

Abstract
Background
NR5A1 loss-of-function mutations are increasingly found to be the cause of 46,XY disorders of sex development (DSD).
Objective
To determine the presence of NR5A1 mutations in an Australasian cohort of 17 46,XY DSD patients with presumed androgen insensitivity syndrome (AIS) who were negative for androgen receptor gene (AR) mutation.
Design
Exons 2-7 of NR5A1 were PCR amplified and sequenced. Gene expression and cellular localization studies were performed on a novel NR5A1 variant c.74A>G (p.Y25C) identified in this study.
Results
We identified one novel mutation, c.74A>G (p.Y25C) in a patient characterized by penoscrotal hypospadias with bifid scrotum. He had elevated testosterone and gonadotropins in early infancy. Functional analysis of p.Y25C in vitro demonstrated reduced transcriptional activation by SF-1 and partially impaired nuclear localization in a proportion of transfected human adrenal NCI-H295R cells.
Conclusion
This is the first reported case of a DSD patient with a NR5A1 mutation and elevated testosterone levels. Our finding supports evaluation of NR5A1 mutations in 46,XY DSD patients with a range of testosterone levels.
Introduction
Disorders of sex development (DSD) encompass all the congenital conditions in which development of chromosomal, gonadal or anatomic sex is atypical. One in 4500 births requires genetic and endocrine studies because of abnormalities of their external genitalia. Definitive diagnosis is made in only 50% of 46,XY children with DSD.1
Recently, several investigators have reported heterozygous loss-of-function mutations in the nuclear receptor subfamily five group A member 1 gene (NR5A1) in patients with clinical features of androgen insensitivity syndrome (AIS) but without androgen receptor gene (AR) mutations.2 Mutations in AR, which is located on chromosome Xq11-q12, is responsible for AIS. The detection rate for AR mutations in Complete AIS (CAIS) range from 66·7% to 83%, whereas for Partial AIS (PAIS) patients, the detection rate for AR mutations range from 13·6% to 28%.3,4 AIS is characterized by a clinical spectrum ranging from phenotypically female patients (CAIS) to decreased virilization (PAIS) in 46,XY individuals with normal or elevated androgen levels.5
The NR5A1 gene is mapped to 9q33 and consists of seven exons spanning approximately 30 kb. Exon 1 is untranslated. It encodes Steroidogenic Factor-1 (SF-1) also known as Adrenal 4-Binding Protein (Ad4BP). SF-1 is a 461 amino acid protein belonging to the NR5A subfamily of nuclear receptors. It is a transcription factor that binds to specific DNA sequences in target genes and regulates transcription.6 The target genes are expressed throughout the hypothalamic-pituitary–adrenal/gonadal axis and include the steroid hydroxylase genes, luteinizing hormone receptor, adrenocorticotrophin receptor, StAR protein and SOX9.6,7 Thus, SF-1 has a central role in regulating adrenal development, gonadal determination and differentiation and in the hypothalamic-pituitary control of reproduction and metabolism.6
In XY mice with homozygous NR5A1 deletions, there is impaired adrenal development, complete testicular dysgenesis with Müllerian structures and female external genitalia. Two human patients with these clinical features and NR5A1 mutations have been reported.8,9 Varying degrees of functional impairment in SF-1 can be associated with a wide range of reproductive phenotypes without adrenal involvement, extending from ambiguous genitalia, hypospadias to male infertility.2,10–13
We evaluated the presence of NR5A1 mutations in an Australasian cohort of 17 46,XY DSD patients with presumed AIS who were negative for AR mutations.
Materials and methods
Patients
NR5A1 testing was performed in a cohort of 17 patients who had presumed androgen insensitivity syndrome but who were negative for AR mutations. Six of these patients have been previously reported.3 Of the 17 patients, 15 were diagnosed with PAIS and two with CAIS.
Molecular analysis of NR5A1
Molecular testing was performed at the National Association of Testing Authorities (NATA) accredited Molecular Laboratory, Mater Pathology, Mater Health Services, Brisbane, QLD.
Oligonucleotide primers were designed with Primer 3 software (http://frodo.wi.mit.edu/primer3/primer3_code.html) and checked for the presence of SNPs using the SNPCheck2 program (https://ngrl.manchester.ac.uk/SNPCheckV2/snpcheck.htm). Primer sequences are available on request.
Genomic DNA was extracted from peripheral blood leucocytes, and exons 2-7 of the NR5A1 gene were amplified using a GC-Rich PCR system (Roche/Boehringer Mannheim, Mannheim, Germany). PCR products were then purified with the High Pure PCR Product Purification Kit (Roche/Boehringer Mannheim) and sequenced using the Big Dye Terminator V3·1 Cycle Sequencing Kit (Applied Biosystems, Scoresby, vic., Australia). Sequences were read on an ABI 3130xl Genetic Analyser (Applied Biosystems) at the Griffith University Sequencing Facility (Griffith University, Nathan, QLD, Australia). All sequences were compared with consensus sequences for NR5A1 genomic and mRNA sequences. DNA mutation numbering is based on GenBank reference DNA sequence NM_004959·4, with the A of the ATG initiation codon designated +1 (http://www.hgvs.org/mutnomen).
Transient gene expression assays
An expression vector containing the p.Y25C change was generated by site-directed mutagenesis (QuikChange, Stratagene, Amsterdam, the Netherlands) using wild-type (WT) pCMXSF-1 as a template. Transient transfection studies were performed in 96-well plates using tsa201 human embryonic kidney cells. These cells do not express SF-1 and have been used widely in studies of SF-1 function in the past.9,11,12 Empty (−), WT or mutant SF-1 expression vectors (2 ng/well; p.G35E, p.Y25C) were co-transfected with the SF-1-responsive minimal promoter of Cyp11a linked to luciferase (100 ng/well) using lipofectamine 2000 (Invitrogen, Paisley, UK). Cells were lysed 24 h later and assayed for luciferase activity (Dual Luciferase Reporter Assay system, Promega; FLUOstar Optima, BMG Labtech, Aylesbury, UK), with standardization for Renilla co-expression.
Studies of cellular localization
A p.Y25C mutant GFP-SF-1 construct was generated by site-directed mutagenesis in a pAcGFP-C1 vector (Clontech, Oxford, UK) to produce a mutant fusion protein with a GFP tag at the amino-terminus of SF-1. Empty (−), WT and mutant p.Y25C pAcGFP-C1SF-1 expression vectors (0·8 μg) were transfected into SF-1 expressing NCI-H295R human adrenal cells. After 24 h, cells were fixed and counterstained with Vectashield containing DAPI (Vector Laboratories, Peterborough, UK). Images were taken on a confocal microscope.
Reproductive hormone assays
Testosterone was analysed on the Spectria Testosterone RIA coated tube (Orion Diagnostica, Finland). Dihydrotestosterone was assayed using an in-house RIA after extraction and partition chromatography. LH and FSH were analysed on Immulite 2000 (Bio-Mediq DPC Pty Ltd, Los Angeles, CA, USA).
Results
Mutation analysis of patients
Molecular analysis revealed four patients to be heterozygous for a c.437G>C (p.G146A) sequence variant and one patient to be heterozygous for a c.74A>G (p.Y25C) sequence variant.
The c.437G>C sequence variant is a previously described variant located in the hinge region.7,14 Some consider it a polymorphism as previous p.G146A in vitro functional studies have demonstrated unaltered SF-1 transactivation activity.15 Therefore, these patients will not be further discussed in this study.
The c.74A>G is a novel variant that has not been found in previous studies of controls.13 This change results in an amino acid change from a highly conserved tyrosine to cysteine at codon 25 (p.Y25C). This amino acid is situated in the first zinc finger DNA-binding domain (See Fig. 1). This appears to be a de novo change as both parents are phenotypically normal and were found to be negative for this variant.
Fig. 1.
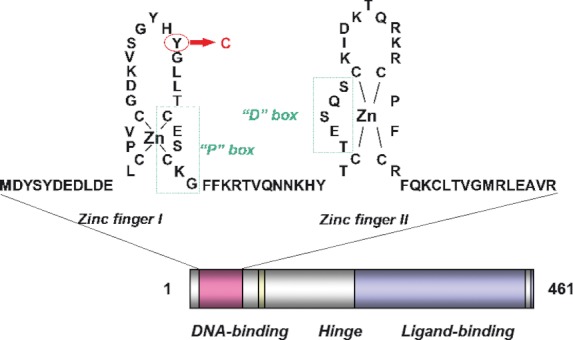
The c.74A>G alteration in this patient causes a tyrosine to cysteine change at codon 25 (p.Y25C). This amino acid is highly conserved and affects a codon located within the first zinc finger DNA-binding domain (DBD).
The proband was the first child for the couple. He was born at 41 + 1 week following an uneventful pregnancy. At birth, he had penoscrotal hypospadias with a small phallus (20 mm long), chordee, a ventrally deficient prepuce, bifid scrotum and two scrotal testes. His pelvic and renal ultrasound demonstrated 0·5–1 ml testes bilaterally in the scrotum with normal kidneys bilaterally and no Müllerian structures. Hormone results shortly after birth showed high testosterone levels (See Table 1).
Table 1.
Hormone results at two periods shortly after birth
| 13 days old | 1 month + 18 days old | |
|---|---|---|
| Testosterone (nm) | 10·8↑ (RI 3·1–10)2 | 25 ↑(RI 1·8–16·6)2 |
| Dihydrotestosterone (nm) | 4·2↑ (RI 0·17–2·1)16 | 5·3 ↑ (RI 0·4–2·9)16 |
| LH (IU/l) | 7·9↑ (RI 2·7–7·8)2 | 1·2 (RI 0·12–4·8)2 |
| FSH (IU/l) | 3·9↑ (RI 1·7–3·5)2 | 1·6 (RI 0·6–5·5)2 |
RI, Reference Interval.
The provisional diagnosis was PAIS in this patient. There was no family history of DSD and/or adrenal insufficiency. At age 4, he had no evidence of adrenal insufficiency on formal short synacthen testing.
Functional analyses
The p.Y25C variant of NR5A1 showed impaired transcriptional activation of the Cyp11a promoter (Fig. 2a). In a substantial proportion of cells, p.Y25C mutant SF-1 showed partially impaired nuclear localization (Fig. 2b).
Fig. 2.
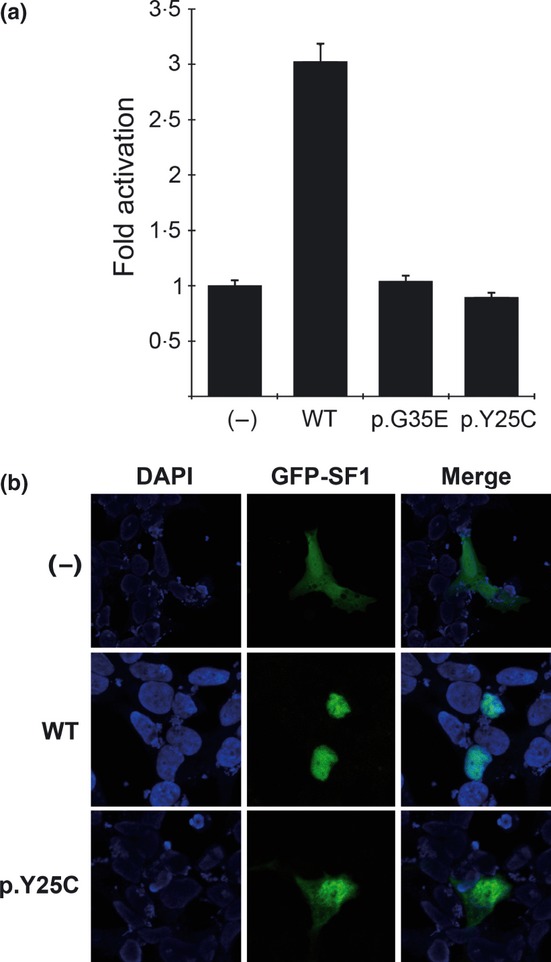
(a) Assays showing activation of a Cyp11a1 promoter by wild-type (WT) SF-1 and impaired transcriptional activity by the p.Y25C mutant. Relative luciferase activity was measured and expressed as fold activation above baseline (empty vector). Results are shown as the mean ± sem of three independent experiments, each performed in triplicate. (−), empty vector; WT, wild-type; p.G35E, a known loss-of-function mutation. (b) Immunocytochemistry to show cellular localization of SF-1 linked to GFP. Wild-type (WT) SF-1 shows strong nuclear localization, whereas the p.Y25C mutant GFP-SF-1 construct shows partially impaired nuclear localization in a proportion of cells.
Discussion
Currently, over 50 different NR5A1 mutations have been reported in humans.15,17,18 These mutations include missense mutations, nonsense mutations caused by nucleotide deletions and duplications, a 3 Mb deletion, a 970 kb deletion and a deletion in exon 2 and 3. The degree of gonadal dysgenesis and underandrogenization varies from mild to severe, with three mutations also causing adrenal failure. All cases of 46,XY DSD (except for two homozygous mutations) are heterozygous for the mutation, indicating that SF-1 dosage is critical.8,9 The varying locations of the different NR5A1 mutations suggest the absence of a mutation hot spot and the lack of a founder effect.
In our study of 17 patients with presumed androgen insensitivity but with no AR mutation, one patient was found to have a de novo mutation in NR5A1 which caused reduced SF-1 transcriptional activation with partially impaired nuclear localization in a significant proportion of transfected cells. To our knowledge, this is the first patient described with a NR5A1 loss-of-function mutation and elevated testosterone shortly after birth. It is known that within 24 h after birth and then at 1–2 months of life, there are physiologically high levels of testosterone that can be as informative as the hCG stimulation test.19 Despite one of our patient's levels being taken outside this period, his level was still high. Although NR5A1 mutations are known to cause dysgenetic testes and/or impaired androgen synthesis, there have been previously published cases of affected 46, XY infants with normal testosterone levels.2,20 In the first described case, the baby had a perineal urogenital sinus, a genital tubercle measuring 12 mm in length, palpable gonads (12 × 5 mm) in the bilateral inguinal regions and a hypotropic scrotum or corrugation of major labia.2 The phenotype in the second case described may be due to other genes in addition to NR5A1 contained in the deletion of chromosome 9q33.3 identified in this child. The phenotypically female infant was noted to have clitoromegaly at 10 weeks of age. She was then found to have a shallow vaginal entrance with a 1 ml gonad palpable in the left labium. Abdominal ultrasonography showed no uterus or Müllerian structures.20 Recently, two more NR5A1 cases with normal testosterone at infancy were published in a cohort study. One patient had scrotal hypospadias, bilateral cryptorchidism and no Müllerian structures. Another had scrotal hypospadias, intraabdominal testes and presence of Müllerian duct remnants.15 Our patient's phenotype suggests that there was a sufficient amount of anti-Müllerian hormone (AMH/MIS) for Müllerian regression and testosterone for Wolffian development. There must have been low dihydrotestosterone or dihydrotestosterone insensitivity, however, at least during the critical period of external genitalia formation (between 8 and 14 weeks gestation).12,21 Elevated testosterone in this patient is unexpected; however, the mechanisms underlying the variable effect of NR5A1 mutations are still not clearly understood. This is illustrated by the previously mentioned study of a baby with normal testosterone levels whose sibling (a 46, XY female) had the same mutation but was less virilized and had low testosterone production.2 It therefore appears that differences in phenotypes and Leydig cell function exist even within a family with the same mutation and other genetic or environmental factors may affect testosterone production.2 It may be that in some cases disruption of SF-1 is associated with functional androgen resistance22 or altered Leydig cell maturation and hyper-responsiveness to postnatal LH stimulation. These possible explanations for the relatively high androgen levels found here are speculative, but it will be of great interest to know whether NR5A1 changes can be found in further systematic studies of children with high or normal androgen concentrations who have been diagnosed as having AR negative PAIS.
This case illustrates that mutations of NR5A1 may be an important genetic cause in 46, XY DSD patients with a range of testosterone levels, and all patients negative for AR mutations should be considered for NR5A1 gene analysis even if testosterone biosynthesis is elevated. It will be of interest to note whether the capacity to produce testosterone is maintained to enable normal progression of puberty and development of secondary sexual characteristics. Initial evidence suggested that with increasing age, there will be decreasing testosterone as one study found that 4% of otherwise healthy men with unexplained azoospermia carried mutations in NR5A1 and that these men may also be at risk of endocrine dysfunction with failing testosterone with increasing age.13 However, recently, two studies presented in total four 46,XY patients with mutations in NR5A1 who entered puberty spontaneously with normal testosterone levels.18,23 It will be interesting to document future testosterone levels in these patients to elucidate whether there is premature failure of testosterone production and to determine their fertility potential.
Both parents were found to be negative for the mutation found in their son. De novo mutations have been documented in the majority of reported NR5A1 cases where the mode of inheritance is known.24 Interestingly, around 30% are documented to be inherited from the mother in a sex-limited dominant fashion, in that mothers may carry the mutation without ovarian dysfunction and pass it onto sons who are affected (mimicking X-linked inheritance).17 For the remainder, the inheritance is unknown, autosomal recessive or from their phenotypically normal fathers in rare instances.15,24 Thus, the absence of a family history should not detract from the possibility of NR5A1 mutations.
The finding of an NR5A1 mutation is important in the management of an infant with 46,XY DSD, as this diagnosis has implications for genetic counselling and future treatment. Unlike AIS, children with NR5A1 mutations should theoretically respond to normal levels of exogenous testosterone. It is unclear whether 46,XY DSD patients with NR5A1 mutations with the presence of testes and raised as males will be at risk of gonadal malignancy; however, it is likely that they will have progressive testicular changes over time with implications for fertility.11,13 This knowledge will influence sex assignment and family planning. As patients can have adrenal insufficiency, this knowledge will prepare patients and allow health professionals to initiate closer evaluation. Mutation carrier mothers and sisters of 46,XY DSD patients may also be at risk of premature ovarian insufficiency and possibly adrenal insufficiency.25 Females might also appear to be asymptomatic but pass on the mutation with the potential of having 46,XY DSD children. Being aware of these possibilities will help with genetic counselling and prepare the families and allow closer follow-up.
For the remainder of 46,XY DSD patients still without a molecular diagnosis other genes should be investigated. Evidence suggests that molecular analysis of SRD5A2 for 5α-reductase deficiency and HSD17B3 for 17β-hydroxysteroid dehydrogenase-3 deficiency are important as endocrine testing are potentially unreliable.26,27 This might also be the case with Leydig cell hypoplasia caused by inactivating mutation of the LH receptor.28 With the rapid advancements in genome sequencing, it is likely that simultaneous testing for all these genes will become more common in future.
In summary, we identified one novel NR5A1 mutation in an Australasian cohort of 17 46,XY DSD patients who had previously been assessed for AR mutations. As AR mutations are only detected in 13·6–28% of patients with clinical diagnosis of PAIS, it is important for other diagnoses, including NR5A1 mutations, to be considered.3,4 The high testosterone measurement in this case highlights the biochemical variability associated with SF-1/NR5A1 mutations.
Acknowledgments
We are grateful to Ron Evans, Meera Ramayya, Masafumi Ito and Larry Jameson for plasmids. JCA holds a Wellcome Trust Senior Research Fellowship in Clinical Science (WT079666 and WT098513).
We are grateful to Lilian Tan, Head of Endocrine Laboratory at South Eastern Area Laboratory Services, for the hormone assay details. We are grateful to Dr Anne Turner, Department Head at Sydney Children's Hospital Randwick, for follow-up of the patient.
References
- 1.Hughes IA, Nihoul-Fékété C, Thomas B, et al. Consequences of the ESPE/LWPES guidelines for diagnosis and treatment of disorders of sex development. Best Practice and Research Clinical Endocrinology and Metabolism. 2007;21:351–365. doi: 10.1016/j.beem.2007.06.003. [DOI] [PubMed] [Google Scholar]
- 2.Coutant R, Mallet D, Lahlou N, et al. Heterozygous mutation of steroidogenic factor-1 in 46,XY subjects may mimic partial androgen insensitivity syndrome. Journal of Clinical Endocrinology and Metabolism. 2007;92:2868–2873. doi: 10.1210/jc.2007-0024. [DOI] [PubMed] [Google Scholar]
- 3.Jeske YW, McGown IN, Cowley DM, et al. Androgen receptor genotyping in a large australasian cohort with androgen insensitivity syndrome; identification of four novel mutations. Journal of Pediatric Endocrinology and Metabolism. 2007;20:893–908. doi: 10.1515/jpem.2007.20.8.893. [DOI] [PubMed] [Google Scholar]
- 4.Ahmed SF, Cheng A, Dovey L, et al. Phenotypic features, androgen receptor binding, and mutational analysis in 278 clinical cases reported as androgen insensitivity syndrome. Journal of Clinical Endocrinology and Metabolism. 2000;85:658–665. doi: 10.1210/jcem.85.2.6337. [DOI] [PubMed] [Google Scholar]
- 5.Hughes IA. Androgen resistance. Best Practice & Research Clinical Endocrinology & Metabolism. 2006;20:577–598. doi: 10.1016/j.beem.2006.11.003. [DOI] [PubMed] [Google Scholar]
- 6.Lin L, Achermann JC. Steroidogenic factor-1 (SF-1, Ad4BP, NR5A1) and disorders of testis development. Sexual Development. 2008;2:200–209. doi: 10.1159/000152036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.WuQiang F, Yanase T, Wei L, et al. Functional characterization of a new human Ad4BP/SF-1 variation, G146A. Biochemical and Biophysical Research Communications. 2003;311:987–994. doi: 10.1016/j.bbrc.2003.10.096. [DOI] [PubMed] [Google Scholar]
- 8.Achermann JC, Ito M, Ito M, et al. A mutation in the gene encoding steroidogenic factor-1causes XY sex reversal and adrenal failure in humans. Nature Genetics. 1999;22:125–126. doi: 10.1038/9629. [DOI] [PubMed] [Google Scholar]
- 9.Achermann JC, Ozisik G, Ito M, et al. Gonadal determination and adrenal development are regulated by the orphan nuclear receptor steroidogenic factor-1, in a dose-dependent manner. Journal of Clinical Endocrinology and Metabolism. 2002;87:1829–183. doi: 10.1210/jcem.87.4.8376. [DOI] [PubMed] [Google Scholar]
- 10.Hasegawa T, Fukami M, Sato N, et al. Testicular dysgenesis without adrenal insufficiency in a 46,XY patient with a heterozygous inactive mutation of steroidogenic factor-1. Journal of Clinical Endocrinology and Metabolism. 2004;89:5930–5935. doi: 10.1210/jc.2004-0935. [DOI] [PubMed] [Google Scholar]
- 11.Lin L, Philibert P, Ferraz-de-Souza B, et al. Heterozygous missense mutations in steroidogenic factor 1 (SF1/Ad4BP, NR5A1) are associated with 46,XY disorders of sex development with normal adrenal function. Journal of Clinical Endocrinology and Metabolism. 2007;92:991–999. doi: 10.1210/jc.2006-1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kohler B, Lin L, Mazen I, et al. The spectrum of phenotypes associated with mutations in steroidogenic factor 1 (SF-1, NR5A1, Ad4BP) includes severe penoscrotal hypospadias in 46,XY males without adrenal insufficiency. European Journal of Endocrinology. 2009;161:237–242. doi: 10.1530/EJE-09-0067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bashamboo A, Ferraz-de-Souza B, Lourenco D, et al. Human male infertility associated with mutations in NR5A1 encoding steroidogenic factor 1. American Journal of Human Genetics. 2010;87:505–512. doi: 10.1016/j.ajhg.2010.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reuter AL, Goji K, Bingham NC, et al. A novel mutation in the accessory DNA-binding domain of human steroidogenic factor 1 causes XY gonadal dysgenesis without adrenal insufficiency. European Journal of Endocrinology. 2007;157:233–238. doi: 10.1530/EJE-07-0113. [DOI] [PubMed] [Google Scholar]
- 15.Camats N, Pandey AV, Fernandez-Cancio M, et al. Ten novel mutations in the NR5A1 gene cause disorders sex development in 46,XY and ovarian insufficiency in 46,XX individuals. Journal of Clinical Endocrinology and Metabolism. 2012;97:E1294–E1306. doi: 10.1210/jc.2011-3169. [DOI] [PubMed] [Google Scholar]
- 16.Esoterix. Endocrinology Expected Values and SI Unit Conversion Tables. CA: Esoterix Laboratory Services Inc; 2011. http://www.esoterix.com/files/Inside-Expected-Values_ESO.pdf (accessed Dec 2011) [Google Scholar]
- 17.Ferraz-de-Souza B, Lin L, Achermann JC. Steroidogenic factor-1 (SF-1, NR5A1) and human disease. Molecular and Cellular Endocrinology. 2011;336:198–205. doi: 10.1016/j.mce.2010.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tantawy S, Lin L, Akkurt I, et al. Testosterone production during puberty in two 46,XY DSD patients with novel NR5A1 (SF-1) mutations. European Journal of Endocrinology. 2012;167:125–130. doi: 10.1530/EJE-11-0944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nicolino M, Bendelac N, Jay N, et al. Clinical and biological assessments of the undervirilized male. British Journal of Urology International. 2004;93(Suppl):20–25. doi: 10.1111/j.1464-410X.2004.04705.x. [DOI] [PubMed] [Google Scholar]
- 20.van Silfhout A, Boot AM, Dijkhuizen T, et al. A unique 970kb microdeletion in 9q33.3 including the NR5A1 gene in a 46,XY female. European Journal of Medical Genetics. 2009;52:157–160. doi: 10.1016/j.ejmg.2009.02.009. [DOI] [PubMed] [Google Scholar]
- 21.Krone N, Hanley NA, Arlt W. Age-specific changes in sex steroid biosynthesis and sex development. Best Practice and Research Clinical Endocrinology and Metabolism. 2007;21:393–401. doi: 10.1016/j.beem.2007.06.001. [DOI] [PubMed] [Google Scholar]
- 22.Jorgensen JS, Nilson JH. AR suppresses transcription of the LHβ subunit by interacting with steroidogenic factor-1. Molecular Endocrinology. 2001;15:1505–1516. doi: 10.1210/mend.15.9.0691. [DOI] [PubMed] [Google Scholar]
- 23.Cools M, Hoebeke P, Wolffenbuttel KP, et al. Pubertal androgenization and gonadal histology in two 46,XY adolescents with NR5A1 mutations and predominantly female phenotype at birth. European Journal of Endocrinology. 2012;166:341–349. doi: 10.1530/EJE-11-0392. [DOI] [PubMed] [Google Scholar]
- 24.Köhler B, Achermann JC. Update – Steroidogenic factor 1 (SF-1, NR5A1) Minerva Endocrinologica. 2010;35:73–86. [PubMed] [Google Scholar]
- 25.Lourenco D, Brauner R, Lin L, et al. Mutations in NR5A1 Associated with Ovarian Insufficiency. The New England Journal of Medicine. 2009;360:1200–1210. doi: 10.1056/NEJMoa0806228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Choi J, Kin G, Seo E, et al. Molecular analysis of the AR and SRD5A2 genes in patients with 46,XY disorders of sex development. Journal of Pediatric Endocrinology and Metabolism. 2008;21:545–553. [PubMed] [Google Scholar]
- 27.Boehmer ALM, Brinkmann AO, Sandkuijl LA, et al. 17β-hydroxysteroid dehydrogenase-3 deficiency: diagnosis, phenotypic variability, population genetics, and worldwide distribution of ancient and de novo mutations. The Journal of Clinical Endocrinology and Metabolism. 1999;84:4713–4721. doi: 10.1210/jcem.84.12.6174. [DOI] [PubMed] [Google Scholar]
- 28.Wu S, C W. Male pseudohermaphroditism due to inactivating luteinizing hormone receptor mutations. Archives of Medical Research. 1999;30:495–500. doi: 10.1016/s0188-4409(99)00074-0. [DOI] [PubMed] [Google Scholar]