

Abstract
Genetic disorders of the Ras/MAPK pathway, termed RASopathies, produce numerous abnormalities, including cutaneous keratodermas. The desmosomal cadherin, desmoglein-1 (DSG1), promotes keratinocyte differentiation by attenuating MAPK/ERK signaling and is linked to striate palmoplantar keratoderma (SPPK). This raises the possibility that cutaneous defects associated with SPPK and RASopathies share certain molecular faults. To identify intermediates responsible for executing the inhibition of ERK by DSG1, we conducted a yeast 2-hybrid screen. The screen revealed that Erbin (also known as ERBB2IP), a known ERK regulator, binds DSG1. Erbin silencing disrupted keratinocyte differentiation in culture, mimicking aspects of DSG1 deficiency. Furthermore, ERK inhibition and the induction of differentiation markers by DSG1 required both Erbin and DSG1 domains that participate in binding Erbin. Erbin blocks ERK signaling by interacting with and disrupting Ras-Raf scaffolds mediated by SHOC2, a protein genetically linked to the RASopathy, Noonan-like syndrome with loose anagen hair (NS/LAH). DSG1 overexpression enhanced this inhibitory function, increasing Erbin-SHOC2 interactions and decreasing Ras-SHOC2 interactions. Conversely, analysis of epidermis from DSG1-deficient patients with SPPK demonstrated increased Ras-SHOC2 colocalization and decreased Erbin-SHOC2 colocalization, offering a possible explanation for the observed epidermal defects. These findings suggest a mechanism by which DSG1 and Erbin cooperate to repress MAPK signaling and promote keratinocyte differentiation.
Introduction
As a barrier, skin protects organisms from environmental pathogens and dehydration, a function that relies heavily upon the unique characteristics of suprabasal, cornified keratinocytes. Mechanical insult or the normal process of desquamation results in constant shedding of the superficial keratinocytes, requiring, in turn, replenishment by a proliferative, basal layer of cells (1). Furthermore, in order to form a functional cornified layer, keratinocytes exiting the basal layer must undergo a program of morphological and biochemical transformations. Repression of ERK signaling constitutes a critical component of this process, as evidenced by cutaneous abnormalities associated with the MAPK-activating mutations linked to RASopathy disorders (2–7). However, the origin of inhibitory signals in keratinocytes is still not well understood (8, 9).
Our recent work suggested that desmosomes — intercellular adhesive structures that play a crucial role in epidermal integrity — may serve as an important hub for ERK inhibition (10). Desmosomes contain an array of cadherins divided into 2 subfamilies, desmocollins (DSC1–DSC3) and desmogleins (DSG1–DSG4). Isotypes of each subfamily demonstrate a differentiation-dependent distribution pattern (11). For example, whereas DSG3 is most abundant in the basal proliferating layer, DSG1 is first expressed as keratinocytes begin transiting to the suprabasal layers and concentrates in the superficial epidermis (12–14). In addition to a well-established role in mediating intercellular adhesion, we recently showed that DSG1 promotes differentiation by inhibiting EGFR/ERK signaling. Yet, this DSG1-dependent function does not require extracellular regions of DSG1 needed for adhesion (10).
These data led us to investigate the DSG1 C terminus as a potential scaffold for intracellular signaling events. A subset of patients with striate palmoplantar keratoderma (SPPK) are DSG1 deficient, with keratinization defects resembling, to an extent, cutaneous symptoms associated with RASopathies (2, 6, 7, 15–18). That these genetic disorders often arise from mutations in ancillary regulators of MAPK signaling highlights the importance of identifying proteins that provide the physical link between DSG1 cytoplasmic domains and the core MAPK machinery. Desmoglein cytoplasmic domains contain 2 membrane proximal domains, which are homologous to the intracellular anchor (IA) and intracellular cadherin sequence (ICS) of classic cadherins responsible for binding plakoglobin (Pg), a β-catenin relative. Beyond the ICS domain extends a relatively uncharacterized C terminus containing a proline-rich linker (PL), a set of repeating unit domains (RUDs), and a desmoglein terminal domain (TD) (11). A DSG1 mutant incapable of binding Pg retains the ability to induce differentiation, suggesting that a novel binding partner links DSG1 to ERK inhibition and, in turn, differentiation (10).
To address how DSG1 engages the EGFR/ERK signaling pathway, we used a yeast 2-hybrid CytoTrap screen using the DSG1 cytoplasmic domain as bait to identify possible signaling intermediates. The screen identified a modulator of ERK signaling known to localize at epidermal cell borders in a differentiation-dependent manner named Erbin (also known as ERBB2IP) (19, 20). Initially described as both a binding partner and mediator of proper ERBB2 localization, Erbin belongs to the LAP family of proteins that harbor leucine-rich repeats and PDZ domains (21). Earlier reports map ERBB2 binding sites to the C-terminal PDZ domain, while the central domains mediate an inhibitory interaction with SMAD3 (21, 22). The N terminus contains sites for fatty acid attachment thought to promote membrane localization as well as leucine-rich repeat domains responsible for interactions with Sur8 (also known as SHOC2) (23, 24). In addition to a previously identified ERBB2-dependent phosphorylation event (21), large-scale phosphoproteomic studies have identified numerous phosphorylated Erbin residues (25–33). Though several of these phosphorylation events are cell cycle dependent (26), the impact of phosphorylation on Erbin function remains unclear.
Prior studies indicate that Erbin supports ERK signaling through its interaction with ERBB2 in neuronal and cardiac tissue (34, 35). It has also been reported to dampen ERK by binding and inhibiting the scaffolding function of SHOC2, which activates ERK by facilitating upstream Ras-Raf interactions (36–39). However, a potential role for Erbin in regulating ERK signaling in the epidermis has not been addressed. Here, we show that DSG1 aids the disruption of Ras-SHOC2 complexes by Erbin, thus attenuating ERK signaling and driving differentiation. Illustrating the importance of this pathway, DSG1-deficient patients diagnosed with SPPK (MIM 148700) exhibited increased Ras-SHOC2 colocalization and decreased SHOC2-Erbin colocalization, as assessed by proximity ligation assays. These data support a model in which DSG1, by concentrating Erbin at the plasma membrane, inhibits ERK and supports differentiation via disruption of upstream SHOC2-Ras interactions.
Results
Erbin interacts and colocalizes with DSG1.
CytoTrap yeast 2-hybrid screening was used to identify DSG1 binding partners that could mediate its promotion of keratinocyte differentiation. This technique entails cotransforming yeast with a Sos-tagged bait (Sos-Dsg1-cyto) and a HeLa cell cDNA library containing constructs with 5′ sequences that translate into N-terminal myristoylation signals. Among the positives displaying galactose-dependent growth at 37°C, the screen identified an ERK regulator known to localize at epidermal cell borders in a differentiation-dependent manner named Erbin (19–21, 23, 34, 35). The Erbin clone encoded residues 1,119–1,371, corresponding to the PDZ-containing C terminus (Figure 1A), indicating that DSG1 binding does not require the N-terminal or central domains of Erbin previously shown to interact with SHOC2 and SMAD3, respectively (22, 23, 40). A truncation of DSG1 lacking the unique PL, RUD, and TD regions, though capable of mediating previously mapped interactions with Pg (41), failed to bind Erbin.
Figure 1. DSG1 interacts with Erbin in yeast 2-hybrid and in vitro binding assays.

(A) Yeast 2-hybrid analysis of interactions between the DSG1 cytoplasmic domain and the Erbin C terminus. Growth on galactose (GAL) at 37°C indicates an interaction; incubation at 37°C on glucose (GLU) serves as a control for temperature reversion; and incubation at 25°C on GLU represents a permissive growth condition. Interactions with a Myr-tagged Pg were used as positive controls. Plus and minus signs indicate the presence or absence of an interaction, respectively. (B) Increasing concentrations of purified GST-Dsg1(cyto) protein carrying the entire DSG1 cytoplasmic domain or a GST-only negative control were incubated with nickel agarose prebound to purified, His-tagged Erbin, corresponding to the C-terminal domain identified in the yeast 2-hybrid screen. Proteins precipitated with centrifuged beads were eluted with SDS and analyzed by Western blot. Sos, son of sevenless fusion.
Erbin and DSG1 both exhibit an affinity for members of the catenin family, raising the formal possibility that previously described catenin-like yeast proteins, such as Vac8 or importin-α/Srp1, mediate the DSG1-Erbin interaction (42–47). To address this possibility, we tested whether recombinant DSG1 and Erbin associate directly in vitro. When bound to nickel beads and incubated with GST or GST-Dsg1(cyto) encoding the DSG1 cytoplasmic domain, His-tagged Erbin (C terminus) specifically precipitated GST-Dsg1(cyto) (Figure 1B). This result suggests that DSG1 binds the C terminus of Erbin directly without an intervening catenin.
We next tested whether the DSG1-Erbin interaction occurs in normal human epidermal keratinocytes (NHEKs), a cell type that expresses DSG1 in vivo. Based on earlier studies that localized Erbin to cell junctions in the suprabasal epidermis, we predicted that DSG1 and Erbin would colocalize in keratinocytes (20). Accordingly, by immunofluorescence, DSG1 and Erbin colocalized in both cultured keratinocytes expressing exogenous DSG1 and human plantar skin, indicating that functional complexes could potentially form under physiological conditions (Figure 2, A–D). Since immunoprecipitation buffers solubilize only small amounts of endogenous DSG1, we immunoprecipitated ectopically expressed full-length FLAG-tagged Dsg1 (FL-Dsg1) to confirm a biochemical interaction (Figure 2E). Erbin specifically coprecipitated from NHEK lysate, with ectopic FL-Dsg1 isolated by anti-FLAG agarose (Figure 2F).
Figure 2. DSG1 colocalizes with and binds Erbin in human keratinocytes.

(A) Immunofluorescent staining of ectopic DSG1 and endogenous Erbin in cultured normal NHEKs. Scale bars: 10 μm. (B) Structured illumination microscopy (SIM) of ectopic DSG1 (red) and Erbin (green) in cultured NHEKs. Scale bars: 2 μm. (C) Staining of endogenous Erbin and DSG1 in human skin. The dotted line approximates the basement membrane. Scale bars: 20 μm. (D) SIM of DSG1 (red) and Erbin (green) in suprabasal keratinocytes of human skin. Arrowheads indicate sites where Erbin and DSG1 stains overlap. Scale bars: 1 μm. (E) DSG1 constructs used in immunoprecipitation studies. (F and G) Lysates from NHEKs transduced with retrovirus (RV) carrying GFP or DSG1 constructs were incubated with M2 (anti-FLAG) beads to precipitate ectopic DSG1 and associated proteins, followed by Western blot analysis. Stars mark putative degradation products. (H) Densitometric quantitation of G. Normalized to FLAG-tagged DSG1 input, Erbin precipitation with ectopic DSG1 is represented as fold change with respect to the amount precipitated with Δ381(WT). Blue samples represent data points from separate experiments. P values (Student’s t test) are indicated; mean ± SEM. EC, extracellular domain; TM, transmembrane domain; FLD, full-length DSG1; Δ381(WT), truncated DSG1 mimicking the exfoliative toxin cleavage product; Δ381(AAA), Pg binding-deficient version of Δ381(WT); Δ569, encodes only the DSG1 cytoplasmic domain; CoIP, coimmunoprecipitation.
To begin addressing which DSG1 domains are important for Erbin binding, we tested several previously published DSG1 truncation mutants (10, 48). These included an N-terminal truncation designed to mimic cleavage of DSG1 adhesive domains by bacterial toxins associated with bullous impetigo [Δ381(WT)] (14, 48). Truncated DSG1 retained an affinity for Erbin, as did a second construct combining the truncation with mutations engineered to disrupt DSG1-Pg interactions [Δ381(AAA)] (Figure 2, G and H). In contrast, ectopic DSG1 consisting only of the cytoplasmic domain (Δ569) did not interact with Erbin. The Δ569 construct demonstrates cytoplasmic localization and fails to induce differentiation according to prior studies (10). DSG1 then relies upon its membrane localization to both bind Erbin and induce differentiation. DSG1-Erbin interactions, however, do not depend on domains required for intercellular adhesion or Pg binding.
DSG1-induced keratinocyte differentiation requires Erbin.
If Erbin mediates DSG1-dependent promotion of keratinocyte differentiation, silencing Erbin should also impair this process. To test this hypothesis, NHEKs were treated with Erbin siRNA and assayed for their ability to differentiate in both 2-dimensional and 3-dimensional stratified organotypic cultures. Compared with cells transfected with nontargeting siRNA, lysates obtained from monolayer cultures treated with Erbin siRNA-1 or Erbin siRNA-2 contained lower levels of DSC1, a desmosomal cadherin with a differentiation-dependent expression pattern resembling that of DSG1 (Figure 3A and Supplemental Figure 1, A and B; supplemental material available online with this article; doi: 10.1172/JCI65220DS1). Additional markers of epidermal differentiation, including loricrin and keratin-10 (K10), responded similarly to Erbin knockdown, demonstrating reduced protein expression (Figure 3B and Supplemental Figure 1B). Erbin knockdown did not affect expression levels of keratin-5 (K5), a marker of basal keratinocytes, or E-cadherin, a core adherens junction component expressed throughout the epidermis (49). Erbin siRNA-1 was the primary reagent used for knockdown and is referred to here and in the figures simply as Erbin siRNA.
Figure 3. Efficient induction of keratinocyte differentiation requires Erbin.

(A) NHEKs treated with control (siNeg) or Erbin siRNA (siErbin) were differentiated by addition of CaCl2. Protein expression levels of desmosomal cadherins expressed during epidermal differentiation DSG1 and DSC1. GAPDH serves as a loading control. (B) Western blot of differentiation markers loricrin (Lor), K10, and DSC1. (C–H) NHEKs treated with siRNA were seeded on collagen plugs and raised to an air-liquid interface for 3 days (unless otherwise noted) to induce stratification and differentiation. (C) Costaining of K10, Erbin (gt anti-Erbin), and nuclei (DAPI). Arrowheads mark cells that escaped Erbin knockdown and retained K10. (D) Western blot of differentiation markers DSC1, involucrin (Inv), and K1. (E) H&E staining of cultures stratified for 6 days. (F) Western blot analysis of cultures with or without the MEK/ERK inhibitor, U0126. (G) Staining of pERK. (H) pERK staining intensity with respect to distance above the collagen substrate. Points represent the mean gray value plotted against distance from the NHEK-collagen border, pooled from 3 experiments. SEM is indicated every 1.67 μm on the graph. *P < 0.05. The dashed lines mark borders between NHEKs and the collagen substrate. Scale bars: 25 μm. E-cad, E-cadherin.
Three-dimensional cultures treated with Erbin siRNA were analyzed at 3 to 4 days following the initiation of stratification, a time when siRNA-mediated knockdown is maximal. Though this represents a relatively early time point in the development of stratified cultures, Erbin knockdown resulted in a detectable loss of K10 staining (Figure 3C). Consistent with our model, cell patches that evaded Erbin knockdown retained K10. Goat anti-Erbin (gt anti-Erbin) was used to assess Erbin knockdown in these experiments. Previously cited by Wilkes et al. (50) and Zhang et al. (51), this reagent recognizes a more restricted region within the Ab139 antigen and preferentially recognized suprabasal Erbin compared with the broader distribution revealed by Ab139 in Figure 2 (see below and Supplemental Figure 1A). It is possible that gt anti-Erbin stains a subset of protein detected by Ab139 because of greater sensitivity to masking by posttranslational modifications or incomplete reversal of formaldehyde-linked protein interactions. Three residues, for example, within the fifty-residue region containing the proprietary gt anti-Erbin epitope have been identified as phosphorylated, one of which (Ser934) was identified in a screen for cell cycle–dependent phosphorylation events (26, 28, 30). Nonetheless, at this stage of culture stratification, gt anti-Erbin signal was found throughout the cells. As the 3-dimensional cultures mature, the signal became more distinct and concentrated at cell borders in the intermediate layers (Supplemental Figure 2A).
Western blot analysis of 3-dimensional cultures corroborated the immunofluorescence and monolayer results, linking Erbin knockdown to reduced expression of multiple differentiation markers, including keratin-1 (K1), involucrin, and DSC1 (Figure 3D). Histological indicators of differentiation, such as granulation, stratum corneum formation, cell flattening, and denucleation, are best assessed at later time points when the cultures have more thoroughly stratified. Though the efficacy of Erbin siRNA typically fades by this point (~6 days after initiating stratification), lasting histological defects were observed in cultures for which knockdown at an earlier time point was confirmed in a parallel culture (Figure 3E and Supplemental Figure 2B). Decreased granulation and poor formation of the nascent stratum corneum were the most commonly observed defects, with a possible increase in intracellular vacuolation. Notably, addition of U0126, a MEK-ERK inhibitor, to 3-dimensional cultures in the background of Erbin knockdown restored differentiation markers (loricrin and DSC1 shown, Figure 3F), suggesting that, like DSG1, the contribution of Erbin to differentiation involves ERK inhibition. While Erbin is known to inhibit ERK, it has also been linked to TGF-β inhibition. However, treatment of monolayer cultures with the Alk5/TGF-β inhibitor, SB431542, failed to rescue differentiation defects stemming from Erbin knockdown (Supplemental Figure 1B).
While ERK inhibition restored differentiation, increases in phosphorylated active ERK1/2 (pERK) were not consistently observed in lysates from Erbin-deficient organotypic cultures. However, compared with controls, immunofluorescence analysis revealed that Erbin knockdown led to significant ERK activation in a restricted region of the intermediate suprabasal layers. Staining intensity of pERK was measured along lines drawn vertically from the position at which keratinocytes contact the collagen substrate (the in vitro equivalent of a basement membrane). Consistent with data linking ERK activation to proliferation in basal keratinocytes (52–56), both control and knockdown cultures exhibited high pERK staining intensity within the first 10 microns, encompassing the basal layer (mean distance to center of basal layer nuclei = 8.1 μm ± 0.7 μm [SEM]) (Figure 3, G and H). Erbin knockdown, though, was specifically associated with an additional peak of ERK activity in suprabasal cells located 10–40 microns above the substrate, corresponding, on average, to the second and third layers (mean distance to center of nuclei ± SEM: second layer = 18.9 μm ± 1.9 μm, third layer = 28.8 μm ± 2.2 μm).
An unexpected signal was detected in the most superficial cells of control and knockdown cultures. Though poorly understood, suprabasal staining of pERK has been reported in several cases, including normal retroauricular human skin, mouse cornea, and reepithelializing skin wounds (57–60). This signal and the spatially restricted nature of ERK activation upon Erbin knockdown may have contributed to the failure of immunoblot analysis to detect significant, reproducible pERK increases.
As Erbin knockdown activated ERK primarily in the layers immediately apical to basal keratinocytes and thus coincided with the initiation of DSG1 expression, we tested the extent to which Erbin participates in DSG1-dependent ERK inhibition and differentiation. The ability of ectopic DSG1 to accelerate differentiation in response to elevated calcium was exploited (10). Western blot analysis demonstrated that expression of FL-DSG1 elevated DSC1 and loricrin expression compared with that in GFP-infected cells (Figure 4, A and B). However, a DSG1 mutant (Dsg1-ICS) lacking the PL, RUD, and TD domains important for Erbin interactions failed to induce differentiation markers as effectively as FL-Dsg1. ERK inhibition, an essential component of the DSG1 phenotype, also required these domains, illustrated by the diminished capacity of Dsg1-ICS to reduce ERK phosphorylation (Figure 4C). The upregulation or stabilization of endogenous DSG1 by Dsg1-ICS, observed in Figure 4B, likely accounts for the partial inhibition of ERK presented in Figure 4C. Notably, treatment with Erbin siRNA blunted the inhibition of ERK phosphorylation and induction of K10 by FL-Dsg1, providing further evidence that DSG1 relies upon both the expression of Erbin and those domains that participate in Erbin binding to promote differentiation (Figure 4, C and D).
Figure 4. Inhibition of ERK and promotion of differentiation by ectopic DSG1 requires Erbin and cytoplasmic DSG1 domains involved in Erbin interactions.

(A) Diagram of FLAG-tagged, FLD and truncated DSG1 (ICS) constructs used in these experiments. (B) GFP, ICS, or FLD-infected NHEKs were induced to differentiate by switching cells from 0.07 to 1.2 mM CaCl2 for 48 hours, and whole cell lysates collected in urea/SDS buffer were analyzed by Western blot for differentiation markers DSC1 and loricrin. GAPDH serves as a loading control. (C) Confluent NHEKs carrying GFP, ICS, or FLD were treated with negative control or Erbin siRNA and induced to differentiate for 24 hours. Cells were lysed in urea/SDS buffer and analyzed by Western blot. Numbers in parentheses represent the fold change of phosphorylated ERK levels compared with the GFP control following normalization to total ERK. (D) Cells treated as in C were lysed in urea/SDS buffer and analyzed for the differentiation marker K10. GAPDH serves as a loading control.
DSG1 promotes differentiation by modulating the association of Erbin, SHOC2, and Ras.
Erbin interacts with ERBB2 as well as downstream Ras complexes. We previously reported that DSG1 knockdown results in elevated ERBB2 phosphorylation (10), raising the possibility that DSG1 inhibits ERK by disrupting upstream, Erbin-dependent ERBB2 function. However, while Erbin-null mice demonstrate defects in ERBB2 signaling, these defects stem from diminished ERBB2 stability rather than alterations in the phosphorylation status (34). Thus, the commonality between Erbin and DSG1 phenotypes likely lies not in the ability to regulate ERBB2 phosphorylation but downstream at the level of ERK inhibition.
To examine the mechanism by which DSG1 modulates or harnesses Erbin function, we first tested whether DSG1 expression affected Erbin distribution within the cell. Immunofluorescence analysis of Erbin localization in 3-dimensional NHEK cultures expressing control, Lamin shRNA, or DSG1 shRNA indicated that DSG1 deficiency resulted in mislocalized Erbin. In control cultures, Erbin was concentrated at intercellular junctions in the suprabasal layers. In DSG1-deficient cultures, Erbin localization became more diffuse, less concentrated at the cell periphery, and more cytoplasmic (Figure 5A). Furthermore, DSG1 expression altered the biochemical pool occupied by Erbin and associated protein complexes. Sucrose gradient centrifugation of lysate obtained from GFP- and FL-Dsg1–infected NHEKs treated with dithiobis(succinimidyl) propionate cross-linker prior to lysis indicated that ectopic expression of DSG1 altered the sedimentation rate of Erbin. Full-length DSG1 expression shifted Erbin away from the dense fractions toward the least dense fractions (Figure 5B). In contrast, the Dsg1-ICS mutant, which lacks domains required for binding Erbin in yeast 2-hybrid assays, failed to alter the Erbin fractionation pattern. As lysis and sedimentation took place in buffers containing urea and SDS, the shifted fractionation pattern likely indicates alterations in cross-linked protein-protein interactions driven by DSG1 expression. Together, these results indicate that DSG1 could modulate the capacity of Erbin to block ERK signaling by regulating its cellular localization and/or microenvironment.
Figure 5. DSG1 expression alters the localization and sedimentation rate of Erbin complexes.

(A) 3-dimensional NHEK cultures expressing either Lamin shRNA or DSG1 shRNA were analyzed by immunofluorescence (IF) for Erbin and DSG1 localization using Ab139 and 4B2, respectively. Large dashed lines represent the boundary between NHEKs and the collagen substrate; small dashed lines represent the upper tissue surface. The bottom right panel was disproportionately brightened to show small amounts of residual DSG1. Scale bars: 25 μm. (B) NHEKs expressing GFP, truncated DSG1 (ICS), or FLD were cross-linked with dithiobis succinimidyl propionate (DSP), lysed in urea/SDS buffer, and layered on top of a 5% to 20% sucrose gradient. Following centrifugation at 150,000 g for 18 hours, fractions were collected and treated with β-mercaptoethanol to reverse cross-links, and then analyzed for Erbin and ectopic FLAG-tagged DSG1 by Western blot. White lines separate additional fractions which were run on a parallel gel, transferred, and exposed simultaneously.
Erbin has been shown to inhibit ERK activation by disrupting complexes containing Ras and Raf. It does so through binding and blocking SHOC2, a scaffolding protein that facilitates ERK by accelerating the formation of upstream Ras-Raf complexes (36). We hypothesized that by positioning Erbin locally, DSG1 might enhance the capacity of Erbin to block recruitment of cytoplasmic SHOC2 to membrane-associated Ras, ultimately, reducing Ras-SHOC2 complexes in favor of Erbin-SHOC2 interactions. We carried out immunoprecipitation of endogenous SHOC2 from RIPA lysates of GFP- and FL-Dsg1–infected NHEKs to test this hypothesis. In addition, NHEKs infected with the Δ569 DSG1 mutant were analyzed to determine whether its poor capacity for binding Erbin would translate into deficient modulation of SHOC2-Erbin interactions. Expression of FLAG-tagged constructs was confirmed by Western blot analysis of the RIPA lysate. In addition, and in contrast to Δ569, FL-Dsg1 was detected by anti-FLAG and anti-DSG1 antibodies in the insoluble pellet commonly occupied by desmosomal components (refs. 61, 62, and Supplemental 2C). Consistent with our hypothesis, forced expression of DSG1 elevated the coimmunoprecipitation of Erbin with SHOC2 (Figure 6A). DSG1 overexpression also consistently reduced the association of KRAS with SHOC2. The Δ569 construct, conversely, failed to impact Erbin-SHOC2-Ras interactions to the extent observed with FL-Dsg1. These results support a model in which keratinocytes, through expression of DSG1, tilt the balance of SHOC2 interactions away from ERK-activating Ras complexes toward ERK-dampening Erbin complexes (Figure 6B).
Figure 6. Overexpression of DSG1 enhances the interaction of Erbin with the Ras-Raf scaffolding protein SHOC2 and diminishes the formation of SHOC2-Ras complexes.

(A) RIPA lysates collected from NHEK cultures infected with GFP, FLAG-tagged DSG1 cytoplasmic domains (Δ569), or FLD were incubated with anti-SHOC2–coated sepharose beads. A combination of the 3 lysates (Pool) was incubated with nonspecific rabbit IgG–coated (rIgG-coated) beads as a negative control. Immunoprecipitated protein and corresponding lysate were analyzed by SDS-PAGE and Western blotting for Erbin, SHOC2, FLAG, and K-Ras. Stars mark putative degradation products. Numbers in parentheses represent densitometric measurement of the specified proteins found in the immunoprecipitations of endogenous SHOC2. Values were converted to fold change over/under the GFP value after subtracting background found in the rabbit IgG immunoprecipitation and normalization to the amount of precipitated SHOC2. (B) Diagram of the proposed formation and Erbin-mediated disruption of Ras-Raf-SHOC2 complexes in the presence or absence of DSG1.
DSG1 deficiency disrupts differentiation, ERK signaling, and Erbin-SHOC2-Ras scaffolds in SPPK.
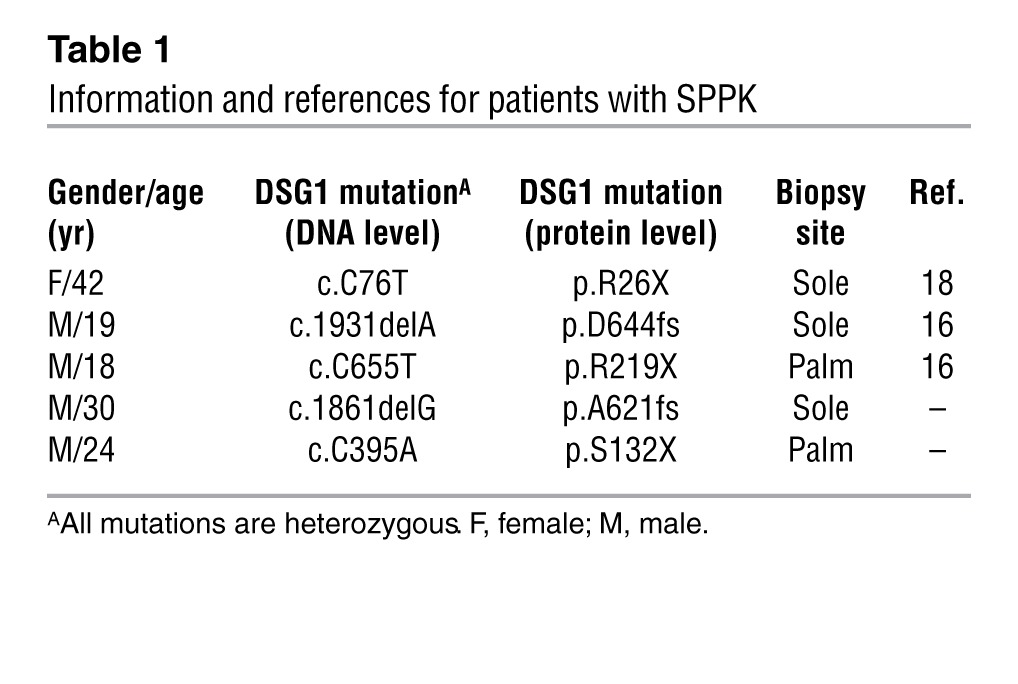
In addressing the extent to which Erbin/SHOC2 signaling contributes to DSG1 function in tissue, we built upon earlier analysis of DSG1-deficient patients diagnosed with SPPK. DSG1 mutations underlie a portion of SPPK cases that, in effect, render patients haploinsufficient with respect to DSG1 expression and prone to structural abnormalities, including hyperkeratosis, widening of intercellular spaces, intracellular vacuolation, hyperproliferation, and thickening of palmoplantar epidermis (16, 63, 64). A previous report demonstrated reduced DSG and K10 expression in a patient carrying mutant DSG1 (p.Y365X) (65). Genetic analysis of a patient with SPPK by Kljuic et al. (66) identified another underlying DSG1 mutation (p.S132X). Analyzing palmar tissue from an unrelated patient with the same mutation, we found that DSG1, K10, and loricrin staining were all perturbed, representing the first molecular characterization of this potentially recurrent mutation to our knowledge (Figure 7A). Though not addressed in earlier studies, our examination also revealed that phosphorylated ERK staining was elevated in the diseased tissue, providing in vivo support for the in vitro model of DSG1-driven ERK inhibition (Figure 7B). Elevated phospho-ERK staining was similarly observed in plantar tissue from a separate patient with the S132X mutation, and diminished K10 staining was found in 2 additional samples from patients harboring p.R26X and p.D644fs DSG1 mutations (Table 1), respectively (data not shown).
Figure 7. Epidermal tissue from patients with SPPK arising from mutant DSG1 displays heightened ERK activity and diminished differentiation marker expression.

(A) Immunofluorescent staining of DSG1 and differentiation markers (K10 and Lor) in control plantar epidermis or palmar epidermis isolated from a patient with SPPK carrying mutant DSG1 (p.S132X). Sections are costained with DAPI to mark nuclei. (B) Tissue from the same patient compared with control plantar tissue, with respect to phosphorylated ERK staining. Boxed regions are shown at higher magnification to the right. Scale bars: 20 μm. Dotted lines were drawn below the row of basal nuclei to distinguish the dermis (D) from the epidermis. Likewise, a dotted line was drawn above the uppermost nucleated cells to approximate the boundary between living keratinocytes and the stratum corneum (SC).
Table 1.
Information and references for patients with SPPK
To assess the possible misregulation of Ras-SHOC2-Erbin interactions upstream of ERK and differentiation defects in SPPK, the 3 relevant proteins were first localized by microscopy in control plantar epidermis. Anti-pan-Ras stained brightly throughout the epidermis but dimly in the stratum corneum and the underlying dermis (Supplemental Figure 3A). At the subcellular level, Ras appears in the cytoplasm but also concentrates at the cell periphery, the latter localization becoming more visible in the upper epidermis as the cytoplasmic staining subsides. Punctate, Ras-positive structures in the cytoplasm may represent late endosomes to which both Ras and SHOC2 relocate upon EGF treatment (39, 67). As in 3-dimensional cell culture, gt anti-Erbin stained the intermediate layers and appeared primarily localized to cell borders, with cytoplasmic staining also present (Supplemental Figure 3, B and C). In control tissue, SHOC2 was present throughout cells but demonstrated a differentiation-dependent distribution, being primarily expressed in the intermediate epidermal layers, with diminished staining intensity in the most basal and most suprabasal keratinocytes (Supplemental Figure 4, A and B).
Next, colocalization studies were conducted to establish whether DSG1 deficiency in SPPK skews Erbin-SHOC2-Ras scaffolding toward promotion of ERK activity. Given the relatively diverse subcellular distribution of the 3 proteins in question, standard colocalization images proved difficult to quantitate. Instead, we used proximity ligation analysis (PLA), based on the ligation of nucleotide adducts present on adjacent secondary antibodies targeting distinct primary antibodies and antigens. The technique is reviewed by Weibrecht et al. (68). As hybridization and ligation require close proximity, a subsequent amplification step in the presence of fluorescent nucleotides produces distinct, countable fluorescent spots at sites of colocalization between 2 proteins of interest.
Compared with that of control tissue, PLA of SPPK skin samples indicated a moderate reduction in the density of sites at which SHOC2 and Erbin exist in close proximity to each other, visible as fluorescent red spots (pseudocolored yellow for presentation) (Figure 8A and Supplemental Table 1). Conversely, SPPK skin samples were more densely populated by sites of Ras-SHOC2 colocalization (Figure 8B). On average, we observed an approximately 30% reduction in potential interactions between SHOC2 and Erbin, which coincided with an approximately 3-fold induction of potential SHOC2-Ras interactions in samples from patients with SPPK (Figure 8C). Traditional immunofluorescent staining of SPPK tissue revealed an altered SHOC2 distribution pattern. Specifically, the apparent downregulation in the uppermost nucleated layers failed to occur, perhaps indicating an incomplete execution of differentiation (Supplemental Figure 4, A and B). The expansion of SHOC2-positive tissue in SPPK samples would, in effect, create more sites for aberrant signaling to produce Ras-SHOC2 scaffolds, potentially exacerbating the elevated Ras-SHOC2 colocalization detected by PLA.
Figure 8. PLA of DSG1-deficient epidermis indicates reduced Erbin-SHOC2 colocalization and enhanced SHOC2-Ras colocalization.

(A) Erbin-SHOC2 PLA. Palmoplantar tissue from controls or patients with SPPK with mutant DSG1 (p.D644fs shown) was incubated with indicated primary antibody pairs. Colocalization allows for hybridization, ligation, and amplification of oligonucleotide adducts fused to secondary antibodies, ultimately producing a fluorescent spot in situ (pseudocolored yellow). Blue DAPI staining marks nuclei. (B) SHOC2-Ras PLA was performed as in A (DSG1 mutation: R219X shown). mIgG, mouse IgG. (C) Quantitation of PLA. PLA spots were counted and divided by area analyzed (mm2) to determine PLA density. Analysis included all nucleated epidermal layers (bounded basally and suprabasally by large and small dashed lines, respectively, in A and B). Each data point represents a ratio of the SPPK PLA density (DSG1 mutation indicated) over the value for 1 out of 3 control samples analyzed in parallel. All control values equal 1. Error bars represent SEM. Unanalyzed areas outside of the nucleated epidermis, including the dermis and stratum corneum, are dimmed in A and B for clarity. PLA spots in A and B were enlarged to enhance visibility at low magnification. Scale bars: 30 μm.
Nonetheless, the shifting of SHOC2 from Erbin to Ras offers a possible mechanism by which DSG1 deficiency leads to ERK activation, deadened differentiation, and aberrant epidermal architecture in patients with SPPK. Our results suggest a model in which DSG1 promotes differentiation by exploiting the ability of Erbin to block ERK activity. More specifically, DSG1 enhances the ability of Erbin to bind SHOC2, disrupt SHOC2-Ras complexes, and, in turn, block the transmission of ERK-activating signals (Figure 9).
Figure 9. Proposed model of differentiation signals initiated by DSG1-Erbin interactions.

Proper keratinocyte differentiation requires the inhibition of ERK signaling by DSG1 in an Erbin-dependent manner. DSG1 expression supports the concentration of Erbin at plasma membranes where its capacity to serve as a SHOC2 sink is enhanced, effectively blocking the formation of ERK activating interactions between SHOC2 and membrane-bound Ras, in favor of Erbin-SHOC2 complexes. Aberrant SHOC2-Ras-Erbin complexing in SPPK, as assessed by PLA, may be compounded by the extension of SHOC2 expression, normally restricted to the intermediate epidermal layers, into the cells bordering the stratum corneum.
Discussion
ERK inhibition constitutes a major component of DSG1-mediated contributions to differentiation. Inhibition of upstream ERBB-EGFR receptors rescues defects stemming from DSG1 deficiency. However, analysis of inhibitors directed at EGFR targets known to influence differentiation, like ERK, PI3K, PKC, and p38MAPK, found that only ERK inhibitors rescue differentiation in DSG1-deficient cultures (8–10, 69, 70). The question as to what dictates the preference for one branch of downstream EGFR signaling over another continues to evoke numerous hypotheses, which highlight the diverse regulatory mechanisms guiding Ras activity (71–74). Alternative Ras signaling outputs often hinge upon the availability and nature of Ras scaffolding proteins at given locations. Sur-8, the C. elegans homolog of SHOC2, for example, specifically converts Ras activity into downstream Raf/MEK/ERK signals, as opposed to activating Akt or Jnk (75). These earlier findings, together with our model, imply a possible relationship between DSG1 and SHOC2, in which DSG1 through Erbin, a protein known to disrupt SHOC2-Ras scaffolding, inhibits and exploits the specificity of SHOC2 for ERK signaling.
How does DSG1 regulate this aspect of Erbin activity? Immunostaining of differentiated keratinocytes in 3-dimensional culture indicated that DSG1 plays a role in concentrating Erbin at the plasma membrane, as DSG1-deficient cultures displayed a more dispersed Erbin pattern reminiscent of that seen in basal keratinocytes or basal cell carcinomas, as shown by Lebeau et al. (20). Whether this mislocalization represents a loss of stability at the plasma membrane or improper trafficking of Erbin to the membrane awaits further study. Regardless, reduction of the presence or time spent at the membrane by Erbin could account for increased Ras-SHOC2 colocalization in SPPK tissue as well as elevated ERK signaling observed in both DSG1 knockdown cultures and diseased tissue.
Erbin-SHOC2 complexes detected by PLA were primarily found in the intermediate layers of control skin. This distribution integrates well with the canonical model of epidermal MAPK signaling, in which, proliferative ERK activity in basal cells must be dampened, presumably in the intermediate layers, to allow differentiation. Ras-SHOC2 complexes, conversely, did not clearly adhere to a predicted, indirect relationship with differentiation. There are a number of potential explanations for this observation. The PLA, for example, does not account for several factors that could affect the distribution or potency of detectable Ras-SHOC2 complexes, such as (a) the activation state of SHOC2-bound Ras, (b) possible alternative functions of the Ras-SHOC2 scaffold, and (c) the half-life or rate of complex degradation in response to upstream signaling. The nature of these molecular processes and their regulation during physiological processes like keratinocyte differentiation await future investigation.
In addition to the mechanism outlined here, it is possible that DSG1 could modulate a number of published Erbin functions, including, for example, support of the ERK-inhibitory protein Merlin (NF2) (50, 76, 77). SHOC2 has also been shown to form an M-Ras effector complex with the phosphatase PP1c capable of dephosphorylating an inhibitory Raf site (78). Though unstudied in keratinocytes and specific to M-Ras, it is possible that DSG1 and Erbin modulate a similar, though currently unidentified, SHOC2 complex in the epidermis. Likewise, a parallel mechanism based on the competition of DSG1 with ERBB2 for the Erbin C terminus could impact downstream ERK activity and remains a possibility. DSG1, Erbin, and ERBB2 all accumulate at the periphery of suprabasal keratinocytes. In turn, DSG1 would have the opportunity to dislodge and alter the distribution of ERBB2 within the membrane, an attribute known to affect receptor signaling (20, 79, 80). Such a mechanism, though, would have to reconcile previous observations that DSG1 loss leads to elevated ERBB2 phosphorylation, while loss of Erbin results in a reduction of ERBB2 stability rather than altered phosphorylation (10, 34).
Removing the unique C-terminal domains (PL, RUD, TD) of DSG1 abrogates the interaction with Erbin in yeast 2-hybrid assays. Likewise, removal of these domains blunted the capacity of ectopic DSG1 to inhibit ERK signaling and promote differentiation. These data, and earlier work that identified DSG1 residue 888 as a caspase-3 cleavage site during UV-induced apoptosis, provide the first experimental evidence to our knowledge that the unique domains (PL, RUD, and TD) participate in epidermal signaling (81–83). In addition, a recent investigation of comparable domains in DSG2 indicates an important role in mediating homodimerization, internalization, and protein stability (84). Though questions remain regarding the purpose and origin of the nonconserved regions, this study contributes to a developing body of evidence that they impart distinct functional characteristics to the desmoglein subfamily.
Implicated by Lebeau et al. (20) in basal cell carcinoma, the possible participation of Erbin in genetic skin disorders remains relatively unstudied. One reported case of dystrophic epidermolysis bullosa describes a family with an inherited collagen (COL7A) mutation. The authors found that 2 out of 3 affected family members also carried a chromosomal translocation that disrupted Erbin and glypican-6 (85). Erbin associates with hemidesmosomal components, and, in turn, it was hypothesized that its disruption might contribute to deficient cell-matrix adhesion and blistering in dystrophic epidermolysis bullosa (86). The study concluded that the production of fused or dysfunctional Erbin products did not worsen the disease phenotype. However, the severity of the condition produced by mutant COL7A alone may have masked contributions made by mutant Erbin. Ultimately, the study suggests that a role for Erbin in blistering conditions cannot be ruled out. This study also poses questions for future research regarding the possible contribution of aberrant Erbin-SHOC2 interactions to other skin diseases, like psoriasis, characterized by elevated MAPK signaling, hyperproliferation, and hyperkeratosis reminiscent of SPPK (55, 87).
At the level of expression defects, Stojadinovic and colleagues (88–90) indicate that Erbin is downregulated 8 fold in the epidermis adjacent to chronically wounded, venous ulcers. Interestingly, these studies conclude that epidermis at the edge of nonhealing wounds is also marked by deregulated expression of desmosomal components and decreased expression of differentiation markers, is hyperproliferative and hyper/parakeratotic, and contains mitotically active suprabasal keratinocytes. The similarities between the descriptions of tissue adjacent to venous ulcers and those given for SPPK tissue warrant further investigation into a potential link between Erbin-desmosome interactions and the process of reepithelializing wounded epidermis.
The involvement of Erbin in DSG1-mediated phenotypes, by extension, implicates SHOC2 in epidermal differentiation. Though recently tied to melanoma drug resistance, prior SHOC2 investigations do not directly address its potential role in keratinocyte biology (91). Highly conserved and linked to the genetic disorder Noonan-like syndrome with loose anagen hair (NS/LAH; MIM: 607721), molecular investigations of SHOC2 in mammalian systems have only recently appeared. Genetic studies identified a gain-of-function SHOC2 mutation responsible for NS/LAH that creates an aberrant myristoylation site, resulting in constitutive localization to the plasma membrane (92). The abnormally high concentration of SHOC2 now available to membrane-bound Ras translates into heightened ERK activity, an event believed to belie NS/LAH pathogenesis. Characterized by a range of cognitive and skeletal abnormalities, patients with NS/LAH also present with a number of cardiac and dermatological conditions, in many ways resembling those identified with genetic disorders stemming from mutant desmosomal proteins (93–95). NS/LAH symptoms like hyperkeratosis and abnormalities of palm and sole skin, in particular, are reminiscent of features associated with SPPK. The reduced association of SHOC2 with Ras in DSG1 overexpression cultures and heightened colocalization of SHOC2 with Ras observed in SPPK palmoplantar tissue raise the intriguing possibility that epidermal defects stemming from both DSG1 deficiency and NS/LAH share a common fault in SHOC2 abnormalities.
In summary, we have identified a binding partner of the DSG1 cytoplasmic tail, Erbin, which mediates the inhibition of ERK and promotion of epidermal differentiation by DSG1. Our data indicate that DSG1 exploits the ability of Erbin to disrupt interactions between SHOC2 and Ras, which lie upstream of ERK activation. Thus, we propose a model in which DSG1, through Erbin, blocks the formation of Ras-SHOC2 complexes, in turn, dampening ERK activity and promoting differentiation.
Methods
Antibodies.
The following primary antibodies were used in this study: M2 mouse anti-FLAG, rabbit anti-FLAG, and rabbit anti-GAPDH (Sigma-Aldrich); 1646 rabbit anti-Erbin and 139 rabbit anti-Erbin were gifts from L. Fontao (Hôpitaux Universitaires de Genève, Geneva, Switzerland); K13 gt anti-Erbin and F234 mouse anti-KRas (Santa Cruz Biotechnology Inc.); goat anti-GST (GE Healthcare Biosciences); 4B2 mouse anti-DSG1 is described in Dusek et al. (83); 27B2 mouse anti-DSG1 (Invitrogen); U100 anti-DSC1 (Progen); LH2 anti-K10 was a gift from I. Leigh (University of Dundee, Dundee City, United Kingdom); rabbit anti-K10, rabbit anti-K1, rabbit anti-K5, and rabbit anti-loricrin were gifts from J. Segre (National Human Genome Research Institute, Bethesda, Maryland, USA); AF62 rabbit anti-loricrin (Covance); HECD1 mouse anti–E-cadherin (gift from M. Takeichi and O. Abe, RIKEN Center for Developmental Biology, Kobe, Japan); rabbit anti-p44/42 MAPK (ERK1/2) (Thr202/Tyr204) (D13.14.4E) XP (Cell Signaling); rabbit anti-total ERK1/2 (Promega); rabbit anti-SHOC2 (ProteinTech); and Ab3 mouse anti-pan-Ras (Calbiochem).
Secondary antibodies for immunofluorescent staining were purchased from Invitrogen. HRP-linked secondary antibodies for Western blot analysis were purchased from Kirkegaard Perry Labs.
DNA constructs.
pMyr and pSos were purchased from Stratagene as components of the CytoTrap yeast 2-hybrid system. pGEX4T-1 was used for cloning GST fusions and was obtained from GE Healthcare Life Sciences. His-tagged Erbin C terminus was cloned into pDEST17 using Gateway Technology (Invitrogen). LZRS-GFP, LZRS-Flag Dsg1, LZRS-FLAG Dsg1(AAA), LZRS-FLAG Dsg1(Δ569), LZRS-FLAG Dsg1[Δ381(WT)], LZRS-FLAG Dsg1[Δ381(AAA)], LZRS-miR Dsg1, and LZRS-miR Lamin were generated and described by Getsios et al. (10) and Simpson et al. (48). LZRS-FLAGDsg1(ICS) was generated by ligating a PCR product encoding nucleotides 21–2,537 of NM_001942.2 into LZRS (provided by M. Denning, National Cancer Institute, Bethesda, Maryland, USA).
Cell culture and retroviral infection.
Primary human keratinocytes were isolated from neonatal foreskin by the Skin Disease Research Core of the Northwestern University Feinberg School of Medicine. Fresh keratinocyte isolates were cultured in M154 media adjusted to 0.07 mM CaCl2 and supplemented with human keratinocyte growth supplement (HKGS) and gentamicin/amphotericin B (Invitrogen). All data presented were collected from confluent keratinocyte monolayers induced to differentiate by addition of CaCl2 to 1.2 mM in the absence or, in the case of FLAG-Dsg1/Erbin coimmunoprecipitations, the presence of HKGS for 1 to 5 days. Keratinocytes were transduced with retroviral supernatants produced from Phoenix cells (provided by G. Nolan, Stanford University, Stanford, California, USA), as previously described (12). For stratified, 3-dimensional keratinocyte cultures, cells were expanded and grown at an air-medium interface according to published protocols (96). These cultures were grown for 3 to 10 days, at which time they were lysed for protein analysis or fixed in 10% neutral-buffered formalin, and embedded in paraffin. For some experiments, cultures were treated with DMSO (Thermo Fisher Scientific), 5 μM U0126 (Cell Signaling Technology), or 5 μM SB431542 (Sigma-Aldrich).
siRNA treatment and transfections.
Primary human keratinocytes at approximately 90% confluency were transfected in a mixture of DharmaFECT and random or Erbin Stealth siRNA (Invitrogen) directed at the following sequences: Erbin siRNA-1, 5′-CCACACTGTTGTATGATCAACCATT-3′, and Erbin siRNA-2, 5′-GCCAGACAATGATTTAACAACGTTA-3′, corresponding to nucleotides 3,055–3,079 and 536–560 of NM_018695.2. Transfections were carried out following manufacturer instructions. For Erbin knockdown, cells were allowed to incubate for at least 3 days or more after transfection, depending on the extent of differentiation required by the experiment. Alternatively, Erbin siRNA was also delivered to cells using the Amaxa electroporation system, according to manufacturer’s instructions, using solution V and program X-001 (Lonza).
Yeast 2-hybrid screening and interaction assays.
Screening was performed using the CytoTrap system (Stratagene). DSG1 bait inserts were generated by ligation of PCR products encompassing nucleotides 1,921–3,363, 1,921–2,318, and 1,921–2,537 of NM_001942.1 into pSos(Stratagene), resulting in Sos fusions of the full DSG1 cytoplasmic domain, a truncated version containing only the IA domain, and a mutant containing only the IA and ICS domains, respectively. Cdc25H yeast cotransformation of the Sos-fused, full cytodomain with a HeLa cell cDNA library carried in pMyr were conducted as described by Stratagene. Cotransformants capable of growth at 37°C in a galactose-dependent manner were considered putative positives and subjected to further analysis. For testing interactions between pairs of proteins, a similar protocol was followed using specific pMyr constructs instead of the pMyr library. These constructs included Myr-Pg, created by ligating a PCR insert encompassing nucleotides 120–2,357 of NM_021991 into pMyr(Stratagene). The Myr-Erbin used for mapping studies is the clone pulled from the screen that includes nucleotides 3,663–4,421 of NM_018695.2, encoding residues 1,119–1,371 of the C terminus and a stop codon.
In vitro binding assays.
Protein expression of His-tagged Erbin was induced by adding arabinose to cultured BL21A1 bacteria (Invitrogen). The bacteria were subsequently lysed, and protein purification was carried out by incubating bacterial lysate with nickel-coated, Ni-NTA beads (Qiagen), followed by washing with 300 mM NaCl, 20 mM imidazole, 50 mM NaH2PO4, pH 8.0. To elute protein, the imidazole concentration was raised to 250 mM. Protein was subjected to anion exchange chromatography and eluted in Tris-buffered saline. Purified GST-Dsg1(cyto) was obtained from isopropyl β-D-1-thiogalactopyranoside–induced BL21 bacteria transformed with pGEX4T-1 carrying a DSG1 insert encoding nucleotides 1,921–3,363 of NM_001942.1 encompassing the entire cytoplasmic domain. Bacterial lysate was subject to affinity and anion exchange chromatography to purify GST-Dsg1(cyto), using glutathione-agarose (Sigma-Aldrich) with excess glutathione elution and DEAE sepharose (Sigma-Aldrich) with NaCl elution, respectively. All elution and washing steps were conducted in tris-buffered saline. A GST-only control protein was purified in parallel. His-Erbin–coated beads were resuspended in RIPA buffer and incubated with various amounts of GST-Dsg1(cyto) or GST-only protein at 37°C, while rotating for 30 minutes in the presence of BSA. Beads were pelleted and washed 3 times with RIPA buffer. Precipitated protein was eluted with SDS/Laemmli buffer and analyzed by Western blot.
Immunoprecipitation.
Immunoprecipitation of FLAG-tagged, ectopic DSG1 constructs from 400 μg of RIPA protein lysate was carried out with M2-agarose (Sigma-Aldrich) by rotating at 4°C overnight, followed by washing and elution in SDS/Laemmli. SHOC2 immunoprecipitations were conducted by adding 4 μg of rabbit anti-SHOC IgG to 20 μl GammaBind Sepharose per subsequent reaction (GE Healthcare Life Sciences), rotated for 4 hours in Tris-buffered saline containing 1% BSA and 0.05% Tween-20 at 4°C, and then washed. 400 μg of precleared RIPA lysate (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 0.1% SDS, 0.5% Na-deoxycholate, 1% Triton X-100, 5% glycerol, with protease and phosphatase inhibitors) was added to IgG-coated beads and rotated 4–16 hours at 4°C. After washing, immunoprecipitates were eluted using SDS/Laemmli. Nonspecific rabbit IgG was used as a parallel control.
Sucrose gradient analysis.
Primary keratinocytes were grown to confluency, switched to 1.2 mM CaCl2, and incubated overnight. Cells were then cross-linked with 100 μM dithiobis(succinimidyl) propionate on ice for 2 hours. Cross-linked cells were lysed in urea/SDS/Tris/glycerol buffer and layered on top of a 5% to 20% sucrose gradient prepared in lysis buffer. Samples were spun at 150,000 g for 18 hours at 20°C. Fractions were collected and were subsequently analyzed by Western blot.
Immunofluorescence analysis.
Keratinocytes cultured on glass coverslips were fixed in either 4% paraformaldehyde (Sigma-Aldrich) in PBS for 5 to 10 minutes followed by Triton X-100 (Sigma-Aldrich) permeabilization or paraformaldehyde followed by 2 minutes in ice-cold acetone. Fixed cells were incubated with 1%BSA/PBS for 30 minutes followed by 1 hour incubation with primary antibodies at 37°C or overnight incubation at 4°C. Washing by rapid dipping in PBS was followed by incubation with secondary antibodies conjugated to Alexa Fluor 350, Alexa Fluor 488, or Alexa Fluor 568 from Invitrogen. After secondary antibody incubation, coverslips were dipped rapidly in distilled H20 and mounted on slides in polyvinyl alcohol (Sigma-Aldrich). For analysis of tissue or 3-dimensional cultures, formaldehyde-fixed, paraffin-embedded tissue sections were processed by the Northwestern University Skin Disease Research Core. Slides were baked, deparaffinized by xylene, dehydrated with ethanol, rehydrated in PBS, and permeabilized by 0.5% Triton X-100 in PBS. Antigen retrieval was performed by incubation in pH 6.0 citrate buffer or Tris (pH 8.0)/10 mM EDTA buffer (for stains incorporating either anti-pan-Ras or gt anti-Erbin) at 95°C for 15 minutes. Sections were blocked in 2% BSA/10% normal goat serum/PBS for 30 minutes at 37°C or overnight at 4°C. Goat serum was omitted when primary antibodies included goat IgG. Primary antibody incubation was carried out overnight at 4°C in blocking buffer followed by washing in chambers of PBS. Secondary antibody incubation was carried out at 37°C for 30 to 60 minutes, followed by washing in PBS. The final wash was done with H20, and a coverslip was mounted on top of the section with polyvinyl alcohol.
Wide-field images were taken using a microscope (DMR; ×10, ×20, or ×40 objective lenses Plan Fluotar) and a digital camera (ORCA-100 model C4742-95; Hamamatsu Photonics). Subdiffraction resolution images were taken using a structured illumination superresolution microscope (N-SIM; Nikon). For N-SIM analysis, the samples were illuminated with spatially high-frequency patterned excitation light (×100 objective lens, NA 1.49; TiE N-SIM microscope [Nikon] and iXon X3 897 camera [Andor Technology]). The moiré patterns were produced and analytically processed (Elements version 4 software; Nikon) to reconstruct the subresolution structure of the samples.
Proximity ligation assay.
Reagents for the PLA of tissue were purchased and utilized using instructions from OLink Biosciences through Axxora LLC. Briefly, sections were prepared for staining with primary antibodies as described above with no goat serum used in the blocking buffer. Anti-pan-Ras, anti-SHOC2, and gt anti-Erbin were all used at 1:500 dilutions. Instead of fluorescently tagged secondary antibodies, specimens were incubated with nucleotide-tethered secondary antibodies of donkey origin targeting various species. Close proximity of target antigens allows their respective secondary antibody nucleotide sequences to hybridize. Following a 30-minute 37°C incubation with ligase and additional oligonucleotides, a closed DNA circle is formed. A subsequent step involving polymerase-driven rolling circle amplification incorporates fluorescently labeled nucleotides. Fluorescent nucleotides used in this experiment fluoresce in the red channel when excited. A fluorescent spot appears at sites of Ras-SHOC2 or SHOC2-Erbin colocalization, analyzable by fluorescence microscopy and quantifiable by ImageJ (NIH, http://imagej.nih.gov/ij/). Data were normalized by dividing the total number of PLA spots counted by the total area of tissue analyzed in square millimeters. Area analyzed encompassed all nucleated layers of epidermis. Raw values for each paired experiment (an SPPK sample versus a control sample) were then converted to fold change with respect to the control. In total, the PLA experiments used tissue samples from 3 control individuals and 5 patients with SPPK (see Tissue samples below).
Quantitation of phospho-ERK staining.
Phospho-ERK1/2 staining of organotypic NHEK cultures was quantified by taking 3–5 images of each culture and drawing 15–25 vertical lines from the site at which basal keratinocytes contacted the underlying collagen substrate to the top of the culture every 40 microns horizontally. Each pixel along a line, corresponding to 0.167-micron increments, was measured and plotted against its vertical position. The results of 3 separate experiments comparing control to Erbin knockdown cultures were pooled. Prior to pooling the data, differences in overall staining intensity from experiment to experiment were accounted for by determining the average pixel intensity of the control culture from each experiment, setting the first experiment as a reference and dividing or multiplying the pixel intensity values of the second and third experiments, accordingly. Erbin knockdown pixel values were adjusted by the same factor as the corresponding control culture. A mean value for each 0.167-micron increment was calculated from 55 to 65 points collected from the 3 experiments. Distance from the collagen substrate to the center of DAPI-stained nuclei was measured to estimate the average vertical position of each cell layer (10 nuclei randomly selected from the 3 control cultures were measured per layer).
Western blot analysis.
Samples separated by SDS-PAGE were transferred to PVDF, blocked in 5% milk/TBS/Tween, probed with primary antibody either in blocking buffer or 2%BSA/TBS/Tween in the case of phospho-antibodies and SHOC2. Secondary, HRP-conjugated antibodies diluted 1:5,000 in blocking solution were added to blots after washing with 0.05% Tween/TBS. Chemiluminescent detection was performed after another set of 0.05% Tween/TBS washes. Unless otherwise noted, whole cell lysates were collected in urea-SDS buffer (8 M urea/1% SDS/60 mM Tris, pH 6.8/5% β-mercaptoethanol/10% glycerol).
Tissue samples.
Biopsies of control plantar skin were taken from the side of the heel. Control biopsies were collected from 3 individuals at either the Tel Aviv Sourasky Medical Center or the Northwestern University Skin Disease Research Center. Biopsies from the heel (side) or palms from 5 patients carrying DSG1 mutations associated with SPPK were collected at the Tel Aviv Sourasky Medical Center. See Table 1 for information pertaining to samples from patients with SPPK as well as references to previously published SPPK samples used in this study.
Statistics.
Where indicated, P values were calculated using a 2-tailed, 2-sample equal variance Student’s t test. P values less than 0.05 were considered statistically significant.
Study approval.
Human tissue samples were collected in a deidentified manner in accordance with Declaration of Helsinki principles. All participants or their legal guardians provided written and informed consent. These studies were approved by the local Helsinki Committee and the Committee for Genetic Studies of the Israeli Ministry of Health or the Institutional Review Board for the Office for the Protection of Research Subjects at Northwestern University.
Supplementary Material
Acknowledgments
The authors would like to thank Spiro Getsios for comments on the paper. Gifts were provided by Birgitte Lane (K10 and 14 antibodies), I. Leigh (K10 antibody), Julie Segre (K1, K10, loricrin antibodies), and Lionel Fontao (1646 rabbit anti-Erbin and 139 rabbit anti-Erbin). Keratinocytes were obtained from the Keratinocyte Core of the Northwestern University Skin Disease Research Center, and histological analysis was conducted either by the Mouse Phenotyping Core of the R.H. Lurie Comprehensive Cancer Center supported by NIH/NCI (P30 CA060553-159026) or by the Pathology Core of the Northwestern University Skin Disease Research Center (P30AR057216), Chicago, Illinois, USA, with support from the NIH/NIAMS. Any opinions, findings, and conclusions or recommendations expressed in this material are those of the author(s) and do not necessarily reflect the views of the Northwestern University Skin Disease Research Center or the NIH/NIAMS. This work was supported by NIH R01 AR041836, with partial support from AR43380 and CA122151 to K.J. Green. Additional support was provided by the JL Mayberry endowment to K.J. Green. C.L. Simpson was supported by a Kirschstein predoctoral fellowship from NIH-NIEHS (F30 ES14990), a Malkin Scholarship from the R.H. Lurie Cancer Center, and a Presidential Fellowship from Northwestern University. J.L. Johnson was supported by a Kirschstein postdoctoral training fellowship from NIH-NCI (T32 CA070085-14) and a Dermatology Foundation Career Development Award. A.D. Dubash received funding from the American Heart Association (11POST7380001). R.M. Harmon was supported by an NIH T32 Training Grant (T32 GM08061) and an American Heart Association predoctoral fellowship.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Citation for this article: J Clin Invest. 2013;123(4):1556–1570. doi:10.1172/JCI65220.
See the related Commentary beginning on page 1419.
References
- 1.Khavari TA, Rinn J. Ras/Erk MAPK signaling in epidermal homeostasis and neoplasia. Cell Cycle. 2007;6(23):2928–2931. doi: 10.4161/cc.6.23.4998. [DOI] [PubMed] [Google Scholar]
- 2.Siegel DH, Mann JA, Krol AL, Rauen KA. Dermatological phenotype in Costello syndrome: consequences of Ras dysregulation in development. Br J Dermatol. 2012;166(3):601–607. doi: 10.1111/j.1365-2133.2011.10744.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tidyman WE, Rauen KA. Noonan, Costello and cardio-facio-cutaneous syndromes: dysregulation of the Ras-MAPK pathway. Expert Rev Mol Med. 2008;10:e37. doi: 10.1017/S1462399408000902. [DOI] [PubMed] [Google Scholar]
- 4.Tartaglia M, Zampino G, Gelb BD. Noonan syndrome: clinical aspects and molecular pathogenesis. Mol Syndromol. 2010;1(1):2–26. doi: 10.1159/000276766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rauen KA, et al. Proceedings from the 2009 genetic syndromes of the Ras/MAPK pathway: From bedside to bench and back. Am J Med Genet A. 2010;152A(1):4–24. doi: 10.1002/ajmg.a.33183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Allanson JE, et al. Cardio-facio-cutaneous syndrome: does genotype predict phenotype? Am J Med Genet C Semin Med Genet. 2011;157(2):129–135. doi: 10.1002/ajmg.c.30295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zenker M. Clinical manifestations of mutations in RAS and related intracellular signal transduction factors. Curr Opin Pediatr. 2011;23(4):443–451. doi: 10.1097/MOP.0b013e32834881dd. [DOI] [PubMed] [Google Scholar]
- 8.Gazel A, Nijhawan RI, Walsh R, Blumenberg M. Transcriptional profiling defines the roles of ERK and p38 kinases in epidermal keratinocytes. J Cell Physiol. 2008;215(2):292–308. doi: 10.1002/jcp.21394. [DOI] [PubMed] [Google Scholar]
- 9.Kolev V, et al. EGFR signalling as a negative regulator of Notch1 gene transcription and function in proliferating keratinocytes and cancer. Nat Cell Biol. 2008;10(8):902–911. doi: 10.1038/ncb1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Getsios S, et al. Desmoglein 1-dependent suppression of EGFR signaling promotes epidermal differentiation and morphogenesis. J Cell Biol. 2009;185(7):1243–1258. doi: 10.1083/jcb.200809044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dusek RL, Godsel LM, Green KJ. Discriminating roles of desmosomal cadherins: beyond desmosomal adhesion. J Dermatol Sci. 2007;45(1):7–21. doi: 10.1016/j.jdermsci.2006.10.006. [DOI] [PubMed] [Google Scholar]
- 12.Getsios S, et al. Coordinated expression of desmoglein 1 and desmocollin 1 regulates intercellular adhesion. Differentiation. 2004;72(8):419–433. doi: 10.1111/j.1432-0436.2004.07208008.x. [DOI] [PubMed] [Google Scholar]
- 13.Lin S, Gordon K, Kaplan N, Getsios S. Ligand targeting of EphA2 enhances keratinocyte adhesion and differentiation via desmoglein 1. Mol Biol Cell. 2010;21(22):3902–3914. doi: 10.1091/mbc.E10-03-0242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hanakawa Y, et al. Molecular mechanisms of blister formation in bullous impetigo and staphylococcal scalded skin syndrome. J Clin Invest. 2002;110(1):53–60. doi: 10.1172/JCI15766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dua-Awereh MB, Shimomura Y, Kraemer L, Wajid M, Christiano AM. Mutations in the desmoglein 1 gene in five Pakistani families with striate palmoplantar keratoderma. J Dermatol Sci. 2009;53(3):192–197. doi: 10.1016/j.jdermsci.2008.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hershkovitz D, Lugassy J, Indelman M, Bergman R, Sprecher E. Novel mutations in DSG1 causing striate palmoplantar keratoderma. Clin Exp Dermatol. 2009;34(2):224–228. doi: 10.1111/j.1365-2230.2008.02733.x. [DOI] [PubMed] [Google Scholar]
- 17.Hunt DM, et al. Spectrum of dominant mutations in the desmosomal cadherin desmoglein 1, causing the skin disease striate palmoplantar keratoderma. Eur J Hum Genet. 2001;9(3):197–203. doi: 10.1038/sj.ejhg.5200605. [DOI] [PubMed] [Google Scholar]
- 18.Keren H, Bergman R, Mizrachi M, Kashi Y, Sprecher E. Diffuse nonepidermolytic palmoplantar keratoderma caused by a recurrent nonsense mutation in DSG1. Arch Dermatol. 2005;141(5):625–628. doi: 10.1001/archderm.141.5.625. [DOI] [PubMed] [Google Scholar]
- 19.Huang YZ, Zang M, Xiong WC, Luo Z, Mei L. Erbin suppresses the MAP kinase pathway. J Biol Chem. 2003;278(2):1108–1114. doi: 10.1074/jbc.M205413200. [DOI] [PubMed] [Google Scholar]
- 20.Lebeau S, et al. Comparative analysis of the expression of ERBIN and Erb-B2 in normal human skin and cutaneous carcinomas. Br J Dermatol. 2005;152(6):1248–1255. doi: 10.1111/j.1365-2133.2005.06687.x. [DOI] [PubMed] [Google Scholar]
- 21.Borg JP, et al. ERBIN: a basolateral PDZ protein that interacts with the mammalian ERBB2/HER2 receptor. Nat Cell Biol. 2000;2(7):407–414. doi: 10.1038/35017038. [DOI] [PubMed] [Google Scholar]
- 22.Dai F, Chang C, Lin X, Dai P, Mei L, Feng XH. Erbin inhibits transforming growth factor beta signaling through a novel Smad-interacting domain. Mol Cell Biol. 2007;27(17):6183–6194. doi: 10.1128/MCB.00132-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dai P, Xiong WC, Mei L. Erbin inhibits RAF activation by disrupting the sur-8-Ras-Raf complex. J Biol Chem. 2006;281(2):927–933. doi: 10.1074/jbc.M507360200. [DOI] [PubMed] [Google Scholar]
- 24.Izawa I, Nishizawa M, Hayashi Y, Inagaki M. Palmitoylation of ERBIN is required for its plasma membrane localization. Genes Cells. 2008;13(7):691–701. doi: 10.1111/j.1365-2443.2008.01198.x. [DOI] [PubMed] [Google Scholar]
- 25.Brill LM, Salomon AR, Ficarro SB, Mukherji M, Stettler-Gill M, Peters EC. Robust phosphoproteomic profiling of tyrosine phosphorylation sites from human T cells using immobilized metal affinity chromatography and tandem mass spectrometry. Anal Chem. 2004;76(10):2763–2772. doi: 10.1021/ac035352d. [DOI] [PubMed] [Google Scholar]
- 26.Dephoure N, et al. A quantitative atlas of mitotic phosphorylation. Proc Natl Acad Sci U S A. 2008;105(31):10762–10767. doi: 10.1073/pnas.0805139105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Heibeck TH, et al. An extensive survey of tyrosine phosphorylation revealing new sites in human mammary epithelial cells. J Proteome Res. 2009;8(8):3852–3861. doi: 10.1021/pr900044c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mayya V, et al. Quantitative phosphoproteomic analysis of T cell receptor signaling reveals system-wide modulation of protein-protein interactions. Sci Signal. 2009;2(84):ra46. doi: 10.1126/scisignal.2000007. [DOI] [PubMed] [Google Scholar]
- 29.Olsen JV, et al. Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell. 2006;127(3):635–648. doi: 10.1016/j.cell.2006.09.026. [DOI] [PubMed] [Google Scholar]
- 30.Rush J, et al. Immunoaffinity profiling of tyrosine phosphorylation in cancer cells. Nat Biotechnol. 2005;23(1):94–101. doi: 10.1038/nbt1046. [DOI] [PubMed] [Google Scholar]
- 31.Tao WA, et al. Quantitative phosphoproteome analysis using a dendrimer conjugation chemistry and tandem mass spectrometry. Nat Methods. 2005;2(8):591–598. doi: 10.1038/nmeth776. [DOI] [PubMed] [Google Scholar]
- 32.Wolf-Yadlin A, Hautaniemi S, Lauffenburger DA, White FM. Multiple reaction monitoring for robust quantitative proteomic analysis of cellular signaling networks. Proc Natl Acad Sci U S A. 2007;104(14):5860–5865. doi: 10.1073/pnas.0608638104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yu LR, Zhu Z, Chan KC, Issaq HJ, Dimitrov DS, Veenstra TD. Improved titanium dioxide enrichment of phosphopeptides from HeLa cells and high confident phosphopeptide identification by cross-validation of MS/MS and MS/MS/MS spectra. J Proteome Res. 2007;6(11):4150–4162. doi: 10.1021/pr070152u. [DOI] [PubMed] [Google Scholar]
- 34.Tao Y, et al. Erbin regulates NRG1 signaling and myelination. Proc Natl Acad Sci U S A. 2009;106(23):9477–9482. doi: 10.1073/pnas.0901844106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shi M, et al. Int J Cardiol. β2-AR-induced Her2 transactivation mediated by Erbin confers protection from apoptosis in cardiomyocytes [published online ahead of print May 5, 2012]. doi: 10.1016/j.ijcard.2012.04.093 . [DOI] [PubMed] [Google Scholar]
- 36.Matsunaga-Udagawa R, et al. The scaffold protein Shoc2/SUR-8 accelerates the interaction of Ras and Raf. J Biol Chem. 2010;285(10):7818–7826. doi: 10.1074/jbc.M109.053975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Moon BS, et al. Sur8/Shoc2 involves both inhibition of differentiation and maintenance of self-renewal of neural progenitor cells via modulation of extracellular signal-regulated kinase signaling. Stem Cells. 2011;29(2):320–331. doi: 10.1002/stem.586. [DOI] [PubMed] [Google Scholar]
- 38.Yoshiki S, Matsunaga-Udagawa R, Aoki K, Kamioka Y, Kiyokawa E, Matsuda M. Ras and calcium signaling pathways converge at Raf1 via the Shoc2 scaffold protein. Mol Biol Cell. 2010;21(6):1088–1096. doi: 10.1091/mbc.E09-06-0455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Galperin E, Abdelmoti L, Sorkin A. Shoc2 is targeted to late endosomes and required for Erk1/2 activation in EGF-stimulated cells. PLoS One. 2012;7(5):e36469. doi: 10.1371/journal.pone.0036469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sflomos G, et al. ERBIN is a new SARA-interacting protein: competition between SARA and SMAD2 and SMAD3 for binding to ERBIN. J Cell Sci. 2011;124(pt 19):3209–3222. doi: 10.1242/jcs.062307. [DOI] [PubMed] [Google Scholar]
- 41.Troyanovsky SM, Troyanovsky RB, Eshkind LG, Krutovskikh VA, Leube RE, Franke WW. Identification of the plakoglobin-binding domain in desmoglein and its role in plaque assembly and intermediate filament anchorage. J Cell Biol. 1994;127(1):151–160. doi: 10.1083/jcb.127.1.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chitaev NA, Averbakh AZ, Troyanovsky RB, Troyanovsky SM. Molecular organization of the desmoglein-plakoglobin complex. J Cell Sci. 1998;111(pt 14):1941–1949. doi: 10.1242/jcs.111.14.1941. [DOI] [PubMed] [Google Scholar]
- 43.Izawa I, Nishizawa M, Tomono Y, Ohtakara K, Takahashi T, Inagaki M. ERBIN associates with p0071, an armadillo protein, at cell-cell junctions of epithelial cells. Genes Cells. 2002;7(5):475–485. doi: 10.1046/j.1365-2443.2002.00533.x. [DOI] [PubMed] [Google Scholar]
- 44.Laura RP, et al. The Erbin PDZ domain binds with high affinity and specificity to the carboxyl termini of delta-catenin and ARVCF. J Biol Chem. 2002;277(15):12906–12914. doi: 10.1074/jbc.M200818200. [DOI] [PubMed] [Google Scholar]
- 45.Pulliam KF, Fasken MB, McLane LM, Pulliam JV, Corbett AH. The classical nuclear localization signal receptor, importin-alpha, is required for efficient transition through the G1/S stage of the cell cycle in Saccharomyces cerevisiae. Genetics. 2009;181(1):105–118. doi: 10.1534/genetics.108.097303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ress A, Moelling K. The PDZ protein erbin modulates beta-catenin-dependent transcription. Eur Surg Res. 2008;41(3):284–289. doi: 10.1159/000148241. [DOI] [PubMed] [Google Scholar]
- 47.Wang YX, Kauffman EJ, Duex JE, Weisman LS. Fusion of docked membranes requires the armadillo repeat protein Vac8p. J Biol Chem. 2001;276(37):35133–35140. doi: 10.1074/jbc.M103937200. [DOI] [PubMed] [Google Scholar]
- 48.Simpson CL, Kojima S, Cooper-Whitehair V, Getsios S, Green KJ. Plakoglobin rescues adhesive defects induced by ectodomain truncation of the desmosomal cadherin desmoglein 1: implications for exfoliative toxin-mediated skin blistering. Am J Pathol. 2010;177(6):2921–2937. doi: 10.2353/ajpath.2010.100397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Maretzky T, Scholz F, Koten B, Proksch E, Saftig P, Reiss K. ADAM10-mediated E-cadherin release is regulated by proinflammatory cytokines and modulates keratinocyte cohesion in eczematous dermatitis. J Invest Dermatol. 2008;128(7):1737–1746. doi: 10.1038/sj.jid.5701242. [DOI] [PubMed] [Google Scholar]
- 50.Wilkes MC, et al. Erbin and the NF2 tumor suppressor Merlin cooperatively regulate cell-type-specific activation of PAK2 by TGF-beta. Dev Cell. 2009;16(3):433–444. doi: 10.1016/j.devcel.2009.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang G, Fenyo D, Neubert TA. Screening for EphB signaling effectors using SILAC with a linear ion trap-orbitrap mass spectrometer. J Proteome Res. 2008;7(11):4715–4726. doi: 10.1021/pr800255a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang Z, et al. Rac1 is crucial for Ras-dependent skin tumor formation by controlling Pak1-Mek-Erk hyperactivation and hyperproliferation in vivo. Oncogene. 2010;29(23):3362–3373. doi: 10.1038/onc.2010.95. [DOI] [PubMed] [Google Scholar]
- 53.Takahashi H, Ibe M, Nakamura S, Ishida-Yamamoto A, Hashimoto Y, Iizuka H. Extracellular regulated kinase and c-Jun N-terminal kinase are activated in psoriatic involved epidermis. J Dermatol Sci. 2002;30(2):94–99. doi: 10.1016/S0923-1811(02)00064-6. [DOI] [PubMed] [Google Scholar]
- 54.Zhu AJ, Haase I, Watt FM. Signaling via beta1 integrins and mitogen-activated protein kinase determines human epidermal stem cell fate in vitro. Proc Natl Acad Sci U S A. 1999;96(12):6728–6733. doi: 10.1073/pnas.96.12.6728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Haase I, Hobbs RM, Romero MR, Broad S, Watt FM. A role for mitogen-activated protein kinase activation by integrins in the pathogenesis of psoriasis. J Clin Invest. 2001;108(4):527–536. doi: 10.1172/JCI12153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Muller EJ, Williamson L, Kolly C, Suter MM. Outside-in signaling through integrins and cadherins: a central mechanism to control epidermal growth and differentiation? J Invest Dermatol. 2008;128(3):501–516. doi: 10.1038/sj.jid.5701248. [DOI] [PubMed] [Google Scholar]
- 57.Ou J, et al. Chronic wound state exacerbated by oxidative stress in Pax6+/– aniridia-related keratopathy. . J Pathol. 2008;215(4):421–430. doi: 10.1002/path.2371. [DOI] [PubMed] [Google Scholar]
- 58.Huisman MA, De Heer E, Grote JJ. Terminal differentiation and mitogen-activated protein kinase signaling in human cholesteatoma epithelium. Otol Neurotol. 2006;27(3):422–426. doi: 10.1097/00129492-200604000-00021. [DOI] [PubMed] [Google Scholar]
- 59.Han G, Li F, Ten Dijke P, Wang XJ. Temporal smad7 transgene induction in mouse epidermis accelerates skin wound healing. Am J Pathol. 2011;179(4):1768–1779. doi: 10.1016/j.ajpath.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tscharntke M, et al. Impaired epidermal wound healing in vivo upon inhibition or deletion of Rac1. J Cell Sci. 2007;120(pt 8):1480–1490. doi: 10.1242/jcs.03426. [DOI] [PubMed] [Google Scholar]
- 61.Mao X, Choi EJ, Payne AS. Disruption of desmosome assembly by monovalent human pemphigus vulgaris monoclonal antibodies. J Invest Dermatol. 2009;129(4):908–918. doi: 10.1038/jid.2008.339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jennings JM, et al. Desmosome disassembly in response to pemphigus vulgaris IgG occurs in distinct phases and can be reversed by expression of exogenous Dsg3. J Invest Dermatol. 2011;131(3):706–718. doi: 10.1038/jid.2010.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Barber AG, Wajid M, Columbo M, Lubetkin J, Christiano AM. Striate palmoplantar keratoderma resulting from a frameshift mutation in the desmoglein 1 gene. J Dermatol Sci. 2007;45(3):161–166. doi: 10.1016/j.jdermsci.2006.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zamiri M, et al. Mutation in DSG1 causing autosomal dominant striate palmoplantar keratoderma. Br J Dermatol. 2009;161(3):692–694. doi: 10.1111/j.1365-2133.2009.09316.x. [DOI] [PubMed] [Google Scholar]
- 65.Wan H, et al. Striate palmoplantar keratoderma arising from desmoplakin and desmoglein 1 mutations is associated with contrasting perturbations of desmosomes and the keratin filament network. Br J Dermatol. 2004;150(5):878–891. doi: 10.1111/j.1365-2133.2004.05996.x. [DOI] [PubMed] [Google Scholar]
- 66.Kljuic A, Gilead L, Martinez-Mir A, Frank J, Christiano AM, Zlotogorski A. A nonsense mutation in the desmoglein 1 gene underlies striate keratoderma. Exp Dermatol. 2003;12(4):523–527. doi: 10.1034/j.1600-0625.2003.00017.x. [DOI] [PubMed] [Google Scholar]
- 67.Lu A, et al. A clathrin-dependent pathway leads to KRas signaling on late endosomes en route to lysosomes. J Cell Biol. 2009;184(6):863–879. doi: 10.1083/jcb.200807186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Weibrecht I, et al. Proximity ligation assays: a recent addition to the proteomics toolbox. Expert Rev Proteomics. 2010;7(3):401–409. doi: 10.1586/epr.10.10. [DOI] [PubMed] [Google Scholar]
- 69.Calautti E, Li J, Saoncella S, Brissette JL, Goetinck PF. Phosphoinositide 3-kinase signaling to Akt promotes keratinocyte differentiation versus death. J Biol Chem. 2005;280(38):32856–32865. doi: 10.1074/jbc.M506119200. [DOI] [PubMed] [Google Scholar]
- 70.Denning MF, Dlugosz AA, Williams EK, Szallasi Z, Blumberg PM, Yuspa SH. Specific protein kinase C isozymes mediate the induction of keratinocyte differentiation markers by calcium. Cell Growth Differ. 1995;6(2):149–157. [PubMed] [Google Scholar]
- 71.Ashery U, Yizhar O, Rotblat B, Kloog Y. Nonconventional trafficking of Ras associated with Ras signal organization. Traffic. 2006;7(9):119–126. doi: 10.1111/j.1600-0854.2006.00459.x. [DOI] [PubMed] [Google Scholar]
- 72.Chiu VK, et al. Ras signalling on the endoplasmic reticulum and the Golgi. Nat Cell Biol. 2002;4(5):343–350. doi: 10.1038/ncb783. [DOI] [PubMed] [Google Scholar]
- 73.Omerovic J, Prior IA. Compartmentalized signalling: Ras proteins and signalling nanoclusters. FEBS J. 2009;276(7):1817–1825. doi: 10.1111/j.1742-4658.2009.06928.x. [DOI] [PubMed] [Google Scholar]
- 74.Onken B, Wiener H, Philips MR, Chang EC. Compartmentalized signaling of Ras in fission yeast. Proc Natl Acad Sci U S A. 2006;103(24):9045–9050. doi: 10.1073/pnas.0603318103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Li W, Han M, Guan KL. The leucine-rich repeat protein SUR-8 enhances MAP kinase activation and forms a complex with Ras and Raf. Genes Dev. 2000;14(8):895–900. [PMC free article] [PubMed] [Google Scholar]
- 76.Rangwala R, Banine F, Borg JP, Sherman LS. Erbin regulates mitogen-activated protein (MAP) kinase activation and MAP kinase-dependent interactions between Merlin and adherens junction protein complexes in Schwann cells. J Biol Chem. 2005;280(12):11790–11797. doi: 10.1074/jbc.M414154200. [DOI] [PubMed] [Google Scholar]
- 77.Gladden AB, Hebert AM, Schneeberger EE, McClatchey AI. The NF2 tumor suppressor, Merlin, regulates epidermal development through the establishment of a junctional polarity complex. Dev Cell. 2010;19(5):727–739. doi: 10.1016/j.devcel.2010.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Rodriguez-Viciana P, Oses-Prieto J, Burlingame A, Fried M, McCormick F. A phosphatase holoenzyme comprised of Shoc2/Sur8 and the catalytic subunit of PP1 functions as an M-Ras effector to modulate Raf activity. Mol Cell. 2006;22(2):217–230. doi: 10.1016/j.molcel.2006.03.027. [DOI] [PubMed] [Google Scholar]
- 79.Lambert S, Ameels H, Gniadecki R, Herin M, Poumay Y. Internalization of EGF receptor following lipid rafts disruption in keratinocytes is delayed and dependent on p38 MAPK activation. J Cell Physiol. 2008;217(3):834–845. doi: 10.1002/jcp.21563. [DOI] [PubMed] [Google Scholar]
- 80.Nagy P, et al. Lipid rafts and the local density of ErbB proteins influence the biological role of homo- and heteroassociations of ErbB2. J Cell Sci. 2002;115(pt 22):4251–4262. doi: 10.1242/jcs.00118. [DOI] [PubMed] [Google Scholar]
- 81.Kami K, Chidgey M, Dafforn T, Overduin M. The desmoglein-specific cytoplasmic region is intrinsically disordered in solution and interacts with multiple desmosomal protein partners. J Mol Biol. 2009;386(2):531–543. doi: 10.1016/j.jmb.2008.12.054. [DOI] [PubMed] [Google Scholar]
- 82.Rutman AJ, Buxton RS, Burdett ID. Visualisation by electron microscopy of the unique part of the cytoplasmic domain of a desmoglein, a cadherin-like protein of the desmosome type of cell junction. FEBS Lett. 1994;353(2):194–196. doi: 10.1016/0014-5793(94)01049-8. [DOI] [PubMed] [Google Scholar]
- 83.Dusek RL, et al. The differentiation-dependent desmosomal cadherin desmoglein 1 is a novel caspase-3 target that regulates apoptosis in keratinocytes. J Biol Chem. 2006;281(6):3614–3624. doi: 10.1074/jbc.M508258200. [DOI] [PubMed] [Google Scholar]
- 84.Chen J, et al. The C-terminal unique region of desmoglein 2 inhibits its internalization via tail-tail interactions. J Cell Biol. 2012;199(4):699–711. doi: 10.1083/jcb.201202105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Stefanova M, et al. Disruption of ERBB2IP is not associated with dystrophic epidermolysis bullosa in both father and son carrying a balanced 5;13 translocation. J Invest Dermatol. 2005;125(4):700–704. doi: 10.1111/j.0022-202X.2005.23875.x. [DOI] [PubMed] [Google Scholar]
- 86.Favre B, et al. The hemidesmosomal protein bullous pemphigoid antigen 1 and the integrin beta 4 subunit bind to ERBIN. Molecular cloning of multiple alternative splice variants of ERBIN and analysis of their tissue expression. J Biol Chem. 2001;276(35):32427–32436. doi: 10.1074/jbc.M011005200. [DOI] [PubMed] [Google Scholar]
- 87.Wang S, Uchi H, Hayashida S, Urabe K, Moroi Y, Furue M. Differential expression of phosphorylated extracellular signal-regulated kinase 1/2, phosphorylated p38 mitogen-activated protein kinase and nuclear factor-kappaB p105/p50 in chronic inflammatory skin diseases. J Dermatol. 2009;36(10):534–540. doi: 10.1111/j.1346-8138.2009.00696.x. [DOI] [PubMed] [Google Scholar]
- 88.Brem H, et al. Molecular markers in patients with chronic wounds to guide surgical debridement. Mol Med. 2007;13(1–2):30–39. doi: 10.2119/2006-00054.Brem. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Stojadinovic O, et al. Molecular pathogenesis of chronic wounds: the role of beta-catenin and c-myc in the inhibition of epithelialization and wound healing. Am J Pathol. 2005;167(1):59–69. doi: 10.1016/S0002-9440(10)62953-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Stojadinovic O, et al. Deregulation of keratinocyte differentiation and activation: a hallmark of venous ulcers. J Cell Mol Med. 2008;12(6B):2675–2690. doi: 10.1111/j.1582-4934.2008.00321.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kaplan FM, Kugel CH 3rd, Dadpey N, Shao Y, Abel EV, Aplin AE. SHOC2 and CRAF mediate ERK1/2 reactivation in mutant NRAS-mediated resistance to RAF inhibitor. J Biol Chem. 2012;287(50):41797–41807. doi: 10.1074/jbc.M112.390906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Cordeddu V, et al. Mutation of SHOC2 promotes aberrant protein N-myristoylation and causes Noonan-like syndrome with loose anagen hair. Nat Genet. 2009;41(9):1022–1026. doi: 10.1038/ng.425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Bazzi H, Christiano AM. Broken hearts, woolly hair, and tattered skin: when desmosomal adhesion goes awry. Curr Opin Cell Biol. 2007;19(5):515–520. doi: 10.1016/j.ceb.2007.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Komatsuzaki S, et al. Mutation analysis of the SHOC2 gene in Noonan-like syndrome and in hematologic malignancies. J Hum Genet. 2010;55(12):801–809. doi: 10.1038/jhg.2010.116. [DOI] [PubMed] [Google Scholar]
- 95.Thomason HA, Scothern A, McHarg S, Garrod DR. Desmosomes: adhesive strength and signalling in health and disease. Biochem J. 2010;429(3):419–433. doi: 10.1042/BJ20100567. [DOI] [PubMed] [Google Scholar]
- 96.Simpson CL, Kojima S, Getsios S. RNA interference in keratinocytes and an organotypic model of human epidermis. Methods Mol Biol. 2010;585:127–146. doi: 10.1007/978-1-60761-380-0_10. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.