

Abstract
Reverse cholesterol transport (RCT) refers to the mobilization of cholesterol on HDL particles (HDL-C) from extravascular tissues to plasma, ultimately for fecal excretion. Little is known about how HDL-C leaves peripheral tissues to reach plasma. We first used 2 models of disrupted lymphatic drainage from skin — 1 surgical and the other genetic — to quantitatively track RCT following injection of [3H]-cholesterol–loaded macrophages upstream of blocked or absent lymphatic vessels. Macrophage RCT was markedly impaired in both models, even at sites with a leaky vasculature. Inhibited RCT was downstream of cholesterol efflux from macrophages, since macrophage efflux of a fluorescent cholesterol analog (BODIPY-cholesterol) was not altered by impaired lymphatic drainage. We next addressed whether RCT was mediated by lymphatic vessels from the aortic wall by loading the aortae of donor atherosclerotic Apoe-deficient mice with [2H]6-labeled cholesterol and surgically transplanting these aortae into recipient Apoe-deficient mice that were treated with anti-VEGFR3 antibody to block lymphatic regrowth or with control antibody to allow such regrowth. [2H]-Cholesterol was retained in aortae of anti–VEGFR3-treated mice. Thus, the lymphatic vessel route is critical for RCT from multiple tissues, including the aortic wall. These results suggest that supporting lymphatic transport function may facilitate cholesterol clearance in therapies aimed at reversing atherosclerosis.
Introduction
Atherosclerosis is a major cause of morbidity and mortality in the world. At the cellular level, atherosclerotic plaques are characterized by macrophages loaded with cholesterol esters. Indeed, ongoing recruitment of monocyte-derived macrophages into plaque is a key event in disease progression. Removal of macrophages involves a cessation of recruitment that correlates with the mobilization of cholesterol stored within macrophages as cholesterol esters (1). Cholesterol ester–loaded macrophages are not inherently proinflammatory (2). Thus, it is not yet understood how cholesterol drives monocyte recruitment to sustain plaque progression. Deposition of cholesterol crystals (3) or undesirable immunity to cholesterol-associated apoproteins (4) are possible means by which inflammation can be sustained in an environment rich in macrophages and cholesterol. Regardless of our gaps in understanding the links between cholesterol in the artery wall and inflammation that drives disease progression, it seems clear that removal of cholesterol, which is largely stored in macrophages as cholesterol esters, from the aortic wall would comprise a step toward disease reversal, as observed in primate models of atherosclerosis regression (5).
Removal of cholesterol from macrophages requires hydrolysis, mobilization, and efflux of stored cholesterol ester from macrophages to lipoprotein acceptors, especially apoA-I, by virtue of the formation of HDL. Cholesterol solubilized by apoA-I and phospholipids (HDL-C) moves through the bloodstream to the liver for eventual fecal elimination (6), or through a nonbiliary pathway (7, 8). This entire transport process is called reverse cholesterol transport (RCT), and macrophage RCT refers specifically to the removal of cholesterol from macrophage cholesterol ester stores (9). The fraction of cholesterol disposed of through macrophages is small overall compared with the amounts in the entire body; nonetheless, as cells that are active in tissue remodeling and clearance of cellular debris, macrophages may take in and need to dispose of more cholesterol than many other cell types (9, 10). Although much is known about cholesterol efflux from macrophages and subsequent uptake of cholesterol in the liver and its secretion into bile, some steps are poorly understood. In particular, little is known about how HDL or apoA-I enters or leaves plaques, or indeed any other tissues, to return to the bloodstream.
The lymphatic system generally governs the transport of macromolecules from the extravascular space into the blood (11). Accordingly, human peripheral lymph contains HDL-C (12–14), and estimates indicate a lymph transport of more than 300 mg per day (15). Placed into the context of estimates of overall neutral and acidic sterol excretion per day per kilogram in humans (16), the flux through lymph could represent a substantial pathway for removal of cholesterol from tissues body wide. However, since apoA-1, which drives the formation of HDL, is thought to enter extravascular tissues by crossing the blood endothelium (17), it is unclear whether clearance of HDL-C from tissues by traversing vascular endothelium may also occur and diminish the need for lymphatic transport to remove HDL-C from tissues. Thus, we set out to address whether lymphatic vessels participate critically in RCT from peripheral tissues, including skin and the aortic wall, where macrophage RCT is especially relevant.
Results
Surgical ablation of lymphatic vessels in the skin blocks RCT without impairing cholesterol efflux from macrophages.
To determine whether lymphatic vessels have a quantitatively important role in the clearance of HDL, we first adapted an in vivo macrophage RCT assay (6, 18, 19). In this assay, RCT is measured by injection of [3H]-cholesterol–loaded macrophages into peripheral tissues of mice to track the mobilization of [3H]-cholesterol to plasma, liver, and feces (6, 18). In preparation for this assay, we surgically separated dermal lymphatic vessels to disrupt lymph flow from the tail dermis of mice without damaging the blood circulation (20). Intradermal injection of Evans blue dye into the tail tip, upstream of the separation, revealed very little dye retrieved in draining lymph nodes compared with the sham control (Figure 1A). This finding allowed us to set up a model to examine whether RCT is intact or severely disrupted after [3H]-cholesterol–loaded macrophages are injected into the tail upstream of the lymphatic separation. In control mice without lymphatic separation, the RCT response to [3H]-cholesterol–loaded macrophages injected into the tail skin was much less robust than in the lower back skin or the previously studied peritoneal cavity (Figure 1B and ref. 18), importantly revealing that different body sites support macrophage RCT to differing extents. As expected (18), macrophage RCT from the tail increased markedly in Apoa1 transgenic mice (Figure 1B), which bear higher levels of HDL (21). Therefore, we utilized Apoa1 transgenic mice in ensuing macrophage RCT assays in the tail following lymphatic separation versus sham operation to improve the magnitude of our readout.
Figure 1. Surgical separation of lymphatic vessels and its impact on RCT.

(A) Image of the mouse tail following surgical separation of lymphatic vessels (red arrow). White boxes depict approximate location of tail-draining lymph nodes. These nodes (lower photomicrographs) were removed after Evans blue dye was injected into the tail; enlarged images of the 2 lymph nodes are shown from a control mouse receiving a sham operation (Sham) and 1 that underwent lymphatic separation (Surgery). Similar results were obtained in 4 other analyses (n = 5) using Evans blue dye to verify repeated success of the procedure. (B) [3H]-Cholesterol–loaded macrophages were injected into the peritoneum (i.p.), in skin of the tail, or the lower back of WT or Apoa1 transgenic mice. Tritium counts in the plasma are charted. (C) Tritium counts in plasma in control mice (solid line) or in those subjected to lymphatic separation (dotted line) after [3H]-cholesterol–loaded macrophages were injected into the tail upstream (with respect to lymph flow) of the lymphatic separation as shown in A. (D) Compiled tritium counts in plasma, liver, and feces from the experimental design used in C. (E) Cholesterol efflux was monitored as the loss of fluorescence from gated CD45.1 macrophages loaded ex vivo with BODIPY-cholesterol and retrieved 24 hours later from the tails of WT or Apoa1 transgenic mice with or without lymphatic separation. All data represent the mean ± SEM from at least 2 experiments performed with 5 replicates per experimental group. *P < 0.05; **P < 0.01. CPM, counts per minute.
Surgical separation of tail lymphatic vessels did not alter plasma lipoprotein profiles (Supplemental Figure 1; supplemental material available online with this article; doi: 10.1172/JCI63685DS1). [3H]-Cholesterol did not appear in plasma for more than 8 hours after [3H]-cholesterol–loaded macrophage injection, but counts peaked by 24 hours (Figure 1C). [3H]-Cholesterol levels were reduced by 50% in plasma in mice with surgically separated lymphatic vessels (Figure 1C). When normalized to the weight or volume of each compartment, there was a net reduction of [3H]-cholesterol in plasma, liver, and feces (Figure 1D). Surgical separation of lymphatic vessels in the tail reduced net RCT by 54% at 24 hours and by 44% at 48 hours (Figure 1D). These data suggest that lymphatic vessels are important conduits for RCT.
However, it is possible that cholesterol efflux from macrophages, rather than egress of HDL from tissue, was reduced in the skin with lymphatic separation. To examine this possibility, we loaded macrophages ex vivo with BODIPY-cholesterol, a fluorescent analog of cholesterol in which a BODIPY moiety is covalently linked to C24 of the aliphatic side chain (22, 23), allowing us to monitor cholesterol efflux from individual macrophages using flow cytometry. Like native cholesterol, we found that BODIPY-cholesterol efflux required the expression of ABCA1/ABCG1 (Supplemental Figure 2). Therefore, we prepared macrophages from CD45.1+ congenic C57BL/6 mice, loaded them with BODIPY-cholesterol, and injected them into the tails of CD45.2+ WT or Apoa1 transgenic mice with or without lymphatic vessel separation. At 24 hours, we digested the tail tissue to retrieve injected cells and gated on CD45.1+ cells to identify the transferred macrophages. Macrophages injected into Apoa1 transgenic mice had reduced fluorescence intensity (shift to the left), indicative of increased BODIPY-cholesterol unloading, compared with injection into WT recipient mice (Figure 1E). Most importantly, there was no difference in this shift to the left in Apoa1 transgenic mice where lymphatic vessels had been separated (Figure 1E), as the mean fluorescence intensity in WT recipients was approximately 600 in WT mice and dropped to 350 in both groups of Apoa1 transgenic mice. Thus, plasma lipoprotein profiles and cholesterol efflux from macrophages were not significantly affected by lymphatic separation, indicating that the substantial reduction in RCT observed after surgically blocking lymphatic transport is attributable to a critical role of lymphatic vessels in transporting HDL-C during RCT.
Genetic ablation of lymphatic vessels disrupts RCT from skin.
So-called “Chy mice” carry 1 mutant allele of the VEGF-C receptor VEGFR3 that controls lymphatic vessel development but does not affect blood vessels (11, 24). Experiments in these mice would avoid the need for surgical separation of lymphatics and would instead comprise a genetic approach to addressing the quantitative importance of lymphatic vessels in RCT. Chy mice are reported to selectively lack lymphatic capillaries in skin (25). We observed that the loss of lymphatic vessels was complete in the limbs and ears of Chy mice, but that the body trunk skin retains some lymphatic vasculature (26). This retention in the body trunk resulted in normal immune cell trafficking through lymph, with molecular transport that was partially reduced, but less substantially so, than in the footpads or the ear (26). Chy mice, whether on their original mixed-strain background or backcrossed 10 generations onto the C57BL/6 background, had profoundly reduced RCT when they served as recipients of [3H]-cholesterol–loaded macrophages injected into the rear footpad. Mean reductions in plasma at 24 hours were 77% and 74% for the 2 Chy strains (Figure 2A), with a net reduction in RCT of 69% when we compiled [3H]-cholesterol counts in the liver, plasma, and feces (Figure 2B). By contrast, injection of [3H]-cholesterol macrophages in the body trunk skin where there were residual, though still reduced, lymphatic vessels led to only a mean 25% inhibition of RCT (Figure 2C), indicating that the impaired RCT correlated with the extent of lymphatic vessel loss of transport. Plasma lipoprotein profiles were similar (Figure 2D), but HDL was actually increased in Chy mice on the C57BL/6 background relative to their littermate controls (Figure 2D), making the impaired RCT in these mice even more striking. Thus, these data using a genetic model confirm the critical importance of lymphatic transport in RCT. The overall greater reduction in RCT in Chy mice compared with the surgical model may be due to substantial, but incomplete, blockade of lymphatic drainage in the surgical model (Figure 1A) relative to the genetic model.
Figure 2. RCT in Chy mutant mice.

(A) [3H]-Cholesterol–loaded macrophages were injected into the rear footpads of Chy mice or control littermates and tritium counts were assessed in plasma at 24 hours. Two background strains were compared; injected macrophages were made from the bone marrow of mice on the same background as the recipients into which they were injected. (B) Compiled tritium counts in plasma (black), liver (white), and feces (gray) from C57BL/6 Chy and WT mice euthanized 24 hours after [3H]-cholesterol–loaded macrophages were injected into the rear footpad. (C) Compiled tritium counts in plasma (black), liver (white), and feces (gray) from C57BL/6 Chy and WT mice euthanized 24 hours after [3H]-cholesterol–loaded macrophages were injected into the scapular area of the back skin. (D) Lipoprotein profiles in the plasma of Chy mice and littermate controls from the mixed or C57BL/6 backgrounds. Each panel represents 2 experiments performed with 5 replicates per experimental group (mean ± SEM). Statistics compared Chy mice with their control littermates in each case. *P < 0.05; **P < 0.01; ***P < 0.001.
Vascular leakiness does not obviate the importance of lymphatic vessels in RCT.
We next examined whether lymphatic vessels would still retain an important role in RCT at sites where the blood vasculature was leaky, potentially increasing direct transfer of HDL into the vasculature. To set up a tractable model with a leaky vasculature as a defining feature, we took advantage of the fact that tumors have a leaky vasculature. Lewis Lung Carcinoma (LLC) cells were allowed to form tumors in the ear pinnae of Chy mice or their littermate controls (Figure 3A). Extravasated Evans blue dye within the ear, measured 30 minutes after i.v. injection, confirmed enhanced vascular leakage in the tumor and surrounding tissue (Figure 3B). [3H]-Cholesterol–loaded macrophages were injected adjacent to the tumor, upstream with respect to lymphatic flow through the tumor-bearing skin, as indicated in Figure 3A. A marked reduction in RCT (54%) was observed in Chy mice (Figure 3C), indicating that lymphatic vessels remain dominant conduits for RCT, even in tissues characterized by a leaky vasculature.
Figure 3. Impact of vascular leakage on RCT through lymphatic vessels.

(A) LLC cells were injected into the ear pinnae of Chy and control littermates (mice on a mixed background were used). Tumors grew for 3 weeks before [3H]-cholesterol–loaded macrophages were injected as shown. (B) Increased vascular permeability was documented using Evans blue dye injected i.v. with extraction of ear tissue 30 minutes later to quantify dye recovery using a spectrophotometer. (C) Analysis of tritium counts in the plasma 24 and 48 hours later. Tumor-bearing mice were housed by requirement under biohazard containment that precluded the use of individual caging needed to collect feces. Each panel represents 2 experiments performed with 5 replicates per experimental group. Data represent the mean ± SEM comparing indicated conditions (B) or Chy and control mice at each time point (C). *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001.
RCT from atherosclerotic aortae is impaired by anti-VEGFR3 mAb that attenuates lymphatic vessel growth.
To assess the potential relevance of findings in skin to the mode of cholesterol transport in the aortic wall, lymphatic vessels were first identified in the adventitia and surrounding adipose tissue of the entire aorta of WT mice and in Apoe–/– mice with atherosclerosis (Figure 4A). These vessels were absorptive lymphatic capillaries, as opposed to lymphatic collecting vessels that transport lymph but do not absorb, since they stained for both LYVE-1 and podoplanin (11, 27, 28). The identified lymphatic capillaries wove in and out of the adjacent adipose tissue and were therefore difficult to keep completely intact when dissecting the aorta. A lymphatic capillary in the adventitia of the lesser curvature proximal to the aortic sinus was consistently observed (Figure 4, B and C, arrow). Lymphatic vessels around the arteries that branch off the aortic arch were observed, but their locations appeared more inconsistent, as in Figure 4, B and C (arrowheads), which shows 2 specimens with lymphatic vessels around different branching arteries. Lymphatic vessels were richly abundant beneath plaques in the aortic sinus (Figure 4D), again surrounded by adipose tissue.
Figure 4. Lymphatic vessel distribution in the mouse aorta and aortic sinus.
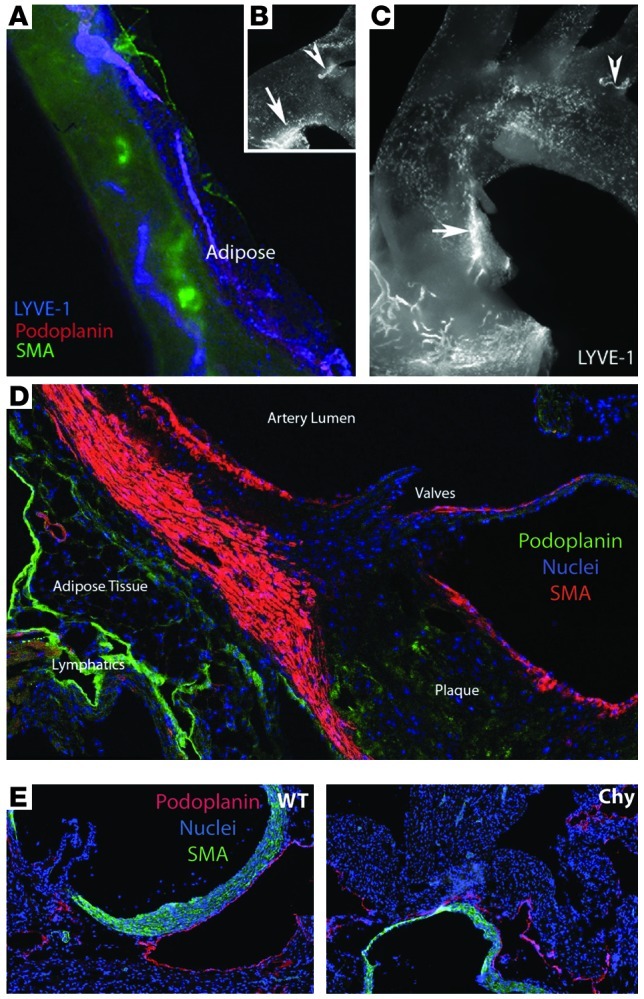
(A) The descending aorta is seen through its autofluorescence in the green channel. Vessels staining positively for podoplanin and LYVE-1 are seen on the adventitial side of the aorta, weaving in and out of adjacent adipose tissue. These vessels lack smooth muscle actin (SMA) coverage, though some nearby blood vessels are positive. LYVE-1+ SMA– lymphatic vessels are absorptive lymphatic capillaries, rather than lymphatic collecting vessels. (B and C) LYVE-1 staining in the region of the aortic arch. Arrows point to a lymphatic capillary along the lesser curvature; arrowheads point out lymphatic capillaries near arteries that branch off the arch. (D) Cross-sectional analysis of the aortic sinus in an Apoe–/– mouse reveals lymphatic vessels under the areas where plaque typically develops. (E) Both WT littermates and Chy mice develop lymphatic vessels in the aorta as depicted in these cross sections of the aortic sinus.
The surgical and genetic approaches used above are unsuitable for assessing whether lymphatic vessels mediate RCT from experimental atherosclerotic plaques. First, it is not technically feasible to surgically disconnect lymphatic vessels from the intact aorta. Second, in the genetic model of Chy mice, lymphatic vessels were not absent in the aortic sinus (Figure 4E) or the arterial tree (data not shown), but were absent in the skin. Indeed, Chy mice and other genetic models of lymphatic vessel loss appear to have relatively normal lymphatic vessels at non-skin sites (25, 29). To generate a tractable model for the study of RCT, we turned to a surgical model in which the aortic arch is transplanted into the abdominal cavity of a recipient mouse. When the recipient is WT and, therefore, has low LDL, this model supports robust disease regression (30–32). The appeal of the model in our case was that the surgery itself disrupts lymphatic connections and thus could be used to determine whether reestablishing lymphatic connections led to differences in RCT.
We have reported that lymphatic vessels are regenerated within 3 days or less in the adventitia of the surgical site, which drains lymph to the iliac lymph nodes (32). To better control the timing of cholesterol efflux in this model, we surgically transplanted aortic arches from Apoe–/– mice containing atherosclerotic plaques into the abdominal aortae of Apoe–/– recipients, rather than in WT recipients that will undergo spontaneous regression. In Apoe–/– recipients, which do not spontaneously regress (31), we could synchronously induce cholesterol efflux, as a key step in regression and a prerequisite for RCT, by administering an apoE expression vector at a given time point (1, 33). Immediately after surgery, one-half of the recipient mice were treated with neutralizing anti-VEGFR3 mAb AFL4 (34, 35) to selectively inhibit reformation of an intact draining lymphatic vasculature, or with isotype-matched control mAb that would allow lymphatics to reform. Prior to transplantation, donor aortae were loaded with stable isotope–labeled (deuterium-labeled) cholesterol ([2H]6-cholesterol; D6-cholesterol) using D6-cholesterol–rich liposomes delivered i.v. Delivery of D6-cholesterol to mice i.v. led to its rapid accumulation in multiple organs, including high levels in the spleen and liver initially (Figure 5A). D6-cholesterol was found mostly as unesterified cholesterol in spleen and liver (Figure 5A), suggesting that it either remained packaged in liposomes or was transiently incorporated into cell membranes. However, in the aorta, where D6-cholesterol levels remained stable for up to 2 weeks, D6-cholesterol increasingly entered the cholesterol ester pool, indicating that it was incorporating into foam cells (Figure 5A). In plasma, high levels of D6-cholesterol were observed as early as 10 minutes after D6-cholesterol delivery. By 2 weeks, most of the D6-cholesterol had cleared from the circulation and much of what remained was found in the pool of esterified cholesterol (Figure 5A), which in this case is most likely because cholesterol was esterified by plasma lecithin:cholesterol acyltransferase (LCAT).
Figure 5. Role of lymphatic vessels in RCT from aortae.

(A) Kinetics of D6-cholesterol were analyzed in duplicate 1 or 14 days after liposomes containing D6-cholesterol were administered i.v. Solid lines correspond to the left-hand y-axis that plots ng of D6-cholesterol detected in aortae. Data depicted by dotted lines correspond to the right-hand y-axis that plots the esterified/total D6-cholesterol ratio. (B) Schematic diagram of the aortic transplant positioned in the abdominal aorta. Green box depicts the portion of tissue collected for mass spectrometric (MS) analysis; orange box shows the portion of tissue collected for imaging lymphatic regrowth. (C) Images of 2 independent pairings of mice transplanted on the same day, 1 treated with control mAb and the other with anti-VEGFR3 mAb; stained for LYVE-1 (white vessels). Stars depict location of the lumen of the transplanted aortic arch where it connects with the original aorta. Arrowheads point to surgical sutures that demarcate the boundary between the transplant (Tx) and the original aorta (OA). Upper images show a pairing from 2 different orientations and lower images show a side view of a different surgical pair. Scale bars: 250 μm. (D) Ester/total D6-cholesterol achieved at baseline prior to transplantation and ratios recovered in 5 pairs of transplants 4 weeks after surgery, treated with apoE vector at 2 weeks and with control or anti-VEGFR3 throughout (E and F). Total (E) and esterified (F) D6-cholesterol recovered in pairs treated with control or anti-VEGFR3 mAb. D and E depict data as the mean ± SD (n = 5–9 for each bar). *P ≤ 0.05.
Two weeks after transplantation and treatment with anti-VEGFR3 or control mAb, we injected Apoe–/– recipients with an apoE-encoding adenoviral vector (36) to induce cholesterol efflux from plaque macrophages while mAb treatment continued. Previous results suggest that within 2 weeks cholesterol esters would be reduced within plaques as they undergo mobilization from macrophages (1). Therefore, 2 weeks after apoE vector treatment, we harvested transplanted aortae and quantified D6-cholesterol by mass spectrometry. The transplanted aortic segment was collected for this analysis, with most of the transplanted segment used for mass spectrometric analysis and only a margin of the transplant and its connection to the original aorta was retained for imaging (Figure 5B). Two transplants were carried out per day with 1 recipient in the pair receiving control mAb and the other receiving anti-VEGFR3 mAb. Images of the transplant interface near the original aorta, shown here for 2 of the transplant pairs, revealed vigorous lymphatic growth associated with transplantation (Figure 5C). The new vessels were differently oriented than the preexisting vessels (compare Figure 4A with Figure 5C). This difference likely reflects altered blood flow associated with the transplant, as blood flow would determine patterns of interstitial fluid flow through the aortic wall, in turn influencing lymphangiogenesis patterning (37). Lymphatic regrowth after transplantation was inhibited, albeit not completely, with anti-VEGFR3 mAb (Figure 5C). Anti-VEGFR3 mAb treatment did not lead to impaired lymphatic transport when assessed in skin (Supplemental Figure 3), suggesting that its main action was on the redevelopment of lymphatics rather than on the transport function of existing vessels. However, it was not possible to inject lymph flow tracers into the aortic wall to quantify lymph transport locally.
As for the fate of D6-cholesterol, a very high proportion of D6-cholesterol was located in the ester pool prior to transplantation (baseline) (Figure 5D), indicating that it was largely confined to foam cells or intact lipoproteins in the aortic wall, rather than less relevant compartments such as the medial smooth muscle wall where it would be found as free D6-cholesterol. Even with partial inhibition of lymphatic regrowth, anti-VEGFR3 mAb treatment led to a greater retention of D6-cholesterol in the transplanted aortae compared with those treated with control mAb (Figure 5E), especially within the ester pool of D6-cholesterol (Figure 5, D and F). Since Vegfr3 mRNA was not expressed in plaque macrophages (Supplemental Figure 4 and Supplemental Methods), these data indicate that VEGFR3-mediated reformation of lymphatic connections in the aorta mediates RCT.
Discussion
With only a handful of clinical or experimental observations, a few physicians as far back as the early 1980s have discussed the logic of considering that a connection between atherosclerosis and decreased lymph transport of cholesterol deserves attention (38–40). However, to date, the quantitative importance of lymphatic vessels in RCT has not been examined, aside from a compelling study that estimated that the net flux of HDL-C through human lymph is substantial (14). With respect to atherosclerosis, the process of macrophage RCT is most relevant, and many details have emerged as to how cholesterol is mobilized from macrophages (6). Removal of cholesterol from tissues where macrophages reside for subsequent return to the plasma and then the liver en route to fecal secretion seems to logically involve lymphatic vessels. Indeed, lymphatic vessels have widespread importance in macromolecular transport from peripheral tissues in general.
Here, we provide experimental evidence that lymphatic vessels are critical during macrophage RCT to bring lipidated apoA-I or HDL-C to the plasma. We started our analysis in skin and chose to employ a model where we could surgically disrupt lymphatic transport from the mouse tail. This procedure necessitated the injection of cholesterol-loaded macrophages into the mouse tail, a site distinct from the peritoneal cavity where this assay for macrophage RCT was originally developed (6). It became clear that macrophage RCT from the tail site was far less efficient than RCT from the peritoneum and that other body sites, like the footpad, were intermediate. This finding illustrates that different tissue environments vary in their ability to support RCT and raises important considerations that have yet to be explored, including whether RCT from atherosclerotic plaque is quantitatively more similar in the mouse tail or in the peritoneum; whether the rather low RCT in the tail is due to tight macrophage packing in the tail (as occurs in atherosclerotic plaque) that might limit apoA-1 access to the cells compared with dispersion of macrophages in the peritoneum; or whether apoA1 levels in interstitial fluid vary between organs or body sites.
We were able to counter poor macrophage RCT from the tail by using Apoa1 transgenic mice and went on to uncover a quantitatively important role for lymphatic vessels in RCT that could not be attributed to altered cholesterol efflux from macrophages. When tail lymphatic vessels were surgically separated, we observed a reduction in RCT of 50%. This reduction in RCT compared similarly to the magnitude of inhibition observed from compound inhibition of ABCA1 and ABCG1 (19), key transporters for the donation of free cholesterol to apoA-I during macrophage cholesterol efflux. An even more dramatic inhibition of RCT was observed in the Chy mouse strain, which bears 1 copy of mutant VEGFR3 with attenuated signaling. This loss of only 1 functional VEGFR3 receptor is sufficient to prevent lymphatic vessels from developing in many skin sites (29), most notably the extremities (26). However, at other body sites, lymphatic vessels are not attenuated unless both copies of VEGFR3 are lost, a condition that is incompatible with survival during embryogenesis (41). Indeed, even in some skin sites, Chy mice have residual lymphatic vessels (26), allowing us to show that the magnitude of inhibited RCT correlated with the extent to which lymphatic vessels were absent from the skin surrounding the site where cholesterol-loaded macrophages were injected.
We next considered the role of lymphatic vessels in RCT in the aortic wall. Technically, this issue is challenging and none of the models used in skin were valuable in the atherosclerosis-affected aorta. We reasoned that a previous surgical model used to study plaque regression had features necessary to address the main technical obstacles (confining the D6-cholesterol tracer to the aortic wall and disrupting the lymphatics, a natural consequence of the surgery). The lymphatic regrowth that occurred in the controls was impressive, and this robust response may contribute to why the surgical model allows for what appears to be more aggressive regression than other models (1, 42). Unfortunately, it is not technically possible to quantify lymphatic transport from the aorta to determine whether anti-VEGFR3 mAb also affected lymph transport in the aortic wall, but transport was not affected systemically. Indeed, it has been established in a variety of scenarios that anti-VEGFR3 blocks the growth of new lymphatic vessels but does not affect existing lymphatic vessels (35, 43–46). Thus, the partial inhibition of lymphatic regrowth that we observed in the aortae of anti-VEGFR3 likely accounts for its effect on retaining cholesterol in the wall of the transplanted aortic segments relative to the controls. As we took measures to ensure that the D6-cholesterol was largely in the esterified pool at the time of transplantation, the cholesterol we traced originated either in macrophages or in intact lipoproteins, which are the two dominant pools of cholesterol ester directly relevant to our study. Cholesterol-laden macrophages and trapped lipoproteins are far more abundant in intimal plaques than in adventitia. Indeed, past studies have measured cholesterol esters in the aorta as a surrogate for atherosclerotic plaque (47, 48). Thus, our findings support the concept that clearance of cholesterol by RCT from the arterial intima relies on lymphatic vessels.
Except in the most advanced plaques, where lymphatic capillaries can invade the intima along with the vasa vasorum (49), this lymphatic vessel–driven mode of RCT would likely require that HDL-C cross the media to gain access to the adventitia. The literature suggests that such movement may preferentially occur through pressure-driven convection (50, 51) and that hydraulic conductivity across the media is increased in the context of hypercholesterolemia (52). Future studies are needed to continue to investigate the role of lymphatic vessels in atherosclerosis and RCT. However, the present findings indicate that the notion of lymphatic vessels as a conduit for cholesterol transport from atherosclerotic plaques is a viable concept and that lymphatic vessel biology in the area of atherosclerosis needs increased attention.
In past studies, we found that lymphatic transport is decreased by hypercholesterolemia in Apoe–/– mice (27), and studies in humans revealed that the rate of lymphatic transport was inversely proportional to cholesterol traversing the lymph (12). This reduced transport that occurs in the presence of high cholesterol may promote cholesterol accumulation in the artery wall, as well as in tissues like skin that are affected in hypercholesterolemia. Even during conditions that support disease regression, cholesterol removal from plaques is not as rapid as other steps in disease reversal. In particular, we were surprised to find that monocyte recruitment is greatly reduced at a time when cholesterol clearance from plaques is just getting underway (1). While the observation may suggest that removal of bulk cholesterol is only part of what is needed to reverse the progression of atherosclerosis, we view the sluggish removal of cholesterol from plaques as a risk for allowing retained cholesterol to readily return to a state that supports plaque progression. We argue that a more desirable disease-protective state is one in which cholesterol is mobilized from the aortic wall in concert with treatments that support lesion stabilization or regression. Thus, our next step is to identify strategies to restore the impaired lymphatic transport that results from hypercholesterolemia and to determine whether such strategies can serve to enhance the mobilization of cholesterol out of plaques when disease-reversing regimens are concurrently applied.
Methods
Mice.
WT and Apoa1 transgenic mice (both C57BL/6) were obtained from the National Cancer Institute (Frederick, Maryland, USA) and The Jackson Laboratories (Bar Harbor, Maine, USA), respectively. Chy embryos were purchased from the Medical Research Council (MRC, Harwell, United Kingdom) on a mixed C3H/HeH and 101/H background. A colony was established and backcrossed to the C57BL/6 background. Since 1 mutant allele is sufficient to confer loss of skin lymphatics (25), all control mice were littermates bearing no copies of the mutations. Macrophages lacking Abca1 and Abcg1 were obtained from mice housed at Columbia University. Experiments were approved by the animal oversight committees at Mount Sinai School of Medicine or Washington University.
Separation of lymphatic vessels in the mouse tail.
We utilized a surgical technique to separate the dermal lymphatics while leaving the primary blood circulation intact along the tail core (20). Briefly, tails were incised circumferentially at 2 locations spaced 2 mm apart, with the proximal incision 1 cm from the base of the tail through the skin. Skin between the 2 incisions was removed sufficiently to separate the lymphatic vasculature. Extreme caution was taken to avoid damaging the underlying vasculature. As a sham control, the tip of the tail was clipped to create an acute-phase reaction without lymphedema. Plasma lipoproteins were analyzed as described (1). In some mice, Evans blue dye was injected immediately after surgery to verify the block in lymph drainage. For RCT studies, macrophages were injected immediately after the surgical separation of lymphatics or sham, avoiding significant lymphedema during the 48-hour assay.
Cholesterol-loaded macrophages and efflux using BODIPY-cholesterol.
Total bone marrow was obtained from mice by flushing femurs and tibiae of mice with RPMI-1640. L929-conditioned medium was used to generate bone marrow–derived macrophages (BMDMs) over a 6-day culture period. To induce foam cell formation and label the intracellular cholesterol pool, BMDMs were loaded on the sixth day of culture for 24 or 48 hours in the presence of 100 μg/ml acetylated LDL (acLDL) that was previously incubated for 30 minutes at 37°C with 5 μM BODIPY-cholesterol (23) (dissolved in ethanol) or 2.5 μCi/ml [3H]-cholesterol (PerkinElmer), respectively. After 24 or 48 hours, respectively, cells were removed from the culture dish using nonenzymatic cell dissociation medium (Cellgro, Corning), washed twice with RPMI-1640, and equilibrated in 0.2% BSA for 4 hours before injection. When cholesterol efflux was assessed in vitro, BODIPY-cholesterol–loaded macrophages were cultured for 20 hours in 5% mouse serum. Cholesterol efflux was assessed directly on a single-cell level by fluorescence microscopy or flow cytometry. After implantation in vivo, transferred macrophages were retrieved from the skin by digesting the skin with Liberase TM (Roche Applied Science) and staining for CD45.1 with exclusion of dead cells using propidium iodide.
In vivo RCT assay.
To assess in vivo RCT, a total of 2.0 × 106 [3H]-cholesterol–labeled BMDMs containing 7 × 105 cpm were injected per mouse. Cells were injected either i.p. (500 μl) or intracutaneously (20 μl) into the footpad, back skin, or tail skin as indicated in Figures 1–3. Then mice were housed individually in clean cages to allow for the collection of fresh feces 24 and 48 hours later. As in previous work (18), blood was collected at 24 and 48 hours, and plasma volume was measured and used for liquid scintillation counting. Feces were collected at 24 and 48 hours, as indicated in Figures 1–3. Feces and livers were also weighed and subjected to liquid scintillation counting.
Lipoprotein analysis.
Total cholesterol levels were determined using a Wako kit (catalog no. 439-17501) according to the manufacturer’s protocol. Lipoprotein cholesterol distributions were determined using plasma samples diluted 1:1 in cold PBS centrifuged at 10,000 g for 2 minutes at 4°C. Approximately 15 mg of cholesterol was injected into an HPLC system with online mixing of enzymatic reagent (Cholesterol Liquid Reagent Set, Pointe Scientific) with effluent from the column. The HPLC system consisted of a Superose 6 10/300 GL column (GE Healthcare); a LaChrom Elite HPLC system (Hitachi High Technologies) consisting of an L-2200 Autosampler with Peltier cooling, an L-2420 UV-Vis Detector, and two L-2100 SMASH pumps — 1 for delivering 0.9% saline with 0.01% EDTA and 0.01% sodium azide at 0.4 ml/min to the column, and the other for delivering cholesterol reagent (or water) at 0.125 ml/min; a 5 m (length) × 0.5 mm (internal diameter) knitted reaction coil (KRC 5-50; Aura Industries) in a water jacket (CJB-10; Aura Industries) at 37°C; an Isotemp circulating water bath (Fisher Scientific); a PEEK Tee (Upchurch part no. P-712) at the junction of column effluent and total cholesterol reagent; and a personal computer running ChromPerfect Spirit Chromatography Data System software, version 5.5 (Justice Laboratory Software) for data acquisition and analysis.
Tumors and vascular leakage assay.
LLC cells (1.0 × 106 or 5.0 × 105) were injected into the skin of the ear and allowed to grow for 3 weeks. A total of 5.0 × 106 [3H]-cholesterol–labeled foam cells containing 1.2 × 106 cpm were injected intradermally in a single injection of 10 μl in the tip of the ear near the tumor. To assess vascular leakage, mice were anesthetized and 200 μl of 2% Evans blue dye (Sigma-Aldrich) was injected into the orbital vein. Thirty minutes later, the mice were euthanized and their ears were removed, and the blue dye was extracted from skin using N,N-dimethylformamide (Sigma-Aldrich) for 24 hours at 56°C and measured with a spectrophotometer (630 nm).
Aortic transplant procedure.
Liposomes were prepared using cholesterol-26,26,26,27,27,27-d6 (D6-cholesterol) obtained from Sigma-Aldrich and 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) obtained from Avanti Polar Lipids. Ten milligrams (0.0255 mmol) of D6-cholesterol and 200 mg (0.263 mmol) of POPC were dissolved in chloroform, mixed, dried onto the bottom of 13 × 100 mm glass tubes, and dried under vacuum overnight. The next day, 6 ml of sterile saline was added to the tubes, which were incubated for 1 hour at 40°C. The tubes were vortexed on a mechanical agitator for approximately 2–3 hours. Liposomes were stored at room temperature until ready for injection.
Nineteen to 21-week-old Apoe–/– donor mice, placed on a Western diet at 6 weeks of age (Harlan Teklad 88137), received a 250 μl bolus injection of a suspension of liposomes containing D6-cholesterol. Following treatment, these donors were housed for 2 weeks to allow esterification of D6-cholesterol in plaques. Then heterotopic aortic transplants of the aortic arch (53) were carried out in similarly aged and Western diet–fed Apoe–/– recipients. Two surgeries were conducted each day. Immediately after surgery, the 2 mice were given 1.2 mg of AFL4 anti-VEGFR3 mAb (LEAF Purified; BioLegend) or isotype-matched control mAb (rat IgG2a, LEAF Purified; Biolegend) i.p., respectively, and 0.6 mg every other day for a total of 4 weeks. Two weeks after surgery, pairs were treated with apoE-encoding adenoviral vector (SignaGen) (1, 33). Pairs were euthanized 2 weeks after vector treatment, and the transplant tissue was harvested for mass spectrometry or imaging as specified. Liver and plasma samples were also collected, but D6-cholesterol levels were too low for detection at the time of collection in these tissues.
Cholesterol analysis by GC-MS.
Sample preparation was conducted by a modification of a previously reported procedure (54). Briefly, lipids were extracted according to Bligh and Dyer (55) after adding 4 μg of [3,4-13C2]-labeled cholesterol as the internal standard. Extracts were evaporated under a stream of argon, dissolved in 1 ml of chloroform/methanol (1:1) and stored at –80°C until analyzed. To measure free cholesterol, a 50-μl aliquot of each extract was evaporated under argon, and the residue was dissolved in toluene and analyzed using a Finnigan TSQ Quantum XLS mass spectrometer interfaced to a Trace gas chromatograph. To measure the total cholesterol, another 50-μl aliquot was saponified at room temperature for 2 hours with methanolic KOH, cooled, and then extracted with hexane. The hexane was evaporated under argon, and the residue was dissolved in toluene and analyzed.
Samples of 1 μl each were injected onto a 250 μm × 10 m DB-1 capillary column (J&W Scientific, Agilent Technologies). Helium was the carrier gas at a constant flow rate of 1.0 ml per minute. The SSL injector temperature was 280°C, the interface 260°C, and the source 260°C. An oven temperature program was used: 150°C for 1 minute, increased at 25°C per minute to 215°C, followed by a 50-minute hold, then increased at 35°C per minute to 260°C, followed by a 3.8-minute hold. The thermo triple quadrupole (TSQ) was operated with the electron impact source in the positive-ion mode with a potential of 42 eV. Data were acquired in the selective ion-reaction mode monitoring the following decompositions for cholesterol: 386.3 m/z > 301.3 m/z, for [3,4-13C2]-cholesterol; 388 m/z > 301.3 m/z and 392.3 m/z > 307.3 m/z for [26,26,26,27,27,27-d6]-cholesterol. The collision cell energy was 10 V and the gas pressure was 1.0 mTorr. Corrections were made for the 13C natural abundance. Ester cholesterol is reported as the difference between total cholesterol and free cholesterol.
Statistics.
Data are expressed as the mean ± SEM or SD, as indicated. Statistical differences were assessed using a 2-tailed Student’s t test (when comparing 2 datasets) or ANOVA (3 or more datasets) with Prism software, version 4.2 (GraphPad).
Supplementary Material
Acknowledgments
We thank Willfried Le Goff and Fei Wang for assistance and technical advice. This work was funded by NIH grant R01 HL-096539 (to G.J. Randolph and M.A. Swartz with a subcontract to M.G. Sorci-Thomas), with additional support from NIH grant R01 HL-64163 (to M.G. Sorci-Thomas), and an American Heart Association Established Investigator Award (to G.J. Randolph). M. Westerterp was supported by a grant from the Netherlands Organization of Scientific Research (NWO VENI 916.11.072) and holds an additional affiliation with the Academic Medical Center, Department of Medical Biochemistry, University of Amsterdam. Analyses performed in the Mass Spectrometer Facility of the Comprehensive Cancer Center of Wake Forest University School of Medicine were supported in part by NCI Center Grant 5P30CA12197. The Finnigan TSQ Quantum XLS GC/MS/MS was purchased with funds from NIH Shared Instrumentation Grant 1S10RR027940MS (to M.J. Thomas). We thank the Genome Technology Access Center in the Department of Genetics at Washington University School of Medicine for help with the genomic analysis. The Center is partially supported by NCI Cancer Center Support Grant P30 CA91842 to the Siteman Cancer Center and by ICTS/CTSA Grant UL1RR024992 from the National Center for Research Resources (NCRR), a component of the NIH and NIH Roadmap for Medical Research. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NCRR or NIH.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Citation for this article: J Clin Invest. 2013;123(4):1571–1579. doi:10.1172/JCI63685.
See the related Commentary beginning on page 1417.
References
- 1.Potteaux S, et al. Suppressed monocyte recruitment drives macrophage removal from atherosclerotic plaques of Apoe–/– mice during disease regression. . J Clin Invest. 2011;121(5):2025–2036. doi: 10.1172/JCI43802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Spann NJ, et al. Regulated accumulation of desmosterol integrates macrophage lipid metabolism and inflammatory responses. Cell. 2012;151(1):138–152. doi: 10.1016/j.cell.2012.06.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Duewell P, et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. 2010;464(7293):1357–1361. doi: 10.1038/nature08938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hermansson A, et al. Inhibition of T cell response to native low-density lipoprotein reduces atherosclerosis. J Exp Med. 2010;207(5):1081–1093. doi: 10.1084/jem.20092243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Small DM, Bond MG, Waugh D, Prack M, Sawyer JK. Physicochemical and histological changes in the arterial wall of nonhuman primates during progression and regression of atherosclerosis. J Clin Invest. 1984;73(6):1590–1605. doi: 10.1172/JCI111366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang X, Rader DJ. Molecular regulation of macrophage reverse cholesterol transport. Curr Opin Cardiol. 2007;22(4):368–372. doi: 10.1097/HCO.0b013e3281ec5113. [DOI] [PubMed] [Google Scholar]
- 7.Temel RE, et al. Biliary sterol secretion is not required for macrophage reverse cholesterol transport. Cell Metab. 2010;12(1):96–102. doi: 10.1016/j.cmet.2010.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Temel RE, Brown JM. Biliary and nonbiliary contributions to reverse cholesterol transport. Curr Opin Lipidol. 2012;23(2):85–90. doi: 10.1097/MOL.0b013e3283508c21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cuchel M, Rader DJ. Macrophage reverse cholesterol transport: key to the regression of atherosclerosis? Circulation. 2006;113(21):2548–2555. doi: 10.1161/CIRCULATIONAHA.104.475715. [DOI] [PubMed] [Google Scholar]
- 10.Hamon Y, et al. ABC1 promotes engulfment of apoptotic cells and transbilayer redistribution of phosphatidylserine. Nat Cell Biol. 2000;2(7):399–406. doi: 10.1038/35017029. [DOI] [PubMed] [Google Scholar]
- 11.Alitalo K. The lymphatic vasculature in disease. Nat Med. 2011;17(11):1371–1380. doi: 10.1038/nm.2545. [DOI] [PubMed] [Google Scholar]
- 12.Cooke CJ, Nanjee MN, Stepanova IP, Olszewski WL, Miller NE. Variations in lipid and apolipoprotein concentrations in human leg lymph: effects of posture and physical exercise. Atherosclerosis. 2004;173(1):39–45. doi: 10.1016/j.atherosclerosis.2003.07.004. [DOI] [PubMed] [Google Scholar]
- 13.Nanjee MN, Cooke CJ, Olszewski WL, Miller NE. Concentrations of electrophoretic and size subclasses of apolipoprotein A-I-containing particles in human peripheral lymph. Arterioscler Thromb Vasc Biol. 2000;20(9):2148–2155. doi: 10.1161/01.ATV.20.9.2148. [DOI] [PubMed] [Google Scholar]
- 14.Nanjee MN, Cooke CJ, Olszewski WL, Miller NE. Lipid and apolipoprotein concentrations in prenodal leg lymph of fasted humans. Associations with plasma concentrations in normal subjects, lipoprotein lipase deficiency, and LCAT deficiency. J Lipid Res. 2000;41(8):1317–1327. [PubMed] [Google Scholar]
- 15.Nanjee MN, Cooke CJ, Wong JS, Hamilton RL, Olszewski WL, Miller NE. Composition and ultrastructure of size subclasses of normal human peripheral lymph lipoproteins: quantification of cholesterol uptake by HDL in tissue fluids. J Lipid Res. 2001;42(4):639–648. [PubMed] [Google Scholar]
- 16.Dietschy JM, Turley SD. Control of cholesterol turnover in the mouse. J Biol Chem. 2002;277(6):3801–3804. doi: 10.1074/jbc.R100057200. [DOI] [PubMed] [Google Scholar]
- 17.Rohrer L, Cavelier C, Fuchs S, Schluter MA, Volker W, von Eckardstein A. Binding, internalization and transport of apolipoprotein A-I by vascular endothelial cells. Biochim Biophys Acta. 2006;1761(2):186–194. doi: 10.1016/j.bbalip.2006.01.009. [DOI] [PubMed] [Google Scholar]
- 18.Zhang Y, Zanotti I, Reilly MP, Glick JM, Rothblat GH, Rader DJ. Overexpression of apolipoprotein A-I promotes reverse transport of cholesterol from macrophages to feces in vivo. Circulation. 2003;108(6):661–663. doi: 10.1161/01.CIR.0000086981.09834.E0. [DOI] [PubMed] [Google Scholar]
- 19.Wang X, et al. Macrophage ABCA1 and ABCG1, but not SR-BI, promote macrophage reverse cholesterol transport in vivo. J Clin Invest. 2007;117(8):2216–2224. doi: 10.1172/JCI32057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rutkowski JM, Moya M, Johannes J, Goldman J, Swartz MA. Secondary lymphedema in the mouse tail: lymphatic hyperplasia, VEGF-C upregulation, and the protective role of MMP-9. Microvasc Res. 2006;72(3):161–171. doi: 10.1016/j.mvr.2006.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Plump AS, Scott CJ, Breslow JL. Human apolipoprotein A-I gene expression increases high density lipoprotein and suppresses atherosclerosis in the apolipoprotein E- deficient mouse. Proc Natl Acad Sci U S A. 1994;91(20):9607–9611. doi: 10.1073/pnas.91.20.9607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Holtta-Vuori M, et al. BODIPY-cholesterol: a new tool to visualize sterol trafficking in living cells and organisms. Traffic. 2008;9(11):1839–1849. doi: 10.1111/j.1600-0854.2008.00801.x. [DOI] [PubMed] [Google Scholar]
- 23.Li Z, Mintzer E, Bittman R. First synthesis of free cholesterol-BODIPY conjugates. J Org Chem. 2006;71(4):1718–1721. doi: 10.1021/jo052029x. [DOI] [PubMed] [Google Scholar]
- 24.Saharinen P, Tammela T, Karkkainen MJ, Alitalo K. Lymphatic vasculature: development, molecular regulation and role in tumor metastasis and inflammation. Trends Immunol. 2004;25(7):387–395. doi: 10.1016/j.it.2004.05.003. [DOI] [PubMed] [Google Scholar]
- 25.Makinen T, et al. Inhibition of lymphangiogenesis with resulting lymphedema in transgenic mice expressing soluble VEGF receptor-3. Nat Med. 2001;7(2):199–205. doi: 10.1038/84651. [DOI] [PubMed] [Google Scholar]
- 26.Platt AM, et al. Normal dendritic cell mobilization to mouse lymph nodes under conditions of severe lymphatic hypoplasia. J Immunol. doi: 10.4049/jimmunol.1202600. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lim HY, et al. Hypercholesterolemic mice exhibit lymphatic vessel dysfunction and degeneration. Am J Pathol. 2009;175(3):1328–1337. doi: 10.2353/ajpath.2009.080963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Baluk P, et al. Functionally specialized junctions between endothelial cells of lymphatic vessels. J Exp Med. 2007;204(10):2349–2362. doi: 10.1084/jem.20062596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Karkkainen MJ, et al. A model for gene therapy of human hereditary lymphedema. Proc Natl Acad Sci U S A. 2001;98(22):12677–12682. doi: 10.1073/pnas.221449198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rong JX, et al. Elevating high-density lipoprotein cholesterol in apolipoprotein E-deficient mice remodels advanced atherosclerotic lesions by decreasing macrophage and increasing smooth muscle cell content. Circulation. 2001;104(20):2447–2452. doi: 10.1161/hc4501.098952. [DOI] [PubMed] [Google Scholar]
- 31.Reis ED, et al. Dramatic remodeling of advanced atherosclerotic plaques of the apolipoprotein E-deficient mouse in a novel transplantation model. Vasc Surgery. 2001;34(3):541–547. doi: 10.1067/mva.2001.115963. [DOI] [PubMed] [Google Scholar]
- 32.Llodra J, Angeli V, Liu J, Trogan E, Fisher EA, Randolph GJ. Emigration of monocyte-derived cells from atherosclerotic lesions characterizes regressive, but not progressive, plaques. Proc Natl Acad Sci U S A. 2004;101(32):11779–11784. doi: 10.1073/pnas.0403259101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tsukamoto K, Smith P, Glick JM, Rader DJ. Liver-directed gene transfer and prolonged expression of three major human ApoE isoforms in ApoE-deficient mice. J Clin Invest. 1997;100(1):107–114. doi: 10.1172/JCI119501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kubo H, et al. Involvement of vascular endothelial growth factor receptor-3 in maintenance of integrity of endothelial cell lining during tumor angiogenesis. Blood. 2000;96(2):546–553. [PubMed] [Google Scholar]
- 35.Karpanen T, et al. Lymphangiogenic growth factor responsiveness is modulated by postnatal lymphatic vessel maturation. Am J Pathol. 2006;169(2):708–718. doi: 10.2353/ajpath.2006.051200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tsukamoto K, Tangirala R, Chun SH, Pure E, Rader DJ. Rapid regression of atherosclerosis induced by liver-directed gene transfer of ApoE in ApoE-deficient mice. Arterioscler Thromb Vasc Biol. 1999;19(9):2162–2170. doi: 10.1161/01.ATV.19.9.2162. [DOI] [PubMed] [Google Scholar]
- 37.Boardman KC, Swartz MA. Interstitial flow as a guide for lymphangiogenesis. Circ Res. 2003;92(7):801–808. doi: 10.1161/01.RES.0000065621.69843.49. [DOI] [PubMed] [Google Scholar]
- 38.Lemole GM. The role of lymphstasis in atherogenesis. Ann Thorac Surg. 1981;31(3):290–293. doi: 10.1016/S0003-4975(10)60949-6. [DOI] [PubMed] [Google Scholar]
- 39.Miller AJ, DeBoer A, Palmer A. The role of the lymphatic system in coronary atherosclerosis. Med Hypotheses. 1992;37(1):31–36. doi: 10.1016/0306-9877(92)90009-2. [DOI] [PubMed] [Google Scholar]
- 40.Solti F, Lengyel E, Jellinek H, Schneider F, Juhasz-Nagy A, Kekesi V. Coronary arteriopathy after lymphatic blockade: an experimental study in dogs. Lymphology. 1994;27(4):173–180. [PubMed] [Google Scholar]
- 41.Oliver G. Lymphatic vasculature development. Nat Rev Immunol. 2004;4(1):35–45. doi: 10.1038/nri1258. [DOI] [PubMed] [Google Scholar]
- 42.Williams KJ, Feig JE, Fisher EA. Rapid regression of atherosclerosis: insights from the clinical and experimental literature. Nat Clin Pract Cardiovasc Med. 2008;5(2):91–102. doi: 10.1038/ncpcardio1086. [DOI] [PubMed] [Google Scholar]
- 43.Yin N, et al. Lymphangiogenesis is required for pancreatic islet inflammation and diabetes. PLoS One. 2011;6(11):e28023. doi: 10.1371/journal.pone.0028023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pytowski B, et al. Complete and specific inhibition of adult lymphatic regeneration by a novel VEGFR-3 neutralizing antibody. J Natl Cancer Inst. 2005;97(1):14–21. doi: 10.1093/jnci/dji003. [DOI] [PubMed] [Google Scholar]
- 45.Rutkowski JM, Boardman KC, Swartz MA. Characterization of lymphangiogenesis in a model of adult skin regeneration. Am J Physiol Heart Circ Physiol. 2006;291(3):H1402–H1410. doi: 10.1152/ajpheart.00038.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chung ES, Saban DR, Chauhan SK, Dana R. Regulation of blood vessel versus lymphatic vessel growth in the cornea. Invest Ophthalmol Vis Sci. 2009;50(4):1613–1618. doi: 10.1167/iovs.08-2212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sparrow CP, et al. Simvastatin has anti-inflammatory and antiatherosclerotic activities independent of plasma cholesterol lowering. Arterioscler Thromb Vasc Biol. 2001;21(1):115–121. doi: 10.1161/01.ATV.21.1.115. [DOI] [PubMed] [Google Scholar]
- 48.Hermanowski-Vosatka A, et al. 11beta-HSD1 inhibition ameliorates metabolic syndrome and prevents progression of atherosclerosis in mice. J Exp Med. 2005;202(4):517–527. doi: 10.1084/jem.20050119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kholova I, et al. Lymphatic vasculature is increased in heart valves, ischaemic and inflamed hearts and in cholesterol-rich and calcified atherosclerotic lesions. Eur J Clin Invest. 2011;41(5):487–497. doi: 10.1111/j.1365-2362.2010.02431.x. [DOI] [PubMed] [Google Scholar]
- 50.Meyer G, Merval R, Tedgui A. Effects of pressure-induced stretch and convection on low-density lipoprotein and albumin uptake in the rabbit aortic wall. Circ Res. 1996;79(3):532–540. doi: 10.1161/01.RES.79.3.532. [DOI] [PubMed] [Google Scholar]
- 51.Kim WS, Tarbell JM. Macromolecular transport through the deformable porous media of an artery wall. J Biomech Eng. 1994;116(2):156–163. doi: 10.1115/1.2895714. [DOI] [PubMed] [Google Scholar]
- 52.Baldwin AL, Wilson LM, Gradus-Pizlo I, Wilensky R, March K. Effect of atherosclerosis on transmural convection an arterial ultrastructure. Implications for local intravascular drug delivery. Arterioscler Thromb Vasc Biol. 1997;17(12):3365–3375. doi: 10.1161/01.ATV.17.12.3365. [DOI] [PubMed] [Google Scholar]
- 53.Chereshnev I, et al. Mouse model of heterotopic aortic arch transplantation. J Surg Res. 2003;111(2):171–176. doi: 10.1016/S0022-4804(03)00039-8. [DOI] [PubMed] [Google Scholar]
- 54.Wilhelm AJ, et al. Apolipoprotein A-I and its role in lymphocyte cholesterol homeostasis and autoimmunity. Arterioscler Thromb Vasc Biol. 2009;29(6):843–849. doi: 10.1161/ATVBAHA.108.183442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bligh EG, Dyer WJ. A rapid method of total lipid extraction and purification. Can J Biochem Physiol. 1959;37(8):911–917. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.