

Abstract
The molecular mechanisms that control the balance between antiangiogenic and proangiogenic factors and initiate the angiogenic switch in tumors remain poorly defined. By combining chemical genetics with multimodal imaging, we have identified an autocrine feed-forward loop in tumor cells in which tumor-derived VEGF stimulates VEGF production via VEGFR2-dependent activation of mTOR, substantially amplifying the initial proangiogenic signal. Disruption of this feed-forward loop by chemical perturbation or knockdown of VEGFR2 in tumor cells dramatically inhibited production of VEGF in vitro and in vivo. This disruption was sufficient to prevent tumor growth in vivo. In patients with lung cancer, we found that this VEGF:VEGFR2 feed-forward loop was active, as the level of VEGF/VEGFR2 binding in tumor cells was highly correlated to tumor angiogenesis. We further demonstrated that inhibition of tumor cell VEGFR2 induces feedback activation of the IRS/MAPK signaling cascade. Most strikingly, combined pharmacological inhibition of VEGFR2 (ZD6474) and MEK (PD0325901) in tumor cells resulted in dramatic tumor shrinkage, whereas monotherapy only modestly slowed tumor growth. Thus, a tumor cell-autonomous VEGF:VEGFR2 feed-forward loop provides signal amplification required for the establishment of fully angiogenic tumors in lung cancer. Interrupting this feed-forward loop switches tumor cells from an angiogenic to a proliferative phenotype that sensitizes tumor cells to MAPK inhibition.
Introduction
Several receptor tyrosine kinases, such as VEGFR2, Flt1/VEGFR1, VEGFR3, or PDGFR, have been implicated in tumor angiogenesis. When activated through binding of the respective ligand, they regulate related, yet distinct, processes involved in neoangiogenesis. Endothelial cell survival, migration, and proliferation of blood vessels is regulated primarily by VEGF-A binding to VEGFR2 (1). In the canonical model, tumor cells express VEGF, which binds to VEGFR2 on endothelial cells (2), thus stimulating tumor vessel formation. This change from prevascular hyperplasia to highly vascularized tumors has been referred to as the angiogenic switch (1). In recent years, several studies have reported expression of VEGFR1 as well as VEGFR2 on tumor cells (3–5). An intriguing finding related to tumor cell expression of Flt1 was the observation that VEGF secreted by tumor cells triggers tumor cell proliferation by binding Flt1 (6). Moreover, in patients with lung cancer, an increase in VEGFR2 gene copy number is associated with chemoresistance and shorter survival (7). Here, we have sought to analyze the impact of VEGFR2 inhibition on tumor cells by combining genetically controlled pharmacological perturbations with multimodal PET imaging.
Results
VEGFR2 is differentially expressed on non-small-cell lung cancer – VEGFR2 inhibition alters amino acid transport in tumor cells but does not affect tumor cell proliferation.
We used the human lung cancer cell lines H441 and H1975, which each expressed high levels of VEGFR2 (Figure 1A and Supplemental Figure 1A; supplemental material available online with this article; doi: 10.1172/JCI65385DS1). We treated mice engrafted with these 2 cell lines with the dual VEGFR2/EGFR inhibitor ZD6474, which has a 40-fold lower activity against VEGFR1 (8). Both cell lines are resistant to EGFR inhibition, due either to a KRAS mutation (H441) or to the presence of the T790M gatekeeper mutation of EGFR (H1975) (ref. 9 and Supplemental Figure 1B). Thus, any therapeutic impact on subcutaneous tumor growth of these cell lines is primarily due to VEGFR2 inhibition and cannot be attributed to inhibition of EGFR. In established tumors, ZD6474 treatment completely abolished methionine uptake after 7 days of treatment, as determined by methyl-l-[11C]-methionine ([11C]MET) PET (Figure 1B and Supplemental Figure 1C). By contrast, uptake of 3′-deoxy-3′-[18F]-fluoro-l-thymidine ([18F]FLT), a marker of proliferation (10), was slightly increased (Figure 1B and Supplemental Figure 1C), suggesting that the cells continued to progress through the cell cycle. Thus, VEGFR2 inhibition seems to inhibit a VEGFR2-dependent signaling pathway in tumor cells that affects amino acid transport without affecting cellular proliferation (Figure 1B). To investigate whether the reduction in [11C]MET uptake is specifically due to ZD6474-mediated VEGFR2 inhibition, we used a chemical genetic approach, which introduces a gatekeeper resistant mutation against ZD6474-induced VEGFR2 inhibition. The substitution of Val916 to Met at the gatekeeper position of VEGFR2 creates a steric clash with the inhibitor that specifically prevents ZD6474 from binding to VEGFR2 (Figure 1C and ref. 11). The introduction of this resistant gatekeeper mutation was sufficient to abrogate the inhibitory effect of ZD6474 on [11C]MET uptake (Supplemental Figure 1D). The cellular uptake of methionine is facilitated by the LAT1 transporter that is regulated by mTOR (12). Hence, we hypothesized that VEGF-bound VEGFR2 on tumor cells induces a VEGFR2 signaling pathway via mTOR. Moreover, as VEGF secretion is regulated by mTOR, we sought to investigate whether VEGF/VEGFR2 signaling induces a feed-forward loop via mTOR.
Figure 1. A VEGF:VEGFR2 feed-forward loop in tumor cells boosts VEGF secretion.

(A) VEGFR2 expression data from the 53 NSCLC cell lines were obtained using Affymetrix U133A arrays (21). (B) Mice with established tumors (H1975) were treated with ZD6474, and PET imaging was performed on day 0 (before start of therapy) and at the indicated time points after treatment (left panels, [18F]FLT-PET; right panels, [11C]MET-PET). (C) Substitution of Val916 by Met at the gatekeeper position of VEGFR2 creates a steric clash with the inhibitor and prevents ZD6474 from binding. (D) Secretion of VEGF by H1975, H441, and (E) H1975 mutant cells (VEGFR2V916M) was determined in vitro by ELISA (D) following stimulation with increasing amounts of recombinant VEGF. H1975 and H441 cells were stimulated with the indicated concentrations of VEGF (V) and pretreated with the indicated compounds (ZD6474 [Z], 1 μM; rapamycin [R], 100 nM). (F and G) Cells were treated with VEGF and the indicated dose of ZD6474, and phosphorylation of VEGFR2 was determined by immunoprecipitation. (F) The impact on activation of downstream signaling was determined by immunoblotting. (H) H1975 mutant cells (VEGFR2V916M) or the wild-type control cells were injected into nude mice and treated with ZD6474 or vehicle on day 1 after tumor cell injections.
VEGF/VEGFR2 signaling induces a feed-forward loop that is mediated by a VEGFR2/PI3K/mTOR/VEGF signaling cascade.
Consistent with a feed-forward loop stimulating VEGF secretion in a VEGFR2-dependent fashion, VEGF secretion was strongly induced by addition of exogenous VEGF in the VEGFR2-expressing tumor cell lines H1975, H441, and HCC1359 (Figure 1D and Supplemental Figure 2B), and this induction was blunted by treatment of cells with the VEGFR2 inhibitor ZD6474 (Figure 1D and Supplemental Figure 2B). To validate the specificity of VEGFR2 as the relevant target for inhibition of VEGFR2-dependent VEGF secretion, we used the gatekeeper resistant mutation against ZD6474-induced VEGFR2 inhibition (V916M) (Figure 1C). Introducing this mutation in H1975 cells was sufficient to abrogate the ZD6474-induced inhibition of VEGF secretion (Figure 1E). Under hypoxic conditions, induction of VEGF secretion was similarly inhibited by VEGFR2 inhibition (Supplemental Figure 3C), suggesting that this autocrine loop is also active in the physiological response to hypoxia. Rapamycin treatment similarly blunted VEGF-induced VEGF secretion of tumor cells, supporting the hypothesis that VEGFR2-dependent secretion of VEGF is under the control of mTOR (13), which also regulates methionine uptake via the LAT-1 transporter (ref. 14 and Figure 1D). Moreover, VEGF-mediated stimulation of VEGFR2 induced S6 phosphorylation in tumor cells expressing high levels of VEGFR2 (Figure 1F, Supplemental Figure 2A, and Supplemental Figure 3A). Phosphorylation of S6 coincided with the activation of PDK1, which might provide an alternative route for mTOR activation (Supplemental Figure 2A and ref. 15). In accordance with the induction of PI3K/mTOR/VEGF signaling, PI3K inhibition resulted in reduced VEGF secretion (Supplemental Figure 2B). We next formulated a general mathematical model describing the postulated VEGF:VEGFR2 feed-forward mechanism. We then demonstrated that the behavior of the model was consistent with the experimental results obtained (Supplemental Figure 4 and Supplemental Note).
Additionally, we detected a consistent reduction in phosphorylation of ERK and of AKT in H1975, H441, and HCC1359 cells (Figure 1F and Supplemental Figure 3A). Thus, in the setting of autocrine VEGF/VEGFR2 signaling, the slight reduction in tumor growth observed in response to VEGFR2 inhibition is likely to be independent of ERK-mediated proliferation. These results agree with those of our PET experiments, showing a continuous uptake of [18F]FLT (Figure 1B). Finally, we confirmed that the observed ZD6474-mediated effects were due to inhibition of VEGFR2 in tumor cells by ectopically expressing the ZD6474-resistant mutant of VEGFR2 (V916M) in H1975 cells (Figure 1, E and G, and ref. 11). This mutant abolished ZD6474-mediated VEGFR2 dephosphorylation and activation of mTOR (Figure 1G). Most strikingly, this VEGFR2 gatekeeper mutant against ZD6474 was sufficient to rescue the tumor growth–inhibiting effects of ZD6474 in vivo (Figure 1H). Thus, in concordance with the [11C]MET PET data, the ZD6474-mediated effects on tumor VEGF/VEGFR2/mTOR signaling are predominantly due to inhibition of VEGFR2 on the tumor cells and not due to inhibition of other kinases or VEGFR2 on endothelial cells.
We next sought to validate our findings on inhibition of VEGF/VEGFR2/mTOR signaling with an additional VEGFR2 inhibitor, PTK787. As with ZD6474, treatment with PTK787 inhibited phosphorylation of S6 and reduced the level of secreted VEGF (Supplemental Figure 2, A and B). Further, we found that, in the absence of VEGF, ZD6474 had only a little effect on the activation on ERK and S6 kinase (Supplemental Figure 3E).
VEGF/VEGFR2 signaling is needed for the induction of tumor angiogenesis and tumor formation in vivo.
To validate our finding with an alternative approach, we stably silenced VEGFR2 with lentiviral shRNA in H1975 and H441 non-small-cell lung cancer (NSCLC) cells (Figure 2A and Supplemental Figure 5B). Consistent with the [18F]FLT PET data, selective silencing of VEGFR2 by shRNA did not reduce tumor cell proliferation in vitro (Supplemental Figure 5A). By contrast, silencing of VEGFR2 dramatically reduced secretion of VEGF by tumor cells in response to hypoxia (Figure 2B), thereby confirming that binding of VEGF to VEGFR2 amplifies VEGF secretion in a VEGFR2-dependent manner. Most strikingly, knockdown of VEGFR2 in tumor cells alone was sufficient to almost entirely abolish initiation of tumor growth in vivo (Figure 2C and Supplemental Figure 5B), suggesting that autocrine VEGF/VEGFR2 signaling in tumor cells is required for the establishment of tumors in vivo. While the large tumors transduced with empty control vector (H1975eV tumors) exhibited a highly angiogenic phenotype with many CD31-positive blood vessels, the small residual tumors in which VEGFR2 had been stably silenced (H1975VEGFR2KD tumors) almost completely lacked blood vessels (Figure 2D). In concordance with the tissue culture results, these tumors lacked expression of VEGF, while H1975eV tumors expressed high amounts of VEGF (Figure 2D). Using bevacizumab (Avastin), we specifically stained for (human) tumor cell-derived VEGF. We herewith could demonstrate that silencing VEGFR2 on tumor cells dramatically reduced the secretion of tumor cell-derived VEGF (Figure 2D). We next analyzed whether the raised levels of VEGF in the VEGFR2 WT cells were due to elevated hypoxia. HIF-1α was equally expressed in wild-type (H1975WT), H1975eV, and H1975VEGFR2KD tumors (Supplemental Figure 5C). By staining with an antibody that specifically recognizes human VEGF bound to human VEGFR2, we confirmed that tumor cell-derived VEGF binds to VEGFR2 on tumor cells in H1975eV tumors but to a much lesser degree in H1975VEGFR2KD xenograft tumors (16), supporting the presence of an autocrine loop in vivo (Figure 2D). Furthermore, H1975VEGFR2KD tumors had a markedly reduced uptake of [15O]H2O, a measure of tumor blood flow as a functional readout of angiogenesis (17), when compared with that of H1975eV tumors (Supplemental Figure 5D). As an alternative approach to determine whether VEGFR2 inhibition on tumor cells can prevent tumor formation, we treated mice with the VEGFR2 inhibitor ZD6474 immediately after tumor cell inoculation. In concordance with the results obtained with VEGFR2-knockdown cells, concomitant VEGFR2 inhibition in NSCLC-H1975 tumors expressing high levels of VEGFR2 completely abrogated the establishment of tumors in vivo (Figure 1H), paralleled by a strong reduction in tumor vessel density (Supplemental Figure 5F). This blocking of tumor growth can be attributed to inhibition of VEGFR2, as the introduction of the resistance mutation VEGFR2V916M in the H1975 cells was sufficient to abrogate the ZD6474-mediated treatment effect (Figure 1H). In contrast, tumor cells with low levels of VEGFR2 expression, such as H1650 and A549, were unaffected by concomitant VEGFR2 inhibition (Supplemental Figure 5E). In the same manner, treatment of mice with bevacizumab at the same time of tumor cell inoculation abrogated the establishment of tumors in vivo (Figure 2E). In summary, the VEGF:VEGFR2 feed-forward loop is active in tumor cells expressing high levels of VEGFR2 in vivo and is required for the establishment of fully angiogenic tumors, and its disruption (either by VEGFR2 inhibition or blockade of VEGF) is sufficient to completely prevent tumor formation in vivo.
Figure 2. Requirement of autocrine VEGF/VEGFR2 signaling for induction of angiogenesis and tumor formation in vivo.

(A) H1975 cells (WT cells) were stably transduced with lentiviral shRNA vectors targeting VEGFR2 (shVEGFR2) or with empty vector control (eV). Knockdown efficiency was determined by Western blotting (top panel, Flt1; bottom panel, VEGFR2). (B) Stable cell lines were cultured over time and quantified under normoxic conditions or exposed to hypoxia (1% O2). VEGF secretion was determined over time by ELISA. (C) Stable cell lines were injected into nude mice, and tumor growth was monitored over time. (D) Tumors were harvested and stained for pan VEGF, CD31, human VEGF (using GFP-labeled Avastin), human VEGF complexed with VEGFR2, Ki67, pERK, and an antibody binding VEGF to VEGFR2. (E) H1975 cells were injected into nude mice and treated with Avastin or vehicle on day 1 and day 10 after tumor cell injections. Scale bar: 50 μm (VEGF, human VEGF); 100 μm (pERK); 200 μm (CD31, Ki67, VEGF:VEGFR2).
VEGF:VEGFR2 feed-forward loop is active in primary human lung adenocarcinomas.
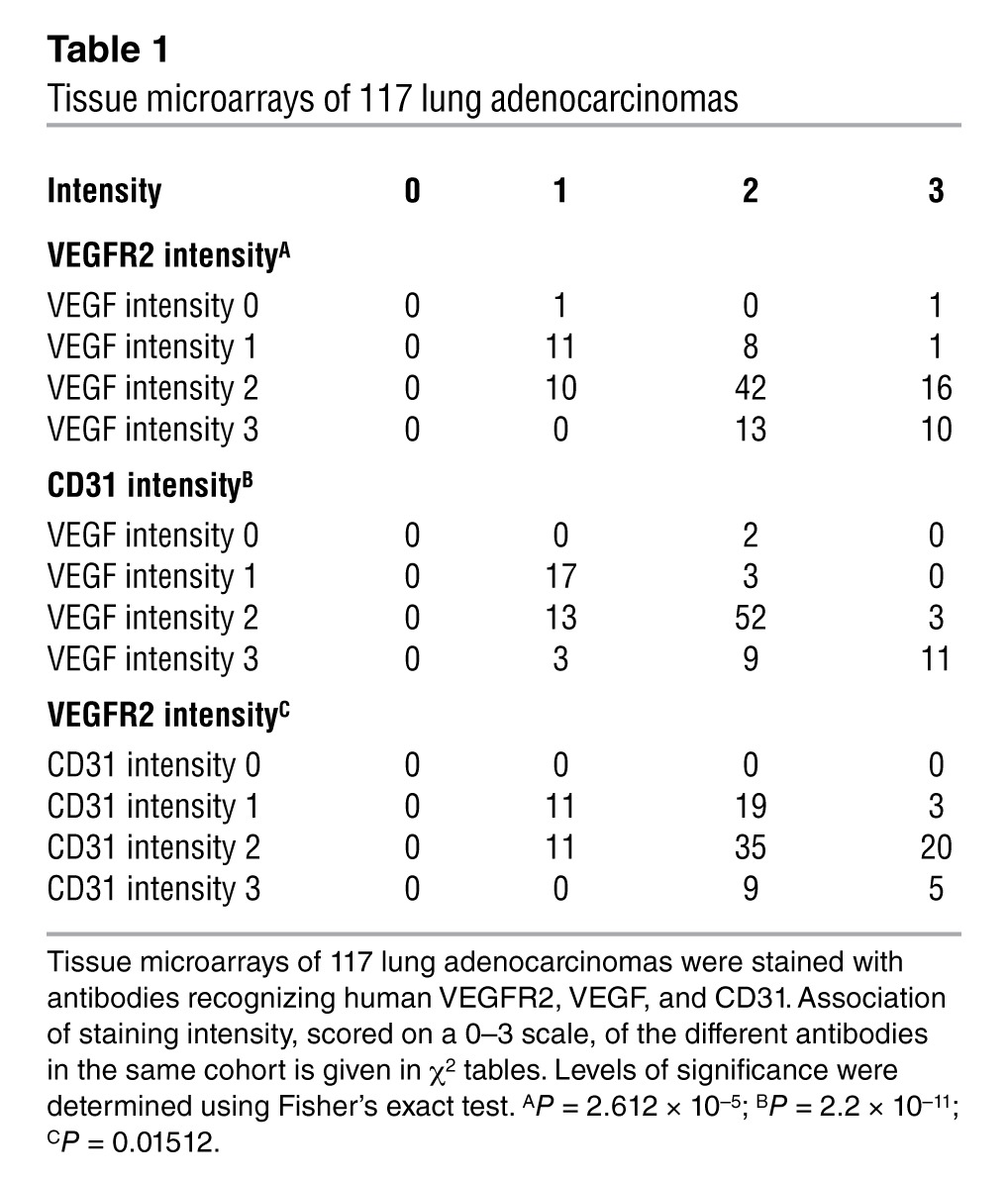
We next performed immunohistochemical analysis of VEGF and VEGFR2 in 117 surgically resected primary human lung adenocarcinomas (Figure 3A), revealing that VEGF expression correlated significantly with expression of VEGFR2 on the same tumor cells (P = 2.612 × 10–5) as well as with microvessel density (P = 2.2 × 10–11; Table 1 and Supplemental Figure 6). This indicates that activation of the autocrine VEGF/VEGFR2 signaling loop is a feature of highly angiogenic lung adenocarcinomas. Finally, staining of representative tumors with an antibody specifically recognizing VEGF bound to VEGFR2 confirmed that, in the tumors with coexpression of VEGF and VEGFR2, VEGF was indeed bound to VEGFR2 on tumor cells (Figure 3B). Moreover, adenocarcinomas that exhibited coexpression and autocrine binding of VEGF and VEGFR2 presented a highly angiogenic phenotype (Figure 3B). These results support a critical function for the VEGF:VEGFR2 feed-forward loop in primary lung tumors in patients.
Figure 3. The VEGF:VEGFR2 feed-forward loop in primary human lung adenocarcinomas.

(A) Human adenocarcinomas were immunofluorescently stained to reveal coexpression of VEGF and VEGFR2 by the same tumor cell population. Scale bar: 50 μm. (B) A representative patient (Pat 1) with an high angiogenic phenotype represented by high VEGF:VEGFR2 staining and high levels of CD31-positive cells. In contrast, another patient (Pat 2) presents a low angiogenic phenotype, with only moderate levels of VEGF:VEGFR2–positive tumor cells, corresponding to a low density of CD31-positive cells. Scale bar: 100 μm (top panels); 200 μm (bottom panels).
Table 1.
Tissue microarrays of 117 lung adenocarcinomas
Inhibition of VEGFR2 induces ERK signaling and sensitizes to MAPK inhibition.
To our surprise, we found that H1975VEGFR2KD tumors exhibited increased levels of Ki67- and pERK-positive cells compared with H1975eV tumors (Figure 2D). In the same manner, VEGF stimulation in vitro reduced pERK signaling that was again increased upon ZD6474 treatment (Figure 1F and Supplemental Figure 3A). Consistent with the in vitro data presented above, ZD6474 treatment induced inhibition of pS6 and an increase in pERK in vivo (Supplemental Figure 7E). We thus hypothesized that inhibiting the VEGF:VEGFR2 feed-forward loop results in activation of the ERK signaling pathway and thereby appears to induce a ERK-dependant proliferative phenotype. Recently, Rosen and colleagues described a negative feedback regulation of IGF signaling via mTOR and its transcriptional regulation of FOXO transcription factors (18). In line with these findings, we observed that inhibiting the VEGF/VEGFR2/mTOR autocrine feed-forward loop enhanced ERK signaling, as a result of activation of the insulin growth factor receptor (IGFR) signaling pathway via IRS-1 (Figure 4, C and D). This activation of ERK was mediated by increased FOXO levels upon inhibition of VEGFR2/mTOR signaling (Figure 4D). Remarkably, combined inhibition of ERK signaling by PD0325901 and VEGFR2 resulted in a dramatic reduction of tumor cell proliferation, as indicated by [18F]FLT PET data, and, finally, in complete tumor shrinkage in vivo (Figure 4, A and B, and Supplemental Figure 7, A and B). In order to confirm our findings on tumor response after combined PD0325901 and ZD6474 treatment in an orthotopic tumor model, we applied a murine Ras-mutated lung cancer model expressing VEGFR2 on tumor cells (Figure 5 and Supplemental Figure 7H). In accordance with our xenograft data, combined PD0325901 and ZD6474 treatment resulted in substantial tumor regression, as detected by bioluminescence imaging (BLI) (Figure 5). These findings support the hypothesis of a recent study that combined MEK and VEGFR inhibition enhances inhibition of tumor growth (19). Of note, complete tumor shrinkage was associated with the expression levels of VEGFR2 on tumor cells. In line with our hypothesis, NSCLC-H1650 and A549, which express only low levels of VEGFR2, did not respond to combined ZD6474 and PD0325901 treatment (Supplemental Figure 7, C and D).
Figure 4. Combined inhibition of VEGFR2 and ERK signaling results in dramatic tumor shrinkage.

(A) H1975 cells were engrafted subcutaneously in nude mice; mice with established tumors were treated with a combination of ZD6474 plus PD0325901, and [18F]FLT-PET imaging was performed on day 0 (before treatment) and at the indicated time points after treatment. Representative imaging results are shown. ID, injected dose. (B) Tumor size of subcutaneously grown H1975 tumors was determined at the indicated time points under treatment with vehicle, ZD6474 alone, PD0325901 (12 mg/kg) alone, or a combination of PD0325901 plus ZD6474. (C and D) Impact of ZD6474 treatment on feed-forward activation of insulin receptor signaling was determined (C) by IHC and (D) by Western blots using the indicated antibodies. Scale bar: 200 μm.
Figure 5. Combined inhibition of VEGFR2 and ERK signaling induces tumor shrinkage in an orthotopic KrasVal12-driven murine lung lung cancer model (RasLo).

(A–C) KrasVal12-driven orthotopic murine lung tumors were induced; mice with Luc-positive tumors were treated, and BL imaging was performed on day 0 (before treatment) and at the indicated time points after treatment. Mice were treated with vehicle, ZD6474 alone, PD0325901 (12 mg/kg) alone, or a combination of PD0325901 plus ZD6474. The scale bar represents the luciferase activity in detected counts (photons/s/cm2/steradian × 10; red, high; blue, low). Graphs show quantification of change in BLI based on ROI analysis. (D–F) Images show representative lungs ex vivo, with macroscopic lung tumors in mice treated with (E) PD0325901 or (F) ZD6474 only.
Discussion
We have identified a VEGF:VEGFR2 feed-forward loop in NSCLC cells expressing VEGFR2 that leads to a signal amplification and a boost in VEGF secretion, which is required for establishment of fully angiogenic tumors in vivo. This VEGF/VEGFR2 signaling cascade via VEGFR2/PI3K/mTOR induces a mTOR-dependent regulation of VEGF secretion (13). VEGF secretion is induced by the upregulation of HIF-1α, which has been shown to be specific to mTORC1 and unaffected by the status of mTORC2 (20). This is supported by our observation that mTORC1 inhibition with rapamycin can prevent elevated VEGF expression induced by the feed-forward loop. Treatment with Torin1, an inhibitor of TORC1 and TORC2, did not further reduce VEGF levels in comparison to rapamycin (Supplemental Figure 2B). Thus, our data strongly support that VEGFR2/mTOR-induced VEGF secretion is mediated via mTORC1.
Our study supports a model wherein tumor cell-autonomous autocrine VEGF signaling loops form an integral part of the early phase of tumor development, promoting blood supply through angiogenesis. Consistent with this notion, we hypothesize that tumor cells reduce the proliferation in a VEGF/VEGFR2–dependent manner as long as the nutrient supply remains decreased. This hypothesis is confirmed by our finding that inhibition of VEGFR2/mTOR signaling results in a time-dependent feedback activation of IRS/MAPK signaling (18). Interrupting the VEGF:VEGFR2 feed-forward loop induces a therapeutically relevant dependency on MAPK signaling in vivo. As a consequence, combined inhibition of VEGFR2 and ERK signaling results in substantial tumor shrinkage in tumors in which this VEGF:VEGFR2 feed-forward loop is active. Our findings provide a potential mechanism for synergistic treatment effects of combined VEGFR2 and MAPK inhibition by PD0325901 and ZD6474. Finally, we have found that about 20% of patients with lung cancer show high tumor cell VEGFR2 expression, which correlates with a highly angiogenic phenotype. We therefore propose that high expression of VEGF:VEGFR2 on tumor cells could serve as a predictive marker for therapeutic efficacy of dual VEGFR2/MEK inhibition in patients with lung cancer. In summary, this study demonstrates the existence of a novel autocrine VEGF/VEGFR2 signaling loop, which amplifies VEGF secretion by tumor cells and is required to provide a switch for development of fully angiogenic tumors in vivo.
Methods
VEGFR2 expression in NSCLC cell lines.
VEGFR2 expression data for the 53 NSCLC cell lines were obtained using Affymetrix U133A arrays ( http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE14925). RNA extraction, hybridization, and scanning of arrays were performed using standard procedures. CEL files from U133A arrays were preprocessed as described previously (21).
Cell lines and reagents.
NSCLC cell lines, H441, H1975, A549, and H1650, were maintained in RPMI medium with 10% FCS and 1% (penicillin plus streptomycin) antibiotic. VEGF was purchased from Tebu-bio GmbH, ZD6474 was purchased from AstraZeneca, rapamycin was purchased from LC Labs, Torin 1 was purchased from Tocris Bioscience, and PK90 was purchased from Axon Medchem. Compound stocks were stored at –20°C and dissolved in DMSO or vehicle solution in a rotating device at 4°C for animal therapy.
Lentiviral RNAi, retroviral expression, and stable transduction.
The VEGFR2 V916M mutation was introduced into H1975 cells using a pBABE vector by site-directed mutagenesis. Replication-incompetent retroviruses were produced by cotransfection with the pCL ampho plasmid in HEK 293T cells (Orbigen) using TRANS-IT (Mirus). Hairpins targeting the different genes were ordered from Sigma-Aldrich. Replication-incompetent lentiviruses were produced from pLKO.1 vector ( www.broad.mit.edu/genome_bio/trc/) by cotransfection with Δ8.9 and pMGD2 in HEK 293T cells ( www.broadinstitute.org/rnai/trc/lib) using TRANS-IT. Cells were transduced in the presence of polybrene. After transduction, cells were selected with puromycin.
Western blotting.
Western blotting was performed using the following antibodies: β-actin (clone C4) (MPBiomedicals LLC); pAKT (S473), AKT, pS6K, S6K, IRS-1, pERK, ERK, pVEGFR2, VEGFR2, VEGFR1, and pFoxO3a (all from Cell Signaling Technology); and anti-rabbit-HRP and anti-mouse-HRP antibodies (all from Millipore).
Immunoprecipitation.
Protein A/G PLUS-Agarose Beads (Santa Cruz Biotechnology Inc.) were washed twice in PBS and resuspended in 500 μl of lysis buffer. Anti-phosphotyrosine antibody (clone 4G10) (Millipore), diluted 1:50, was added and incubated overnight in a rotating chamber at 4°C. Tubes were centrifuged at 1,000 g for 1 minute and washed 3 times in ice-cold PBS. 500 μg of cell lysate was added, and the volume of the tube was filled up to 1 ml with lysis buffer. Tubes were incubated in a similar manner and were centrifuged and washed as described above. After removing the supernatant, beads were resuspended in ×4 NuPage LDS buffer (Invitrogen) and heated at 80°C for 10 minutes. Supernatant was carefully pipetted and loaded in a gel for Western blotting. pVEGFR2 was used as the primary antibody.
ELISA assay.
Cells were plated in 6-well plates and incubated for 24 hours in starving media. Cells were then stimulated with 40 ng VEGF-A 165 either alone or after pretreatment with the VEGFR2 inhibitor ZD6474 (1 μM) or with rapamycin (100 nM) for 4 hours. Secretion of VEGF into cell culture supernatants was measured with the VEGF Human ELISA Kit from Tebu-Bio GmbH (catalog no. ELH-VEGF-001) according to the manufacturer’s instructions.
Flow cytometry.
Cells were plated in 6-well plates and incubated for 24 hours in starving media. Cells were then either treated with DMSO or stimulated with 40 ng VEGF-A 165 alone or after pretreatment with ZD6474 (0.5 and 1 μM) for 4 hours. The incorporated BrdU was stained with specific anti-BrdU fluorescent antibodies. All protocols were performed according to instructions for the BrdU Flow Kit from BD Pharmingen (catalog no. 559619). The levels of cell-associated BrdU were then measured on a Gallios Flow Cytometer from Beckman Coulter. Results were calculated using Gallios FACS software.
PET and BLI.
Nude mice bearing macroscopic H1975WT tumors were treated daily with an oral gavage of 75 mg/kg ZD6474 and investigated using a FOCUS microPET Scanner (Concord Microsystems Inc.). [18F]FLT and [11C]MET were synthesized as described previously (22). No-carrier-added [18F]FLT and [11C]MET were administered i.v. (tail vein) into experimental animals with a dose of 200 μCi per mouse and 400 μCi per mouse, respectively. [18F]FLT PET and [11C]MET PET imaging were performed 60 minutes and 20 minutes after injection, respectively. Evaluation of data was based on a ROI analysis of the entire tumor using in-house software (VINCI). For data analysis, we used the maximal voxel radioactivity within the tumors. The mediastinum was chosen as a reference for determination of uptake ratio, since we observed constant uptake for [18F]FLT and [11C]MET in this region. The heart was used as a reference for calculation of the [15O]H2O. All data were decay corrected.
Analysis of luciferase gene expression was performed using an optical imaging system (Biospace). For bioluminescence detection, mice were injected intraperitoneally with d-luciferin (4 mg per animal in 200 μl PBS), and images were acquired 10 minutes after luciferin injection. Evaluation of data was performed using ROI analysis of BLI images to determine maximum values in photons. Data were background subtracted.
Mouse models.
All animal procedures were approved by the local animal protection committee and the local authorities. For each tumor xenograft, 5 × 106 cells from individual cell lines suspended in plain RPMI were injected subcutaneously into male nude mice. Mice (with established tumors [70 mm3] or 1 day after tumor cell inoculation) were treated daily by oral gavage of ZD6474 (75 mg/kg, dissolved in sterile, deionized water with 1% Tween 80), PD0325901 (12 mg/kg, dissolved in propylene glycol/water [1:1]), the combination of ZD6474 (75 mg/kg) and PD0325901 (12 mg/kg), or vehicle alone. Avastin treatment was given i.p. (twice a week, 5 mg/kg). Tumor size was monitored by measuring perpendicular diameters. Tumor volumes were calculated by determination of the largest diameter and its perpendicular according to the following equation: tumor volume = a × (b2/2), where a represents the largest diameter and b represents the perpendicular diameter.
The RasLO construct is under the β-actin promoter followed by a STOP codon flanked by LoxP sites. Human mutated KrasVal12 and a fusion molecule consisting of ovalbumin, S-tag, and luciferase are expressed after excision of the STOP codon by Cre-recombinase encoded in adenoVirus. Tumor formation was then noninvasively monitored by BLI (Biospace). In order to induce tumor growth specifically in the lung, 107 PFUs of adeno-Cre were applied intranasally in RasLO genotyped positive mice between 6 to 8 weeks old that had been previously anesthetized with ketamine.
Tumor samples and immunohistochemistry.
All tumor samples were received from the CIO Biobank at the Institute of Pathology, University of Bonn, Bonn, Germany. All tumors were clinically and pathologically identified as being the primary and only neoplastic lesion and classified according to WHO guidelines. Briefly, 3-μm-thick sections of FFPE tumors were deparaffinized, and antigen retrieval was performed by boiling the section in citrate buffer at pH 6 or EDTA at pH 9 for 20 minuets. Primary antibodies used were as follows: VEGF (sc-152, 1:100, pH 6, Santa Cruz Biotechnology Inc.), VEGF (bevacizumab, 1:100, pH 6, Roche; secondary anti-human IgG-FITC, Dako), CD31 (SZ31, 1:50, pH 6, Dianova), VEGFR2 (no. 2479, 1:200, pH 9, Cell Signaling Technology), Ki67 (mib-1, 1:100, pH 6, Thermo Scientific), pERK (no. 4376, 1:50, pH 6, Cell Signaling Technology), pMAPK (no. 4631, 1:50, pH 6, Cell Signaling Technology), IRS-1 (ab40777, 1:50, pH 6, Abcam), VEGF:VEGFR 2 (GV39M, 1:2, culture supernatant) (16), and HIF-1α (1:300, Cell Signaling Technology). Corresponding secondary antibody detection kits for reduced background on murine tissue were used (Histofine Simple Stain Mouse MAX PO, medac) and stained on an automated stainer (LabVision Autostainer 480S, Thermo Scientific). VEGF and VEGFR2 immunofluorescence was performed using the same primary antibodies and secondary antibodies. Staining intensities were individually evaluated by 3 independent observers, using a 4-tier scoring system. Statistical analysis was performed using a Fisher’s exact test.
Statistics.
Fisher’s exact tests were performed using R version 2.7.1 ( http://www.r-project.org/). A level of significance of P < 0.05 was chosen. Data are presented as mean ± SD in all figure panels in which error bars are shown.
Study approval.
All animal procedures were in accordance with the German laws for animal protection and were approved by the local animal care committee and local governmental authorities (Recklinghausen, Germany).
Supplementary Material
Acknowledgments
We wish to thank Ryan Anderson and Paul Elvin for helpful discussions and for providing ZD6474 and A. Florin and J. Czerwitzki for their expert technical assistance. This study was supported by AstraZeneca, by the German Ministry of Science and Education (BMBF) as part of the NGFNplus program (grant 01GS08100 to R.K. Thomas), by the Fritz-Thyssen-Stiftung (grant 10.08.2.175), by the Max Planck Society (M.IF.A.NEUR8061 to R.K. Thomas), and by the Deutsche Forschungsgemeinschaft (DFG) through SFB 832 (Z2 to R.T. Ullrich and B. Neumaier; TP6 to R.K. Thomas and R.T. Ullrich; TP5 to L.C. Heukamp and R. Buettner; and Z1 to L.C. Heukamp and R. Buettner).
Footnotes
Conflict of interest: Roman K. Thomas received consulting and lecture fees from Sanofi-Aventis, Merck KGaA, Bayer, Lilly, Roche, Boehringer Ingelheim, Johnson & Johnson, AstraZeneca, ATLAS Biolabs, Daiichi-Sankyo, and Blackfield as well as research support from AstraZeneca, Merck, and EOS. Roman K. Thomas is a founder and shareholder of Blackfield, a company involved in cancer genome services and cancer genomics-based drug discovery.
Citation for this article: J Clin Invest. 2013;123(4):1732–1740. doi:10.1172/JCI65385.
Roman K. Thomas, Martin Peifer, and Mirjam Koker’s present address is: Department of Translational Genomics, University of Cologne, Cologne, Germany.
References
- 1.Baeriswyl V, Christofori G. The angiogenic switch in carcinogenesis. Semin Cancer Biol. 2009;19(5):329–337. doi: 10.1016/j.semcancer.2009.05.003. [DOI] [PubMed] [Google Scholar]
- 2.Carmeliet P. Mechanisms of angiogenesis and arteriogenesis. Nat Med. 2000;6(4):389–395. doi: 10.1038/74651. [DOI] [PubMed] [Google Scholar]
- 3.Lee TH, et al. Vascular endothelial growth factor mediates intracrine survival in human breast carcinoma cells through internally expressed VEGFR1/FLT1. PLoS Med. 2007;4(6):e186. doi: 10.1371/journal.pmed.0040186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chung GG, et al. Vascular endothelial growth factor, FLT-1, and FLK-1 analysis in a pancreatic cancer tissue microarray. Cancer. 2006;106(8):1677–1684. doi: 10.1002/cncr.21783. [DOI] [PubMed] [Google Scholar]
- 5.Silva SR, et al. VEGFR-2 expression in carcinoid cancer cells and its role in tumor growth and metastasis. Int J Cancer. 2011;128(5):1045–1056. doi: 10.1002/ijc.25441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lichtenberger BM, Tan PK, Niederleithner H, Ferrara N, Petzelbauer P, Sibilia M. Autocrine VEGF signaling synergizes with EGFR in tumor cells to promote epithelial cancer development. Cell. 2010;140(2):268–279. doi: 10.1016/j.cell.2009.12.046. [DOI] [PubMed] [Google Scholar]
- 7.Yang F, et al. Increased VEGFR-2 gene copy is associated with chemoresistance and shorter survival in patients with non-small-cell lung carcinoma who receive adjuvant chemotherapy. Cancer Res. 2011;71(16):5512–5521. doi: 10.1158/0008-5472.CAN-10-2614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wedge SR, et al. ZD6474 inhibits vascular endothelial growth factor signaling, angiogenesis, and tumor growth following oral administration. Cancer Res. 2002;62(16):4645–4655. [PubMed] [Google Scholar]
- 9.Pao W, et al. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005;2(3):e73. doi: 10.1371/journal.pmed.0020073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shields AF, et al. Imaging proliferation in vivo with [F-18]FLT and positron emission tomography. Nat Med. 1998;4(11):1334–1336. doi: 10.1038/3337. [DOI] [PubMed] [Google Scholar]
- 11.Blencke S, et al. Characterization of a conserved structural determinant controlling protein kinase sensitivity to selective inhibitors. Chem Biol. 2004;11(5):691–701. doi: 10.1016/j.chembiol.2004.02.029. [DOI] [PubMed] [Google Scholar]
- 12.Fuchs BC, Bode BP. Amino acid transporters ASCT2 and LAT1 in cancer: partners in crime? Semin Cancer Biol. 2005;15(4):254–266. doi: 10.1016/j.semcancer.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 13.Brugarolas JB, Vazquez F, Reddy A, Sellers WR, Kaelin WG. TSC2 regulates VEGF through mTOR-dependent and -independent pathways. Cancer Cell. 2003;4(2):147–158. doi: 10.1016/S1535-6108(03)00187-9. [DOI] [PubMed] [Google Scholar]
- 14.Kanai Y, Segawa H, Miyamoto K, Uchino H, Takeda E, Endou H. Expression cloning and characterization of a transporter for large neutral amino acids activated by the heavy chain of 4F2 antigen (CD98). J Biol Chem. 1998;273(37):23629–23632. doi: 10.1074/jbc.273.37.23629. [DOI] [PubMed] [Google Scholar]
- 15.Vasudevan KM, et al. AKT-independent signaling downstream of oncogenic PIK3CA mutations in human cancer. Cancer Cell. 2009;16(1):21–32. doi: 10.1016/j.ccr.2009.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brekken RA, Huang X, King SW, Thorpe PE. Vascular endothelial growth factor as a marker of tumor endothelium. Cancer Res. 1998;58(9):1952–1959. [PubMed] [Google Scholar]
- 17.Laking GR, Price PM. Positron emission tomographic imaging of angiogenesis and vascular function. Br J Radiol. 2003;1:S50–S59. doi: 10.1259/bjr/30399077. [DOI] [PubMed] [Google Scholar]
- 18.O’Reilly KE, et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 2006;66(3):1500–1508. doi: 10.1158/0008-5472.CAN-05-2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Takahashi O, et al. Combined MEK and VEGFR inhibition in orthotopic human lung cancer models results in enhanced inhibition of tumor angiogenesis, growth, and metastasis. Clin Cancer Res. 2012;18(6):1641–1654. doi: 10.1158/1078-0432.CCR-11-2324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Duvel K, et al. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol Cell. 2010;39(2):171–183. doi: 10.1016/j.molcel.2010.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sos ML, et al. Predicting drug susceptibility of non-small cell lung cancers based on genetic lesions. J Clin Invest. 2009;119(6):1727–1740. doi: 10.1172/JCI37127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ullrich R, et al. Glioma proliferation as assessed by 3’-fluoro-3’-deoxy-L-thymidine positron emission tomography in patients with newly diagnosed high-grade glioma. Clin Cancer Res. 2008;14(7):2049–2055. doi: 10.1158/1078-0432.CCR-07-1553. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.