (TGGGCAATTGTATTCTCTGC), BLoli3563 (AAAAGCCAACGTAAATGCTG), and oligo-dT were used for RNaseH cleavage of ATP23 mRNA. The mix was heated to 65°C in a heat block, and a second heat block at 75°C was placed on top of the tubes to reduce condensation. After 5 min, the heat block sandwich was transferred onto a Styrofoam box to allow slow cooling to room temperature. After 45 min of slow cooling, RNaseH buffer (final concentration 1×; New England Biolabs) and RNaseH enzyme (5 U) were added, and samples were incubated for 30 min at 37°C. RNaseH-treated samples were ethanol-precipitated for at least 1 h at −20°C. RNA pellets were recovered by centrifugation, dissolved in 1× formamide loading buffer, and run on a polyacrylamide gel for Northern analysis as described below.
Northern blot
Samples were run on 4% polyacrylamide gels in Tris-borate-EDTA (TBE) containing 7 M urea and transferred to a Biodyne Nylon transfer membrane (Pall Corporation) in TBE buffer. Hybridizations with radiolabeled probes were carried out in Church-Gilbert buffer at 55°C (TER1 probe) or 42°C (reporter and sn101 probes). A TER1 probe recognizing the RNaseH cleavage products was generated by nick translation of a PCR fragment (nucleotides 536–998 of TER1) in the presence of 32P-α-dCTP. The ura4 ORF was detected using BLoli2072 (GTCTTTGCTGATATGCCTTCCAACCAGCTTC), and the 3′ UTR was detected with BLoli2318 (GAAACCGGAAACGGAATTCAGCATGTTTTAATAAAAAGAT), each labeled with polynucleotide kinase in the presence of 32P-γ-ATP. BLoli1136 (CGCTATTGTATGGGGCCTTTAGATTCTTA) complementary to snRNA101 was used to visualize a loading control. ATP23 mRNA was detected on Northern blots using a nick-translated PCR fragment generated with primers BLoli3693 (TTTGACGACCACAGGTTTGA) and BLoli3592 (AGGAATTGAACACTTCTTCAACGAT) as probe and a hybridization temperature of 55°C.
RT–PCR
Semiquantitative RT–PCR for TER1 was performed as described (Box et al. 2008). For 3′ end sequencing, oligo-dT primer was used for the RT reaction, and PBoli517 (GCTTTTGGCTGAAATGTCTTCC) was used as forward primer in the PCR step.
Spotting assay
Cultures were grown to log phase (5 × 106 cells per milliliter) in EMM supplemented with uracil (150 mg/L), leucine (150 mg/L), and adenine (75 mg/L). Threefold dilutions of samples were plated onto PMG (pombe minimal medium with glutamate) in triplicate using an epMotion automated pipetting system (Eppendorf), and plates were incubated at 22°C, 32°C, and 36°C, respectively. Plates were scanned after 3 d at 32°C and 36°C and after 7 d at 22°C.
Computational analysis
To create a database of S. pombe introns, we first compiled the sequences corresponding to the 4862 introns based on a 2007 release of the S. pombe genome. The sequence corresponding to the TER1 intron was added manually. We then applied a filter to remove duplicate sequences and annotated introns that did not begin with “GT/C” and end with “AG,” which reduced the number to 4824 putative intronic sequences.
A Gibb's sampler (http://bayesweb.wadsworth.org/gibbs/gibbs.html) was then used to construct PWM. The first 7 and last 3 nt were excluded, as they represent the 5′ and 3′ SSs, respectively. The recursive sampling algorithm was used with the default settings for eukaryotes. The top five motifs of lengths 7, 9, and 10 were evaluated. All PWMs generated by this analysis resembled BSs. The Gibb's sampler was rerun to construct the best PWM found at least once in all of the sequences. The final BS PWM (represented as a sequence logo in Fig. 6A) was 10 nt long.
BS scores were calculated as follows. At each position, the BS score SBS was calculated by applying the following equation to a 10-nt window starting at that position:
In this equation, 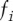 is the frequency for the nucleotide found in the PWM at position
is the frequency for the nucleotide found in the PWM at position 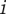 , and
, and 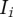 is the Shannon information (in bits) for position
is the Shannon information (in bits) for position 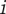 as calculated from the PWM for that position (the Shannon information was calculated as described in Schneider 2010). The score SBS is therefore the total number of bits for the sequence contained in the window as defined by the BS PWM. Using this scoring method, the top scoring sequence is “TTACTAACTT,” which has a SBS = 7.856.
as calculated from the PWM for that position (the Shannon information was calculated as described in Schneider 2010). The score SBS is therefore the total number of bits for the sequence contained in the window as defined by the BS PWM. Using this scoring method, the top scoring sequence is “TTACTAACTT,” which has a SBS = 7.856.
The average SBS for all positions was 3.1, but the average score for the highest-scoring position in each intron was 7.1 (Supplemental Fig. 5D). Approximately 96% of all introns had at least one site with a score >6 (Supplemental Fig. 5C), and this was therefore used as the cutoff for predicting putative BSs in each intron. Nearly half (42%) of the introns had only a single site with a score >6 (Supplemental Fig. 5D). In the cases where there was more than one site with a score >6, the site that was closest to the acceptor site but >5 nt from the 3′ SS was chosen as the putative BS.
Acknowledgments
We thank T. Tani for the U2AF mutant strain, Richard Law and Madelaine Gogol for technical assistance, E. Janzen for characterizing prp43 mutants, the Molecular Biology Core Facility for site-directed mutagenesis and sequencing, and C.-K. Cheng, D. Semlow, and members of the Baumann laboratory for discussions and comments on the manuscript. This work was funded in part by the Stowers Institute for Medical Research, a predoctoral fellowship from the American Heart Association to R.K., and grants from the National Institutes of Health to J.A.B. (AR059833) and J.P.S. (GM062264). P.B. is an Early Career Scientist with the Howard Hughes Medical Institute. R.K and P.B designed the overall study; R.K performed all experiments except for initial prp22 and prp43 mutant characterization, which was done by S.H.; R.B.V. and J.A.B. performed the computational analysis shown in Figure 6 and Supplemental Figure 5; J.P.S. motivated and advised on discard pathway mutants and mechanism of splicing; and R.K and P.B analyzed the data and wrote the manuscript.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.212738.112.
References
- Agafonov DE, Deckert J, Wolf E, Odenwalder P, Bessonov S, Will CL, Urlaub H, Luhrmann R 2011. Semiquantitative proteomic analysis of the human spliceosome via a novel two-dimensional gel electrophoresis method. Mol Cell Biol 31: 2667–2682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baillat D, Hakimi MA, Naar AM, Shilatifard A, Cooch N, Shiekhattar R 2005. Integrator, a multiprotein mediator of small nuclear RNA processing, associates with the C-terminal repeat of RNA polymerase II. Cell 123: 265–276 [DOI] [PubMed] [Google Scholar]
- Baumann P, Cech TR 2000. Protection of telomeres by the Ku protein in fission yeast. Mol Biol Cell 11: 3265–3275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentley DL 2005. Rules of engagement: Co-transcriptional recruitment of pre-mRNA processing factors. Curr Opin Cell Biol 17: 251–256 [DOI] [PubMed] [Google Scholar]
- Bessonov S, Anokhina M, Will CL, Urlaub H, Luhrmann R 2008. Isolation of an active step I spliceosome and composition of its RNP core. Nature 452: 846–850 [DOI] [PubMed] [Google Scholar]
- Blackburn EH, Collins K 2011. Telomerase: An RNP enzyme synthesizes DNA. Cold Spring Harb Perspect Biol 3: a003558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Box JA, Bunch JT, Tang W, Baumann P 2008. Spliceosomal cleavage generates the 3′ end of telomerase RNA. Nature 456: 910–914 [DOI] [PubMed] [Google Scholar]
- Brun C, Dubey DD, Huberman JA 1995. pDblet, a stable autonomously replicating shuttle vector for Schizosaccharomyces pombe. Gene 164: 173–177 [DOI] [PubMed] [Google Scholar]
- Burke JD, Gould KL 1994. Molecular cloning and characterization of the Schizosaccharomyces pombe his3 gene for use as a selectable marker. Mol Gen Genet 242: 169–176 [DOI] [PubMed] [Google Scholar]
- Cellini A, Felder E, Rossi JJ 1986. Yeast pre-messenger RNA splicing efficiency depends on critical spacing requirements between the branch point and 3′splice site. EMBO J 5: 1023–1030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiara MD, Palandjian L, Kramer RF, Reed R 1997. Evidence that U5 snRNP recognizes the 3′ splice site for catalytic step II in mammals. EMBO J 16: 4746–4759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chua K, Reed R 2001. An upstream AG determines whether a downstream AG is selected during catalytic step II of splicing. Mol Cell Biol 21: 1509–1514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coolidge CJ, Seely RJ, Patton JG 1997. Functional analysis of the polypyrimidine tract in pre-mRNA splicing. Nucleic Acids Res 25: 888–896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corvelo A, Hallegger M, Smith CWJ, Eyras E 2010. Genome-wide association between branch point properties and alternative splicing. PLoS Comput Biol 6: e1001016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fouser LA, Friesen JD 1986. Mutations in a yeast intron demonstrate the importance of specific conserved nucleotides for the two stages of nuclear mRNA splicing. Cell 45: 81–93 [DOI] [PubMed] [Google Scholar]
- Fu XD, Katz RA, Skalka AM, Maniatis T 1991. The role of branchpoint and 3′-exon sequences in the control of balanced splicing of avian retrovirus RNA. Genes Dev 5: 211–220 [DOI] [PubMed] [Google Scholar]
- Gao K, Masuda A, Matsuura T, Ohno K 2008. Human branch point consensus sequence is yUnAy. Nucleic Acids Res 36: 2257–2267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gooding C, Clark F, Wollerton MC, Grellscheid SN, Groom H, Smith CW 2006. A class of human exons with predicted distant branch points revealed by analysis of AG dinucleotide exclusion zones. Genome Biol 7: R1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunisova S, Elboher E, Nosek J, Gorkovoy V, Brown Y, Lucier JF, Laterreur N, Wellinger RJ, Tzfati Y, Tomaska L 2009. Identification and comparative analysis of telomerase RNAs from Candida species reveal conservation of functional elements. RNA 15: 546–559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haraguchi N, Andoh T, Frendewey D, Tani T 2007. Mutations in the SF1–U2AF59–U2AF23 complex cause exon skipping in Schizosaccharomyces pombe. J Biol Chem 282: 2221–2228 [DOI] [PubMed] [Google Scholar]
- Harigaya Y, Parker R 2012. Global analysis of mRNA decay intermediates in Saccharomyces cerevisiae. Proc Natl Acad Sci 109: 11764–11769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez N, Weiner AM 1986. Formation of the 3′ end of U1 snRNA requires compatible snRNA promoter elements. Cell 47: 249–258 [DOI] [PubMed] [Google Scholar]
- Hilleren PJ, Parker R 2003. Cytoplasmic degradation of splice-defective pre-mRNAs and intermediates. Mol Cell 12: 1453–1465 [DOI] [PubMed] [Google Scholar]
- Horning H, Aebi M, Weissmann C 1986. Effect of mutations at the lariat branch acceptor site on β-globin pre-mRNA splicing in vitro. Nature 324: 589–591 [DOI] [PubMed] [Google Scholar]
- Hoskins AA, Friedman LJ, Gallagher SS, Crawford DJ, Anderson EG, Wombacher R, Ramirez N, Cornish VW, Gelles J, Moore MJ 2011. Ordered and dynamic assembly of single spliceosomes. Science 331: 1289–1295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang T, Vilardell J, Query CC 2002. Pre-spliceosome formation in S. pombe requires a stable complex of SF1–U2AF(59)–U2AF(23). EMBO J 21: 5516–5526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacquier A, Rosbash M 1986. RNA splicing and intron turnover are greatly diminished by a mutant yeast branch point. Proc Natl Acad Sci 83: 5835–5839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konarska MM, Vilardell J, Query CC 2006. Repositioning of the reaction intermediate within the catalytic center of the spliceosome. Mol Cell 21: 543–553 [DOI] [PubMed] [Google Scholar]
- Leonardi J, Box JA, Bunch JT, Baumann P 2008. TER1, the RNA subunit of fission yeast telomerase. Nat Struct Mol Biol 15: 26–33 [DOI] [PubMed] [Google Scholar]
- Luukkonen BG, Seraphin B 1997. The role of branchpoint-3′ splice site spacing and interaction between intron terminal nucleotides in 3′ splice site selection in Saccharomyces cerevisiae. EMBO J 16: 779–792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayas RM, Maita H, Staley JP 2006. Exon ligation is proofread by the DExD/H-box ATPase Prp22p. Nat Struct Mol Biol 13: 482–490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayas RM, Maita H, Semlow DR, Staley JP 2010. Spliceosome discards intermediates via the DEAH box ATPase Prp43p. Proc Natl Acad Sci 107: 10020–10025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer M, Plass M, Perez-Valle J, Eyras E, Vilardell J 2011. Deciphering 3′ss selection in the yeast genome reveals an RNA thermosensor that mediates alternative splicing. Mol Cell 43: 1033–1039 [DOI] [PubMed] [Google Scholar]
- Moldon A, Malapeira J, Gabrielli N, Gogol M, Gomez-Escoda B, Ivanova T, Seidel C, Ayte J 2008. Promoter-driven splicing regulation in fission yeast. Nature 455: 997–1000 [DOI] [PubMed] [Google Scholar]
- Patterson B, Guthrie C 1991. A U-rich tract enhances usage of an alternative 3′ splice site in yeast. Cell 64: 181–187 [DOI] [PubMed] [Google Scholar]
- Potashkin J, Naik K, Wentz-Hunter K 1993. U2AF homolog required for splicing in vivo. Science 262: 573–575 [DOI] [PubMed] [Google Scholar]
- Reed R 1989. The organization of 3′ splice-site sequences in mammalian introns. Genes Dev 3: 2113–2123 [DOI] [PubMed] [Google Scholar]
- Romfo CM, Wise JA 1997. Both the polypyrimidine tract and the 3′ splice site function prior to the first step of splicing in fission yeast. Nucleic Acids Res 25: 4658–4665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roscigno RF, Weiner M, Garcia-Blanco MA 1993. A mutational analysis of the polypyrimidine tract of introns. Effects of sequence differences in pyrimidine tracts on splicing. J Biol Chem 268: 11222–11229 [PubMed] [Google Scholar]
- Schneider TD 2010. A brief review of molecular information theory. Nano Commun Netw 1: 173–180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semlow DR, Staley JP 2012. Staying on message: Ensuring fidelity in pre-mRNA splicing. Trends Biochem Sci 37: 263–273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh R, Valcarcel J, Green MR 1995. Distinct binding specificities and functions of higher eukaryotic polypyrimidine tract-binding proteins. Science 268: 1173–1176 [DOI] [PubMed] [Google Scholar]
- Smith DJ, Query CC, Konarska MM 2007. Trans-splicing to spliceosomal U2 snRNA suggests disruption of branch site–U2 pairing during pre-mRNA splicing. Mol Cell 26: 883–890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DJ, Konarska MM, Query CC 2009. Insights into branch nucleophile positioning and activation from an orthogonal pre-mRNA splicing system in yeast. Mol Cell 34: 333–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spingola M, Grate L, Haussler D, Ares M Jr 1999. Genome-wide bioinformatic adn molecular analysis of introns in Saccharomyces cerevisiae. RNA 5: 212–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang W, Kannan R, Blanchette M, Baumann P 2012. Telomerase RNA biogenesis involves sequential binding by Sm and Lsm complexes. Nature 484: 260–264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng CK, Liu HL, Cheng SC 2011. DEAH-box ATPase Prp16 has dual roles in remodeling of the spliceosome in catalytic steps. RNA 17: 145–154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umen JG, Guthrie C 1995. The second catalytic step of pre-mRNA splicing. RNA 1: 869–885 [PMC free article] [PubMed] [Google Scholar]
- Wahl MC, Will CL, Luhrmann R 2009. The spliceosome: Design principles of a dynamic RNP machine. Cell 136: 701–718 [DOI] [PubMed] [Google Scholar]
- Wassarman DA, Steitz JA 1992. Interactions of small nuclear RNA's with precursor messenger RNA during in vitro splicing. Science 257: 1918–1925 [DOI] [PubMed] [Google Scholar]
- Webb CJ, Zakian VA 2008. Identification and characterization of the Schizosaccharomyces pombe TER1 telomerase RNA. Nat Struct Mol Biol 15: 34–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilusz CJ, Wilusz J 2005. Eukaryotic Lsm proteins: Lessons from bacteria. Nat Struct Mol Biol 12: 1031–1036 [DOI] [PubMed] [Google Scholar]
- Xu YZ, Query CC 2007. Competition between the ATPase Prp5 and branch region-U2 snRNA pairing modulates the fidelity of spliceosome assembly. Mol Cell 28: 838–849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamore PD, Green MR 1989. Identification, purification, and biochemical characterization of U2 small nuclear ribonucleoprotein auxiliary factor. Proc Natl Acad Sci 86: 9243–9247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamore PD, Green MR 1991. Biochemical characterization of U2 snRNP auxiliary factor: An essential pre-mRNA splicing factor with a novel intranuclear distribution. EMBO J 10: 207–214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang MQ, Marr TG 1994. Fission yeast gene structure and recognition. Nucleic Acids Res 22: 1750–1759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang Y, Goldstein AM, Weiner AM 1989. UACUAAC is the preferred branch site for mammalian mRNA splicing. Proc Natl Acad Sci 86: 2752–2756 [DOI] [PMC free article] [PubMed] [Google Scholar]