

Abstract
Autism spectrum disorders (ASD) are believed to have genetic and environmental origins, yet in only a modest fraction of individuals can specific causes be identified1,2. To identify further genetic risk factors, we assess the role of de novo mutations in ASD by sequencing the exomes of ASD cases and their parents (n= 175 trios). Fewer than half of the cases (46.3%) carry a missense or nonsense de novo variant and the overall rate of mutation is only modestly higher than the expected rate. In contrast, there is significantly enriched connectivity among the proteins encoded by genes harboring de novo missense or nonsense mutations, and excess connectivity to prior ASD genes of major effect, suggesting a subset of observed events are relevant to ASD risk. The small increase in rate of de novo events, when taken together with the connections among the proteins themselves and to ASD, are consistent with an important but limited role for de novo point mutations, similar to that documented for de novo copy number variants. Genetic models incorporating these data suggest that the majority of observed de novo events are unconnected to ASD, those that do confer risk are distributed across many genes and are incompletely penetrant (i.e., not necessarily causal). Our results support polygenic models in which spontaneous coding mutations in any of a large number of genes increases risk by 5 to 20-fold. Despite the challenge posed by such models, results from de novo events and a large parallel case-control study provide strong evidence in favor of CHD8 and KATNAL2 as genuine autism risk factors.
In spite of the substantial heritability, few genetic risk factors for ASD have been identified1,2. Copy number variants (CNVs), often de novo and covering multiple adjacent genes, have been identified as conferring risk3,4. While these CNVs provide important leads to underlying biology, they rarely implicate single genes, are rarely fully penetrant, and many confer risk to a broad range of conditions including intellectual disability(ID), epilepsy, and schizophrenia5. There are also documented instances of rare single nucleotide variants (SNVs) that are highly penetrant for ASD6.
Large-scale genetic studies make clear that the origins of ASD risk are multifarious, and recent estimates based on CNV data put the number of independent risk loci in the hundreds4. Yet knowledge regarding specific risk-determining genes and the overall genetic architecture for ASD remains incomplete. Although new sequencing technologies provide a catalog of most variation in the genome, the profound locus heterogeneity of ASD makes it challenging to distinguish variants that confer risk from the background noise of inconsequential SNVs. De novo variation, being less frequent and potentially more deleterious, could offer insights into risk-determining genes. For this reason we sought to carefully evaluate the observed rate and consequence of de novo point mutations in the exomes of ASD subjects.
We performed exome sequencing of 175 ASD probands and their parents across five centers with multiple protocols and validation techniques (Supplementary Information). We used a sensitive and specific analytic pipeline based on current best practices7-9 to analyze all data and observed no heterogeneity of mutation rate among centers.
In the entire sample, we observed 161 coding region point mutations (101 missense, 50 silent and 10 nonsense), with an additional 2 conserved splice site (CSS) SNVs and 6 frameshift indels validated and included in pathway analyses (Supplementary Table 1).
To determine whether the rate of coding region point mutations was elevated, we estimated the mutation rate in light of coverage and base context using two parallel approaches (Supplementary Information). Based on both models, the exome target should have a significantly increased (≈30%) mutation rate compared to the genome. Conservatively, by assuming the low- end of the estimated mutation rate from recent whole-genome data (1.2×10-8)10, we estimate a mutation rate of 1.5×10-8 for the exome sequence captured here. The observed point mutation rate of 0.92/exome is slightly but not significantly elevated versus expectation (Table 1) and is insensitive to adjustment for lower coverage regions (Supplementary Information). Indeed our rate is similar to Sanders et al. (in press).
Table 1.
Distribution of Events Per Family.
| Events per family |
All ASD trios | Random Mut-Exp3 |
|
|---|---|---|---|
| exon DN SNVs |
Exp | ||
| 0 | 71 | 69.7 | 73.2 |
| 1 | 62 | 64.2 | 63.8 |
| 2 | 28 | 29.5 | 27.8 |
| 3 | 10 | 9.1 | 8.1 |
| 4 | 2 | 2.1 | 1.8 |
| 5 | 1 | 0.4 | 0.3 |
| 0.920 | 0.871 | ||
exon DN-SNVs include all single nucleotide variants in coding sequence but excludes indels and intronic variants
exp is the expected distribution of number of trios with a given event count as determined by the Poisson
Random Mut-Exp is the expectation for 175 trios based on the sequence-context mutation rate model M1 (Supplementary Materials) based on the count of the number of trios that have at least 10× coverage.
Per-family events were distributed according to the Poisson distribution (Table 1), yielding no evidence for ASD tracing to high rates of de novo mutation. The relative rates of ‘functional’ (missense, nonsense, CSS and read-through) versus silent changes did not deviate from expectation (Table 2). We did, however, observe 10 nonsense mutations (6.2%), which exceeded expectation (3.3%) (one-tailed P=0.04; Supplementary Information).
Table 2.
Rates of mutation annotation given variant type.
| Type of de novo mutation |
De Novo1 | Random De Novo |
Singletons2 | Doubletons2 | ≥ 32 |
|---|---|---|---|---|---|
| Missense | 62.7% | 66.1% | 59.5% | 55.4% | 48.8% |
| Nonsense | 6.2% | 3.3% | 1.2% | 0.8% | 0.4% |
| Synonymous | 31.1% | 30.6% | 39.3% | 43.8% | 50.8% |
| Benign3 | 35.0% | 35.9% | 46.6% | 51.3% | 63.4% |
| Possibly Damaging3 | 21.0% | 18.9% | 18.8% | 17.7% | 15.1% |
| Probably Damaging3 | 44.0% | 45.2% | 34.7% | 31.0% | 21.4% |
All indels and failing variants were removed.
Singletons, doubletons, and ≥3 (copies) are only those variants called in the parents from Wave 1.
Benign, Possibly Damaging, and Probably Damaging as defined by PolyPhen2
We examined the missense mutations as such variation can cause loss of function11 using PolyPhen2 scores12 to measure mutation severity. These also showed no deviation from random expectation. The observed PolyPhen2 scores clearly deviate from standing variation in the parents (Table 2): Such variation, even the rarest category, has survived selective pressure and is not an appropriate control for de novo events.
We observed 3 genes with two de novo mutations: BRCA2 (2 missense), FAT1 (2 missense) and KCNMA1 (1 missense, 1 silent). A gene with two or more non-synonymous de novo hits across a panel of trios might suggest strong candidacy. However, simulations (Supplementary Information) show that two such hits are inadequate to define a gene as a conclusive risk factor given the number of observed events in the study.
From analyses of secondary phenotypes (Supplementary Tables 2-3), the most striking result is that paternal and maternal age, themselves highly correlated (r2=0.679, P-value<0.0001), each strongly predicts the number of de novo events per offspring (paternal age P=0.0013, maternal age P=0.000365), consistent with aggregating mutations in germ cells in the paternal line13. Consistent with genetic theory, there is an increased rate of de novo mutation in female versus male cases (1.214 for females vs. 0.914 for males); however, the difference is not significant, perhaps owing to limited sample size. Considering phenotypic correlates, we observed no rate difference between subjects with strict autism versus those with a broader ASD diagnosis, between positive and negative family history, or any significant effect of de novo mutation on verbal, nonverbal or full scale IQ (Supplementary Table 3).
While hundreds of loci are apparently involved in autism4 and de novo mutations therein affect ASD risk, modeling of different numbers of risk genes and penetrances (Supplementary Information) shows that a model of hundreds of genes with high penetrance mutations is excluded by our data; however, more modest contributions of de novo variants are not. For example, 10-20% of cases carrying a de novo risk-conferring event and conferring ten to twentyfold increased risk, is consistent with these data (Supplementary Table 4). Thus, our data are consistent with either chance mutation or a modest role for de novo mutations on risk.
We therefore posed two questions of the group of genes harboring de novo functional mutations: do the protein products of these genes interact with each other more than expected, and are they unusually enriched in, or connected to, prior curated lists of ASD-implicated genes? Using an in silico approach (DAPPLE)14,15 the protein-protein connectivity in the set of 113 genes harboring ‘functional’ de novo mutations was evaluated. These analyses (Figure 1) showed significantly greater connectivity amongst the de novo identified proteins than would be expected by chance (P<0.001) (Supplementary Information).
Figure 1. Protein-Protein interaction for genes with an observed functional de novo event.
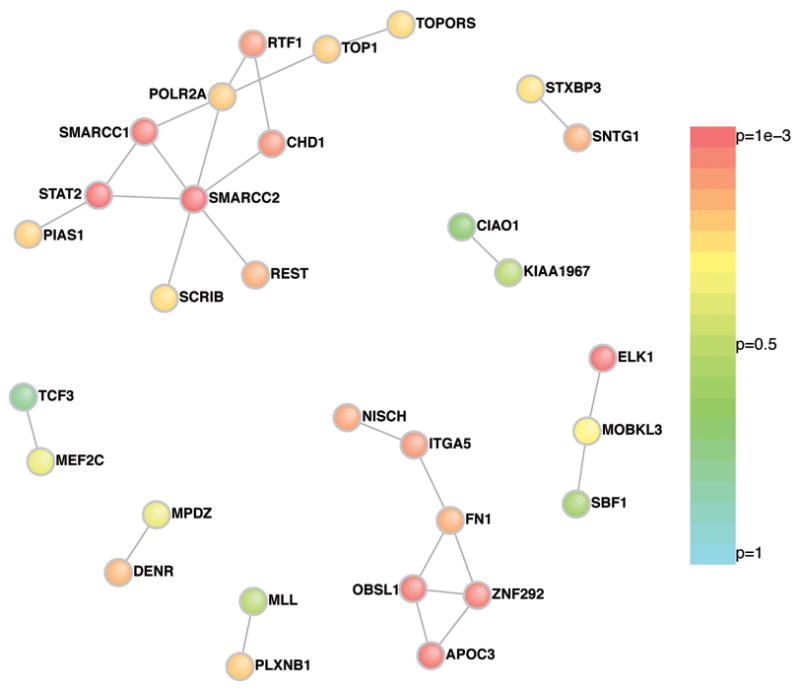
Direct protein connections from InWeb, restricting to genes harboring de novo mutations for DAPPLE analysis. Two extensive networks are identified, the first centered on SMARCC2 with 12 connections across 11 genes and the second centered on FN1 with 7 connections across 6 genes. The P-value for each gene having as many connections as those observed color the nodes of the network.
Querying previously-defined, manually-curated lists of genes6 associated with high risk for ASD with and without ID(Supplementary Tables 5), and high risk ID genes (Supplementary Tables 6), we asked whether there was significant enrichment for de novo mutations in these genes. Five genes with ‘functional’ de novo events were previously associated with ASD and/or ID (STXBP1, MEF2C, KIRREL3, RELN and TUBA1A); for four of these genes (all but RELN) the prior evidence indicated autosomal dominant inheritance.
We then assessed the average distance (Di, Supplementary Figure 2) of the de novo coding variants in brain-expressed genes (see supplement) to the ASD/ID list using a Protein-Protein Interaction background network. To enhance power, data from a companion study (Sanders et al.) were used, including the observed silent de novo variants and de novo variants in unaffected siblings as comparators. The average distance for non-synonymous variants was significantly smaller for the case set than the comparator set (3.66±0.42 versus 3.78±0.59; permutation P=0.033) (Figure 2). Much of this signal comes from 31 synaptic genes identified by three large-scale synaptic proteomic studies (Di=3.47±0.46 versus 3.57±0.60; permutation P=0.084) (Supplemental Figure 3; see also Supplemental Fig. 4 for the complete data). Taken in total, these independent gene set analyses, along with the modest enrichment of de novo variants over background rates in ASD, indicate that a proportion of the de novo events observed in this study likely contribute to autism risk.
Figure 2. Direct and indirect Protein-Protein interaction for genes with a functional de novo event and existing ASD/ID genes.
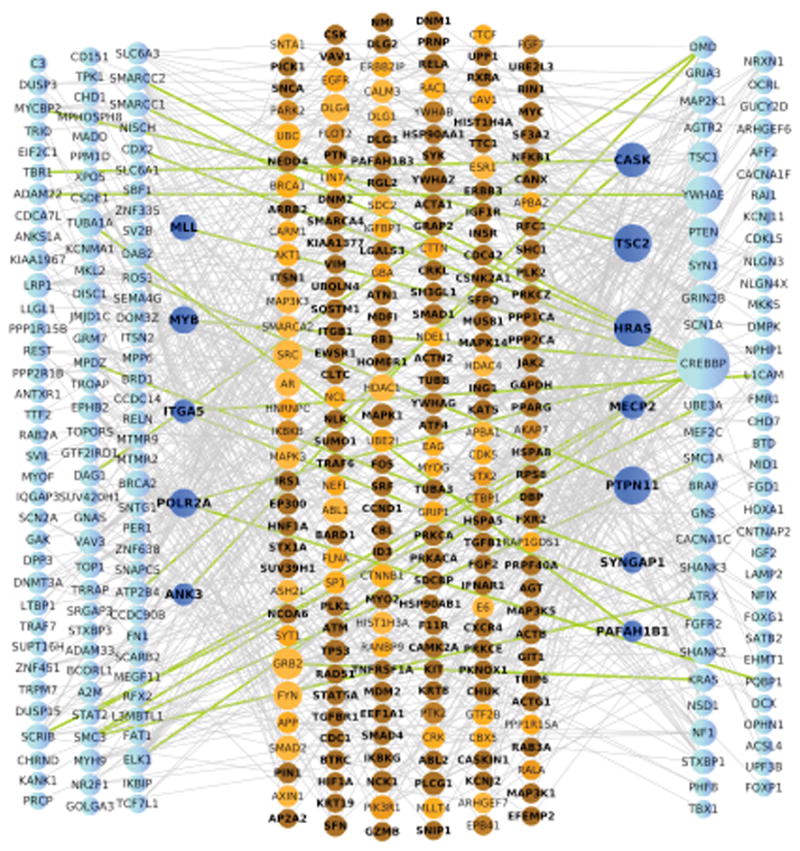
PPI network analysis for de novo variants and prior ASD genes (ASD112). Nodes are sized based on connectivity. Genes harboring de novo variants (left) and prior ASD genes (right) are colored blue with dark blue nodes represent genes that belong to one of these lists and are also intermediate proteins. Intermediate proteins (center) are colored in shades of orange based on a p-value computed using a proportion test where darker color represents a lower p-value. Green edges represent direct connections between genes harboring de novo variants (left) and prior ASD genes. All other edges, connecting to intermediate proteins are shown in grey.
Using whole-exome sequencing of autism trios, we demonstrate a rate, functional distribution and predicted impact of de novo mutation largely consistent with chance mutational processes governed by sequence context. This lack of significant deviation from random mutational processes suggests a more limited role for the contribution of de novo mutations to ASD pathogenesis than has previously been suggested15, and specifically highlights the fact that observing a single de novo mutation, even an apparently ‘severe’ LOF allele, is insufficient to implicate a gene as a risk factor. Yet the pathway analyses presented here assert that the overall set of genes hit with ‘functional’ de novo mutations are not random and are biologically related to each other and to previously identified ASD/ID candidate genes. Modeling the de novo mutational process under a range of genetic models reveals that some models are inconsistent with the observed data – e.g., one hundred rare, fully penetrant Mendelian genes similar to Rett syndrome – while others are not such as spontaneous ‘functional’ mutation in a 1,000 genes that would increase risk by ten or twentyfold (Supplementary Table 4). Models that fit the data are consistent with the relative risks estimated for most de novo CNVs4 and suggest that de novo SNVs, like most CNVs, often combine with other risk factors rather than fully cause disease. Furthermore, these models suggest that de novo SNVs events will likely explain <5% of the overall variance in autism risk (Supplementary Table 4).
Considering the two companion manuscripts, 18 genes with two functional de novo mutations are observed in the complete data. Using simulations, 11.91 genes on average harbor functional mutations by chance (Supplementary Table 7). Thus, a set of 18 genes with two or more hits is not quite significant (p=0.063). Matching loss-of-function variants, however, at SCN2A, KATNAL2 and CHD8 (Supplementary Table 7) are unlikely to occur by chance because the expected very low rate of de novo nonsense, splice and frameshift variants. We evaluated these strong candidates further using exome sequencing on 935 cases and 870 controls and at both KATNAL2 and CHD8, three additional LoF mutations were observed in cases with none in controls (no additional LoF mutations were seen at SCN2A). Using data from more than 5000 individuals in the NHLBI Exome Variant Server as additional controls, 3 LoF mutations were seen in KATNAL2 but none again in CHD8, making the additional observation of 3 CHD8 LoF mutations in our cases significant evidence (p<0.01) of this being a genuine autism susceptibility gene. Not all genes with double hits are nearly so promising (Supplementary Information; Supplementary Tables 8-9) supporting the estimate above that most such observations are simply chance events. Overall, these data underscore the challenge of establishing individual genes as conclusive risk factors for ASD, a challenge that will require larger sample sizes and, likely, deeper analytic integration with inherited variation.
Methods (for online version only)
Phenotype assessment
Affected probands were assessed by research-reliable research personnel using Autism Diagnostic Interview-Revised (ADI-R) and the Autism Diagnostic Observation Schedule-Generic (ADOS) and DSM-IV diagnosis of a pervasive developmental disorder was made by a clinician. All probands met criteria for autism on the ADI-R and either autism or ASD on the ADOS, except for the 3 subjects from AGRE that were not assessed with the ADOS. In all 85% of probands were classified with autism on both the ADI-R and ADOS. All subjects provided informed consent and the research was approved by institutional human subjects board.
Exome sequencing, variant identification, and de novo detection
Exome capture and sequencing was performed at each site using similar methods. Exons were captured using the Agilent 38Mb SureSelect v2 (University of Pennsylvania and Broad Institute n=118), the NimbleGen Seq Cap EZ SR v2 (Mt Sinai School of Medicine, Vanderbilt University n=51), or NimbleGen VCRome 2.1 (Baylor n=6). After capture, another round of LM-PCR was performed to increase the quantity of DNA available for sequencing. All libraries were sequenced using an IlluminaHiSeq2000.
Sequence processing and variant calling was performed using a similar computational workflow at all sites. Data was processed with Picard (http://picard.sourceforge.net/), which utilizes base quality-score recalibration and local realignment at known indels8 and BWA7 for mapping reads to hg19. SNPs were called using GATK8,9 for all trios jointly. The variable sites that we have considered in analysis are restricted to those that pass GATK standard filters. From this set of variants, we identified putative de novo mutations as sites where both parents were homozygous for the reference sequence and the offspring was heterozygous and each genotype call was made confidently (see Supplementary Information).
Validation of de novo events
Putative de novo events were validated by sequencing the carrier and both parents using Sanger sequencing methods (University of Pennsylvania, Mt. Sinai School of Medicine, Vanderbilt University, Baylor Medical College) or by Sequenom MALDI-TOF genotyping of trios (Broad).
Gene annotation
All identified mutations were then annotated using Refseq hg19. The functional impact of variants was assessed for all isoforms of each gene, with the most severe annotation taking priority. Splice site variants were identified as occurring within two basepairs of any intron/exon boundary.
Expectation of de novo mutation calculation
To calculate the expected de novo rate, we assessed the mutability of all possible trinucleotide contexts in the intergenic region of the human genome for variation in two fashions: fixed genomic differences compared to chimpanzee and baboon12 and variation identified from the 1,000 Genomes project. The overall mutation rate for the exome was then determined by summing the probability of mutation for all bases in the exome that were captured successfully. We also determined the probability of each class functional mutation by summing the annotated variants.
Pathway analyses
We applied DAPPLE14, which uses the InWeb database15, to determine whether there is excess protein protein interaction across the genes hit by a functional de novo event. We also assessed whether these genes were more closely connected to a list of ASD genes.6
Modeling de novo events
We modeled a Poisson process consistent with the expected distribution defined by the mutation model and with the observed data. We varied the fraction of genes that influence risk, the probability a variant in a gene would be functional, and the penetrance of functional de novo events. We also simulated a random set of de novo events to estimate the probability of hitting a gene multiple times.
Association Analysis
We performed association tests using SKAT17, a generalization of C-alpha18. Our primary analyses treat case-control data generated at Baylor and Broad separately (23 genes X 2 sites), but we also performed mega and meta-analyses (23 genes X 2 methods).
Supplementary Material
Acknowledgments
This work was directly supported by NIH grants R01MH089208 (MJD), R01 MH089025 (JDB), R01 MH089004 (GS), R01MH089175 (RG) and R01 MH089482 (JSS) and supported in part by NIH grants P50 HD055751 (EHC), RO1 MH057881 (BD), and R01 MH061009 (JSS). YK, GC, and SY are Seaver Fellows, supported by the Seaver Foundation. We thank Thomas Lehner (NIMH), Adam Felsenfeld (NHGRI), and Patrick Bender (NIMH) for their support and contribution to the project. We thank Stephan Sanders and Matthew State for discussions on the interpretation of de novo events. We thank David Reich for comments on the abstract and message of the manuscript. We acknowledge the assistance of Melissa Potter, Anna McGrew and Genea Crockett without whom these studies would not be possible, and Center for Human Genetics Research resources: Computational Genomics Core, Genetic Studies Ascertainment Core and DNA Resources core, supported in part by NIH NCRR grant UL1 RR024975, and the Vanderbilt Kennedy Center for Research on Human Development (P30 HD015052). We acknowledge partial support for U54 HG003273 (RG). JDB, BD, MD, RG, AS, GS, JSS are lead investigators in the Autism Sequencing Consortium (ASC). The ASC is comprised of groups sharing massively parallel sequencing data in autism.
Footnotes
Author Contributions: Laboratory work: AS, CS, GC, OJ, ZP, JDB, DM, IN, YW, LL, YH, SG, ELC, NGC, ETG
Data Processing: BMN, KES, EL, AK, JF, MF, KS, TF, KG, EB, RP, MDP, SG, SY, VM, JL, JDB, AS, CS, UN, JGR, JRW, BEB, SEL, CFL, LSW, OV
Statistical Analysis: BMN, LL, KES, CS, BFV, JM, ER, SS, PP, YK, AM, RD, CFL, LSW, HL, TZ, EB, RAG, JDB, CB, EHC, JSS, GDS, BD, KR, MJD
PIs/Study design: EB, RAG, EHC, JDB, KR, BD, GDS, JSS, MJD; Yan Kou, Li Liu, Avi Ma'ayan, Kaitlin E. Samocha, Aniko Sabo, and Chiao-Feng Lin contributed equally to this work. Eric Boerwinkle, Joseph D. Buxbaum, Edwin H. Cook, Jr., Bernie Devlin, Richard A. Gibbs, Kathryn Roeder, Gerard D. Schellenberg, James S. Sutcliffe, and Mark J. Daly are lead investigators of the ARRA Autism Sequencing Collaboration.
Author information: Data included in this manuscript has been deposited at dbGaP under accession number phs000298.v1.p1 and is available for download at http://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs000298.v1.p1
Reprints and permissions information is available at http://www.nature.com/reprints.
The authors declare no competing financial interests.
Supplementary Information is linked to the online version of the paper at http://www.nature.com/nature
References
- 1.Lichtenstein P, Carlstrom E, Rastam M, Gillberg C, Anckarsater H. The genetics of autism spectrum disorders and related neuropsychiatric disorders in childhood. The American journal of psychiatry. 2010;167:1357–1363. doi: 10.1176/appi.ajp.2010.10020223. [DOI] [PubMed] [Google Scholar]
- 2.Hallmayer J, et al. Genetic Heritability and Shared Environmental Factors Among Twin Pairs With Autism. Archives of General Psychiatry. 2011 doi: 10.1001/archgenpsychiatry.2011.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pinto D, et al. Functional impact of global rare copy number variation in autism spectrum disorders. Nature. 2010;466:368–372. doi: 10.1038/nature09146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sanders SJ, et al. Multiple recurrent de novo CNVs, including duplications of the 7q11.23 Williams syndrome region, are strongly associated with autism. Neuron. 2011;70:863–885. doi: 10.1016/j.neuron.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sebat J, Levy DL, McCarthy SE. Rare structural variants in schizophrenia: one disorder, multiple mutations; one mutation, multiple disorders. Trends in genetics TIG. 2009;25:528–535. doi: 10.1016/j.tig.2009.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Betancur C. Etiological heterogeneity in autism spectrum disorders: more than 100 genetic and genomic disorders and still counting. Brain Research. 2011;1380:42–77. doi: 10.1016/j.brainres.2010.11.078. [DOI] [PubMed] [Google Scholar]
- 7.Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26:589–595. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.DePristo MA, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 43:491–498. doi: 10.1038/ng.806. ng.806 [pii] 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McKenna A, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Research. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Conrad DF, et al. Variation in genome-wide mutation rates within and between human families. Nat Genet. 43:712–714. doi: 10.1038/ng.862. ng.862 [pii] 10.1038/ng.862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7(4):248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kryukov GV, Pennacchio LA, Sunyaev SR. Most rare missense alleles are deleterious in humans: implications for complex disease and association studies. American Journal of Human Genetics. 2007;80:727–739. doi: 10.1086/513473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Crow JF. The origins, patterns and implications of human spontaneous mutation. Nat Rev Genet. 2000;1:40–47. doi: 10.1038/35049558. [DOI] [PubMed] [Google Scholar]
- 14.Rossin EJ, et al. Proteins encoded in genomic regions associated with immune-mediated disease physically interact and suggest underlying biology. PLoS genetics. 2011;7:e1001273. doi: 10.1371/journal.pgen.1001273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lage K, et al. A large-scale analysis of tissue-specific pathology and gene expression of human disease genes and complexes. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:20870–20875. doi: 10.1073/pnas.0810772105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.O'Roak BJ, et al. Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nat Genet. 43:585–589. doi: 10.1038/ng.835. ng.835 [pii] 10.1038/ng.835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wu MC, et al. Rare-variant association testing for sequencing data with the sequence kernel association test. American Journal of Human Genetics. 2011;89:82–93. doi: 10.1016/j.ajhg.2011.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Neale BM, et al. Testing for an unusual distribution of rare variants. PLoS Genet. 7:e1001322. doi: 10.1371/journal.pgen.1001322. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.