

Serrano et al. present a model for the role of Sirtuins in maintaining genome stability and controlling the cell cycle via epigenetic regulation. They establish a functional link between SirT2 and PR-Set7 during mitosis and delineate a mechanism for how cross-talk occurs between active and repressive histone marks. Interestingly, SirT2 loss in mice results in genome instability and an elevated risk of tumorigenesis. This study thus provides new insights into Sirtuin function, chromatin, and tumor suppression pathways.
Keywords: H4K20me1, PR-Set7, SIRT2, epigenetics, genome instability, sirtuins
Abstract
The establishment of the epigenetic mark H4K20me1 (monomethylation of H4K20) by PR-Set7 during G2/M directly impacts S-phase progression and genome stability. However, the mechanisms involved in the regulation of this event are not well understood. Here we show that SirT2 regulates H4K20me1 deposition through the deacetylation of H4K16Ac (acetylation of H4K16) and determines the levels of H4K20me2/3 throughout the cell cycle. SirT2 binds and deacetylates PR-Set7 at K90, modulating its chromatin localization. Consistently, SirT2 depletion significantly reduces PR-Set7 chromatin levels, alters the size and number of PR-Set7 foci, and decreases the overall mitotic deposition of H4K20me1. Upon stress, the interaction between SirT2 and PR-Set7 increases along with the H4K20me1 levels, suggesting a novel mitotic checkpoint mechanism. SirT2 loss in mice induces significant defects associated with defective H4K20me1–3 levels. Accordingly, SirT2-deficient animals exhibit genomic instability and chromosomal aberrations and are prone to tumorigenesis. Our studies suggest that the dynamic cross-talk between the environment and the genome during mitosis determines the fate of the subsequent cell cycle.
The members of the Sir2 family of NAD+-dependent deacetylases, also known as Sirtuins, are crucial factors in the response to metabolic, oxidative, or genotoxic stress (Chalkiadaki and Guarente 2012). The capacity of Sirtuins to sense these compromising conditions appears to be due to the requirement of NAD+ for their enzymatic activity (Imai et al. 2000; Landry et al. 2000; Smith et al. 2000). A second enzymatic activity, a mono-ADP ribosyltransferase activity, has been reported for some Sirtuins, although the relevance of this activity for the Sir2 family remains a matter of debate (Tanny et al. 1999). Sirtuins participate in a myriad of functions directed at facilitating adaptation to these stress conditions at both the cellular and organismal levels. These functions include metabolic homeostasis, survival under stress, the maintenance of genomic stability, and cell differentiation and development (Saunders and Verdin 2007; Finkel et al. 2009).
Sirtuins have been present since early in evolution. From this time, they have apparently played a major role in the cross-talk between environmental stimuli and genomic information (Vaquero 2009). In fact, eukaryotic Sirtuins often exert their functions by affecting chromatin. These functions include gene silencing, chromatin structure modulation, DNA damage signaling, DNA repair, and cell cycle regulation. The mechanisms through which Sirtuins act on chromatin involve the deacetylation of both histone and nonhistone proteins ranging from chromatin machinery enzymes to transcription factors.
SirT2 is among the least understood of the seven mammalian Sirtuins. To date, it has been linked to the regulation of mitotic progression (Dryden et al. 2003), oxidative stress response (Wang et al. 2007), metabolism (Jiang et al. 2011), microtubule dynamics (North et al. 2003), cell migration (Pandithage et al. 2008), apoptosis (Li et al. 2011), neurotoxicity (Outeiro et al. 2007), and the inhibition of differentiation (Jing et al. 2007; Li et al. 2007). The role of SirT2 in cell cycle regulation is particularly intriguing. SirT2 has been found to be up-regulated during mitosis, and its overexpression has been reported to induce the lengthening of mitosis (Dryden et al. 2003) and shortening of G1 (Bae et al. 2004). Additionally, microinjection of either SirT2 or its yeast ortholog, Hst2p, has been shown to inhibit starfish oocyte maturation and embryonic cell division (Borra et al. 2002). Primary mouse embryonic fibroblasts (MEFs) derived from SirT2 knockout mice show a very limited decrease in the length of mitosis but longer G1 and shorter S phases—a finding that confirms that SirT2 has a significant role in the cell cycle and, at least in part, a direct impact on G1/S (Vaquero et al. 2006). Recent studies have suggested a role for SirT2 in the control of mitotic exit by acting on the anaphase-promoting complex; namely, via the deacetylation of its coactivators, APCCDH1 and CDC20 (Kim et al. 2011).
A very interesting link between chromatin regulation and SirT2 that directly points to cell cycle regulation is the acetylation of Lys 16 on histone H4 (H4K16Ac), a mark that has been proposed to directly regulate chromatin structure (Shia et al. 2006). Previous studies have determined that in terms of deacetylase activity, SirT2 is highly specific for H4K16Ac (Vaquero et al. 2006), in contrast to the other major mammalian H4K16Ac deacetylase, the Sirtuin SirT1, which has a wider specificity (Vaquero et al. 2004). Studies have shown that H4K16Ac inhibits the folding of chromatin fiber in vitro and therefore facilitates the formation of higher orders of chromatin organization (Shogren-Knaak et al. 2006; Robinson et al. 2008). The acetylation/deacetylation of H4K16 has been implicated in transcriptional regulation (Dou et al. 2005; Bell et al. 2007), DNA repair (Jazayeri et al. 2004) and recombination (Cesarini et al. 2012), many epigenetic phenomena (Vaquero et al. 2007), aging (Dang et al. 2009), and cancer (Fraga et al. 2005). These findings consistently support an important role for the acetylation/deacetylation of H4K16 in chromatin dynamics. Furthermore, H4K16Ac is tightly regulated during the cell cycle: Its levels peak during S phase and drop dramatically in the G2/M transition. SirT2 is present in the cytoplasm throughout the cell cycle, except during the G2/M transition, when it is transported to the nucleus (Wilson et al. 2006; North and Verdin 2007a), where it globally deacetylates H4K16Ac before mitosis begins. MEFs derived from SIRT2−/− mice show hyperacetylation of H4K16Ac during mitosis, in contrast to those from SIRT1−/− mice, which have apparently normal H4K16Ac levels (Vaquero et al. 2006). Interestingly, the effects of SirT2 on chromatin appear to be restricted to early mitosis, as SirT2 dissociates from the chromosomes during metaphase (Vaquero et al. 2006).
Several levels of interplay have been identified between H4K16Ac and other histone marks. The best characterized of these are linked to the transcriptional role of H4K16Ac at the level of initiation (H3K4me3 and H3S10P) or elongation (H3K36me3) (Dou et al. 2005; Bell et al. 2007; Zippo et al. 2009). Interestingly, an antagonism between H4K16Ac and H4K20 methylation has been proposed. In vitro studies with peptides have shown that H4K16Ac could inhibit the monomethylation of H4K20 (H4K20me1) by the histone methyltransferase (HMT) PR-Set7 (also known as SET8 or KMT5A) (Nishioka et al. 2002a). The relevance of this antagonism lies in the importance of H4K20 mono-, di-, or trimethylation in the control of cell cycle progression, chromosome compaction, development, DNA repair signaling, and genome stability (Sanders et al. 2004; Schotta et al. 2004, 2008; Karachentsev et al. 2005; Oda et al. 2009). H4K20me1 is established in late G2/early M by PR-Set7 and is paramount in metaphasic chromosome compaction and mitotic exit during mitosis (Houston et al. 2008; Oda et al. 2009) and in DNA repair and DNA replication (Jorgensen et al. 2007; Tardat et al. 2007, 2010). During late M/early G1, some H4K20me1 is further methylated into H4K20me2 (required for DNA repair) or H4K20me3 (required for heterochromatin structure formation) by the HMTs Suv4-20h1 or Suv4-20h2, respectively (Sanders et al. 2004; Schotta et al. 2004, 2008). PR-Set7 levels are tightly regulated during the cell cycle, and this protein is clearly detected from late M to early G1 (Oda et al. 2009). Despite the importance of H4K20 methylation to cell cycle progression, little is known about the signals and pathways that regulate this modification or the mechanisms through which the enzymes involved are modulated. The only clue currently is the reported phosphorylation of S29 in PR-Set7 by CDK1, which appears to be important for regulating the binding of PR-Set7 to chromatin and for preventing its degradation by APCCDH1-mediated polyubiquitination (Wu et al. 2010).
While investigating the role of SirT2 and H4K16Ac in cell cycle control and genome stability, we discovered roles for SirT2 in establishing PR-Set7-mediated H4K20me1 during early mitosis and in the maintenance of genome stability. We deciphered an unexpected and close functional relationship between these two enzymes and linked, for the first time, the deposition of H4K20 methylation to a mitotic checkpoint and to the SirT2-dependent stress response. Here we describe an unprecedented functional explanation for the maintenance of genomic stability by Sirtuins and provide strong evidence that supports a dynamic role of Sirtuins in controlling the cell cycle through the modulation of epigenetic regulatory information.
Results
Tissue from SirT2-deficient mice shows H4K16 hyperacetylation
To understand the functional relationship among SirT2, H4K16Ac, and mitosis, we aimed to determine the consequences of SirT2 loss in vivo. SirT2 knockout mice were generated by replacing exons 5 and 6 and part of exon 7 of the SIRT2 gene (Fig. 1A–D) with a Neo gene (Materials and Methods). The resultant mice exhibited a complete lack of the SirT2 protein (Fig. 1C). These mice did not show any obvious developmental defect and postnatally appeared to be normal. In agreement with previous findings involving the SIRT2−/− MEFs (Vaquero et al. 2006), the SIRT2−/− mice showed hyperacetylation of H4K16Ac in different tissues, such as the kidney (Fig. 1E) and liver (Supplemental Fig. 1). Moreover, no significant changes in the levels of another histone H4 mark, H4K12Ac, were observed (Fig. 1E). In agreement with our previous results in primary MEFs, proliferating cells in these tissues showed H4K16 hyperacetylation during mitosis (marked by H3S28P) compared with cells in wild-type tissues (Fig. 1F).
Figure 1.
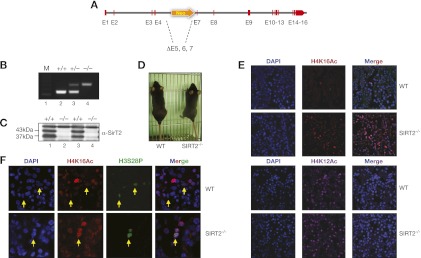
SIRT2−/− mice show H4K16 hyperacetylation in vivo. (A) Schematic of the generation of SIRT2−/− mice. Integration of a neo cassette led to deletion of exons 5 and 6 and part of 7. (B) Genotyping of wild-type (WT), SIRT2+/−, and SIRT2−/− mice. Genomic DNA was extracted from ear punches, as described. (C) SirT2 protein levels measured by Western blot in wild-type or SIRT2−/− MEFs (D) Young SIRT2−/− adults do not show any characteristic phenotype. (E) H4K16Ac and H4K12Ac levels in kidney cryosections derived from wild-type or SIRT2−/− mice, as detected by immunohistochemistry. (F) H4K16Ac levels in kidney mitotic cells, identified by staining with the marker H3S28P, as detected by immunohistochemistry of cryosections.
SirT2 alters H4K20me1 deposition during early mitosis
Once we confirmed the functional link between H4K16Ac and SirT2 in vivo, we aimed to determine the role of this global deacetylation during the cell cycle. We envisaged two different scenarios as the most likely mechanisms. First, considering the proposed role of H4K16Ac in inhibiting chromatin compaction, H4K16Ac hypoacetylation might be required for the proper compaction of metaphase chromosomes. However, as we explain below, the current body of evidence does not completely support this explanation. Second, H4K16Ac deacetylation appears to be a prerequisite for certain processes during mitosis. Considering the proposed interplay between K16 and K20 in the histone H4 tail (Fig. 2A; Nishioka et al. 2002b), we hypothesized that SirT2 could be involved in H4K20 methylation. Interestingly, during mitosis, H4K20me1 is chiefly established between the G2/M transition and metaphase, which is the same time that SirT2 localizes to the chromatin and deacetylates H4K16Ac (Vaquero et al. 2006). To test this hypothesis, we analyzed the levels of H4K16Ac and H4K20me1 in metaphasic chromosomes in cells derived from either wild-type or SIRT2−/− mice. Interestingly, the SIRT2−/− metaphasic chromosomes exhibited H4K16 hyperacetylation and an associated loss of H4K20me1 (Fig. 2B,C), thereby confirming that the antagonism is real—at least during early mitosis. Other findings support a functional link between SirT2 and the establishment of H4K20me1. First, the SIRT2−/− mouse kidney exhibited significantly lower levels of H4K20me1 compared with wild type but did not show any change in the levels of H3K4me3 (Fig. 2D). Second, an analysis of the changes in the H4K20me1 distribution in SIRT2−/− and wild-type primary fibroblasts revealed that the loss of H4K20me1 caused by SirT2 loss occurred chiefly during mitosis and did not affect other cell cycle phases (Fig. 2E). Interestingly, this effect was SirT2-specific, as indicated by the absence of any clear defect in the H4K20me1 levels during mitosis (Fig. 2F) in SIRT1−/− MEFs (Vaquero et al. 2004). This effect was directly dependent on SirT2 activity because the expression of exogenous active SirT2 but not a SirT2 catalytically inactive point mutant rescued the levels of H4K20me1 (Fig. 2G). Considering the relatively low proliferation rate of kidney cells, the global H4K20me1 hypomethylation shown in Figure 2D may indicate that this effect is also important for quiescence. Overall, the aforementioned data establish a specific role for SirT2 in the deposition of H4K20me1 during mitosis.
Figure 2.
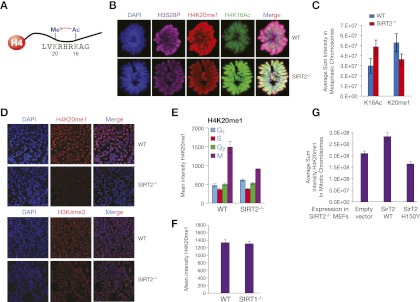
SirT2 regulates H4K20me1 deposition in early mitosis. (A) Interplay between H4K16 and H4K20 in the N-terminal tail of histone H4. (B) IF of metaphase cells from wild-type (WT) or SIRT2−/− MEFs. Cells were stained with antibodies against H3S28P, H4K20me1, and H4K16Ac and then counterstained with DAPI. (C) Quantification of multiple experiments as in B measuring H4K20me1 levels versus H4K16Ac. (D) H4K20me1 and H3K4me3 levels in kidney cryosections prepared from wild-type or SIRT2−/− mice. (E) Quantification of the mean intensity of H4K20me1 in IF assays from wild-type or SIRT2−/− MEFs through the different stages of the cell cycle (n > 20 for each time point). (F) Quantification of mean intensity of H4K20me1 in metaphasic chromosomes of wild-type or SIRT1−/− MEFs (n > 20 for each time point). (G) Quantification of the levels of H4K20me1 in SIRT2−/− MEFs upon overexpression of SirT2 wild type or catalytically inactive point mutant H150Y.
SirT2 interacts with PR-Set7 and modulates its activity, chromatin localization, and distribution during early mitosis
This functional link suggests that SirT2 might modulate the function of PR-Set7. The most obvious explanation is that SirT2 is required for the deacetylation of H4K16Ac, which in turn enables H4K20me1 deposition by the HMT. Previous studies have shown that in contrast to the strict nucleosomal specificity of wild-type PR-Set7, the HMT activity of a PR-Set7 deletion mutant capable of targeting H4 peptides is inhibited by the presence of K16 acetylation in these peptides (Nishioka et al. 2002b). However, whether this inhibitory effect holds in the context of the wild-type enzyme and nucleosomes has not yet been reported. Thus, to answer this question, we performed a PR-Set7 HMT assay using substrates that were recombinant nucleosomes containing an H4 N-terminal tail generated by chemical ligation to contain either unmodified or acetylated K16. A titration of nucleosomes in these HMT assays clearly demonstrated that the interplay between H4K16Ac and PR-Set7 in the context of nucleosomes does indeed hold (Fig. 3A,B). This inhibitory effect of H4K16Ac was not due to an alteration produced by the chemical ligation, since the H4K16Ac-containing recombinant octamers (rOcts) did not inhibit the activity of a bacterially expressed catalytic SET domain of the H3K9me2-specific HMT G9a (Fig. 3A). Preincubation of SirT2 with native nucleosomes and NAD+ significantly increased the specific HMT activity of PR-Set7 (Fig. 3C, lane 4), a finding that supports a positive effect of SirT2 on PR-Set7, at least partially, through H4K16Ac deacetylation. Furthermore, part of this effect by SirT2 on PR-Set7 in vitro was independent of the catalytic activity of SirT2 (Fig. 3C, lane 3). The observation that SirT2 also increased the PR-Set7-specific activity in the absence of NAD+ suggests that the two factors may interact stably. This possibility was supported by the mass spectrometry analysis of elutions from Myc-tagged PR-Set7 affinity purification using Myc resin, in which we unequivocally identified endogenous SirT2 as one of the proteins pulled down by PR-Set7 in the chromatin-insoluble fractions (Fig. 3D). Interestingly, no SirT2 was identified in the PR-Set7 pull-downs from the soluble nuclear extracts (Fig. 3D) or in any other PR-Set7-unrelated Myc immunoprecipitation of the chromatin pellet (data not shown). Further studies clearly demonstrated that PR-Set7 immunoprecipitates SirT2 in HeLa cells (Fig. 3E), and pull-down studies with recombinant proteins confirmed that the two proteins interact directly (Supplemental Fig. 2A). This interaction was drastically reduced with a catalytically inactive SirT2 point mutant, supporting a role for SirT2 activity in these processes (Supplemental Fig. 2B). Interestingly, PR-Set7 immunoprecipitated only the upper band of SirT2 (Fig. 3E, lane 3), which corresponds to its phosphorylated form (SirT2 is phosphorylated chiefly during mitosis) (Supplemental Fig. 2C; North and Verdin 2007b).
Figure 3.
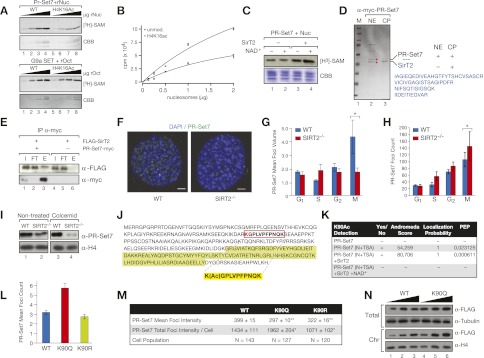
SirT2 regulates H4K20me1 deposition by directly interacting with PR-Set7. (A, top) HMT assay of recombinant PR-Set7 using as substrates increasing amounts (0.125, 0.25, 0.5, and 1 μg) of recombinant nucleosomes either unmodified (wild-type [WT]) or containing symmetrical H4K16Ac assembled by chemical ligation. (Bottom) As a control, 0.125, 0.25, 0.5, and 1 μg of recombinant octamers (rOct) (either wild type or acetylated in the H4K16A by chemical ligation) were tested in HMT assays as substrates of the recombinant catalytic SET domain of G9a. These octamers were previously used to reconstitute the recombinant nucleosomes used at top. In both HMT assays, histone proteins were stained with Coomassie blue (CBB). A representative experiment of a set of three assays is shown in each case. (B) Quantification of similar experiments shown in the top part of A but using 200 ng instead of 50 ng of recombinant PR-Set7 was quantified by scintillation counting. Data shown are means and SEM of two experiments. (C) HMT assay of PR-Set7 using oligonucleosomes purified from HeLa cells as substrates preincubated with or without SirT2 ± NAD+. Histone proteins were stained with Coomassie blue (CBB). (D) Colloidal-stained gel of Myc-PR-Set7 purified with Myc resin from nuclear extract (NE) and chromatin pellet (CP) of Myc-PR-Set7-overexpressing cells. The band corresponding to PR-Set7 was cut and analyzed by mass spectrometry (see the Supplemental Material). Endogenous SirT2 peptide sequences identified only in chromatin pellet are shown in blue. (E) Myc resin immunoprecipitation of extracts from HeLa cells previously transfected with Flag-SirT2 and/or PR-Set7-myc as indicated. (I) Input; (FT) flow-through; (E) elutions. (F) PR-Set7 foci in wild-type and SIRT2−/− fibroblasts detected by IF experiments in G2/M cells (bar, 5 μm) as described previously (Tardat et al. 2010). (G) Quantification of the mean volume of PR-Set7 foci from cells at different stages of the cell cycle (n > 20 for each time point). (H) The number of foci in the same experiment was quantified (data shown). (I) Levels of endogenous PR-Set7 in insoluble chromatin fractions of wild-type or SIRT2−/− MEFs either asynchronous or blocked in mitosis with colcemid. Histone H4 Western blot is shown as a loading control. (J) Primary structure of PR-Set7. The sequence of one of the detected K90-acetylated peptides is indicated in yellow. The catalytic SET domain is shown in green. (K, rows 1,2) Mass spectrometry analysis of PR-Set7 purified from cells in different conditions found an acetylation site in K90 of PR-Set7 (indicated in the PR-Set7 sequence shown above) in cells previously treated with HDAC inhibitors N+TSA but not in untreated cells. Analysis of tandem mass spectrometry parameters in K90 identification indicates a high confidence in the detection. The table includes three parameters: andromeda score (peptide score), localization probability, and PEP (posterior error probability of the identification). (Rows 3,4) Purified PR-Set7 (N+TSA) was incubated with SirT2 ± NAD+. (L,M) Quantification of the number of foci per cell (L) and the mean intensity of PR-Set7 per foci and per nucleus (M) of an experiment similar to that in F but using primary MEFs overexpressing PR-Set7 wild-type or mutant proteins, as indicated. (N) Western blot from whole-cell extract (Total) or chromatin pellet (Chr) prepared from HeLa cells previously transfected with an increased amount of expression vector encoding either Flag-tagged PR-Set7 wild type or K90Q. Tubulin was used as a control of whole-cell extract, and histone H4 was used as control of chromatin pellet.
Thus, our studies show that SirT2 promotes the enzymatic activity of PR-Set7 through the deacetylation of H4K16Ac and through the direct binding of both factors, which most likely involves a conformational change in PR-Set7.
The interplay between SirT2 and PR-Set7 during mitosis apparently includes other levels of regulation, as reflected by the observation that PR-Set7 mitotic foci were dramatically altered in SirT2-deficient cells. The loss of SirT2 induced a higher number of foci, but these were significantly smaller than those in the wild-type cells (Fig. 3F–H). Moreover, SIRT2−/− cells also exhibited significantly decreased levels of PR-Set7 associated with insoluble chromatin, particularly during mitosis (Fig. 3I), whereas the total amount of PR-Set7 was unaffected (Supplemental Fig. 2D). The findings that the activity and localization of PR-Set7 as well as the architecture of its foci were all affected in the absence of SirT2 strongly suggest that SirT2 activity is also required for the proper loading of the PR-Set7 enzyme.
SirT2-mediated PR-Set7 K90 deacetylation regulates its chromatin localization
We also tested the possibility that SirT2 modulates PR-Set7 function via the direct deacetylation of PR-Set7. Our mass spectrometry experiments identified a single residue acetylated in PR-Set7 at K90 (Fig. 3J,K; data not shown). SirT2 can specifically deacetylate this residue given that no K90Ac was detected after the incubation of PR-Set7 with SirT2 plus NAD+ (Fig. 3K). However, the acetylation/deacetylation of K90 did not appear to have a significant effect on the enzymatic activity of PR-Set7. Thus, PR-Set7 purified from cells previously incubated with the histone deacetylase (HDAC) inhibitors nicotinamide and TSA (N+TSA) did not result in any difference in the HMT activity (Supplemental Fig. 2E). Moreover, incubation of this “hyperacetylated” PR-Set7 with SirT2 and NAD+ did not produce any change in the PR-Set7 HMT activity (Supplemental Fig. 2E). A very important clue in determining the role of this modification came from the comparative analysis of PR-Set7 wild type with K90 point mutations to arginine (K90R; to mimic deacetylated lysine) or to glutamine (K90Q; to mimic acetylated lysine). All three forms showed similar HMT-specific activities (Supplemental Fig. 2F; data not shown). Interestingly, the immunofluorescence (IF) analysis of the nuclear levels of the PR-Set7 wild-type, K90R, and K90Q proteins and the features of the foci formed by each protein after overexpression in primary MEFs revealed that the number of K90Q foci per cell was nearly double that formed by the wild-type form, whereas K90R formed significantly fewer foci under the same conditions (Fig. 3L). Moreover, although the intensity level of K90Q in each focus was, on average, lower than that of K90R or wild type (see PR-Set7 mean foci intensity) (Fig. 3M), the total amount of K90Q retained in the foci, and therefore in the chromatin pellet, was significantly higher than that in the wild-type or K90R-overexpressing cells (Fig. 3M). In support of these results, a Western blot of the chromatin pellet from cells under the same conditions clearly showed significantly higher levels of K90Q than wild type in these fractions (Fig. 3N).
Interestingly, pull-down experiments between purified SirT2 and wild-type, K90R, or K90Q PR-Set7 showed that SirT2 binds with a stronger affinity to K90R than to wild type or K90Q (Supplemental Fig. 2G,H). PR-Set7 overexpression in HeLa cells led to a greater incorporation of endogenous or exogenous SirT2 in insoluble chromatin during mitosis, but SirT2 overexpression did not have the same effect on endogenous or exogenous PR-Set7 levels (Fig. 4A; Supplemental Fig. 3A,B). These results suggest that PR-Set7 may first recruit SirT2 to the chromatin and that SirT2 may in turn help stabilize PR-Set7 on the chromatin by deacetylating K90. Altogether, these results support a role for K90 in the regulation of PR-Set7-chromatin dynamics.
Figure 4.

SirT2 and PR-Set7 may participate in a mitotic checkpoint. (A) Experiment similar to that in Figure 3I, but in this case, determining the levels of exogenous SirT2-HA and Flag-PR-Set7 in insoluble chromatin fractions after overexpression of the indicated vectors in HeLa cells. (Lanes 3,6,9) The G2/M-blocked cells were also subjected to H2O2-induced oxidative stress. Whole-cell levels of SirT2-HA and Flag-PR-Set7 are shown in Supplemental Figure 3A. (B) Immunoprecipitation with Flag resin of extracts from HeLa cells previously transfected with Flag-SirT2 and/or PR-Set7-myc under normal conditions or H2O2-induced oxidative stress. (C) H4K20me1 levels in IF experiments from wild-type (WT) and SIRT2−/− primary fibroblasts at different stages of the cell cycle, cultured under normal conditions or under oxidative stress. The values shown represent the ratio of H4K20me1 levels in treated cells (100 μM H2O2 for 1 h) to those of untreated cells. (*) The differences between wild-type and SIRT2−/− cells in S phase and G2/M were significant (P < 0.01). (D) The cells sorted at G2/M were incubated with 100 μΜ H2O2 for 1 h. Cells were harvested either immediately after 1 h of H2O2 treatment (t0) or at 15 h post-treatment (t15), fixed, and then analyzed by FACs. DNA was stained with 7AAD. (E) FACS analysis of mitotic cells shown in D. Wild-type and SIRT2−/− MEFs treated or untreated for 1 h with 100 μM H2O2 sorted by DNA content (7AAD) and a mitotic cell marker (H3 phosphorylation). (F) Quantification of the percentage of mitotic cells in wild-type and SIRT2−/− MEFs shown in E.
SirT2 and PR-Set7 may participate in a mitotic checkpoint mechanism
We then tackled another open question: Considering the role of SirT2 in the response to oxidative and genotoxic stress (Wang et al. 2007), is the functional link between SirT2 and PR-Set7 related to stress response? We found strong evidence to suggest that this is the case. First, oxidative stress increased the levels of both PR-Set7 and SirT2 in insoluble chromatin (Fig. 4A). Second, oxidative stress generated by H2O2 induced a significantly stronger interaction between SirT2 and PR-Set7 (Fig. 4B), even though the stress led to a decrease in the PR-Set7 levels in the whole cell, as was previously reported (Supplemental Fig. 3C; Oda et al. 2010). Third, although wild-type primary fibroblasts exhibited up-regulation of H4K20me1 under conditions of stress compared with control conditions, this up-regulation was abrogated in SIRT2−/− fibroblasts (Fig. 4C). Finally, sorted wild-type cells in G2/M that were treated with H2O2 remained blocked in G2/M 15 h post-treatment. In contrast, SirT2-deficient cells were not blocked: They progressed through G1, where the induced damage resulted in higher levels of apoptosis (Fig. 4D). In a similar experiment focused exclusively on cells undergoing mitosis, oxidative stress treatment led to a significantly greater population increase of mitotic SIRT2−/− cells compared with wild-type cells (Fig. 4E,F). Interestingly, this response was conserved under other types of stress (data not shown). Consistent with a direct role of SirT2 in this response to stress, expression of ectopic SirT2 under the same conditions as in Figure 4D rescued cells from the G2/M block (Supplemental Fig. 3D,E). Altogether, these findings suggest that PR-Set7, SirT2, and H4K20me1 participate together in the mitotic stress response.
SirT2 knockout cells exhibit drastically altered levels of H4K20me2/3 during the cell cycle and show various defects previously associated with a decrease in both marks
The chromatin mark H4K20me1 is not only paramount for cell cycle progression, but also required for the di- and trimethylation of H4K20 by Suv4-20 enzymes in late M/early G1 (Schotta et al. 2004). Interestingly, the decrease in H4K20me1 that occurs in SIRT2−/− MEFs correlates to significantly lower levels of H4K20me2 and H4K20me3 (Fig. 5A–C), which corroborates a role for SirT2 in the regulation of H4K20me1 deposition. Furthermore, and in contrast to H4K20me1, the levels of H4K20me2 and H4K20me3 were also lower in all phases of the cell cycle (Fig. 5B,C). Moreover, the observations that the loss of SirT2 has a progressive effect on the conversion of H4K20me1 to H4K20me2/3 (Fig. 5C) and that SirT2 does not interact or colocalize with any of the Suv4-20h enzymes (Supplemental Fig. 4A; data not shown) support the idea that all of these events stem from the mitotic deposition of H4K20me1 by PR-Set7. The major role of SirT2 as a regulator of H4K20 methylation is also supported by strong functional data. One key observation is that MEFs defective in either SIRT2−/− or both Suv4-20h1 and Suv4-20h2 (Suv4-20DN) exhibit almost identical cell cycle distribution effects: Compared with wild-type MEFs, they have more cells in G1, fewer cells in S phase, and a slight increase in the number of cells progressing through G2/M (Supplemental Fig. 4B) (Vaquero et al. 2006; Schotta et al. 2008). As expected, SIRT2−/− cells exhibit various defects in cell cycle progression and in genome stability that have previously been described for cases of total or partial loss of H4K20me1–H4K20me3. First, SIRT2−/− cells exhibit significantly higher levels of rereplication than wild-type cells (Fig. 5D,E; Tardat et al. 2010), a phenomenon that occurs after S-phase completion, in which some cells already entering G2/M (labeled by cyclin B1) restart the replication process (as measured by EdU incorporation). Thus, not only does the SIRT2−/− cell population contain a higher level of >4n cells positive for EdU (Fig. 5D), but a significant number of these cells are positive for cyclin B1 (Fig. 5E). Second, and in agreement with the previously described accumulation of SIRT2−/− MEF populations in G1/S (Vaquero et al. 2006), these cells suffer from delayed S-phase entry but then undergo accelerated progression through S phase, as has previously been shown for Suv4-20DN (Fig. 5F; Schotta et al. 2008; Tardat et al. 2010). Although these effects are not well understood, one possible explanation is based on the proposed role of H4K20me1 in licensing of replication origins during replication (Tardat et al. 2010) as well as on the role of H4K20me1/2 in DNA repair during replication (Sanders et al. 2004; Tardat et al. 2007). Alterations of these marks may initially delay entry into S phase, but once cells have entered this phase, these alterations may promote faster progression through S phase without any supervision or repair. Third, given the critical role of H4K20me3 in the heterochromatin structure and its intimate relationship with H3K9me3 (Schotta et al. 2004), we reasoned that the decrease in the H4K20me3 levels observed in SIRT2−/− cells should produce defects in pericentromeric constitutive heterochromatin (PCH). Thus, the PCH structure of these cells is defective, as indicated by the derepression of the major heterochromatin γ-satellite and the reduced density of H3K9me3 in the heterochromatin foci (Fig. 5G,H). Finally, the observation that there is a decrease in the number of 53BP1 foci upon DNA damage during S phase, as has previously been reported for the loss of H4K20me1 and H4K20me2 (Sanders et al. 2004; Schotta et al. 2008), confirms that there is a defect in DNA repair during S phase (Fig. 5I,J). Overall, these data support a major role for SirT2 in cell cycle control—specifically in S-phase progression—through the regulation of H4K20me1 deposition during mitosis.
Figure 5.
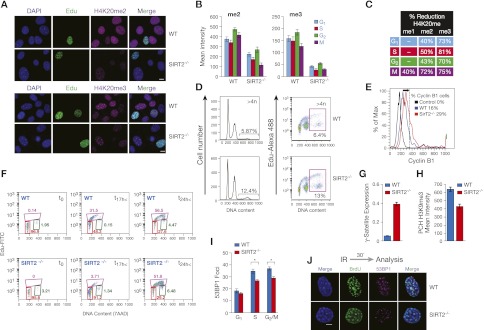
SIRT2−/− cells suffer from drastically low levels of H4K20me2,3 and share common defects with Su(var)4-20 double knockouts. (A) Levels of H4K20me2 and H4K20me3 in wild-type (WT) or SIRT2−/− MEFs determined by IF. S-phase cells were visualized by EdU staining. (B) Quantification of experiment in A through the different stages of the cell cycle. (C) Quantification (as percentage) of the decrease in H4K20me1, H4K20me2, and H4K20me3 levels in SIRT2−/− and wild-type cells at the different stages of the cell cycle. The data shown were generated in the experiment corresponding to B and Figure 2E. (D) Cell cycle profiles of wild-type and SIRT2−/− fibroblasts obtained by FACS analysis using DNA content either alone (left panel) or in combination with EdU (right panel). The values shown are the percentage of polyploid cells (DNA content >4N). (Right panels) EdU staining denotes the presence of polyploid cells with rereplicated DNA. (E) Overlay histograms showing expression of Cyclin B1 in polyploid cells from wild-type and SIRT2−/− fibroblasts. (F) Analysis of S-phase entrance of wild-type and SIRT2−/− fibroblasts. Cells were sorted at G1 and harvested at the indicated times. S-phase progression was measured by FACS as in D. (G) mRNA levels of pericentromeric γ-satellite in wild-type and SIRT2−/− MEFs analyzed by real-time PCR. (H) Average density of H3K9me3 in pericentromeric heterochromatin in wild-type and SIRT2−/− MEFs. (I) Quantitative analysis of 53BP1 focus formation at X-ray (IR)-induced DSBs in wild-type and SIRT2−/− MEFs as shown in J throughout progression of the cell cycle (n > 20 for each time point) (J) Microscopic analyses of DNA damage marked by accumulation of 53BP1. The S-phase cells were stained by EdU incorporation and counterstained with DAPI. Bar, 5 μm.
SirT2−/− mice suffer from greater DNA damage and genome instability
Overall, our data presented above suggest that the loss of SirT2 is associated with greater genome instability. Therefore, we next sought to determine whether adult fibroblasts derived from these animals exhibit significantly higher levels of DNA damage compared with cells from their wild-type littermates. Comet assays clearly showed that this was indeed the case: The SIRT2−/− cells contained high levels of double-strand breaks (DSBs) (Fig. 6A–C), which were further confirmed by measuring the DSB marker γH2AX in IF and Western blot experiments (Fig. 6D; Supplemental Fig. 5A,B). Interestingly, and contrary to what we had expected, we observed only a slightly higher number of DSBs (as measured by γH2AX) in the G1 phase (Fig. 6E; Supplemental Fig. 5C). Considering the previously mentioned possibility that global H4K16Ac deacetylation in G2/M might strongly affect the mitotic compaction of chromatin and given the H4K16 hyperacetylation that we observed in SIRT2−/− cells, we had expected to see major damage in G1. These results challenge the view, based on the in vitro studies, that H4K16Ac deacetylation may not be as important for in vivo chromatin compaction. Interestingly, the greatest DNA damage in these cells was observed in S phase (Fig. 6E; Supplemental Fig. 5C), a finding that supports the premise that the H4K20me1 and H4K20me2 levels in these cells were defective (Tardat et al. 2010). A detailed analysis showed that the damage accumulates throughout S phase (Fig. 6F; Supplemental Fig. 5D). In agreement with these observations, the SIRT2−/− mice exhibited markedly higher levels of γH2AX in their tissues (Fig. 6G; Supplemental Fig. 5E) and significantly higher rates of polyploidy in their cells (Figs. 5D, 6H). The expression of active SirT2, but not of the inactive SirT2 H150Y mutant, was able to rescue the levels of γ-H2AX, confirming a direct role of SirT2 activity in these processes (Supplemental Fig. 5F,G). Considering the results of these experiments, we reasoned that SIRT2−/− mice should contain a significant number of chromosomal aberrations. To test this hypothesis, we performed fluorescence in situ hybridization (FISH) experiments with telomeric and centromeric probes in metaphase chromosome spreads from SIRT2−/− and wild-type cells (Fig. 6I). The analysis of these spreads clearly indicated a greater frequency of defects associated with alterations in constitutive heterochromatin structure (summarized in Fig. 6J), which supported our evidence for the drastic alteration of H4K20me1, H4K20me2, and H4K20me3 in these cells. Quantification of these aberrations demonstrated that SIRT2−/− animals clearly have higher levels of genomic instability than their wild-type counterparts.
Figure 6.
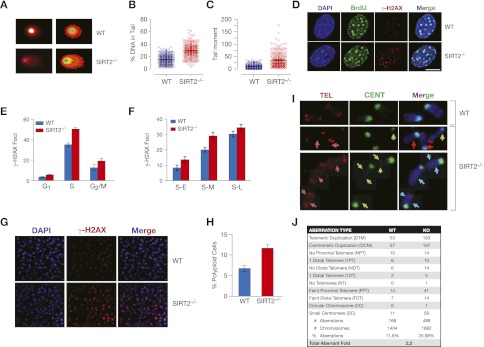
SIRT2−/− mice exhibit major DNA damage and genomic instability. (A) DNA damage in wild-type (WT) and SIRT2−/− primary fibroblasts, as measured by neutral comet assay. (B,C) Quantification of experiments shown in A. The data shown are the percentages of DNA present in the comet tail (B) or the tail moment quantification (C). (D) IF experiments on wild-type and SIRT2−/− MEFs of BrdU incorporation. DAPI staining of cells is shown as a control for DNA. (E) Quantitative analysis of γH2AX levels of wild-type and SIRT2−/− MEFs throughout progression of the cell cycle (n > 20 for each time point). (F) Quantitative analysis as in E, but focused on progression through S phase. (S-E) Early S phase; (S-M) middle S phase; (S-L) late S phase. (G) γH2AX levels in liver cryosections derived from wild-type or SIRT2−/− mice, detected by immunohistochemistry. (H) Quantification of polyploidy levels from the FACS profiles shown in Figure 5D in wild-type and SIRT2−/− fibroblasts. (I) IF of metaphase chromosome spreads from wild-type and SIRT2−/− primary fibroblasts stained with FISH chromosome paints for centromeres (CEN) and telomeres (TEL) and counterstained with DAPI. The arrows indicate chromosomal aberrations relative to lost or duplicated centromeres and telomeres. (J) Quantification of chromosomal aberrations observed in I.
SirT2−/− animals are more prone to develop tumors in an induced skin tumorigenesis assay
The above-described data support the previously formulated hypothesis that SirT2 is a tumor suppressor (Bosch-Presegue and Vaquero 2011; Kim et al. 2011). Given that SIRT2−/− animals suffer from such a high degree of genomic damage, they would be expected to be more prone than wild-type animals to develop tumors. We tested this hypothesis by evaluating our SIRT2−/− mice in a DMBA/TPA-induced skin tumorigenesis assay (Blanco et al. 2007). For this assay, we treated a portion of the skin with the mutagenic agent DMBA once and then treated the same site with the proliferation-inducing agent TPA twice weekly for 15 wk (Fig. 7A). During and after the treatment period, we characterized the formation of papillomas and determined whether any of the animals developed tumors. Our analysis showed that starting at the eighth week of treatment, the SIRT2−/− mice developed larger papillomas at higher frequencies (Fig. 7B–D; Supplemental Fig. 6). Strikingly, 35 wk after the end of the 15-wk treatment, 50% of the SIRT2−/− mice had died, whereas 100% of the wild-type mice were still alive (Fig. 7E). The histopathological study of these animals confirmed that most of them had developed malignant tumors, including different locally aggressive and metastatic forms of squamous cell carcinoma and fibrosarcoma (Fig. 7F; Supplemental Fig. 6). Therefore, our studies confirmed that the SirT2 loss is associated with a greater risk of tumorigenesis, which corroborates a role for SirT2 in cancer and pinpoints it as a bona fide tumor suppressor (Bosch-Presegue and Vaquero 2011). Interestingly, and in contrast to a recent report on another SIRT2 knockout mouse model (Kim et al. 2011), these SIRT2−/− mice did not spontaneously develop tumors within their first year. The difference may lie in the strains: Whereas the earlier study used a mixed-lineage strain, the knockout mice described here were C57-BL6, a strain well known for its high resistance to tumor development.
Figure 7.
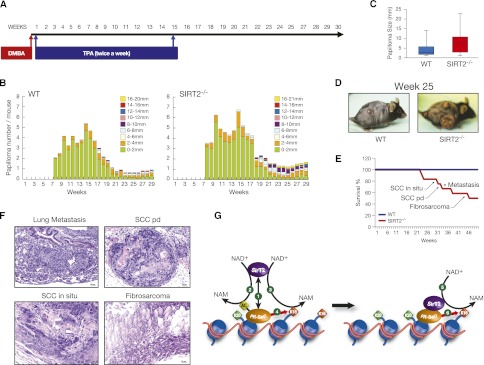
SIRT2−/− mice are more prone to tumorigenesis. (A) Schematic representation of the DMBA/TPA-induced skin tumorigenesis assay. (B) Average number of papillomas per mouse at the indicated number of weeks after starting the DMBA/TPA treatment. The treatment was interrupted at week 15. (C) Papilloma size in wild-type (WT) versus SIRT2−/− mice at week 25 after starting the treatment. (D) Representative wild-type and SIRT2−/− animals in week 25. (E) Survival curve of DMBA/TPA-treated wild-type and SIRT2−/− mice. (F) Representative examples of the neoplasias and metastases indicated in E developed by SIRT2−/− mice. (Top left image) Lung metastasis of a poorly differentiated skin squamous cell carcinoma. (Top right) Poorly differentiated skin squamous cell carcinoma (SCC pd) with pleomorphism and numerous mitotic cells, clearly growing invasively into the dermis, subcutaneous tissue, and subcutaneous muscle. (Bottom left) In situ skin squamous cell carcinoma (SCC in situ) with focal basal membrane disruption. (Bottom right) Cutaneous fibrosarcoma with pleomorphic spindle cells that invade skeletal muscle. (G) Proposed model of the regulation of H4K20me1 deposition by SirT2 based in our results. (1) PR-Set7 recruits SirT2 to specific chromatin regions. SirT2 then deacetylates both PRSet7 (2) and H4K16Ac (3) from neighboring nucleosomes. PR-Set7 deacetylation in K90 induces its mobilization. (4) Then, SirT2-bound PR-Set7 monomethylates H4K20 in the neighbor nucleosome. This is followed by translocation to the following nucleosome, where deacetylation of H4K16Ac (5) and H4K20me1 (6) take place again. Overall, we propose that SirT2 regulates the activation and spreading of PR-Set7 in G2/M.
Overall, our results support a role for SirT2 as a key maintainer of genome stability and as a tumor suppressor.
Discussion
Evidence accumulated during the last decade has challenged the classical view of mitosis as a cell cycle phase focused exclusively on chromatid segregation and nuclear division. Other critical events that occur during mitosis apparently determine events in the subsequent cell cycles. These processes include the establishment of the epigenetic mark H4K20me1, a cornerstone of cell cycle progression and genomic stability. Here, we characterized an unexpected role for SirT2 in the deposition of this mark during early mitosis and the establishment of a previously unknown functional relationship with PR-Set7. Our findings include the first report of an acetylation/deacetylation mechanism by which SirT2 modulates the activity of PR-Set7. We also linked PR-Set7 and H4K20me1 to a SirT2-dependent mitotic checkpoint, suggesting an active role of PR-Set7 in these processes—one that is far more complex than previously anticipated. Furthermore, we presented the first report of a crucial role for Sirtuins in cell cycle control via the modulation of epigenetic information; our findings suggest that each cell cycle undergoes constant and dynamic modulation to protect genome integrity in the subsequent cell cycle. These findings offer new insight into the way that basic cellular mechanisms, such as cell cycle progression, are modulated through constant cross-talk between a cell's environment and epigenetic regulatory memory.
A functional relationship involving SIRT2, H4K16Ac, and PR-Set7
Among the most interesting aspects of this work is the elucidation of a relationship between SirT2 and PR-Set7. Interestingly, both enzymes are phosphorylated around the G2/M transition by CDK1/cyclin B and dephosphorylated around early anaphase by Cdc14a,b phosphatases (North and Verdin 2007b; Wu et al. 2010). As previously described for SirT1 and Suv39h1, SirT2 promotes PR-Set7 activity through a combination of mechanisms, including the deacetylation of H4K16Ac and binding to PR-Set7, which appears to increase the specific activity of the enzyme. However, SirT2 appears to have an even more complex role, as it apparently alters the levels of PR-Set7 on chromatin. In this sense, the deacetylation of PR-Set7 at K90 by SirT2 appears to be important for allowing the proper dynamic binding of the methyltransferase to the chromatin, which most likely also determines the focal architecture. The PR-Set7 point mutant K90Q, which mimics K90Ac, forms more foci that contain a lower density of protein compared with the wild-type or K90R proteins but binds to SirT2 with less affinity than K90R. These observations suggest that K90 acetylation may be required in the early stages of PR-Set7 arrival to the chromatin, whereas the SirT2-mediated deacetylation of K90 may help mobilize PR-Set7, allowing the spread of H4K20me1.
Based on our findings, we therefore hypothesize that during mitosis, SirT2 regulates H4K20me1 deposition via PR-Set7 and propose a model for H4K20me1 spreading. PR-Set7 initially arrives at the chromatin during late G2, likely in an acetylated state at K90, and recruits SirT2 during G2/M. In turn, SirT2 promotes the enzymatic activity of PR-Set7 through the aforementioned mechanism (Fig. 7G) and deacetylates K90. The effects of H4K16Ac deacetylation by SirT2 on neighboring nucleosomes then enables the spread of PR-Set7 binding on chromatin and the subsequent methylation of these nucleosomes. Future studies should determine the validity of this model. Of particular importance is the identification of the regions in H4K20 that are monomethylated by PR-Set7 and those acetylated at H4K16 in wild-type and SIRT2−/− cells during early mitosis.
H4K20 methylation defects and genomic instability
Although the loss of SirT2 also correlates with a global decrease in H4K20me2/3 and the cell cycle distribution of Suv4-20DN MEFs is strikingly similar to that of SIRT2−/− MEFs (Supplemental Fig. 4A; Vaquero et al. 2006; Schotta et al. 2008), our findings support the idea that all of these effects are produced by a decrease in PR-Set7 function. Not only are the partial losses of me1–me3 progressive (see the table in Fig. 5C), but the loss of SirT2 does not associate with any accumulation of H4K20me1, in contrast to what has been shown for Suv4-20DN (Schotta et al. 2008). Instead, this profile fits perfectly with previous reports that PR-Set7 activity leads to the depletion of H4K20me1 and reduced levels of H4K20me2/3 (Oda et al. 2009).
Interestingly, other investigators have previously claimed that SirT2 is involved in the modulation of APCCdh1 activity through the deacetylation of Cdh1 (Kim et al. 2011), an event that might be involved in genomic stability. However, the observations in our mouse model suggest that this activity may not be that functionally relevant and that it cannot be the main cause of genomic instability. Thus, considering that APCcdh1 has been shown to regulate PR-Set7 levels during late mitosis, we would expect that the total levels of PR-Set7 would be altered in SIRT2−/− cells. However, we did not observe any changes in our cells (Supplemental Fig. 2D). Moreover, if the increased DNA damage observed in the SirT2-deficient cells was the result of mitotic defects in general, then we would not expect the bulk of the damage to accumulate in S phase, which is what we observed.
Consistent with a key role of SirT2 in H4K20 methylation in vivo, SIRT2−/− mouse cells exhibit myriad specific phenotypes that have been assigned to defects in the mono-, di-, or trimethylation of H4K20. Therefore, our findings strongly suggest that the main driver of the genomic instability detected in these mice is a global defect in H4K20 methylation—a link that has been well established by multiple reports (Jorgensen et al. 2007; Tardat et al. 2007; Houston et al. 2008; Schotta et al. 2008; Oda et al. 2009). However, although PR-Set7 knockout causes death in the embryonic stage in mice (Oda et al. 2009) and in the third-instar larval stage in Drosophila (Karachentsev et al. 2005), we never observed developmental defects in the SIRT2−/− mice, possibly because SirT2 does not affect PR-Set7 activity during development or because of the existence of some redundant activity during this process. Alternatively, the fact that SirT2 loss leads to drastically lower levels of H4K20me1 but does not eliminate it completely suggests that the reduced levels of H4K20me1 are sufficient to progress through development. Therefore, additional experiments are required to clarify this process.
SirT2, PR-Set7, and a mitotic checkpoint
Our studies have shown that SirT2 and PR-Set7 may function together in a mitotic checkpoint mechanism and suggest a previously unknown role for H4K20me1 in mitosis. In fact, the involvement of SirT2 in a G2/M mitotic checkpoint has previously been proposed (Inoue et al. 2007) as a response to chronic stress produced by microtubule inhibitors. Our evidence suggests that under stress, SirT2 strongly binds to PR-Set7 and that this binding is associated with the enrichment of both proteins in insoluble chromatin, which correlates with the blocking of mitosis and an accumulation of H4K20me1. This scenario is supported by the observation that under stress, SIRT2−/− cells do not experience either blocked G2/M or an increase in H4K20 methylation. A role for H4K16Ac in mitotic entry and control is corroborated by the finding that the depletion of MOF in mouse cells leads to an accumulation of cells in G2/M (Smith et al. 2005; Taipale et al. 2005). However, many questions remain to be addressed in future works, such as: What is the exact role of PR-Set7? Is the H4K20me1 peak part of the mitotic checkpoint or simply a consequence of PR-Set7 activation under stress?
The interplay between H4K16Ac and H4K20me1 in mitosis
Another interesting point is the extent of the antagonism between H4K16Ac and H4K20me1. Although the inhibitory effect of H4K16Ac on PR-Set7 was originally proposed in the context of peptides as substrates and of a PR-Set7 mutant capable of targeting nonnucleosomal substrates (Nishioka et al. 2002b), here we demonstrated that this inhibition is also valid in the context of the wild-type enzyme and nucleosomes (Fig. 3A,B). This interplay is supported by the finding that H4K16Ac inhibits not only the specific activity of PR-Set7, but also the binding of specific factors to these marks, such as 53BP1, a binding factor specific for H4K20me1/2 (Supplemental Fig. 7). Although researchers originally proposed that the presence of both modifications in the same H4 N-terminal tail alone could represent a steric problem, subsequent studies have demonstrated that this is not the case and that H4K16Ac actually coexists with all three levels of H4K20 methylation (Fraga et al. 2005; Pesavento et al. 2008). This coexistence suggests that the antagonism may be unidirectional and that H4K20 methylation might not efficiently inhibit H4K16 acetylation. Given the general role of H4K16Ac in regulating chromatin structure and its association with active transcription, H4K20me1 genome-wide distribution might be expected to be restricted to inactive regions. However, the analysis of nine different human cell lines from different tissues and/or differentiation stages by chromatin immunoprecipitation (ChIP) sequencing has revealed that H4K20me1 is actually present in highly expressed genes in all cell types (Beck et al. 2012)—a finding that argues against a general global interplay between the two modifications. In contrast, our observations do support the existence of an antagonistic effect between these two modifications that is restricted to early mitosis, as the loss of SirT2 correlates with an inversion in the levels of H4K16Ac and H4K20me1 (Fig. 2B,C). Determining the precise conditions and limits for this antagonism should be an exciting and fruitful research focus of future epigenetic studies.
Materials and methods
IF, immunohistochemistry, and comet assays
The IF and immunohistochemistry experiments were performed as described elsewhere (Vaquero et al. 2006). The images were acquired using a Zeiss LSM510 Meta confocal laser-scanning microscope. Three-dimensional reconstructions and image analysis were conducted using Imaris software (Bitplane, A.G.). FISH on metaphase chromosome spreads was performed according to the StarFISH Protocol N Pan Telomeric and Centromeric Chromosome Paints (Cambio 1696). To detect DSBs, the neutral comet assay was performed as described previously (Olive and Banath 2006). The images were acquired using a Leica AF-5000 confocal microscope and analyzed and quantified using the Comet Score program.
Chromatin fractionation
HeLa cells were transfected with SirT2-HA, Flag-PR-Set7, or both and then synchronized using a double-thymidine block as described in the Supplemental Material. The cells were then harvested at different times after G1/S release. The cell cycle stage was determined using FACS analysis. For the chromatin fractionation, after the Dignam protocol extraction, the chromatin pellet was washed once with BC-500 and then dissolved in Laemmli buffer by sonication in a water bath.
Tumor induction experiments and histopathological analysis
Fourteen age-matched (8- to 12-wk-old) mice of each genotype—SIRT2−/− and wild type, respectively—were treated with DMBA and TPA according to Blanco et al. (2007). The number and size of papillomas per mouse were recorded weekly. Skin samples and other mouse tissues were recovered after sacrifice and embedded in paraffin according to standard protocols. For histopathological study, hematoxylin–eosin stain was used.
Acknowledgments
We are grateful to Dr. Fred Alt and Hwei-Ling Cheng (Harvard Medical School) for providing the original mice and embyronic stem cells. We thank Dr. Danny Reinberg (Howard Hughes Medical Institute-New York University) for help and support. We also thank Dr. Peggy L. Olive (BC Cancer Agency Research Centre, Vancouver) for help with comet assay protocols, Dr. Alan Underhill (University of Alberta, Canada) for reagents, Theresa Choiat (Environmental and Occupational Health Sciences Institute/Cancer Institute of New Jersey/Rutgers University) for her expert assistance with FACS , and Josh Trackray for help with the rescue experiment. Thanks to the members of the Vaquero laboratory and to Dr. Dave Monk for stimulating discussions. This work was supported by Spanish Ministry of Science and Innovation grants SAF2008-00923 (to A.V.) and SAF2011-25860 (to A.V.), the Catalan Government Agency AGAUR 2009-SGR-914 (to A.V.), and a grant from the Human Genetics Institute of New Jersey (to L.S. and J.A.T.). Grants GM064844 and R37GM037120 from the National Institute of Health partially supported these studies (P.V. and D.B.B.). B.N.V. was supported by post-doctoral fellowship EX-2010-278 from the Spanish Ministry of Education, Culture, and Sports. P.V. was supported by post-doctoral fellowships from the Deutsche Akademie der Naturforscher Leopoldina (LPDS 2009-5) and the Empire State Training Program in Stem Cell Research (NYSTEM, contract no. C026880).
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.211342.112.
References
- Bae NS, Swanson MJ, Vassilev A, Howard BH 2004. Human histone deacetylase SIRT2 interacts with the homeobox transcription factor HOXA10. J Biochem 135: 695–700 [DOI] [PubMed] [Google Scholar]
- Beck DB, Oda H, Shen SS, Reinberg D 2012. PR-Set7 and H4K20me1: At the crossroads of genome integrity, cell cycle, chromosome condensation, and transcription. Genes Dev 26: 325–337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell O, Wirbelauer C, Hild M, Scharf AN, Schwaiger M, MacAlpine DM, Zilbermann F, van Leeuwen F, Bell SP, Imhof A, et al. 2007. Localized H3K36 methylation states define histone H4K16 acetylation during transcriptional elongation in Drosophila. EMBO J 26: 4974–4984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco R, Munoz P, Flores JM, Klatt P, Blasco MA 2007. Telomerase abrogation dramatically accelerates TRF2-induced epithelial carcinogenesis. Genes Dev 21: 206–220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borra MT, O'Neill FJ, Jackson MD, Marshall B, Verdin E, Foltz KR, Denu JM 2002. Conserved enzymatic production and biological effect of O-acetyl-ADP-ribose by silent information regulator 2-like NAD+-dependent deacetylases. J Biol Chem 277: 12632–12641 [DOI] [PubMed] [Google Scholar]
- Bosch-Presegue L, Vaquero A 2011. The dual role of sirtuins in cancer. Genes Cancer 2: 648–662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cesarini E, D'Alfonso A, Camilloni G 2012. H4K16 acetylation affects recombination and ncRNA transcription at rDNA in Saccharomyces cerevisiae. Mol Biol Cell 23: 2770–2781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalkiadaki A, Guarente L 2012. Sirtuins mediate mammalian metabolic responses to nutrient availability. Nat Rev Endocrinol 8: 287–296 [DOI] [PubMed] [Google Scholar]
- Dang W, Steffen KK, Perry R, Dorsey JA, Johnson FB, Shilatifard A, Kaeberlein M, Kennedy BK, Berger SL 2009. Histone H4 lysine 16 acetylation regulates cellular lifespan. Nature 459: 802–807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dou Y, Milne TA, Tackett AJ, Smith ER, Fukuda A, Wysocka J, Allis CD, Chait BT, Hess JL, Roeder RG 2005. Physical association and coordinate function of the H3 K4 methyltransferase MLL1 and the H4 K16 acetyltransferase MOF. Cell 121: 873–885 [DOI] [PubMed] [Google Scholar]
- Dryden SC, Nahhas FA, Nowak JE, Goustin AS, Tainsky MA 2003. Role for human SIRT2 NAD-dependent deacetylase activity in control of mitotic exit in the cell cycle. Mol Cell Biol 23: 3173–3185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkel T, Deng CX, Mostoslavsky R 2009. Recent progress in the biology and physiology of sirtuins. Nature 460: 587–591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraga MF, Ballestar E, Villar-Garea A, Boix-Chornet M, Espada J, Schotta G, Bonaldi T, Haydon C, Ropero S, Petrie K, et al. 2005. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat Genet 37: 391–400 [DOI] [PubMed] [Google Scholar]
- Houston SI, McManus KJ, Adams MM, Sims JK, Carpenter PB, Hendzel MJ, Rice JC 2008. Catalytic function of the PR-Set7 histone H4 lysine 20 monomethyltransferase is essential for mitotic entry and genomic stability. J Biol Chem 283: 19478–19488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai S, Armstrong CM, Kaeberlein M, Guarente L 2000. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature 403: 795–800 [DOI] [PubMed] [Google Scholar]
- Inoue T, Hiratsuka M, Osaki M, Yamada H, Kishimoto I, Yamaguchi S, Nakano S, Katoh M, Ito H, Oshimura M 2007. SIRT2, a tubulin deacetylase, acts to block the entry to chromosome condensation in response to mitotic stress. Oncogene 26: 945–957 [DOI] [PubMed] [Google Scholar]
- Jazayeri A, McAinsh AD, Jackson SP 2004. Saccharomyces cerevisiae Sin3p facilitates DNA double-strand break repair. Proc Natl Acad Sci 101: 1644–1649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang W, Wang S, Xiao M, Lin Y, Zhou L, Lei Q, Xiong Y, Guan KL, Zhao S 2011. Acetylation regulates gluconeogenesis by promoting PEPCK1 degradation via recruiting the UBR5 ubiquitin ligase. Mol Cell 43: 33–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jing E, Gesta S, Kahn CR 2007. SIRT2 regulates adipocyte differentiation through FoxO1 acetylation/deacetylation. Cell Metab 6: 105–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen S, Elvers I, Trelle MB, Menzel T, Eskildsen M, Jensen ON, Helleday T, Helin K, Sorensen CS 2007. The histone methyltransferase SET8 is required for S-phase progression. J Cell Biol 179: 1337–1345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karachentsev D, Sarma K, Reinberg D, Steward R 2005. PR-Set7-dependent methylation of histone H4 Lys 20 functions in repression of gene expression and is essential for mitosis. Genes Dev 19: 431–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HS, Vassilopoulos A, Wang RH, Lahusen T, Xiao Z, Xu X, Li C, Veenstra TD, Li B, Yu H, et al. 2011. SIRT2 maintains genome integrity and suppresses tumorigenesis through regulating APC/C activity. Cancer Cell 20: 487–499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landry J, Sutton A, Tafrov ST, Heller RC, Stebbins J, Pillus L, Sternglanz R 2000. The silencing protein SIR2 and its homologs are NAD-dependent protein deacetylases. Proc Natl Acad Sci 97: 5807–5811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Zhang B, Tang J, Cao Q, Wu Y, Wu C, Guo J, Ling EA, Liang F 2007. Sirtuin 2, a mammalian homolog of yeast silent information regulator-2 longevity regulator, is an oligodendroglial protein that decelerates cell differentiation through deacetylating α-tubulin. J Neurosci 27: 2606–2616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Matsumori H, Nakayama Y, Osaki M, Kojima H, Kurimasa A, Ito H, Mori S, Katoh M, Oshimura M, et al. 2011. SIRT2 down-regulation in HeLa can induce p53 accumulation via p38 MAPK activation-dependent p300 decrease, eventually leading to apoptosis. Genes Cells 16: 34–45 [DOI] [PubMed] [Google Scholar]
- Nishioka K, Chuikov S, Sarma K, Erdjument-Bromage H, Allis CD, Tempst P, Reinberg D 2002a. Set9, a novel histone H3 methyltransferase that facilitates transcription by precluding histone tail modifications required for heterochromatin formation. Genes Dev 16: 479–489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishioka K, Rice JC, Sarma K, Erdjument-Bromage H, Werner J, Wang Y, Chuikov S, Valenzuela P, Tempst P, Steward R, et al. 2002b. PR-Set7 is a nucleosome-specific methyltransferase that modifies lysine 20 of histone H4 and is associated with silent chromatin. Mol Cell 9: 1201–1213 [DOI] [PubMed] [Google Scholar]
- North BJ, Verdin E 2007a. Interphase nucleo–cytoplasmic shuttling and localization of SIRT2 during mitosis. PLoS ONE 2: e784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- North BJ, Verdin E 2007b. Mitotic regulation of SIRT2 by cyclin-dependent kinase 1-dependent phosphorylation. J Biol Chem 282: 19546–19555 [DOI] [PubMed] [Google Scholar]
- North BJ, Marshall BL, Borra MT, Denu JM, Verdin E 2003. The human Sir2 ortholog, SIRT2, is an NAD+-dependent tubulin deacetylase. Mol Cell 11: 437–444 [DOI] [PubMed] [Google Scholar]
- Oda H, Okamoto I, Murphy N, Chu J, Price SM, Shen MM, Torres-Padilla ME, Heard E, Reinberg D 2009. Monomethylation of histone H4-lysine 20 is involved in chromosome structure and stability and is essential for mouse development. Mol Cell Biol 29: 2278–2295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oda H, Hubner MR, Beck DB, Vermeulen M, Hurwitz J, Spector DL, Reinberg D 2010. Regulation of the histone H4 monomethylase PR-Set7 by CRL4(Cdt2)-mediated PCNA-dependent degradation during DNA damage. Mol Cell 40: 364–376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olive PL, Banath JP 2006. The comet assay: A method to measure DNA damage in individual cells. Nat Protoc 1: 23–29 [DOI] [PubMed] [Google Scholar]
- Outeiro TF, Kontopoulos E, Altmann SM, Kufareva I, Strathearn KE, Amore AM, Volk CB, Maxwell MM, Rochet JC, McLean PJ, et al. 2007. Sirtuin 2 inhibitors rescue α-synuclein-mediated toxicity in models of Parkinson's disease. Science 317: 516–519 [DOI] [PubMed] [Google Scholar]
- Pandithage R, Lilischkis R, Harting K, Wolf A, Jedamzik B, Luscher-Firzlaff J, Vervoorts J, Lasonder E, Kremmer E, Knoll B, et al. 2008. The regulation of SIRT2 function by cyclin-dependent kinases affects cell motility. J Cell Biol 180: 915–929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pesavento JJ, Yang H, Kelleher NL, Mizzen CA 2008. Certain and progressive methylation of histone H4 at lysine 20 during the cell cycle. Mol Cell Biol 28: 468–486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson PJ, An W, Routh A, Martino F, Chapman L, Roeder RG, Rhodes D 2008. 30 nm chromatin fibre decompaction requires both H4-K16 acetylation and linker histone eviction. J Mol Biol 381: 816–825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders SL, Portoso M, Mata J, Bahler J, Allshire RC, Kouzarides T 2004. Methylation of histone H4 lysine 20 controls recruitment of Crb2 to sites of DNA damage. Cell 119: 603–614 [DOI] [PubMed] [Google Scholar]
- Saunders LR, Verdin E 2007. Sirtuins: Critical regulators at the crossroads between cancer and aging. Oncogene 26: 5489–5504 [DOI] [PubMed] [Google Scholar]
- Schotta G, Lachner M, Sarma K, Ebert A, Sengupta R, Reuter G, Reinberg D, Jenuwein T 2004. A silencing pathway to induce H3-K9 and H4-K20 trimethylation at constitutive heterochromatin. Genes Dev 18: 1251–1262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schotta G, Sengupta R, Kubicek S, Malin S, Kauer M, Callen E, Celeste A, Pagani M, Opravil S, De La Rosa-Velazquez IA, et al. 2008. A chromatin-wide transition to H4K20 monomethylation impairs genome integrity and programmed DNA rearrangements in the mouse. Genes Dev 22: 2048–2061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shia WJ, Pattenden SG, Workman JL 2006. Histone H4 lysine 16 acetylation breaks the genome's silence. Genome Biol 7: 217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shogren-Knaak M, Ishii H, Sun JM, Pazin MJ, Davie JR, Peterson CL 2006. Histone H4-K16 acetylation controls chromatin structure and protein interactions. Science 311: 844–847 [DOI] [PubMed] [Google Scholar]
- Smith JS, Brachmann CB, Celic I, Kenna MA, Muhammad S, Starai VJ, Avalos JL, Escalante-Semerena JC, Grubmeyer C, Wolberger C, et al. 2000. A phylogenetically conserved NAD+-dependent protein deacetylase activity in the Sir2 protein family. Proc Natl Acad Sci 97: 6658–6663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith ER, Cayrou C, Huang R, Lane WS, Cote J, Lucchesi JC 2005. A human protein complex homologous to the Drosophila MSL complex is responsible for the majority of histone H4 acetylation at lysine 16. Mol Cell Biol 25: 9175–9188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taipale M, Rea S, Richter K, Vilar A, Lichter P, Imhof A, Akhtar A 2005. hMOF histone acetyltransferase is required for histone H4 lysine 16 acetylation in mammalian cells. Mol Cell Biol 25: 6798–6810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanny JC, Dowd GJ, Huang J, Hilz H, Moazed D 1999. An enzymatic activity in the yeast Sir2 protein that is essential for gene silencing. Cell 99: 735–745 [DOI] [PubMed] [Google Scholar]
- Tardat M, Murr R, Herceg Z, Sardet C, Julien E 2007. PR-Set7-dependent lysine methylation ensures genome replication and stability through S phase. J Cell Biol 179: 1413–1426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tardat M, Brustel J, Kirsh O, Lefevbre C, Callanan M, Sardet C, Julien E 2010. The histone H4 Lys 20 methyltransferase PR-Set7 regulates replication origins in mammalian cells. Nat Cell Biol 12: 1086–1093 [DOI] [PubMed] [Google Scholar]
- Vaquero A 2009. The conserved role of sirtuins in chromatin regulation. Int J Dev Biol 53: 303–322 [DOI] [PubMed] [Google Scholar]
- Vaquero A, Scher M, Lee D, Erdjument-Bromage H, Tempst P, Reinberg D 2004. Human SirT1 interacts with histone H1 and promotes formation of facultative heterochromatin. Mol Cell 16: 93–105 [DOI] [PubMed] [Google Scholar]
- Vaquero A, Scher MB, Lee DH, Sutton A, Cheng HL, Alt FW, Serrano L, Sternglanz R, Reinberg D 2006. SirT2 is a histone deacetylase with preference for histone H4 Lys 16 during mitosis. Genes Dev 20: 1256–1261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaquero A, Sternglanz R, Reinberg D 2007. NAD+-dependent deacetylation of H4 lysine 16 by class III HDACs. Oncogene 26: 5505–5520 [DOI] [PubMed] [Google Scholar]
- Wilson JM, Le VQ, Zimmerman C, Marmorstein R, Pillus L 2006. Nuclear export modulates the cytoplasmic Sir2 homologue Hst2. EMBO Rep 7: 1247–1251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Nguyen M, Qin FX, Tong Q 2007. SIRT2 deacetylates FOXO3a in response to oxidative stress and caloric restriction. Aging Cell 6: 505–514 [DOI] [PubMed] [Google Scholar]
- Wu S, Wang W, Kong X, Congdon LM, Yokomori K, Kirschner MW, Rice JC 2010. Dynamic regulation of the PR-Set7 histone methyltransferase is required for normal cell cycle progression. Genes Dev 24: 2531–2542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zippo A, Serafini R, Rocchigiani M, Pennacchini S, Krepelova A, Oliviero S 2009. Histone crosstalk between H3S10ph and H4K16ac generates a histone code that mediates transcription elongation. Cell 138: 1122–1136 [DOI] [PubMed] [Google Scholar]