

Abstract
Anti-neutrophil cytoplasmic antibody–associated (ANCA-associated) small vessel necrotizing vasculitis is caused by immune-mediated inflammation of the vessel wall and is diagnosed in some cases by the presence of myeloperoxidase-specific antibodies (MPO-ANCA). This multicenter study sought to determine whether differences in ANCA epitope specificity explain why, in some cases, conventional serologic assays do not correlate with disease activity, why naturally occurring anti-MPO autoantibodies can exist in disease-free individuals, and why ANCA are undetected in patients with ANCA-negative disease. Autoantibodies from human and murine samples were epitope mapped using a highly sensitive epitope excision/mass spectrometry approach. Data indicated that MPO autoantibodies from healthy individuals had epitope specificities different from those present in ANCA disease. Importantly, this methodology led to the discovery of MPO-ANCA in ANCA-negative disease that reacted against a sole linear sequence. Autoantibodies against this epitope had pathogenic properties, as demonstrated by their capacity to activate neutrophils in vitro and to induce nephritis in mice. The confounder for serological detection of these autoantibodies was the presence of a fragment of ceruloplasmin in serum, which was eliminated in purified IgG, allowing detection. These findings implicate immunodominant epitopes in the pathology of ANCA-associated vasculitis and suggest that autoantibody diversity may be common to other autoimmune diseases.
Introduction
Myeloperoxidase-specific anti-neutrophil cytoplasmic antibodies (MPO-ANCA) and proteinase 3–specific ANCA (PR3-ANCA) are serologic markers used in routine clinical assays to diagnose small vessel necrotizing vasculitis (e.g., microscopic polyangiitis [MPA] and granulomatosis with polyangiitis [GPA]) (1). In vitro and in vivo studies provide compelling evidence that ANCA play a critical role in the pathogenesis of ANCA-associated vasculitis (AAV) (2, 3). However, 3 clinical observations plague the contention that ANCA are pathogenic. First, conventional serologic assays fail to detect ANCA in some patients with classic clinical and pathologic features of AAV (4). These patients are labeled as having ANCA-negative disease. Second, ANCA titers do not correlate well with disease activity, especially in MPO-ANCA disease (5, 6). Third, naturally occurring anti-MPO and anti-PR3 antibodies exist in healthy individuals (7, 8).
In this study, we used highly sensitive MALDI-TOF/TOF-MS (where MS indicates mass spectrometry) (9) to identify specific epitopes on MPO in an effort to explain these inconsistencies. The findings implicate a role for immunodominant epitopes, and perhaps epitope spreading, in the evolution of AAV and accentuate the diversity of autoantibody specificity that is likely to occur in many other autoimmune diseases. One epitope identified here appears to be a critical immunodominant epitope in that (a) reactive autoantibodies to it are found solely in patients with active disease, (b) surprisingly, it is the immunodominant autoantibody in some patients with ANCA-negative AAV, and (c) it is pathogenic in that the transfer of antibodies from mice immunized with this murine epitope cause glomerulonephritis.
Previously undetected in serum-based assays, the discovery of an immunodominant epitope and corresponding autoantibody whose detection is masked by a natural inhibitor of MPO raises the possibility of a similar phenomenon in other “seronegative” autoimmune diseases.
Results
Epitope diversity in patients with MPO-ANCA.
MPO-ANCA–positive patients (n = 45) from the University of North Carolina cohort (UNC) were analyzed in the initial studies. Demographics of the cohort (Table 1) indicate a group of predominantly mixed European descent. Approximately 40% were diagnosed with MPA, 40% with renal-limited disease, and 20% with GPA. To compare epitope specificities related to disease activity, longitudinal samples were analyzed to include active disease (n = 52) and remission (n = 35) from the UNC cohort (Supplemental Tables 1 and 2; supplemental material available online with this article; doi: 10.1172/JCI65292DS1). Ten healthy subjects were included in the analyses. Purified Ig from sera samples was subjected to analysis by epitope excision MALDI-TOF/TOF MS (depicted in Supplemental Figure 1A) to identify epitope profiles of autoantibodies reactive to MPO. In order to epitope map low-titer anti-MPO antibodies from healthy subjects and from patients in disease remission, an approach was designed using the 16O-to-18O exchange technique, an adaptation of a proteomics-based quantification method (10). Incorporation of H218O on the carboxytermini of proteolytic fragments produces a 4-kDa mass shift in the MS spectra, decipherable from background noise. MPO epitopes unique to Ig from patients with active disease were designated “exclusive to active disease”; epitopes identified in remission samples were designated “persistent during remission; and those present in samples from healthy subjects were designated asymptomatic or “natural.”
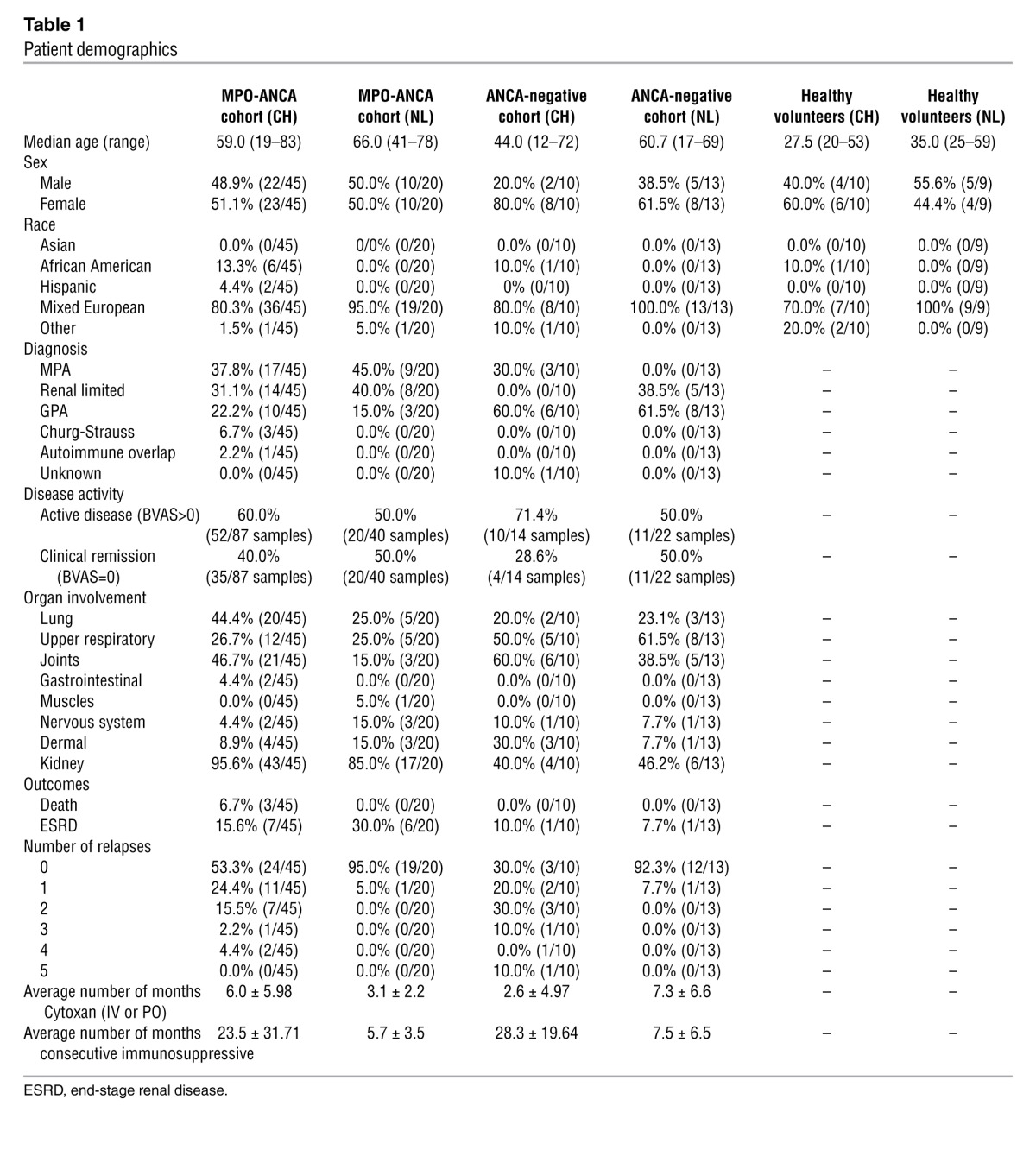
Table 1.
Patient demographics
A total of 25 anti-MPO autoantibody epitopes were identified and categorized by disease association (Figure 1A). Twelve epitopes were unique and exclusive to MPO-ANCA expressed during active disease. Eight epitopes were reactive with Ig from healthy subjects. Notably, asymptomatic or natural autoantibodies were present in all active disease samples. There was no correlation between number of epitope-specific autoantibodies and age of the individual at the time of the sample (R2 = 0.074) (Supplemental Figure 2).
Figure 1. Study of autoantibody epitope specificity within an MPO-ANCA–positive cohort.

(A) Heat map demonstrating high-level positive epitope binding (bright red), weakly positive epitope binding (low titer) detected only with very sensitive H218O labeling (dark red), and negative detection (black). The y axis includes samples from active disease patients (n = 52), patients in remission (n = 35), and healthy subjects (HS) (n = 10). The x axis includes 25 epitopes classified as exclusive to active disease, persistent during remission if detected in active disease, in remission, and rarely, at low level, in healthy subjects. Epitopes were designated asymptomatic or natural if present in healthy subjects (required H218O labeling for detection). (B) Analysis of epitope specificity by MS was performed on Ig from a UNC cohort (n = 97) and an NL cohort (n = 20). (B–D) Distribution of autoantibody epitopes identified. UNC–active disease (n = 52), NL–active disease (n = 20), UNC–clinical remission (n = 35), UNC–healthy subjects (n = 10). Unique epitopes strictly associated with ANCA disease were identified in all 72 active disease samples (B and C). Extremely low-level anti-MPO autoantibodies from healthy subjects were negative for reactivity with disease-specific epitopes (D).
To examine whether the active disease MPO-ANCA epitope profile was universal to ANCA disease, a replication study was performed on a cohort from Groningen, The Netherlands (NL), of patients with active disease (n = 20) (Table 1). The frequencies of the 6 most prominent MPO-ANCA epitopes found in both the UNC and NL cohorts were consistent in specificity, although they differed in distribution (Figure 1, B–D).
Both conformational and linear epitopes are detected by epitope excision MALDI-TOF/TOF MALDI-MS. To distinguish conformational epitopes from linear epitopes, the protocol was modified such that Ig was incubated with predigested MPO protein, with the assumption that loss of epitope reactivity signified a dependence on conformation. Of the 25 epitopes originally identified, 20 were conformational. Synthetic peptides corresponding to the 5 linear peptides were tested by ELISA for validation of MALDI-MS results. A linear epitope located on the heavy chain of MPO (aa 447–459), which was exclusively associated with active disease by MS, was confirmed by ELISA with reactivity in 42.9% (21/49) of UNC patients’ samples and in 52.2% (12/23) of the NL cohort (Figure 2A). Reactivity declined in clinical remission for both the UNC and NL cohorts (Figure 2A). Of note, all samples with borderline or low OD values by ELISA were analyzed MS/MS to determine whether they were negative for the epitope or whether they were low-titer positive. MPO-ANCA patients with values in the range of healthy individuals were found to be negative by MS analysis. MPO peptide (aa 516–524), a second linear epitope identified by MS, was confirmed by ELISA to be reactive with Ig from active disease samples; however, reactivity was found in remission samples in both cohorts (Figure 2B). In an effort to validate conformational MPO epitopes, peptides of the amino acid sequences were synthesized and used as substrates for ELISAs. Ig samples positive by MS were negative by ELISA, confirming that these are conformationally dependent epitopes (Supplemental Figure 3).
Figure 2. MPO-ANCA reactive with epitope aa–447-459 are exclusively associated with active disease.

(A and B) ELISA results testing for reactivity against 2 linear epitopes identified by MS. Anti-MPO447–459 autoantibodies correlated with disease activity in both the UNC and NL cohorts (A). Anti-MPO516–524 autoantibodies were present in active disease and remission but were absent in healthy subjects in both cohorts (B). (Note: ELISAs of NL cohorts were conducted at the UMCG using their specific protocol and reagents, except for synthetic peptides provided by UNC). (C) Location of epitopes on the MPO molecule. Disease-associated epitopes (blue) (including aa 447–459) exist in tandem with epitopes recognized by natural autoantibodies (green). Amino acids predicted to be required for autoantibody binding are highlighted in red.
Intriguingly, visualization of epitopes on the crystal structure of MPO as a monomer, shown in Figure 2C, revealed that epitopes of asymptomatic or natural autoantibodies were spatially adjacent to epitopes “exclusive to active disease.” Essential amino acids within these epitopes were identified by fine epitope mapping using a chymotrypsin enzymatic digestion (for refined epitopes see Supplemental Figure 4). The sequential alignment of epitopes is suggestive of epitope spreading from nonpathogenic to pathogenic epitopes as disease develops.
“ANCA-negative” AAV patients have MPO-ANCA specific for an epitope exclusive to active disease.
A variation of epitope excision MS for MPO-ANCA methodology produced a “discovery” tool used to search for putative autoantigens in ANCA-negative AAV. ANCA-negative AAV patients were identified by retrospective review of medical records insuring histological evidence of vasculitis with negative serological test results from the UNC Hospital clinical laboratory. As an additional measure, sera samples selected for analysis were retested for ANCA reactivity in the research laboratory and externally in the laboratory of John Niles (Massachusetts General Hospital), and all were confirmed ANCA negative. Protein lysate of total peripheral blood leukocytes was probed for antigen or antigens reactive with purified total Ig from ANCA-negative AAV patients’ sera (Figure 3A). A sole peptide was eluted and identified in the MS spectrum analysis at a mass of 1492.141 (Figure 3B), which corresponded to the MPO peptide aa 447–459 (RKIVGAMVQIITY), the linear epitope we identified in approximately 50% of MPO-ANCA–positive AAV patients with active disease. It was found that purified Ig reacted with native MPO by ELISA even though sera from these ANCA-negative patients did not (Figure 3, C and D). By ELISA testing purified Ig, levels of reactivity to native MPO and MPO peptide aa 447–459 were detected in 8 of 10 UNC AAV patients and in 6 of 11 NL AAV patients (cohort demographics, Table 1). Reactivity corresponded well with disease activity, declining to low levels as patients entered disease remission in both the UNC (n = 4) and NL (n = 5) patient cohorts.
Figure 3. Epitope excision/MS detects autoantibodies in patients with an ANCA-negative serology.

Ig from seronegative UNC patients (n = 10) and NL patients (n = 12) was incubated with leukocyte protein lysates as depicted (A). Reactive antigens were captured by autoantibodies. Sites of contact between the autoantigen and the Ig (epitope) were protected from digestion. Peptides remaining bound to Ig after digestion were eluted and analyzed by MALDI-TOF/TOF MS/MS. A search for autoantigens recognized by Ig purified from patients with pauci-immune vasculitis and ANCA-negative serology revealed a single MS peak determined to be an MPO epitope aa 447–459 (B). MS results were validated by ELISA indicating Ig reactivity against native MPO and MPO peptide aa 447–459 (C and D). Analysis of longitudinal samples (UNC cohort, n = 4) (NL cohort, n = 5) indicated a correlation between active disease and the presence of Ig reactive with native MPO and MPO peptide aa 447–459 (C and D).
An epitope-masking factor in sera interferes with conventional serologic testing for MPO-ANCA in ANCA-negative AAV.
To address the question of why the discrepancy of no detectable ANCA reactivity in sera but positive reactivity in purified Ig, an ELISA was performed to directly compare serum to purified Ig from each ANCA-negative patient. Data confirmed that anti-MPO447–459 autoantibodies were not detectable by conventional serologic tests, while purified Ig readily reacted with both native MPO protein (Figure 4A) and MPO peptide aa 447–459 (Figure 4B). Titration of serum into purified Ig blocked reactivity, regardless of whether serum was from a patient with ANCA disease or from a healthy subject (Figure 4C). We hypothesized that a component in sera was obscuring detection of antibodies reactive with MPO aa 447–459.
Figure 4. MPO epitope aa 447–459 is masked by a proteolytic fragment of a common serum protein.

Sera from ANCA-negative vasculitis patients (n = 8) was negative for reactivity against native MPO, while Ig was positive by direct ELISA (A). (B) Similar results when testing for reactivity against MPO peptide aa 447–459 by direct ELISA. (C) Inhibitory effects of serum spiked into purified Ig. (D–F) Data from protein studies to identify the masking factor in serum. Affinity purification of serum proteins that complex with peptide aa 447–459 identified an approximately 50-kDa protein by SDS-PAGE, Coomassie-stained gel (D). MS analysis identified the protein as CP. Identity was confirmed by Western blot (E) probed with an anti-CP antibody. Purified CP was purchased and digested with plasmin in vitro to produce a 50-kDa fragment SDS-PAGE, Coomassie-stained gel (samples were run on the same gel but were not contiguous) (F). ELISA results (G) indicated that full-length CP (151 kDa) did not mask the epitope, while CP cleaved by plasmin was effective in blocking reactivity by 30%–50%. Reactivity appears unaffected by addition of undigested CP to MPO-ANCA IgG (polyclonal) from 4 patients (H), indicating that specificity of the CP fragment effect on aa 447–459. Error bars represent the mean ± SEM.
To isolate the putative serum factor, immobilized MPO aa 447–459 peptide was used as bait to fish out host proteins with binding capacity. Ceruloplasmin (CP) was identified by MS as a binding partner. However, the CP bound to MPO peptide aa 447–459 was primarily a 50-kDa protein on SDS-PAGE, while full-length CP is 151 kDa (Figure 4D). An anti-CP Western blot confirmed the 50 kDa protein was a fragment of CP (Figure 4E). To test whether proteolytically processed CP was required for epitope blocking, a plasmin-generated fragment of CP (11) was tested for epitope-masking capability (Figure 4F). The digested CP decreased anti-MPO447–459 autoantibody reactivity by 30%–50% (n = 8) (Figure 4G), while full-length CP did not (Figure 4G). The effect of the CP-masking fragment was selective for blocking anti-MPO447–459 reactivity and did not affect the overall binding of polyclonal MPO-ANCA Ig to native MPO (Figure 4H).
Pathogenic potential of anti-MPO autoantibodies specific for MPO epitope aa 447–459.
Anti–MPO447–459 autoantibodies induced neutrophilic release of reactive oxygen species similar to total MPO-ANCA in an in vitro activation assay (Figure 5), indicative of a pathogenic potential. For comparison, 3 asymptomatic or natural anti-MPO autoantibodies purified from patients’ sera (n = 4) during disease remission and adjusted for concentration showed essentially no release of reactive oxygen species. This study replicates prior studies of MPO-ANCA activation of neutrophils (2).
Figure 5. In vitro and in vivo pathogenic potential of anti-MPO447–459.

Affinity-purified anti-MPO447–459 autoantibodies were capable of activating neutrophils, as measured by their ability to induce release of reactive oxygen species, while nonpathogenic (MPO516–524) and natural (MPO579–590, MPO237–248, and MPO530–536) anti-MPO autoantibodies were not. Neutrophils isolated from healthy subjects (n = 4) were exposed to purified autoantibodies from unique individuals (n = 4). Results represent the mean ± SEM. No further statistics were done due to the limited sample size.
In collaboration with colleagues at Monash University, the pathogenic potential of anti-MPO autoantibodies targeting epitope aa 447–459 was studied in their established animal model. Mice deficient in murine MHC II and transgenic for HLA-DRB1*15:01 (DR2 Tg mice) were immunized with murine MPO peptide aa 409–428 (PRWNGEKLYQEARKIVGAMV) (bold letters indicate the aa overlap between human and murine peptides), an epitope that induces glomerulonephritis in mice (12). This pathogenic murine T cell epitope overlaps with the human disease–associated B cell epitope aa 447–459 (RKIVGAMVQIITY).
DR2 Tg mice were immunized with a peptide containing the human disease–associated B cell epitope (LYQEARKIVGAMVQITTYR; the human and mouse sequences are identical). Compared with OVA aa 323–339–immunized mice, MPO aa 442–460–immunized mice developed albuminuria and hematuria and had increased blood urea nitrogen (BUN) values and mild glomerular proliferation (Supplemental Figure 5). Pooled sera IgG purified from these mice showed a p-ANCA pattern when applied to normal Mpo+/+, but not Mpo–/– neutrophils (Supplemental Figure 5A). This IgG pool was passively transferred into naive DR2 Tg mice treated with LPS. When studied 1 and 6 days after IgG transfer, recipients of anti-MPO antibodies from MPO aa 442–460–immunized donors developed albuminuria, some early hematuria, and a raised BUN (Figure 6, A–C). IgG induced glomerular injury (Figure 6, D and E) and neutrophil recruitment after transfer to DR2 Tg recipients (Figure 6F). Each recipient mouse developed glomerular histological abnormalities marked by hypercellularity with glomerular neutrophil accumulation, producing a proliferative, albeit not a necrotizing, lesion. Immunizing MHC II humanized mice with aa 442–460 (conserved between human and mice) induced a proliferative glomerular disease and MPO-reactive antibodies after 28 days. A causal role for MPO-reactive antibodies was supported by disease of greater severity upon transfer of IgG from the immunized animals into naive recipients.
Figure 6. Passive transfer of IgG from MPO442–460–immunized mice is nephritogenic.

Independently, a T cell MPO epitope had been identified, and DR2 transgenic mice were injected with the overlapping MPO peptide aa 442–460 (LYQEARKIVGAMVQIITYR) that includes the human MPO epitope aa 447–459 (RKIVGAMVQIITY). Albuminuria and hematuria (A and B) were measured on days 1 and 6 and BUN on day 6 (C). Abnormal glomeruli (day 6, D, E, G, and H) were assessed based on capillary wall thickening and mesangial hypercellularity on formalin-fixed PAS–stained kidney sections. Scale bar: 45 μm). Neutrophil recruitment (F) was assessed based on immunohistochemistry by anti–Gr-1 antibodies on PLP-fixed frozen kidneys.
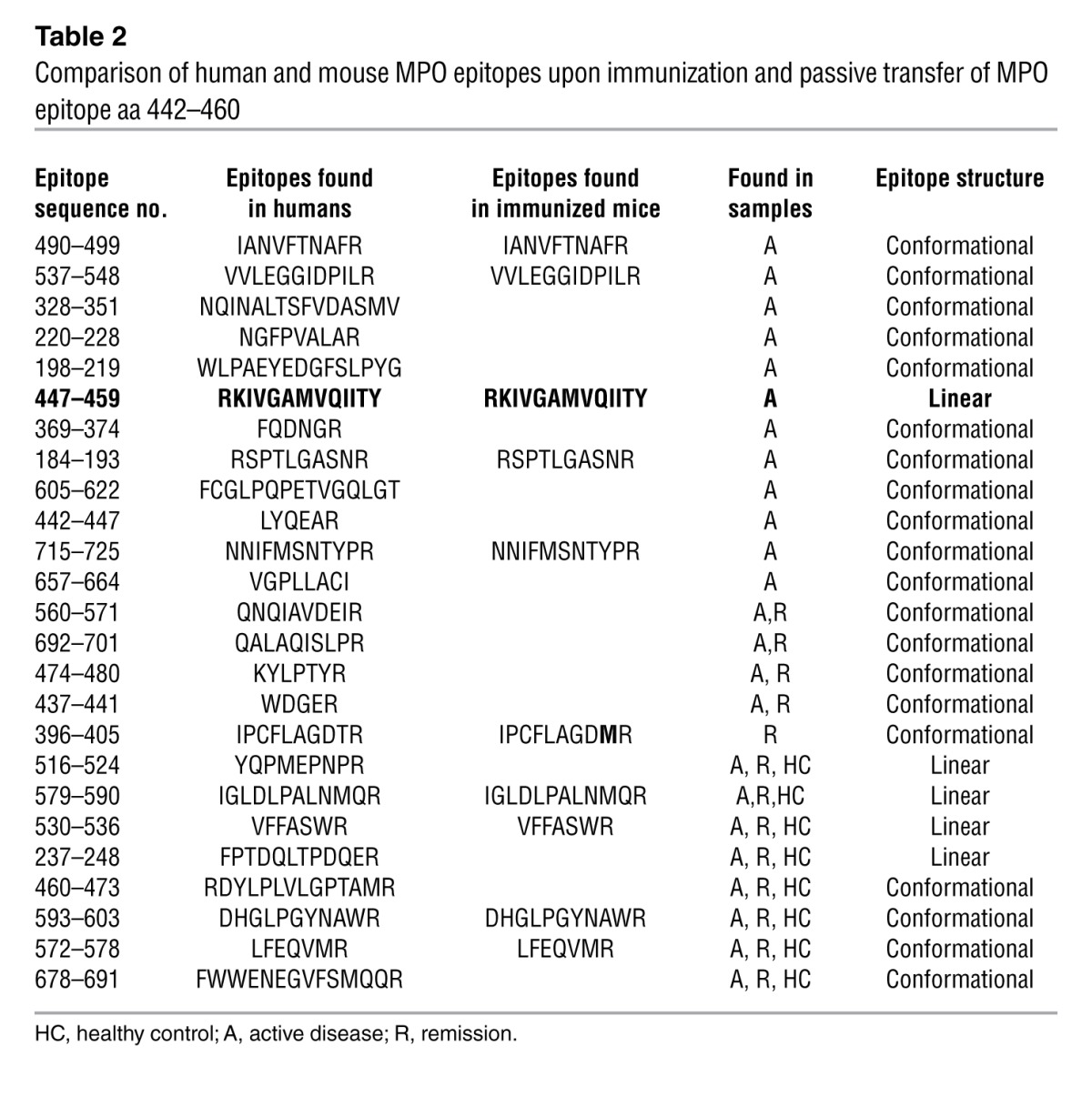
As HLA-MHC II help shape the B and T cell repertoires, and consequently the response to foreign and self antigens, we wanted to define the epitope specificity of the anti-MPO autoantibodies that could be induced experimentally using DR2 Tg mice. In this way, we could assess development of the anti-MPO antibody response after immunization of a single epitope in the context of a human MHC II. IgG from sera of immunized mice was profiled for anti-MPO epitope specificity using epitope excision MS. DR2 Tg mice developed antibodies targeting the human B cell epitope aa 447–459, which is within a conserved region of both murine and human MPO. Unpredictably, immunized DR2 Tg mice initiated a polyclonal response producing autoantibodies against the entire MPO molecule. The epitope profile was astonishingly similar to that of the MPO-ANCA human profile (Table 2), indicative of the critical nature of this epitope.
Table 2.
Comparison of human and mouse MPO epitopes upon immunization and passive transfer of MPO epitope aa 442–460
Discussion
Fundamental questions problematic to the understanding of ANCA vasculitis were resolved, at least in part, by the studies presented here. These studies explain why asymptomatic or natural MPO autoantibodies can exist in individuals free of disease: epitope specificity defines pathogenicity. They explain why there is a lack of correlation between MPO-ANCA titers and active disease: clinical tests do not discriminate between natural anti-MPO autoantibodies and potentially pathogenic ANCA. They explain why some patients with vasculitis are seronegative for ANCA in clinical tests: detection of a monoclonal MPO-ANCA reactive with a small, restricted epitope is obstructed by a protein present in sera.
For years, ANCA-negative small vessel vasculitis (SVV) has posed a clinical and pathogenical dilemma. Our findings suggest that they develop a restricted autoantibody response against a linear epitope on MPO (aa 447–459), an epitope masked by a CP fragment, which is a crucial finding concerning this group of patients. Clinically, patients with ANCA-negative SVV have the same signs and symptoms of disease as those patients who are ANCA-positive (4, 13). Frequently, these patients are diagnosed late in the course of their illness. Many of these patients have reoccurring ear, nose, and throat disease. We were surprised that many, but not all, of the UNC and NL cohorts who were ANCA-negative had an antibody to MPO and not to PR3. From a pathogenic perspective, ANCA-negative patients presented a real quandary to those who propose that ANCA are fundamental to the pathogenesis of pauci-immune SVV. The findings in this study, that a portion of ANCA-negative patients have MPO-ANCA, diminish that concern. The linear epitope identified is the same one that was found to be associated with active disease in the MPO-ANCA–positive patient group, which declined upon clinical remission. These findings underscore the pathogenic capacity of antibodies against this epitope, further supported by their ability to activate neutrophils in vitro and cause nephritogenic disease in a transgenic murine model. A crucial role of an autoantibody to murine MPO epitope aa 409–428 or overlapping human MPO epitope aa 442–460 (12) is further demonstrate by data implying that this initial immune response may be the instigating event that triggers a polyclonal autoantibody profile, similar to an MPO-positive patient’s polyclonal repertoire during active disease (Table 2).
Anti–MPO447–459 autoantibodies are exclusive to active disease. An enzymatically produced fragment of a natural host protein, abundant in serum, shields MPO epitope aa 447–459 from autoantibody binding. It remains a puzzle as to why only enzymatically processed CP fragments have the epitope “blocking” capability, while full-length protein does not. We used plasmin to digest CP mainly because plasmin had been reported to produce a fragment of approximately 50 kDa (11). We found that CP cleaved with urokinase also blocked antibody binding, but thrombin did not, underscoring the specificity of the “blocking” mechanism. Full-length CP is the natural inhibitor of MPO activity, and it was initially suggested that patients diagnosed with MPO-ANCA might be deficient in CP. Subsequent reports determined the converse to be true, that ANCA patients did not have a deficiency in serum CP levels and instead had a marked increase during active disease (14, 15). In addition, ANCA were reported to have the ability to disassociate the MPO/CP complex (16). However, anti–MPO447–459 autoantibodies did not dislodge the complex of MPO peptide aa 447–459 and the CP fragment in our assays, highlighting the high affinity of the complex.
Researchers who have studied anti-MPO–specific epitopes agreed that most epitopes were likely conformationally dependent, although a few linear epitopes were identified. Our data verify and extend the epitope mapping literature. The light chain of MPO was reported to contain immunodominant linear epitopes recognized by MPO-ANCA sera samples (17). We too found a linear epitope in the light chain (aa 237–248) of MPO along with 3 conformational epitopes. Conformationally dependent epitopes were reported to lie within 2 regions of the heavy chain (aa 279–341 and aa 474–512) (18–20), which overlap with 3 disease-associated conformational epitopes we identified here. Using MPO human/mouse chimeras, aa 517–667 and aa 668–774 were identified as important epitopes (21). We also identified 12 epitopes that fall within these 2 segments. Another group used overlapping peptide ELISAs and identified 7 linear immunodominant epitopes (22). One of these, aa 511–522 (RLDNRYQPMEPN), overlapped with one we identified as a disease-associated epitope, aa 516–524 (YQPMEPNPR).
Asymptomatic or natural autoantibodies are known to occur prior to the onset of autoantibody-induced ANCA and anti–glomerular basement membrane (anti-GBM) disease (7, 8, 23). Visualizing epitopes on the 3D crystal structure of MPO (pdb 3f9) revealed an interesting adjacency of natural epitopes to epitopes exclusive to active disease (Figure 2C). This suggests the possibility that an asymptomatic autoantibody response to MPO may precede the onset of autoimmune disease (7, 8, 23) and that epitope spreading produces disease-causing antibodies. It is also conceivable that asymptomatic autoantibodies against MPO could structurally alter the conformation of adjacent epitopes and expose cryptic epitopes that would induce potentially pathogenic autoantibodies.
A limitation of this study is the focus on continuous (or linear) epitopes. Discontinuous (or conformational) epitopes are structurally more complicated to study due to the tertiary and quaternary structure of the protein, and further characterization is beyond our epitope-mapping methodology. We are aware of the likelihood that other critical epitopes may not have been detected using our approach. An anti–MPO447–459 autoantibody was not detected in 2 ANCA-negative sera from the UNC cohort and 5 from the NL cohort, indicating that another autoantigen or masked epitope may be a target. The inherently small sample size of the ANCA-negative cohort limited the statistical power of our study.
The highly sensitive epitope excision MS assay employed in our study could facilitate the discovery of unidentified disease-associated epitopes and autoantibody-autoantigen pairs in other diseases and help elucidate a role for modulation of epitope specificity in the development, progression, and remission of autoimmune diseases. Moreover, it provides a channel to gain information on the contribution of asymptomatic circulating autoantibodies in healthy individuals who eventually develop autoimmune disease (8). The detection of autoantibodies directed against MPO, a previously unrecognized autoantigen target in patients with ANCA-negative AAV, illustrates the power of this method for identifying novel autoantigens and epitopes in other autoimmune diseases. The elucidation of the structural and functional diversity of autoantibodies exclusive to active disease and asymptomatic and natural autoantibodies demonstrates that not all ANCA have equal pathogenic potential.
In summary, these observations demonstrate substantial MPO-ANCA epitope diversity. Three functionally distinct classes of autoantibody/epitope pairs are recognized: epitopes exclusive to active disease, persistent during remission, and asymptomatic or natural. While titers of total MPO-ANCA do not correlate well with disease activity; specific titers of MPO-ANCA directed against epitopes exclusive to active disease correlate extremely well with disease activity. For what we believe is the first time, the perplexing ANCA-negative subset of AAV patients is explained by identifying occult MPO-ANCA that have very restricted specificity for an anti-MPO autoantibody that is blocked in serum by a CP fragment.
Methods
Patient data
UNC cohort.
Patients with ANCA SVV, categorized by the Chapel Hill Consensus Conference nomenclature (24), were included in this study. The UNC cohort (Table 1) included 45 MPO-ANCA–positive patients with MPA (37.8%), GPA (22.2%), renal-limited disease (31.1%), and overlapping pauci-immune vasculitides (2.2%). This cohort was 51.1% female and 80.3% of mixed European descent, with a median age of 59.0 years and a range of 19–83 years. Samples for MS studies were obtained from 52 patients with active disease and 35 patients in remission. Of these samples, there were 22 longitudinal active and remission samples from individual patients. The median follow-up for the cohort was 3.9 years, with 50% of patients followed from 1.2 to 9.3 years (full range was a few days [for those who died early] to as long as 27.9 years). Healthy subjects’ samples for MALDI-MS assays were from 10 volunteers who were carefully screened for autoimmune diseases, hypertension, and inflammatory diseases. Sera from an additional 40 healthy volunteers were used for ELISA. Definitions of disease remission and relapse have been previously described (25).
Criteria for classification of ANCA-negative SVV required biopsy of kidney, lung, or upper respiratory tract, with histologic findings consistent with AAV and persistently negative ANCA tests by antigen-specific ELISA for MPO and PR3. Table 1 shows the 14 blood samples obtained from 10 ANCA-negative patients (80% female and 80% mixed European descent with a median age of 44.0 years). Three of the ten patients had a positive p-ANCA IFA on one occasion, with persistently negative MPO ELISA results. For 4 patients, longitudinal samples were obtained during disease activity and in remission.
ANCA-negative samples from UNC were tested at the UNC Hospital clinical laboratory using both the INOVA direct ELISA kit and IFA (Quanta lite and NOVA lite). Secondly, they were assayed by an in-house direct ELISA (21). In addition, they were confirmed to be ANCA-negative by a radioimmunoassay at Massachusetts General Hospital (26). ANCA-negative samples from the NL cohort were assayed by IFA (27) and in-house capture ELISA (28).
The potential that medications may influence autoantibody detection was examined. There was no obvious bias. The patient cohort was inclusive of all stages of disease with varied treatment regimens. The study included 11 of 52 samples collected at disease onset prior to initiation of treatment. Of the 35 patients in remission, 11 remained positive for anti-MPO reactivity. Three were on no immunosuppressive or maintenance therapy, and the remainder were on a variety of therapies, including prednisone, methylprednisolone, azathioprine, and mycophenolate mofetil (Table 1).
NL replication study.
An independent replication cohort was tested in Groningen, The Netherlands (demographics shown in Table 1). Immunoglobulin samples (n = 49) obtained from UMCG included MPO-ANCA glomerulonephritis patients with active disease (n = 20) and in disease remission (n = 20), in addition to 9 samples from healthy individuals. The 49 samples included patients with MPA (52.2%), renal-limited disease (34.8%), and GPA (13.0%). This cohort was 47.8% female and 95.7% of mixed European descent, with a mean age of 61.9 years.
UMCG had an ANCA-negative cohort (Table 1) of 13 unique individuals (1 individual was positive for elastase) with samples from active disease (n = 11) and disease remission (n = 11) from patients with GPA (61.5%) and renal-limited disease (38.5%). This cohort was 61.5% female and 100% of mixed European descent, with a median age of 60.7 years. Matched, active disease versus remission, samples were obtained from 9 patients. At UMCG, criteria for ANCA-negative SVV required kidney, lung, or nose biopsies showing pauci-immune extracapillary glomerulonephritis with or without fibrinoid necrosis and crescents in the kidney (n = 5) or capillaritis or granuloma of the lung (n = 1) or upper respiratory tract (n = 7), with pathologic evidence of necrotizing vasculitis or leukocytoclastic vasculitis. Sera were routinely tested by indirect immunofluorescence by the Laboratory of Clinical Immunology UMCG as described (27) and were found negative in 10 of 13 patients in the ANCA-negative cohort at the moment of diagnosis (the 3 sera-positive on IIF showed an atypical [n = 2] or perinuclear [n = 1] pattern of fluorescence). All sera were retested in an in-house–developed capture ELISA for antibodies PR3 and MPO performed at the Laboratory of Clinical Immunology UMCG and determined to be negative on all ANCA-negative patients (28). One patient was found positive for antibodies against elastase by capture ELISA.
MALDI-TOF/TOF MS
Total Ig was purified from sera using protein A/G PLUS-Agarose Reagent according to commercial protocol (Santa Cruz Biotechnology Inc.). Purified Ig was immobilized on CNBr-activated Sepharose 4B (GE Healthcare) in compact reaction columns (CRC) (USB Corporation) and exposed to human native MPO protein (Elastin Products Co, Inc) followed by digestion with sequencing grade TPCK-treated trypsin (Worthington). For murine studies, Ig was exposed to recombinant mouse MPO (R&D Systems). MPO peptides remaining bound to Ig after digestion were eluted with 0.1% Trifluoro acetic acid and analyzed on a 4800 Plus MALDI-TOF/TOF-MS/MS (referred to in text as MS) in conjunction with ProteinPilot software (AB SCIEX).
MS analysis of ANCA-negative Ig samples were conducted as above, with the exception that immobilized ANCA-negative Ig were exposed to a leukocyte protein lysate preparation from a healthy donor.
MS analysis for epitopes recognized by MPO autoantibodies from healthy individuals (n = 10) and for detection of low ANCA titers in remission samples (n = 35) required the use of isotope H218O (Cambridge Isotope Laboratories) to increase sensitivity (10). H218O in the diluent during tryptic digestion resulted in incorporation of H218O isotope on the carboxyterminus of each proteolytic fragment of MPO with a 4-kDa shift in mass.
To identify linear epitopes versus conformational epitopes, native MPO was predigested/fragmented with immobilized trypsin (Promega) prior to exposure to Ig fractions. Positive identification of a bound fragment by MS indicated a linear epitope.
ELISA
UNC cohort.
A total of 195 Ig samples were tested by ELISA for native MPO or peptide reactivity. Samples included Ig from healthy controls (n = 59), those with active disease (n = 80), and remission patients (n = 56). Total Ig was added to wells (2 μg/well) precoated with 1–2 μg peptide (UNC-CH peptide synthesis core). Goat anti-human with alkaline phosphatase conjugate secondary antibody specific to Ig (H+L) (Millipore) (1:10,000) was added and detected using 1-Step PNPP substrate (Thermo Scientific) and read at λ = 405 nm after 30 minutes.
NL cohort.
Aloquots of peptides used for ELISAs at UNC were provided to Groningen. All other substrates used to test NL cohort samples by ELISAs were prepared in Groningen. Total Ig (1:500, ∼2 μg/well) was added to precoated wells blocked with 1% BSA. Secondary antibody, goat anti-human conjugated with alkaline phosphatase (A5403; Sigma-Aldrich) (1:5000), was added and detected using p-nitrophenyl-phosphate disodium substrate.
Identification of the epitope-masking factor in sera
Serum proteins that bound immobilized MPO peptide aa 447–459 were eluted and sequenced for identification. A protein present in both patient and healthy sera was observed at ∼50 kDa by SDS-PAGE. Sequenced on a 4800 MALDI-TOF-TOF, the protein was identified as CP and confirmed by Western blot analysis probed with a rabbit anti-human anti-CP polyclonal antibody (Abcam). Native CP was purchased (Enzo Life Science) and proteolytically digested in vitro by plasmin (Haemtech).
In vitro neutrophil activation
Neutrophil activation assays were performed using MPO-ANCA affinity purified to linear MPO epitopes. Reactivity and specificity of purified antibodies were confirmed by ELISA. Human neutrophils isolated from 4 healthy donors were purified (2) and primed with cytochalasin B (Sigma-Aldrich). Activation of human neutrophils was assessed by the amount of reactive oxygen species released using the superoxide dismutase.
In vivo assay
Actively induced anti-MPO–associated glomerulonephritis.
DR2 Tg mice (12, 29) were immunized subcutaneously with 3 × 100 μg of peptide antigen (OVA323–339 [n = 4] or MPO442–460 [n = 10]) first in FCA then FIA on days 0, 7, and 14. Mice were culled on day 28.
MPO-ANCA–induced glomerulonephritis.
Serum Ig purified from DR2 Tg mice immunized with either OVA323–339 or MPO442–460 were passively transferred intravenously (35 μg/g, n = 5 per group) into LPS primed (1 μg/g intraperitoneally) DR2 Tg naive recipient mice, which were culled 6 days later.
Assessment of injury.
For indirect immunofluorescence, ethanol-fixed thioglycollate–induced peritoneal neutrophils were cytospun onto slides, and purified serum Ig (1 mg/ml) was applied for 20 minutes; then anti-mouse Ig Ab was detected using FITC-conjugated anti-mouse Ig Ab (Silenus).(12, 30). Albuminuria was assessed by ELISA (Bethyl Laboratories) and hematuria assessed by urine test strips (Combur) from a 24-hour urine sample. BUN was measured using standard laboratory methods on serum collected at the end of experiment. Glomerular abnormalities assessed on 3-μm thick, PAS-stained, formalin-fixed, paraffin-embedded sections (≥ 50 glomeruli/mouse) were thickening of the capillary walls and mesangial hypercellularity (31) Total glomerular cell nuclei were enumerated (≥20 glomeruli/mouse). Glomerular neutrophils were detected by immunoperoxidase staining of 6-μm thick, periodate lysine paraformaldehyde–fixed, frozen kidney sections using anti–Gr-1 antibodies (RB6-8C5).
Statistics
P values were calculated by Wilcoxon’s 2 sample tests for 2-sample comparisons, Kruskal-Wallis test for 3 groups comparison, and signed rank test for paired group comparisons. Bonferroni’s correction was used, α = 0.05/3 = 0.167. For in vivo mouse studies, P values were calculated using unpaired 2-tailed Student’s t test and a P value of less than 0.05 was considered significant.
Study approval
This study and study protocols were compliant with and approved by the UNC Institutional Review Board. Informed consent was obtained prior to all blood collections. Animal studies were approved by the Monash University Animal Ethics Committee.
Supplementary Material
Acknowledgments
This work was supported by federal grant P01 DK058335-06 (NIH/NIDDK) and the NHMRC Australia Project Grant 1008849. The authors acknowledge the UNC Michael Hooker Proteomic Center for training and access to the 4800 MALDI-TOF/TOF mass spectrometer under the expert guidance of Nely Dicheva. We would like to thank Gary Hess for his programming assistance of the compilation of epitope profiles. Anna Fisher and Gary Hatch, U.S. Environmental Protection Agency, are acknowledged for their excellent internal review of the manuscript. The research described in this article has been reviewed by the National Health and Environmental Effects Research Laboratory, U.S. Environmental Protection Agency, and approved for publication. Approval does not signify that the contents necessarily reflect the views and the policies of the Agency nor does mention of trade names or commercial products constitute endorsement or recommendation for use.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Note regarding evaluation of this manuscript: Manuscripts authored by scientists associated with Duke University, The University of North Carolina at Chapel Hill, Duke-NUS, and the Sanford-Burnham Medical Research Institute are handled not by members of the editorial board but rather by the science editors, who consult with selected external editors and reviewers.
Citation for this article: J Clin Invest. 2013;123(4):1773–1783. doi:10.1172/JCI65292.
Aleeza J. Roth’s present address is: Mt. Sinai Medical Center, New York City, New York, USA.
References
- 1.Tervaert JW, Stegeman CA, Kallenberg CG. Serial ANCA testing is useful in monitoring disease activity of patients with ANCA-associated vasculitides. Sarcoidosis Vasc Diffuse Lung Dis. 1996;13(3):241–245. [PubMed] [Google Scholar]
- 2.Falk RJ, Terrell RS, Charles LA, Jennette JC. Anti-neutrophil cytoplasmic autoantibodies induce neutrophils to degranulate and produce oxygen radicals in vitro. Proc Natl Acad Sci U S A. 1990;87(11):4115–4119. doi: 10.1073/pnas.87.11.4115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Xiao H, et al. Antineutrophil cytoplasmic autoantibodies specific for myeloperoxidase cause glomerulonephritis and vasculitis in mice. J Clin Invest. 2002;110(7):955–963. doi: 10.1172/JCI15918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen M, Kallenberg CG, Zhao MH. ANCA-negative pauci-immune crescentic glomerulonephritis. Nat Rev Nephrol. 2009;5(6):313–318. doi: 10.1038/nrneph.2009.67. [DOI] [PubMed] [Google Scholar]
- 5.Hogan SL, Nachman PH, Wilkman AS, Jennette JC, Falk RJ. Prognostic markers in patients with antineutrophil cytoplasmic autoantibody-associated microscopic polyangiitis and glomerulonephritis. J Am Soc Nephrol. 1996;7(1):23–32. doi: 10.1681/ASN.V7123. [DOI] [PubMed] [Google Scholar]
- 6.Pagnoux C, et al. Predictors of treatment resistance and relapse in antineutrophil cytoplasmic antibody-associated small-vessel vasculitis: comparison of two independent cohorts. Arthritis Rheum. 2008;58(9):2908–2918. doi: 10.1002/art.23800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cui Z, Zhao MH, Segelmark M, Hellmark T. Natural autoantibodies to myeloperoxidase, proteinase 3, and the glomerular basement membrane are present in normal individuals. Kidney Int. 2010;78(6):590–597. doi: 10.1038/ki.2010.198. [DOI] [PubMed] [Google Scholar]
- 8.Olson SW, et al. Asymptomatic autoantibodies associate with future anti-glomerular basement membrane disease. J Am Soc Nephrol. 2011;22(10):1946–1952. doi: 10.1681/ASN.2010090928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Parker CE, Tomer KB. MALDI/MS-based epitope mapping of antigens bound to immobilized antibodies. Mol Biotechnol. 2002;20(1):49–62. doi: 10.1385/MB:20:1:049. [DOI] [PubMed] [Google Scholar]
- 10.Mirza SP, Greene AS, Olivier M. 18O labeling over a coffee break: a rapid strategy for quantitative proteomics. J Proteome Res. 2008;7(7):3042–3048. doi: 10.1021/pr800018g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kingston IB, Kingston BL, Putnam FW. Chemical evidence that proteolytic cleavage causes the heterogeneity present in human ceruloplasmin preparations. Proc Natl Acad Sci U S A. 1977;74(12):5377–5381. doi: 10.1073/pnas.74.12.5377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ooi JD, et al. The immunodominant myeloperoxidase T-cell epitope induces local cell-mediated injury in antimyeloperoxidase glomerulonephritis. Proc Natl Acad Sci U S A. 2012;109(39):E2615–E2624. doi: 10.1073/pnas.1210147109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen M, Yu F, Wang SX, Zou WZ, Zhao MH, Wang HY. Antineutrophil cytoplasmic autoantibody-negative Pauci-immune crescentic glomerulonephritis. J Am Soc Nephrol. 2007;18(2):599–605. doi: 10.1681/ASN.2006091021. [DOI] [PubMed] [Google Scholar]
- 14.Ara J, Pascual J, Mirapeix E, Rodriguez R, Abellana R, Darnell A. Ceruloplasmin in small vessel vasculitis. Nephrol Dial Transplant. 1999;14(2):515–517. doi: 10.1093/ndt/14.2.515b. [DOI] [PubMed] [Google Scholar]
- 15.Baskin E, et al. Ceruloplasmin levels in antineutrophil cytoplasmic antibody-positive patients. Pediatr Nephrol. 2002;17(11):917–919. doi: 10.1007/s00467-002-0969-0. [DOI] [PubMed] [Google Scholar]
- 16.Griffin SV, Chapman PT, Lianos EA, Lockwood CM. The inhibition of myeloperoxidase by ceruloplasmin can be reversed by anti-myeloperoxidase antibodies. Kidney Int. 1999;55(3):917–925. doi: 10.1046/j.1523-1755.1999.055003917.x. [DOI] [PubMed] [Google Scholar]
- 17.Pedrollo E, Bleil L, Bautz FA, Kalden JR, Bautz EK. Antineutrophil cytoplasmic autoantibodies (ANCA) recognizing a recombinant myeloperoxidase subunit. Adv Exp Med Biol. 1993;336:87–92. doi: 10.1007/978-1-4757-9182-2_13. [DOI] [PubMed] [Google Scholar]
- 18.Fujii A, et al. Epitope analysis of myeloperoxidase (MPO) specific anti-neutrophil cytoplasmic autoantibodies (ANCA) in MPO-ANCA-associated glomerulonephritis. Clin Nephrol. 2000;53(4):242–252. [PubMed] [Google Scholar]
- 19.Tomizawa K, et al. A panel set for epitope analysis of myeloperoxidase (MPO)-specific antineutrophil cytoplasmic antibody MPO-ANCA using recombinant hexamer histidine-tagged MPO deletion mutants. J Clin Immunol. 1998;18(2):142–152. doi: 10.1023/A:1023251001261. [DOI] [PubMed] [Google Scholar]
- 20.Suzuki K, et al. Analysis of risk epitopes of anti-neutrophil antibody MPO-ANCA in vasculitis in Japanese population. Microbiol Immunol. 2007;51(12):1215–1220. doi: 10.1111/j.1348-0421.2007.tb04017.x. [DOI] [PubMed] [Google Scholar]
- 21.Erdbrugger U, et al. Mapping of myeloperoxidase epitopes recognized by MPO-ANCA using human-mouse MPO chimers. Kidney Int. 2006;69(10):1799–1805. doi: 10.1038/sj.ki.5000354. [DOI] [PubMed] [Google Scholar]
- 22.Bruner BF, Vista ES, Wynn DM, James JA. Epitope specificity of myeloperoxidase antibodies: identification of candidate human immunodominant epitopes. Clin Exp Immunol. 2011;164(3):330–336. doi: 10.1111/j.1365-2249.2011.04372.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xu PC, Cui Z, Chen M, Hellmark T, Zhao MH. Comparison of characteristics of natural autoantibodies against myeloperoxidase and anti-myeloperoxidase autoantibodies from patients with microscopic polyangiitis. Rheumatology (Oxford). 2011;50(7):1236–1243. doi: 10.1093/rheumatology/ker085. [DOI] [PubMed] [Google Scholar]
- 24.Jennette JC, et al. Nomenclature of systemic vasculitides. Proposal of an international consensus conference. Arthritis Rheum. 1994;37(2):187–192. doi: 10.1002/art.1780370206. [DOI] [PubMed] [Google Scholar]
- 25.Nachman PH, Hogan SL, Jennette JC, Falk RJ. Treatment response and relapse in antineutrophil cytoplasmic autoantibody-associated microscopic polyangiitis and glomerulonephritis. J Am Soc Nephrol. 1996;7(1):33–39. doi: 10.1681/ASN.V7133. [DOI] [PubMed] [Google Scholar]
- 26.Csernok E, et al. Evaluation of capture ELISA for detection of antineutrophil cytoplasmic antibodies directed against proteinase 3 in Wegener’s granulomatosis: first results from a multicentre study. Rheumatology (Oxford). 2004;43(2):174–180. doi: 10.1093/rheumatology/keh028. [DOI] [PubMed] [Google Scholar]
- 27.Tervaert JW, et al. Occurrence of autoantibodies to human leucocyte elastase in Wegener’s granulomatosis and other inflammatory disorders. Ann Rheum Dis. 1993;52(2):115–120. doi: 10.1136/ard.52.2.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Savige J, et al. Addendum to the International Consensus Statement on testing and reporting of antineutrophil cytoplasmic antibodies. Quality control guidelines, comments, and recommendations for testing in other autoimmune diseases. Am J Clin Pathol. 2003;120(3):312–318. doi: 10.1309/WAEPADW0K4LPUHFN. [DOI] [PubMed] [Google Scholar]
- 29.Rich C, et al. Myelin oligodendrocyte glycoprotein-35-55 peptide induces severe chronic experimental autoimmune encephalomyelitis in HLA-DR2-transgenic mice. Eur J Immunol. 2004;34(5):1251–1261. doi: 10.1002/eji.200324354. [DOI] [PubMed] [Google Scholar]
- 30.Lock RJ. ACP Broadsheet No 143: January 1994. Detection of autoantibodies to neutrophil cytoplasmic antigens. J Clin Pathol. 1994;47(1):4–8. doi: 10.1136/jcp.47.1.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ooi JD, Phoon RK, Holdsworth SR, Kitching AR. IL-23, not IL-12, directs autoimmunity to the Goodpasture antigen. J Am Soc Nephrol. 2009;20(5):980–989. doi: 10.1681/ASN.2008080891. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.